
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA new approach for identifying positional isomers of glycans cleaved from monoclonal antibodies†
Irina
Dyukova
 a,
Ahmed
Ben Faleh
a,
Stephan
Warnke
a,
Natalia
Yalovenko
a,
Vasyl
Yatsyna
ab,
Priyanka
Bansal
a and
Thomas R.
Rizzo
*a
a,
Ahmed
Ben Faleh
a,
Stephan
Warnke
a,
Natalia
Yalovenko
a,
Vasyl
Yatsyna
ab,
Priyanka
Bansal
a and
Thomas R.
Rizzo
*a
aLaboratoire de Chimie Physique Moléculaire, École Polytechnique Fédérale de Lausanne, EPFL SB ISIC LCPM, Station 6, CH-1015 Lausanne, Switzerland. E-mail: thomas.rizzo@epfl.ch
bUniversity of Gothenburg, Department of Physics, 412 96 Gothenburg, Sweden
First published on 30th June 2021
Abstract
Glycosylation patterns in monoclonal antibodies (mAbs) can vary significantly between different host cell types, and these differences may affect mAbs safety, efficacy, and immunogenicity. Recent studies have demonstrated that glycan isomers with the terminal galactose position on either the Man α1-3 arm or the Man α1-6 arm have an impact on the effector functions and dynamic structure of mAbs. The development of a robust method to distinguish positional isomers of glycans is thus critical to guarantee mAb quality. In this work, we apply high-resolution ion mobility combined with cryogenic infrared spectroscopy to distinguish isomeric glycans with different terminal galactose positions, using G1F as an example. Selective enzymatic synthesis of the G1(α1-6)F isomer allows us to assign the peaks in the arrival-time distributions and the infrared spectra to their respective isomeric forms. Moreover, we demonstrate the impact of the host cell line (CHO and HEK-293) on the IgG G1F gycan profile at the isomer level. This work illustrates the potential of our approach for glycan analysis of mAbs.
Introduction
Monoclonal antibody (mAb) drugs are used to treat some of the most serious, life-threatening, and chronic diseases, such as cancers,1 immune-mediated inflammatory conditions,2 and diabetes.3 Most of the current therapeutic mAbs are humanized or human immunoglobulins G (IgGs), produced as recombinant glycoproteins in eukaryotic cells.4 IgGs are about 150 kDa in size and comprised of two identical heavy chains of ∼50 kDa and two identical light chains of ∼25 kDa (Fig. 1).5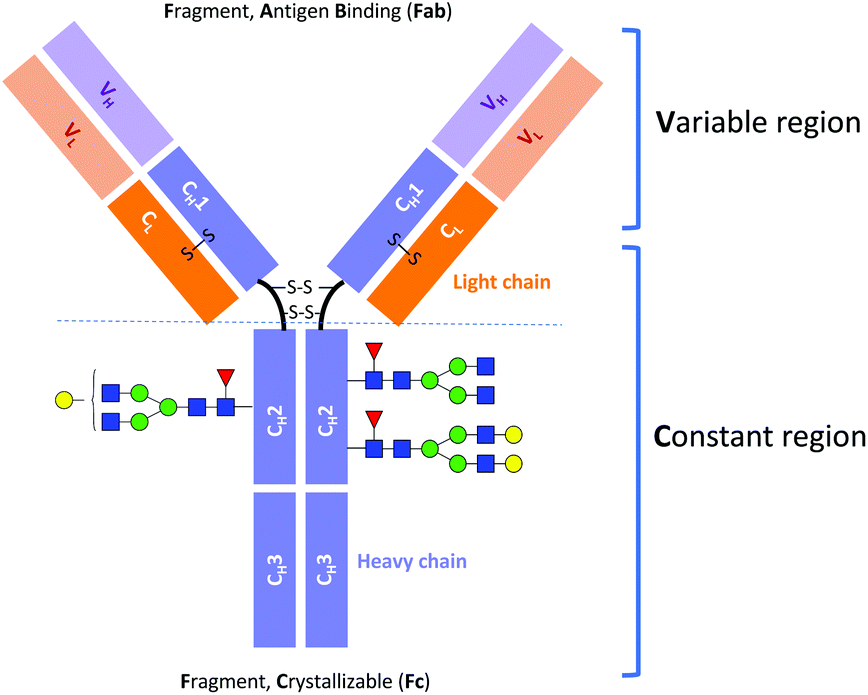 | ||
| Fig. 1 Schematic structure of IgG antibody. | ||
Immunoglobulin G molecules are glycosylated in the CH2 domains of the Fc region (see Fig. 1), with glycans being covalently attached at the Asn297 residue. The N-glycans of the Fc region contribute approximately 2–3% to the total mass of the IgG protein.4,5 Despite this low percentage, the N-glycan moieties can have a significant impact on the effector functions of antibodies, such as the antibody-dependent cell mediated cytotoxicity (ADCC) and the complement-dependent cytotoxicity (CDC).6,7 For example, it has been established that the absence of core fucose (Fuc) residues in the N-glycans of the Fc region substantially increases the ADCC activity.8,9 Moreover, a high sialic acid content reduces ADCC activity but at the same time plays an important role in anti-inflammatory responses.10,11 Terminal galactose is well known to enhance CDC activity, and its impact on ADCC activity has also been reported.12–17 However, it has only recently been demonstrated that the terminal galactose position (i.e., on either the core mannose (Man) α1-6 or α1-3 branch) has a significant effect on the effector functions of mAbs. Aoyama et al. have shown that the G1(α1-6)F mAb has higher complement component 1q (C1q)- and Fc gamma receptor (FcγR)-binding activities and CDC activity than the G1(α1-3)F mAb because of the greater involvement of the galactose on the α1-6 branch in the structural stability of the CH2 domain.18
It is important to note that protein biotherapeutics such as mAbs generally exhibit micro-heterogeneities that can lead to the presence/absence or different ratios between the N-glycans in the Fc region with terminal Gal on the Man α1-6 and α1-3 arms. Effective tools are thus needed to analyse protein glycoforms, even at the isomer level, for both biological mAbs and biosimilars.19–22
Several methods have been implemented to distinguish and identify positional isomers of released N-linked glycans with terminal Gal (α1-6/α1-3). These include tandem mass spectrometry,23 ion mobility spectrometry (IMS),24 and various combinations of selective enzymatic digestion or synthesis with nuclear magnetic resonance (NMR) or high-performance liquid chromatographic (HPLC) analysis.25–28 The most commonly used method currently combines hydrophobic interaction liquid chromatography (HILIC) and mass spectrometry (MS), where the chromatographic peak assignment is based on the previously published work indicating that the glycan with a terminal galactose on the upper Man (α1-6) arm elutes prior to that with galactose on the lower Man (α1-3) arm.29 Despite the potential of this hybrid technique, glycan LC workflows typically involve a derivatization step to label the glycans with a fluorescent tag, since they do not contain a natural chromophore.30 While this improves sensitivity and facilitates quantification, it complicates the workflow, and the labels can be expensive.
Recently there has been a surge in the application of gas-phase spectroscopy together with ion mobility spectrometry for the structural characterization of glycans.24,31–36 In the present work, we use a combination of ultrahigh-resolution IMS with cryogenic infrared spectroscopy as a rapid and reliable technique for glycan isomer identification. Our approach allows one to obtain highly resolved, isomer-specific vibrational spectra, even of larger, more complex glycan ions. We have implemented a chemoenzymatic approach37,38 to synthesize selectively the glycan isomer G1(α1-6)F and characterize it along with the G1(α1-3)F isomer by IMS and vibrational spectroscopy. We then demonstrate the impact of the host cell line (CHO and HEK-293) on the ratio of G1F isomers within the glycan profile of IgG.
Experimental approach
Selective chemoenzymatic synthesis of the G1(α1-6)F isomer
The synthesis of G1(α1-6)F (Fig. 2) was performed in a total volume of 50 μL containing 0.27 mM of an acceptor G-NGA2 N-linked glycan (Dextra Laboratories, UK), 0.54 mM of GDP-Fuc (guanosine 5′-diphospho-β-L-fucose sodium salt) (Sigma-Aldrich), a MES (2-(N-morpholino)ethanesulfonic acid) buffer solution (100 mM, pH 7.0), and 0.12 mg mL−1 of recombinant human α1,6-fucosyltransferase (FUT8) (Creative BioMart, USA). The reaction was incubated overnight at 37 °C and then quenched by adding 30 μL of ice-cold acetonitrile to the mixture.37–39 | ||
| Fig. 2 Positional isomers of G-NGA2 and G1F glycans with a scheme for chemoenzymatic synthesis of the G1(α1-6)F isomer using human α1,6-fucosyltransferase FUT8. | ||
After synthesis, the reaction mixture was analysed by HILIC using an AQUITY UPLC H-Class Plus System (Waters) coupled to a Q-TOF mass spectrometer (Waters Premier). The final product, G1(α1-6)F isomer, with a yield of ∼50% (Table S1, ESI†), was collected using a Waters Fraction Collector III coupled to a UPLC-MS system and concentrated to 15–20 μM (50/50 H2O/acetonitrile mixture).
Enzymatic release of N-linked glycans
Recombinant IgG glycoproteins from CHO and HEK-293 cell lines were produced and purified at the EPFL protein production and structure core facility. One hundred μg of lyophilized mAb was diluted in 16 μL of H2O and incubated with 4 μL of Rapid PNGase F buffer and 1 μL of PNGaseF enzyme (BioConcept, Allschwil, Switzerland) at 50 °C for 10 minutes to detach the N-linked glycans.To remove peptides, protein, and other contaminants present in the glycan samples after digestion, we used Sep-Pak C18 cartridges from Waters. The cartridges were first conditioned with 1 mL of MeOH followed by 1 mL of 5% acetic acid three times before glycan samples were applied directly after the deglycosylation step. Then cartridges were washed three times with 1 mL of 5% acetic acid. The collected fractions with released N-linked glycans were lyophilised and diluted in 80 μL of a 30/70 H2O/acetonitrile mixture. The mixtures of released N-linked glycans from IgG produced in CHO and HEK-293 cell lines were analysed by UPLC-MS (Fig. 3). The obtained glycoprofiles for both mAbs are consistent with previously published data. They include three major glycans: G0F, G1F, G2F.40,41 The glycan G1F appears as a single peak consisting of two positional isomers: G1(α1-6)F and G1(α1-3)F. Using the fraction collection procedure explained above, the peak corresponding to G1F cleaved from four portions of 100 μg of each mAb was collected and concentrated.
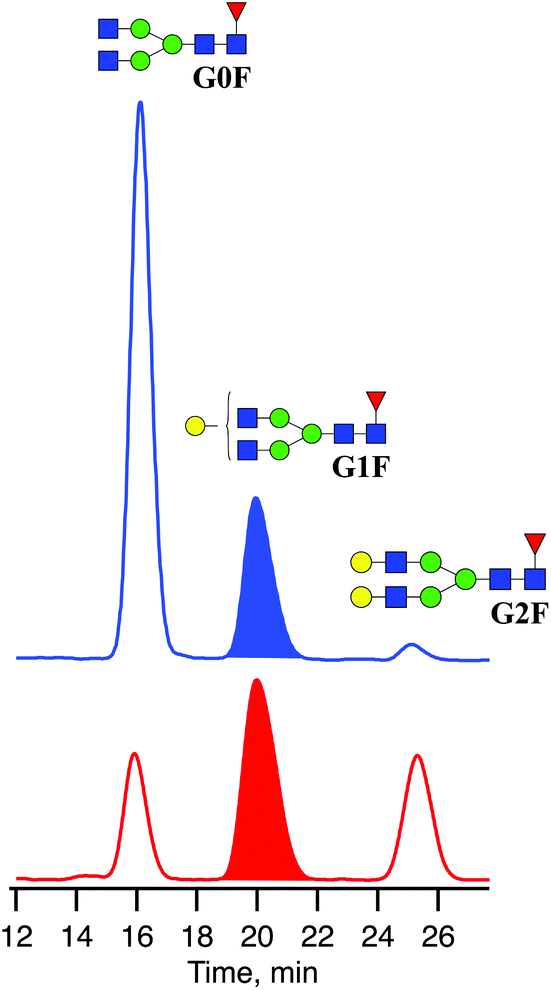 | ||
| Fig. 3 Chromatograms of three major N-linked glycans released from IgGs produced in CHO (blue) and HEK-293 (red) cell lines. | ||
The glycan standard G1F was purchased from Dextra Laboratories (UK) and contains a mixture of the G1(α1-6)F and G1(α1-3)F positional isomers. Solutions were prepared by dissolving the respective powder in 50/50 H2O/acetonitrile mixture for a concentration of 15–20 μM. Sample solutions were stored at −20 °C. We added sodium acetate to all collected fractions to observe the doubly sodiated adduct as the predominant species for ion-mobility and spectroscopy measurements.
Ion-mobility-selective IR spectroscopy
Analysis of the glycan standard and collected fractions was performed on a home-built ultrahigh-resolution ion mobility spectrometer coupled to a cryogenic ion trap and a time-of-flight (TOF) MS, which improves on a previously published design32 and will be described in detail in an upcoming publication.42 Briefly, doubly sodiated glycan ions were produced via nanoelectrospray ionization (nESI) of 15–20 μM solutions (50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :50 water/acetonitrile) and transferred into the instrument through a stainless-steel capillary heated to 120 °C. A dual-stage ion funnel assembly43 focuses and transfers ions into the ion-mobility region, which is based on structures for lossless ion manipulation (SLIM) technology, originally developed by Smith and co-workers.44,45 In the SLIM-IMS device, traveling wave (TW) electric potentials created between electrodes on a pair of printed circuit boards propel ions through the drift gas (2 mbar, N2) on a serpentine path to achieve high mobility resolution. The length of the serpentine separation path is 10 m and can be extended by sending ions for multiple roundtrips along this path. The ion mobility resolving power after a single cycle was determined to be ∼200 using the ion mobility standard peptides GRGDS and SDGRG. The overall resolving power can be increased by a factor of
:50 water/acetonitrile) and transferred into the instrument through a stainless-steel capillary heated to 120 °C. A dual-stage ion funnel assembly43 focuses and transfers ions into the ion-mobility region, which is based on structures for lossless ion manipulation (SLIM) technology, originally developed by Smith and co-workers.44,45 In the SLIM-IMS device, traveling wave (TW) electric potentials created between electrodes on a pair of printed circuit boards propel ions through the drift gas (2 mbar, N2) on a serpentine path to achieve high mobility resolution. The length of the serpentine separation path is 10 m and can be extended by sending ions for multiple roundtrips along this path. The ion mobility resolving power after a single cycle was determined to be ∼200 using the ion mobility standard peptides GRGDS and SDGRG. The overall resolving power can be increased by a factor of  as the number of separation cycles n is increased.46 Prior to separation, ions are accumulated on the SLIM board in a 2 m storage region and released in short packages into the separation region. Following separation, ions are transferred into high vacuum, where the arrival time distributions (ATDs) are measured by means of a nested TOF measurement,47 yielding an ATD with 100 μs time increments (bin size). Compared to the previous design, the new instrument exhibits higher IMS resolving power and higher sensitivity, both as a result of an updated SLIM device incorporating published design features.48
as the number of separation cycles n is increased.46 Prior to separation, ions are accumulated on the SLIM board in a 2 m storage region and released in short packages into the separation region. Following separation, ions are transferred into high vacuum, where the arrival time distributions (ATDs) are measured by means of a nested TOF measurement,47 yielding an ATD with 100 μs time increments (bin size). Compared to the previous design, the new instrument exhibits higher IMS resolving power and higher sensitivity, both as a result of an updated SLIM device incorporating published design features.48
To record cryogenic infrared spectra, ions separated by SLIM-IMS are sent to a cryogenic ion trap, which is held at 45 K. There, the ions are cooled in collisions with a 20:80 mixture of nitrogen and helium, which is pulsed into the trap just before the ions arrive. During the collisional cooling process, ions are tagged with N2 molecules in preparation for performing messenger-tagging spectroscopy.49,50 The nitrogen-tagged ions are then irradiated for 50 ms with a continuous-wave, mid-IR laser (IPG Photonics) operated at 0.7 W output power. When the laser wavenumber is resonant with a vibrational transition of the molecule, energy is absorbed and then redistributed, causing the nitrogen tag to detach. Monitoring the depletion of the N2-tagged ions with the TOF mass analyser as a function of the laser wavenumber gives an IR fingerprint spectrum of the molecule.
Results and discussion
Isomer identification
For the first time, we demonstrate a comprehensive analysis of G1F N-linked glycan positional isomers using a combination of IMS and cryogenic IR spectroscopy. Commercially available G1F is provided as a mixture of two positional isomers, with the terminal galactose sitting on either the Man α1-6 or Man α1-3 arms (Fig. 2).Fig. 4 shows the ATD for the doubly sodiated G1F glycan standard ([G1F + 2Na]2+, m/z 835). Four distinct peaks were observed after six cycles on the SLIM board (60 m drift path) at an IMS resolving power of approximately 500, as determined by our calibration with GRGDS and SDGRG. Since two neighbouring peaks (II and III) are just resolved, we can estimate the difference in their CCS values to be 0.2–0.3%.
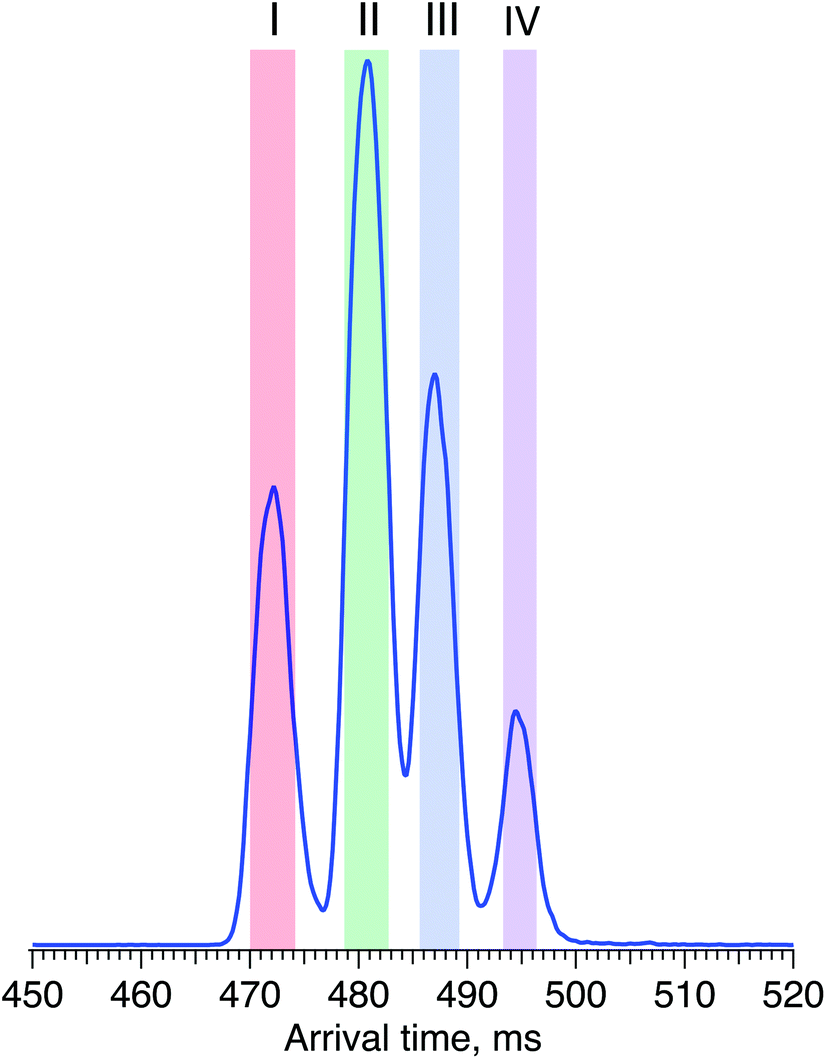 | ||
| Fig. 4 Arrival time distribution of G1F after 60 m SLIM-IMS separation (i.e., six separation cycles), representing a mixture of the G1(α1-6)F and G1(α1-3)F positional isomers. | ||
While SLIM-based ion mobility serves as a powerful, high-resolution tool for glycan separation, it is not ideally suited for identification and database construction. Arrival time distributions determined using SLIM depend sensitively on the traveling wave speed and amplitude as well as the temperature and pressure of the drift gas, and hence need to be calibrated to obtain collision cross section (CCS) values.51 Given a typical deviation of reported CCS values of approximately 2%,52 it would be tenuous to identify isomers that have CCSs as close as the species separated here by this method alone, even if done carefully. In contrast, the vibrational spectrum is an intrinsic property of the molecule and provides an identifying fingerprint that is insensitive to external parameters.
For two positional isomers, we observed four distinct peaks on the ATD. These peaks could arise from the two reducing-end anomers,53–55 which we have previously shown can be separated by ultrahigh-resolution IMS, or from different conformers for each isomer.
Fig. 5 displays cryogenic IR spectra of each peak in the ATD of G1F. Even though G1F consists of nine monosaccharide units, each spectrum exhibits a highly distinct spectral fingerprint that can serve as a unique and robust identifier. We confirmed that regardless of the experimental conditions affecting the arrival time distribution, the spectra of the individual species in the ATD are reproducible.
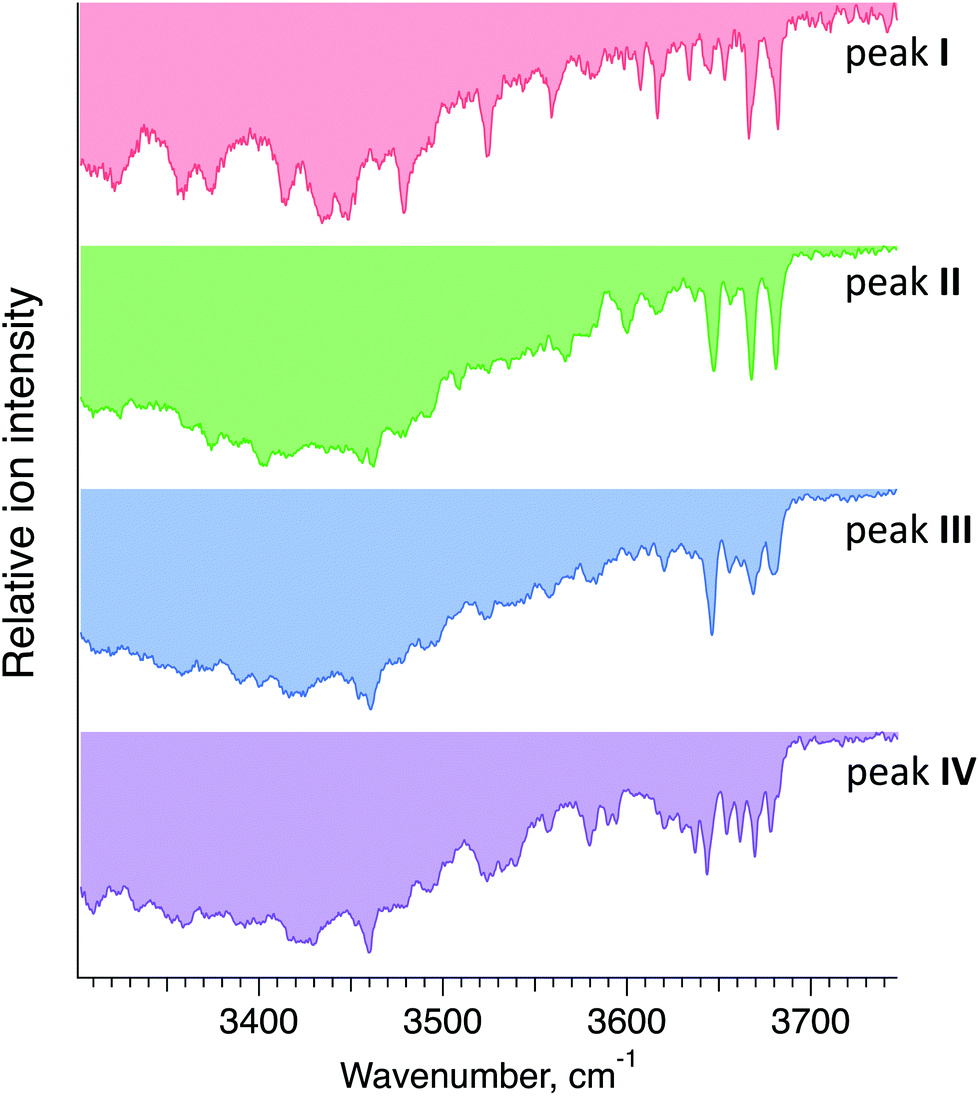 | ||
| Fig. 5 Cryogenic IR spectra of peaks I–IV observed in the ATD of G1F glycan shown in Fig. 4. | ||
Each IR scan was acquired within ∼10 min to allow for extensive signal averaging. However, many fewer spectral data points are needed to uniquely identify each species. Moreover, one can see that the 3600–3700 cm−1 range exhibits sufficiently unique absorption bands in every one of the four spectra to facilitate compound identification. Taken together, this would allow us to identify each of these glycans in less than one minute.
Pure isomers G1(α1-6)F and G1(α1-3)F are difficult to obtain commercially. The samples we purchased from the few manufacturers that offer them were of unsatisfactory isomeric purity, exhibiting the same ATD profile as for the G1F mixture. The two main strategies to produce asymmetrically branched N-linked glycans are to release them from natural sources56 or to synthesize them (either chemically or chemoenzymatically).39,57–59 Isolation of adequate amounts of N-linked glycans with specific structures from natural sources is challenging due to their low abundance. On the other hand, synthetic methods require sufficiently powerful analytical techniques to assess the isomeric purity.
In order to assign each peak in the ATD of G1F to one of two positional isomers (G1(α1-6)F and G1(α1-3)F), we use a chemoenzymatic approach to synthesise the G1(α1-6)F isomer selectively.37 The FUT8 enzyme, which is a recombinant human α1,6-fucosyltransferase, requires a free GlcNAc on the α1,3-Man branch of its N-glycan substrate for activity. The enzyme catalyses the transfer of L-fucose to form an α1,6-linkage to the reducing-end GlcNAc of G(α1-6)-NGA2, leading to the selective synthesis of the G1(α1-6)F isomer.
Fig. 6 displays the ATD obtained for the doubly sodiated G1F glycan mixture (top) and the synthesised G1(α1-6)F isomer (bottom) performed under the same experimental conditions after 6 cycles (60 m) of separation by SLIM-IMS. One can see that the ATD of the G1(α1-6)F isomer is simpler, exhibiting only two distinct peaks (III* and IV*) that match the drift times of peaks III and IV of the G1F mixture, respectively.
 | ||
| Fig. 6 ATDs of G1F glycan representing a mixture of G1(α1-6)F and G1(α1-3)F positional isomers (top) and the synthesised G1(α1-6)F isomer (bottom) after 60 m SLIM-IMS separation. | ||
To confirm that peaks III* and IV* in the ATD of G1(α1-6)F correspond to peaks III and IV in the ATD of the G1F mixture, we measured and compared their respective IR spectra. The good agreement between IR fingerprints of III*/III and IV*/IV shown in Fig. 7 indeed confirms our assignment. As described in the ESI,† these kinds of isomer assignments can be made using a machine learning approach based on principal component analysis combined with a clustering algorithm.
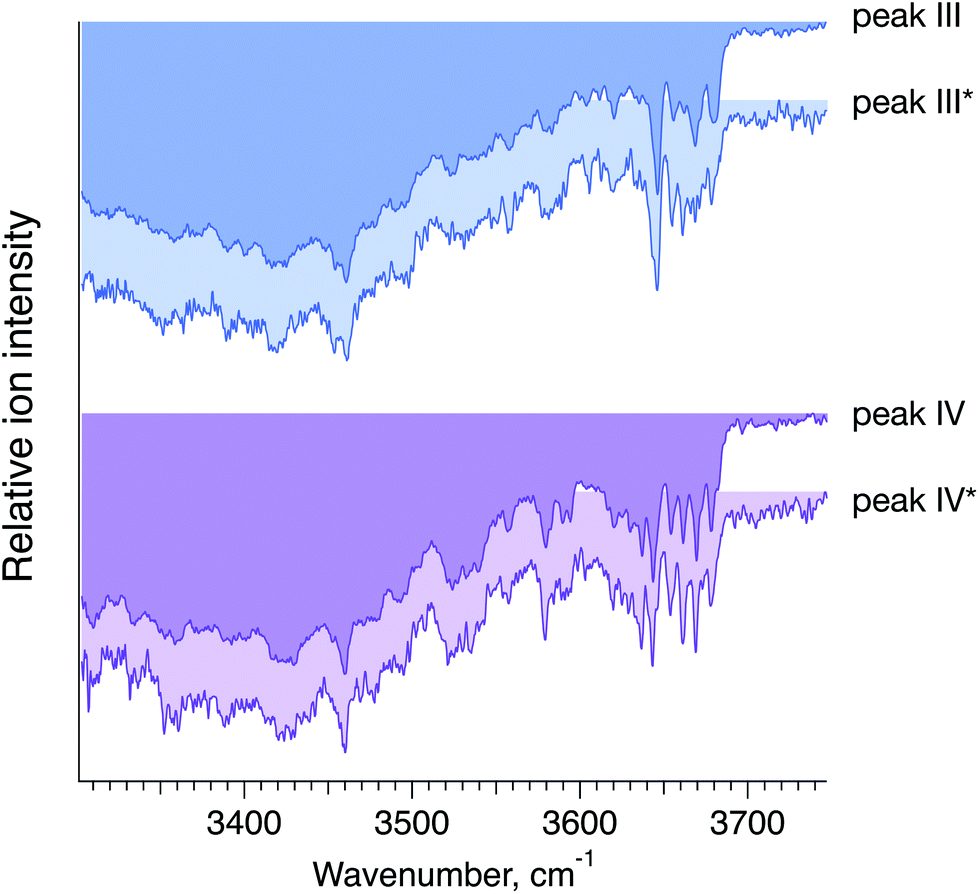 | ||
| Fig. 7 Cryogenic IR spectra of peaks III vs. III* and IV vs. IV* observed in the ATDs of G1F, representing a mixture of the G1(α1-6)F and G1(α1-3)F positional isomers (foreground, peaks III and IV), and the synthesised G1(α1-6)F isomer (background, peaks III* and IV*) (see Fig. 6). | ||
Based on the selectivity of the chemoenzymatic synthesis of the G1(α1-6)F isomer, these results demonstrate that peaks III and IV of the ATD of the G1F mixture correspond to G1(α1-6)F, and thus peaks I and II must correspond to G1(α1-3)F. It is worth noting that in the ATD measured here by ion mobility, G1(α1-3)F arrives prior to G1(α1-6)F, while using HILIC, the opposite is observed.29 This difference results from the different mechanism of separation. In HILIC, G1(α1-3)F has higher affinity to the stationary phase and elutes after G1(α1-6)F. In IMS, G1(α1-3)F has a more compact structure and therefore undergoes fewer collisions with the buffer gas, which makes it arrive sooner than G1(α1-6)F.60
Impact of the host cell line on the glycan profile
The results above show that the combination of high-resolution ion mobility and cryogenic ion spectroscopy provides a fast and reliable method for glycan isomer identification. It can be used to complement, or even replace, existing methods for establishing similarity of glycan profiles between biological drugs and their biosimilars. Different biosimilar suppliers use independently derived cell lines and different manufacturing processes that affect the glycosylation profile, and thus potentially the quality, safety, and efficacy of the final protein product. In the case of G1F, we demonstrate the impact of the host cell line on its ATD profile, which reflects the ratio of positional isomers. Fig. 8 shows the ATD profile of the G1F standard together with those of G1F released from IgG antibodies produced in either CHO or HEK-293 cell lines.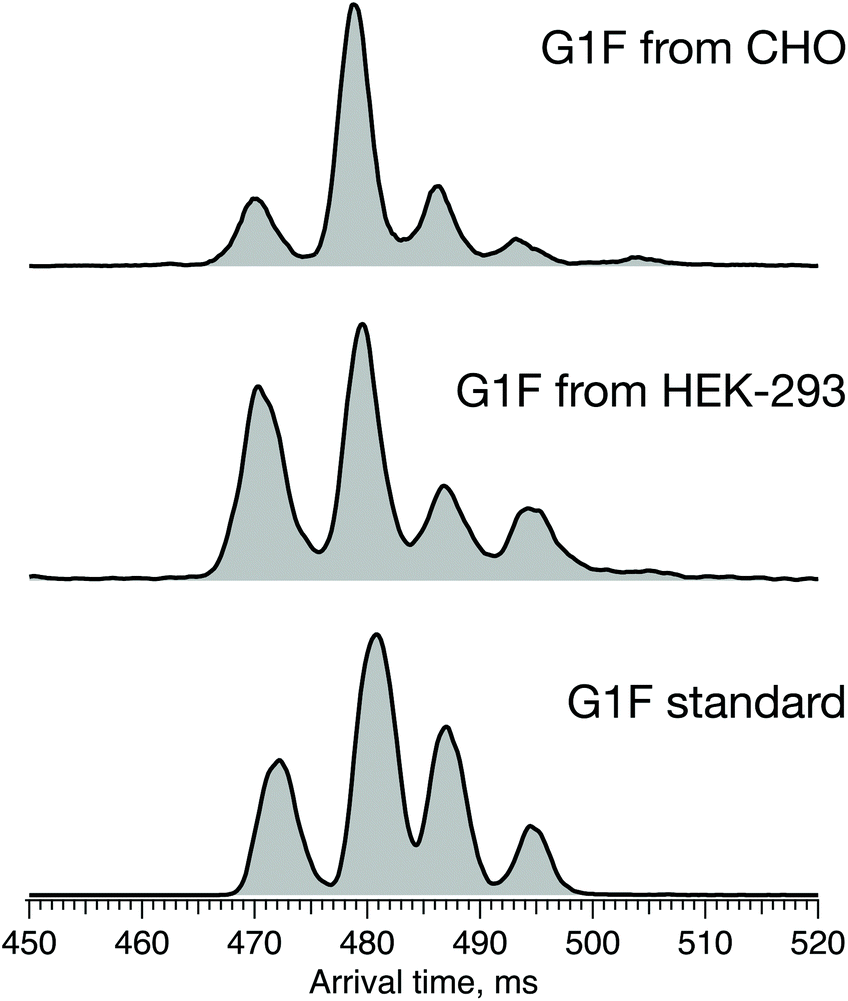 | ||
| Fig. 8 ATDs of G1F standard (bottom) and G1F released from recombinant IgG antibody produced in HEK-293 (middle) and CHO cell lines (top) after 60 m SLIM-IMS separation. | ||
While the ATDs look quite similar in the number of features and their respective drift times, demonstrating the presence of both positional isomers in all three samples, their ratio changes. Based upon the integrated area of each pair of peaks corresponding to the respective positional isomers, the ratio of G1(α1-3)F to G1(α1-6)F increases in the following order: standard (65/35) < HEK-293 (70/30) < CHO (75/25) (Fig. S1, ESI†).
In addition to the different ratios of positional isomers, we observe a difference in the relative intensities of the two peaks in the ATD assigned to each isomer. As discussed above, these peaks could arise from the two reducing-end anomers or from different conformations for each isomer. While we suspect that they may arise from the two anomers, the assignment of these features is the subject of ongoing studies.
Conclusions
Our results demonstrate that the combination of ultrahigh-resolution ion mobility separation with cryogenic vibrational spectroscopy represents a rapid and reliable analytical method to distinguish positional isomers of glycans. We used a chemoenzymatic approach with the FUT8 enzyme for the selective synthesis of G1(α1-6)F, which we employ as a standard for assigning the mobility-separated positional isomers of G1F based on their unique IR fingerprint spectra. With this assignment, we can now identify G1F positional isomers in subsequent experiments based on their vibrational fingerprints alone. One should note that the observed isomers differ by as little as 0.2% in their collision cross section, emphasizing the difficulty to distinguish them by IMS-MS methods alone.We then applied this technique for determining the difference in the ratio of the individual G1F positional isomers from mAbs that have been produced from two different cell lines: CHO and HEK-293. This work demonstrates the power of our approach for monitoring subtle differences in galactosylation of N-glycans cleaved from mAbs.
Author contributions
NY, ID, and TRR were involved in the conceptualization of the project. SW, ABF, ID, and NY developed different aspects of the experimental methodology. ID, ABF, VY, PB, and NY performed the experiments. ID wrote the original draft and NY, SW, and TRR were involved in editing it. TRR supervised the project and acquired the funding.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank the European Research Council (Grant 788697-GLYCANAL), the Swiss National Science Foundation (Grants 200020_184838 and 206021_177004), the Swedish Research Council (international postdoc grant 2019-00512) and the EPFL for their generous support of this work. We also thank Dr David Hacker and Dr Kelvin Lau from the EPFL protein production and structure core facility for providing the glycoproteins and Kristina Makasheva from the LCBM EPFL for providing equipment for sample storage and purification.References
- D. Henry and C. Taylor, Semin. Oncol., 2014, 41(Suppl 3), S13–S20 CrossRef PubMed.
- J. S. Smolen, R. Landewe, J. Bijlsma, G. Burmester, K. Chatzidionysiou, M. Dougados, J. Nam, S. Ramiro, M. Voshaar, R. van Vollenhoven, D. Aletaha, M. Aringer, M. Boers, C. D. Buckley, F. Buttgereit, V. Bykerk, M. Cardiel, B. Combe, M. Cutolo, Y. van Eijk-Hustings, P. Emery, A. Finckh, C. Gabay, J. Gomez-Reino, L. Gossec, J. E. Gottenberg, J. M. W. Hazes, T. Huizinga, M. Jani, D. Karateev, M. Kouloumas, T. Kvien, Z. Li, X. Mariette, I. McInnes, E. Mysler, P. Nash, K. Pavelka, G. Poor, C. Richez, P. van Riel, A. Rubbert-Roth, K. Saag, J. da Silva, T. Stamm, T. Takeuchi, R. Westhovens, M. de Wit and D. van der Heijde, Ann. Rheum. Dis., 2017, 76, 960–977 CrossRef PubMed.
- L. Heinemann, J. Diabetes Sci. Technol., 2015, 10, 457–462 CrossRef PubMed.
- A. Beck, E. Wagner-Rousset, M. C. Bussat, M. Lokteff, C. Klinguer-Hamour, J. F. Haeuw, L. Goetsch, T. Wurch, A. Van Dorsselaer and N. Corvaia, Curr. Pharm. Biotechnol., 2008, 9, 482–501 CAS.
- A. Wright and S. L. Morrison, Trends Biotechnol., 1997, 15, 26–32 CrossRef CAS.
- T. S. Raju, Curr. Opin. Immunol., 2008, 20, 471–478 CrossRef CAS PubMed.
- T. Dashivets, M. Thomann, P. Rueger, A. Knaupp, J. Buchner and T. Schlothauer, PLoS One, 2015, 10, e0143520 CrossRef.
- P. Umana, J. Jean-Mairet, R. Moudry, H. Amstutz and J. E. Bailey, Nat. Biotechnol., 1999, 17, 176–180 CrossRef CAS.
- R. L. Shields, J. Lai, R. Keck, L. Y. O'Connell, K. Hong, Y. G. Meng, S. H. Weikert and L. G. Presta, J. Biol. Chem., 2002, 277, 26733–26740 CrossRef CAS.
- B. J. Scallon, S. H. Tam, S. G. McCarthy, A. N. Cai and T. S. Raju, Mol. Immunol., 2007, 44, 1524–1534 CrossRef CAS PubMed.
- Y. Kaneko, F. Nimmerjahn and J. V. Ravetch, Science, 2006, 313, 670–673 CrossRef CAS.
- J. Hodoniczky, Y. Z. Zheng and D. C. James, Biotechnol. Prog., 2005, 21, 1644–1652 CrossRef CAS.
- L. Liu, J. Pharm. Sci., 2015, 104, 1866–1884 CrossRef CAS PubMed.
- G. Dekkers, R. Plomp, C. A. Koeleman, R. Visser, H. H. von Horsten, V. Sandig, T. Rispens, M. Wuhrer and G. Vidarsson, Sci. Rep., 2016, 6, 36964 CrossRef CAS.
- M. Kiyoshi, J. M. M. Caaveiro, M. Tada, H. Tamura, T. Tanaka, Y. Terao, K. Morante, A. Harazono, N. Hashii, H. Shibata, D. Kuroda, S. Nagatoishi, S. Oe, T. Ide, K. Tsumoto and A. Ishii-Watabe, Sci. Rep., 2018, 8, 3955 CrossRef.
- D. Houde, Y. Peng, S. A. Berkowitz and J. R. Engen, Mol. Cell. Proteomics, 2010, 9, 1716–1728 CrossRef CAS PubMed.
- M. Thomann, K. Reckermann, D. Reusch, J. Prasser and M. L. Tejada, Mol. Immunol., 2016, 73, 69–75 CrossRef CAS PubMed.
- M. Aoyama, N. Hashii, W. Tsukimura, K. Osumi, A. Harazono, M. Tada, M. Kiyoshi, A. Matsuda and A. Ishii-Watabe, MAbs, 2019, 11, 826–836 CrossRef CAS PubMed.
- K. Marino, J. Bones, J. J. Kattla and P. M. Rudd, Nat. Chem. Biol., 2010, 6, 713–723 CrossRef CAS.
- A. Beck, S. Sanglier-Cianferani and A. Van Dorsselaer, Anal. Chem., 2012, 84, 4637–4646 CrossRef CAS PubMed.
- B. J. Geist, D. Davis, T. McIntosh, T. Y. Yang, K. Goldberg, C. Han, C. Pendley and H. M. Davis, MAbs, 2013, 5, 150–161 CrossRef.
- B. L. Duivelshof, W. Jiskoot, A. Beck, J. L. Veuthey, D. Guillarme and V. D'Atri, Anal. Chim. Acta, 2019, 1089, 1–18 CrossRef CAS.
- M. R. Wormald, P. M. Rudd, D. J. Harvey, S. C. Chang, I. G. Scragg and R. A. Dwek, Biochemistry, 1997, 36, 1370–1380 CrossRef CAS.
- E. G. Pallister, M. S. F. Choo, I. Walsh, J. N. Tai, S. J. Tay, Y. S. Yang, S. K. Ng, P. M. Rudd, S. L. Flitsch and T. Nguyen-Khuong, Anal. Chem., 2020, 92, 15323–15335 CrossRef CAS PubMed.
- A. W. Barb and J. H. Prestegard, Nat. Chem. Biol., 2011, 7, 147–153 CrossRef CAS.
- T. Mizuochi, J. Hamako and K. Titani, Arch. Biochem. Biophys., 1987, 257, 387–394 CrossRef CAS.
- N. Takahashi, I. Ishii, H. Ishihara, M. Mori, S. Tejima, R. Jefferis, S. Endo and Y. Arata, Biochemistry, 1987, 26, 1137–1144 CrossRef CAS PubMed.
- Y. Yamaguchi, K. Kato, M. Shindo, S. Aoki, K. Furusho, K. Koga, N. Takahashi, Y. Arata and I. Shimada, J. Biomol. NMR, 1998, 12, 385–394 CrossRef CAS.
- D. Reusch, M. Haberger, B. Maier, M. Maier, R. Kloseck, B. Zimmermann, M. Hook, Z. Szabo, S. Tep, J. Wegstein, N. Alt, P. Bulau and M. Wuhrer, MAbs, 2015, 7, 167–179 CrossRef CAS PubMed.
- E. Largy, F. Cantais, G. Van Vyncht, A. Beck and A. Delobel, J. Chromatogr. A, 2017, 1498, 128–146 CrossRef CAS.
- C. Masellis, N. Khanal, M. Z. Kamrath, D. E. Clemmer and T. R. Rizzo, J. Am. Soc. Mass Spectrom., 2017, 28, 2217–2222 CrossRef CAS PubMed.
- A. Ben Faleh, S. Warnke and T. R. Rizzo, Anal. Chem., 2019, 91, 4876–4882 CrossRef CAS PubMed.
- O. Hernandez, S. Isenberg, V. Steinmetz, G. L. Glish and P. Maitre, J. Phys. Chem. A, 2015, 119, 6057–6064 CrossRef CAS.
- B. Schindler, L. Barnes, C. J. Gray, S. Chambert, S. L. Flitsch, J. Oomens, R. Daniel, A. R. Allouche and I. Compagnon, J. Phys. Chem. A, 2017, 121, 2114–2120 CrossRef CAS.
- C. Manz and K. Pagel, Curr. Opin. Chem. Biol., 2018, 42, 16–24 CrossRef CAS.
- E. Mucha, A. Stuckmann, M. Marianski, W. B. Struwe, G. Meijer and K. Pagel, Chem. Sci., 2019, 10, 1272–1284 RSC.
- A. D. Calderon, Y. Liu, X. Li, X. Wang, X. Chen, L. Li and P. G. Wang, Org. Biomol. Chem., 2016, 14, 4027–4031 RSC.
- A. D. Calderon, J. Zhou, W. Guan, Z. Wu, Y. Guo, J. Bai, Q. Li, P. G. Wang, J. Fang and L. Li, Org. Biomol. Chem., 2017, 15, 7258–7262 RSC.
- L. Li, Y. Liu, C. Ma, J. Qu, A. D. Calderon, B. Wu, N. Wei, X. Wang, Y. Guo, Z. Xiao, J. Song, G. Sugiarto, Y. Li, H. Yu, X. Chen and P. G. Wang, Chem. Sci., 2015, 6, 5652–5661 RSC.
- S. Nallet, L. Fornelli, S. Schmitt, J. Parra, L. Baldi, Y. O. Tsybin and F. M. Wurm, Nat. Biotechnol., 2012, 29, 471–476 CAS.
- D. T. Monteil, V. Juvet, J. Paz, M. Moniatte, L. Baldi, D. L. Hacker and F. M. Wurm, Biotechnol. Prog., 2016, 32, 1174–1180 CrossRef CAS PubMed.
- S. Warnke, A. Ben Faleh and T. R. Rizzo, Meas. Sci., 2021 Search PubMed , submitted.
- T.-C. Chen, T. L. Fillmore, S. A. Prost, R. J. Moore, Y. M. Ibrahim and R. D. Smith, Anal. Chem., 2015, 87, 7326–7331 CrossRef CAS PubMed.
- L. Deng, I. K. Webb, S. V. B. Garimella, A. M. Hamid, X. Zheng, R. V. Norheim, S. A. Prost, G. A. Anderson, J. A. Sandoval, E. S. Baker, Y. M. Ibrahim and R. D. Smith, Anal. Chem., 2017, 89, 4628–4634 CrossRef CAS PubMed.
- A. M. Hamid, S. V. B. Garimella, Y. M. Ibrahim, L. Deng, X. Zheng, I. K. Webb, G. A. Anderson, S. A. Prost, R. V. Norheim, A. V. Tolmachev, E. S. Baker and R. D. Smith, Anal. Chem., 2016, 88, 8949–8956 CrossRef CAS.
- A. A. Shvartsburg and R. D. Smith, Anal. Chem., 2008, 80, 9689–9699 CrossRef CAS.
- C. S. Hoaglund, S. J. Valentine, C. R. Sporleder, J. P. Reilly and D. E. Clemmer, Anal. Chem., 1998, 70, 2236–2242 CrossRef CAS PubMed.
- A. Li, G. Nagy, C. R. Conant, R. V. Norheim, J. Y. Lee, C. Giberson, A. L. Hollerbach, V. Prabhakaran, I. K. Attah, C. D. Chouinard, A. Prabhakaran, R. D. Smith, Y. M. Ibrahim and S. V. B. Garimella, Anal. Chem., 2020, 92(22), 14930–14938 CrossRef CAS PubMed.
- M. Z. Kamrath, E. Garand, P. A. Jordan, C. M. Leavitt, A. B. Wolk, M. J. Van Stipdonk, S. J. Miller and M. A. Johnson, J. Am. Chem. Soc., 2011, 133, 6440–6448 CrossRef CAS PubMed.
- A. B. Wolk, C. M. Leavitt, E. Garand and M. A. Johnson, Acc. Chem. Res., 2014, 47, 202 CrossRef CAS PubMed.
- A. Li, C. R. Conant, X. Zheng, K. J. Bloodsworth, D. J. Orton, S. V. B. Garimella, I. K. Attah, G. Nagy, R. D. Smith and Y. M. Ibrahim, Anal. Chem., 2020, 92(22), 14976–14982 CrossRef CAS.
- S. M. Stow, T. J. Causon, X. Y. Zheng, R. T. Kurulugama, T. Mairinger, J. C. May, E. E. Rennie, E. S. Baker, R. D. Smith, J. A. McLean, S. Hann and J. C. Fjeldsted, Anal. Chem., 2017, 89, 9048–9055 CrossRef CAS.
- S. Warnke, A. Ben Faleh, V. Scutelnic and T. R. Rizzo, J. Am. Soc. Mass Spectrom., 2019, 30, 2204–2211 CrossRef CAS PubMed.
- G. Nagy, I. K. Attah, S. V. B. Garimella, K. Tang, Y. M. Ibrahim, E. S. Baker and R. D. Smith, Chem. Commun., 2018, 54, 11701–11704 RSC.
- J. Ujma, D. Ropartz, K. Giles, K. Richardson, D. Langridge, J. Wildgoose, M. Green and S. Pringle, J. Am. Soc. Mass Spectrom., 2019, 30, 1028–1037 CrossRef CAS PubMed.
- S. Hua, H. J. An, S. Ozcan, G. S. Ro, S. Soares, R. DeVere-White and C. B. Lebrilla, Analyst, 2011, 136, 3663–3671 RSC.
- Y. Maki, R. Okamoto, M. Izumi, T. Murase and Y. Kajihara, J. Am. Chem. Soc., 2016, 138, 3461–3468 CrossRef CAS PubMed.
- Z. Wang, Z. S. Chinoy, S. G. Ambre, W. Peng, R. McBride, R. P. de Vries, J. Glushka, J. C. Paulson and G. J. Boons, Science, 2013, 341, 379–383 CrossRef CAS PubMed.
- I. A. Gagarinov, T. Li, J. S. Torano, T. Caval, A. D. Srivastava, J. A. Kruijtzer, A. J. Heck and G. J. Boons, J. Am. Chem. Soc., 2017, 139, 1011–1018 CrossRef CAS PubMed.
- S. Re, S. Watabe, W. Nishima, E. Muneyuki, Y. Yamaguchi, A. D. MacKerell, Jr. and Y. Sugita, Sci. Rep., 2018, 8, 1644 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1an00780g |
| This journal is © The Royal Society of Chemistry 2021 |