
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCharacterization of a liquid-core waveguide cell for studying the chemistry of light-induced degradation†
Iris
Groeneveld
 *a,
Suzan E.
Schoemaker
a,
Govert W.
Somsen
a,
Freek
Ariese
b and
Maarten R.
van Bommel
cd
*a,
Suzan E.
Schoemaker
a,
Govert W.
Somsen
a,
Freek
Ariese
b and
Maarten R.
van Bommel
cd
aDivision of Bioanalytical Chemistry, Amsterdam Institute of Molecular and Life Sciences, Vrije Universiteit Amsterdam, De Boelelaan 1108, 1081 HZ Amsterdam, The Netherlands. E-mail: i.groeneveld@vu.nl; Tel: +31 (0) 20 59 86 892
bLaserLaB, Vrije Universiteit Amsterdam, The Netherlands
cAnalytical Chemistry Group, van ‘t Hoff Institute for Molecular Sciences, University of Amsterdam, Science Park 904, 1098 XH Amsterdam, The Netherlands
dConservation and Restoration of Cultural Heritage, Amsterdam School for Heritage, Memory and Material Culture, University of Amsterdam, P.O. Box 94552, 1091 GN, Amsterdam, The Netherlands
First published on 7th April 2021
Abstract
Many organic compounds undergo changes under the influence of light. This might be beneficial in, for example, water purification, but undesirable when cultural-heritage objects fade or when food ingredients (e.g., vitamins) degrade. It is often challenging to establish a strong link between photodegradation products and their parent molecules due to the complexity of the sample. To allow effective study of light-induced degradation (LID), a low-volume exposure cell was created in which solutes are efficiently illuminated (especially at low concentrations) while simultaneously analysed by absorbance spectroscopy. The new LID cell encompasses a gas-permeable liquid-core waveguide (LCW) connected to a spectrograph allowing collection of spectral data in real-time. The aim of the current study was to evaluate the overall performance of the LID cell by assessing its transmission characteristics, the absolute photon flux achieved in the LCW, and its capacity to study solute degradation in presence of oxygen. The potential of the LID set-up for light-exposure studies was successfully demonstrated by monitoring the degradation of the dyes eosin Y and crystal violet.
1. Introduction
Light-induced degradation (LID) or photodegradation takes place almost everywhere light reaches. After absorbing light, a molecule is prone to react with other molecules to form new products or degrade, losing particular structural properties. This can be a beneficial process, for instance for production of drinking water when photodegradation is used for the removal of unwanted chemicals by applying ultra-violet (UV) radiation to surface water. However, in some cases, this might lead to the formation of unknown compounds which may be toxic or affect the environment. In addition, chemical degradation after light exposure is unwanted in, for example, cultural heritage or the food industry where properties such as colour and structure or smell and taste should remain stable as long as possible.A straight-forward method for studying photodegradation is irradiation of a solution of the sample in a vial or cell for a fixed time, followed by analysis of solution aliquots by e.g. liquid chromatography (LC). Using such an approach, for example, Confortin studied the degradation of the dye crystal violet (CV), Toohey investigated the effect of iron in presence of UV light on the sweetener rebaudioside A, and Hora examined the degradation of pharmaceutical compounds in wastewater.1–3 Similar approaches have been applied to e.g., pharmaceuticals,4–9 pesticides,10,11 foodstuffs,12 and polymers.13,14
These approaches mostly work well if a single, fairly pure compound is studied and a limited number of degradation products is formed. However, if the sample is a (complex) mixture and potentially contains unknown components, interpretation of the results becomes very difficult. This complexity increases when multiple types of molecules degrade and/or start to react with other sample components or formed fragments. A major challenge in such photodegradation studies is, therefore, to distinguish which degradation products arise from which (parent) molecule.
A way to decrease sample complexities that are met in LID studies, might be to carry out a separation of the sample components prior to light exposure. This preferably would be done in an on-line fashion where a separation technique like LC is coupled to a low-volume irradiation cell in which isolated components can be exposed to light. Ideally, such a cell would also allow real-time spectroscopic monitoring of the content. After exposure, the cell content could be transferred on-line to another LC system for separation and analysis of the degradation products. On-line post-column photoreactors have been reported for reaction detection of target compounds,15–18 however, these reactors are less suited for on-line LID studies. The continuous flow reactors have relatively large volumes, the actual illumination of the sample is hard to control, and they do not allow in situ monitoring of photochemical reactions.
Liquid core waveguides (LCWs) are tubes of small internal diameter that are based on the principle of total internal reflection (TIR) and are typically made of low refractive index (RI) materials, which behave as an optical fibre once filled with a liquid of higher RI. They might offer a solution to the abovementioned challenges faced in on-line coupling of photoreactors. The possibility of coupling LCWs with LC has already been proven to be possible in earlier work where the LCW was used as a post-column (chemically-active) detection cell.19–21 Therefore, we anticipate that on-line coupling of an LCW as an LID cell to LC in a pre- or post-column fashion will be feasible. LCWs facilitate axial illumination of its entire content and the possibility to detect the transmitted light, making the optical path length of an LCW quite significant (up to 1 m), which is an advantage when concentrations of exposed components are low. Furthermore, since the light source is focused inside the capillary, the light dose can be easily controlled. Because of these properties, Teflon AF capillaries or coatings have been used for sensitive UV-Vis absorbance,22,23 fluorescence,24,25 mid-IR,26 and Raman spectroscopic detection.27–31
LCWs are typically made of Teflon AF, which is an amorphous and translucent polymer of tetrafluoroethylene (TFE) and 2,2-bis(trifluoromethyl)-4,5-difluoro-1,3-dioxole (PDD) with an RI of 1.29–1.31. This makes it suitable as an LCW when using core solvents such as water (RI≈1.33), methanol (RI≈1.333) and acetonitrile (RI≈1.34), which all are frequently used in LC eluents. There are currently two types of LCWs that are fabricated using Teflon AF. Type I is a tube entirely made of such a polymer, whereas type II is a silica capillary coated externally with Teflon AF.32 For a type II LCW, TIR occurs both at the core/silica and the silica/polymer interface, making it less suited as LID cell because a significant fraction of the light will travel through the capillary wall rather than through the liquid core where the sample will be located. For type I LCWs, TIR occurs at the core/polymer interface, providing optimal light-exposure of the total content in the LCW. An additional benefit of the use of type I LCWs made of Teflon AF is their permeability to gases, allowing diffusion of e.g. oxygen into the cell required for photooxidation processes.33,34
This study investigates the potential of a Teflon AF2400 LCW as an LID cell coupled to a spectrograph allowing efficient illumination together with collection of real-time transmission and absorption spectra of the cell content. The light transmission characteristics of LCWs of different length were studied as a function of wavelength. The effects of filling procedures and stability throughout the measurements were evaluated, and the diffusion of oxygen gas through the cell walls was verified using methylene blue as indicator. The absolute photon flux in the cell was determined by actinometry. The basic performance of the new LID cell was evaluated by monitoring the degradation of eosin Y (EY) and crystal violet (CV) in the LCW and subsequent analysis of the content of the LID by LC. To the best of our knowledge, the use of an LCW cell for studying photodegradation has not been reported before.
2. Materials and methods
2.1 Chemicals
For the preparation of LC eluents, Ultrapure Milli-Q water (MQ), methanol (MeOH) (Biosolve, UPLC/MS grade), formic acid (FA) (Sigma-Aldrich), sodium hydroxide (NaOH) (Sigma-Aldrich) and triethylamine (TEA) (Sigma-Aldrich) were used. Actinometry experiments were performed using 1,2-bis(2,4-dimethyl-5-phenyl-3-thienyl)-3,3,4,4,5,5-hexafluoro-1-cyclopentene (DAE) obtained from TCI Chemicals (Tokyo, Japan) dissolved in hexane (Sigma-Aldrich). In order to assess gas diffusion, methylene blue hydrate (Sigma-Aldrich, ≥97% purity), NaOH and glucose (Sigma) were used. Degradation experiments were executed using solutions of eosin Y (Sigma-Aldrich, ≥99% purity) and crystal violet (Sigma-Aldrich, ≥90% purity) dissolved in MQ.2.2 LID cell set-up and detection system
A schematic overview of the LID set-up is shown in Fig. 1. Illumination was performed with a Xenon lamp purchased from Hamamatsu. When only a specific part of the spectrum of the Xenon lamp was required, bandpass filters were placed inside the lamp housing. The light from the lamp was guided through an optical fibre (core diameter, 1500 μm) into the LID cell via a coupling piece (Fig. A.1, ESI†). An identical coupling piece was used at the end of the LID cell to measure the transmitted light. The coupling pieces were made of PEEK and designed with three threaded inlets: one in the axial face for fitting the LID cell, and two side inlets at a 45° angle. The back of the pieces were sealed with a quartz window to allow introduction or collection of light. The two side inlets at the first coupling piece can be used for introduction of the sample and (if necessary) another solvent or reagent e.g., a catalyst or inhibitor; details of sample introduction are provided in section 2.3. The side inlets of the coupling piece at the end of the LCW were used for waste or for sample collection. | ||
| Fig. 1 Schematic overview of the design of the LID cell. From left to right: (a) connection with solvent pump, (b) sample delivery by syringe, (c) gas inlet, (d) gas outlet, (e) waste channel. | ||
The LID cell consisted of a type I LCW made of Teflon AF2400 (ID, 800 μm; OD, 1000 μm), obtained from Cambridge Reactor Design (Cottenham, United Kingdom), which was placed inside an outer tubing made of high-density polyethylene (HDPE), creating a tube-in-tube design. T-pieces made of PEEK were glued into the outer tubing to permit addition of gases, such as nitrogen or oxygen (see Fig. 1). Both ends of the LID cell were glued into standard chromatographic finger-tight fittings with Valco thread facilitating the connection of the LID cell to the coupling pieces. To study the effect of gas permeability of the type I LCW, type II LCWs were also used which are not gas permeable. These were made of fused-silica, coated externally with ±15 μm Teflon AF2400 (ID, 100 μm; OD, 363 μm), obtained from CM Scientific Ltd (Silsden, United Kingdom). The type II LCW was fitted with two chromatographic finger-tight fittings using micro sleeves to prevent leaking.
Transmitted light was collected by use of an optical fibre at the other end of the cell that was connected to an SR-163 spectrograph equipped with a DV420A CCD camera, both from Andor Technologies (Belfast, UK). The distance of the optical fibre from the cell could be varied to either increase or decrease the intensity of the measured signal. Generally, a 1% transmission filter was placed in between the end of the LID cell and the optical fibre to reduce the signal and to avoid overloading the detector response. The spectrograph was equipped with a grating of 600 lines per mm blazed at 300 nm, a light entrance slit with adjustable width, and a back illuminated CCD chip of 256 × 1024 pixels with a pixel size of 26 μm2 for detection of the dispersed transmitted light. The spectral range covered by the CCD camera in non-scanning mode was about 263 nm and was calibrated using a Hg calibration lamp. The CCD chip was cooled at −60 °C. Unless stated differently, detection settings used for transmission measurements were: slit width, 10 μm; exposure time, 0.1 s; vertical shift speed, 8.25 μs; horizontal read-out, 33 kHz at 16-bit. Acquired spectra were collected using the Full Vertical Binning mode and were background corrected in the case of absorption measurements. Data acquisition was performed using Solis software from Andor Technologies.
2.3 Injection/filling
The LID cells were filled and flushed with solvent by means of a K-500 solvent pump (Knauer, Berlin, Germany) at a flow rate of 200 μL min−1. The pump was connected to one of the side inlets of the first coupling piece, unless specified otherwise. Samples of EY and CV were directly introduced using a syringe, which was connected to the second inlet, while the solvent pump was turned off. The cell was cleaned in between samples by flushing with 300 μL of MeOH followed by 300 μL of MQ using the solvent pump.2.4 LC system
In order to prepare the LC eluent, a buffer was made by adding FA and NaOH to 1 L of MQ at a final concentration of 0.1 M and 0.02 M respectively, to obtain a pH of 3. The eluent was prepared by mixing buffer and MeOH in ratios of 95/5 [v/v] respectively for mobile phase A, and 5/95 [v/v] for B. TEA was added as an ion pairing agent to both A and B at a final concentration of 5 mM. All analyses were carried out on an Agilent 1100 series LC system equipped with a quaternary solvent delivery system, a column oven, an autosampler and a photodiode array detector (PDA). A ZORBAX Eclipse RRHD C18 column (2.1 × 150 mm; particle size, 1.8 μm) and a security guard column (2.1 × 5 mm) containing the same C18 packing were both obtained from Agilent. Gradient elution was performed going linearly from 5% to 95% B in 10 min. After 5 min at 95% B, the mobile phase changed back to 5% B. Including 15 min of equilibration time, the total analysis time was 30 min. The flow rate was set at 150 μL min−1 and the column oven at 40 °C. Spectral data were recorded from 250 to 800 nm with a resolution of 1.0 nm, a sampling rate of 144 Hz, and analysis monitoring was set at detection the wavelengths 254, 400, 515, 550, and 595 nm. The equipment was controlled by ChemStation Chromatography Data Software from Agilent.3. Results & discussion
The performance of the LCW is first characterized with respect to parameters affecting its transmittance. We then evaluate the photon flux and gas permeability of the Teflon AF2400 LCW. Finally, we demonstrate the applicability of the LCW in a proof-of-concept experiment studying the light-induced degradation of EY and CV.3.1 LCW transmittance
The light transmittance of the LCW inside the LID cell is a critical property: it determines both the photodegradation rate as well as the detection limit when used in combination with spectroscopy. Preferably, light losses should be minimized to achieve a consistent illumination of the entire solution. Transmittance of the LCW can be dependent on, or influenced by several parameters, such as length of the LCW, concentration and molar absorption coefficient of the sample components, scattering losses inside the LCW, pressure applied on the LCW, and temperature. Furthermore, it has recently been reported that when transmittance is sufficient, small internal LCW diameters (<1 mm) allow higher and a more homogeneous irradiance resulting in shorter reaction times of photochemical reactions.35 For this study we used LCWs with an ID of 800 μm and the effects of the above-mentioned parameters were studied.The influence of the cell length was tested by fabricating four LID cells using type I LCWs of 12 cm (cell volume ∼60 μL), 20 cm (∼100 μL), 40 cm (∼200 μL) and 60 cm (∼300 μL) long.
These were filled with water and the intensity of the transmitted light was measured for each cell at different wavelengths using specific bandpass filters. The intensities followed from power measurements in triplicate (each time after realignment, RSD <9%), where the sensor of the power meter was positioned at the excitation fibre outlet or at the exit of the LCW filled with MQ. The light transmittance was calculated by comparing the intensity at the end of the LCW (I) with the intensity at the exit of the excitation fibre (I0). Fig. 2 shows the average (n = 3) transmittance of the four LCWs at different wavelengths, normalized to the highest transmittance value measured for the 12 cm LCW at 630 nm. Fig. 2a shows that the transmittance decreases significantly with increasing length. Light losses are most probably due to reflection losses caused by pores and imperfections in the Teflon capillary surface, as also indicated by Altkorn et al.31 The number of reflections increases with the length of the capillary, causing more light to be lost. Apparently, this effect is strongest in the first part of the LCW; the difference between LCWs of 40 and 60 cm was found to be negligible in our experiment.
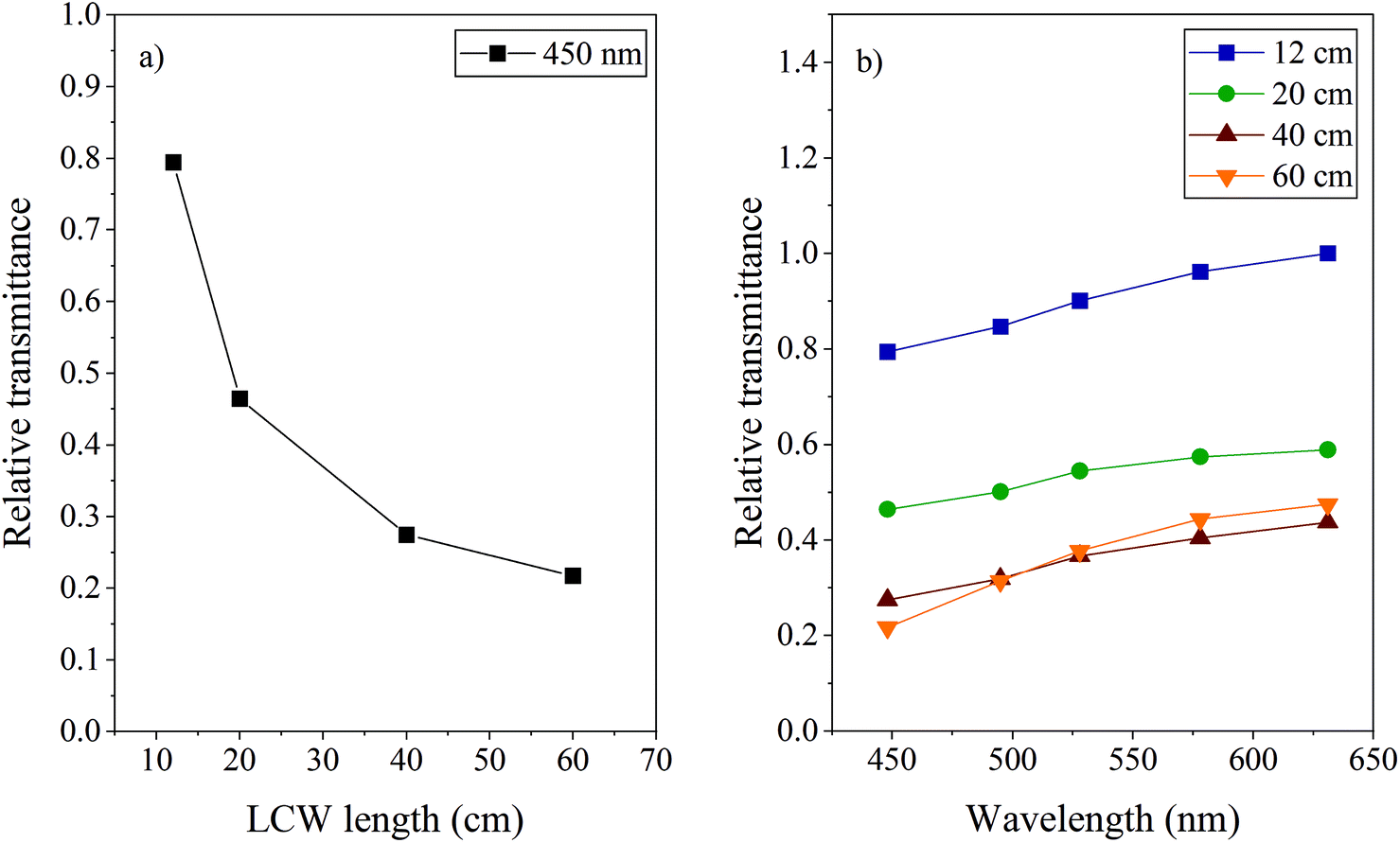 | ||
| Fig. 2 (a) The relative transmittance for light of 450 nm as function of LCW length calculated by comparing the transmitted (I) with the input power (I0); (b) the relative transmittance for light of different wavelengths using LCW lengths of 12, 20, 40 and 60 cm. | ||
The light absorbance by a compound inside the cell is a point of consideration. Increased analytical sensitivity due to a long optical path length might be beneficial, however, from the viewpoint of analyte degradation, the formation of a strong light-intensity gradient inside the LID cell is not favoured. Therefore, for LID studies of samples showing significant light absorbance at the applied wavelength, medium-length LCWs are preferred over very long ones. Another reason to limit the length of the LCWs, is to increase compatibility with LC. LCW volumes larger than LC peak volumes would cause loss of LC resolution when using the cell in a post-column setup.
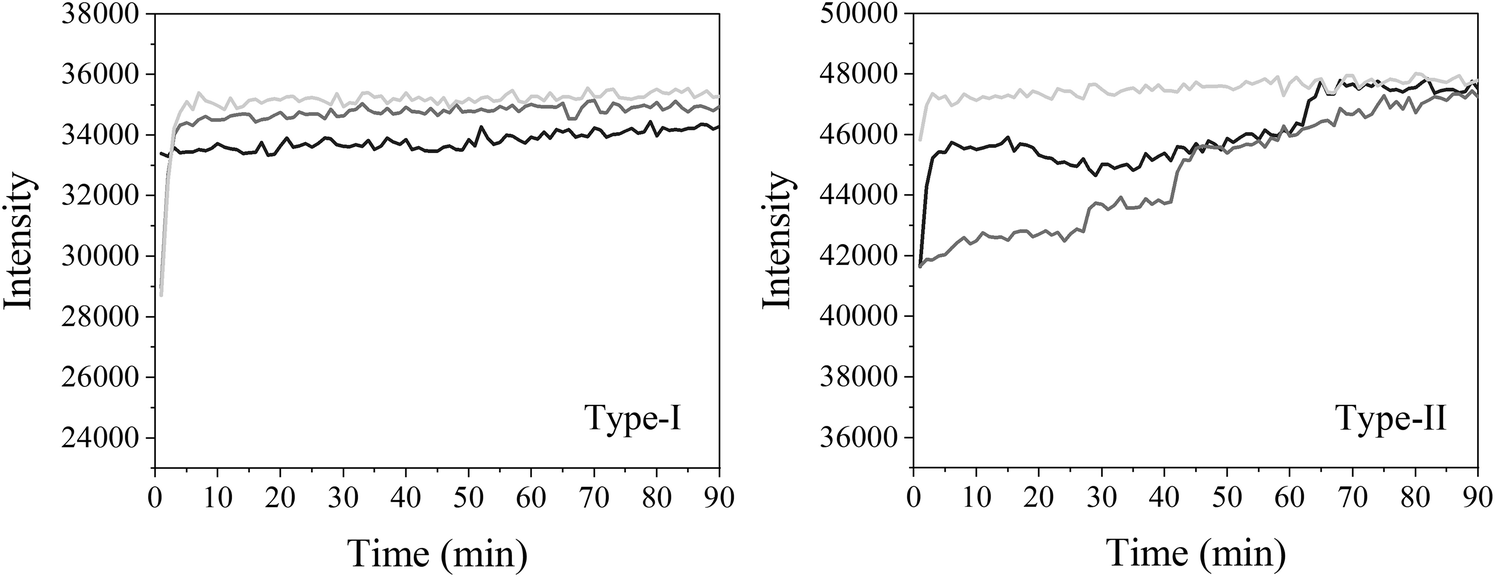 | ||
| Fig. 3 The intensity of the transmitted light measured in triplicate at 462 nm every minute for 90 min for a type I LCW and a type II LCW filled with MQ. | ||
3.2 Light absorption in LCW
 | ||
| Fig. 4 Measured absorbance as function of the concentration of CV measured in a 12 cm long LCW. | ||
A 20 cm LCW was filled with a solution of DAE in hexane and was irradiated with the Xenon lamp. Since this is a polychromatic light source, an equilibrium between the open and closed form will be reached after a certain period of time. Therefore, a UV shortpass filter (<350 nm) was used to turn DAE into the violet closed form (DAE-c), and a bandpass filter of (nominal value) 580 nm was used to allow DAE to turn into the colourless open form (DAE-o). This wavelength was chosen because it is close to the absorption maximum of DAE-c. The actual maximum transmitted wavelength of the bandpass filter was determined to be 578 nm and was taken into account. Calculations and technical aspects regarding the measured photon flux are further elaborated in the ESI.†
Fig. 5 shows the decrease in absorbance in real-time for the reaction from DAE-c to DAE-o at 578 nm. The linear part of the decrease in absorption was plotted and used to calculate the average rate of the conversion. From this, the photon flux at 578 nm was calculated. This was compared to the flux calculated from power measurements, obtained by placing the power meter behind the end of the LID cell filled with water. Using actinometry, a photon flux of 10.3 × 1012 photons per s was calculated, compared to 9.49 × 1012 photons per s obtained from power measurements. The similarity between the obtained results with both methods indicate that the power measurements are a good estimate of the photon flux passing the LCW. However, the power meter measures the number of photons reaching the end of the LID cell, whereas the actinometer measures the effective intensity inside the LCW. Therefore, we presume that the DAE actinometer gives a better representation of the real photon flux inside the LID cell.
 | ||
| Fig. 5 Actinometry of the LCW. (a) Measured change in absorbance in real-time during the isomerization reaction of DAE-c to DAE-o at 578 nm inside a 20 cm long LID cell. (b) Linear part of the decrease in absorbance is plotted; slope was used to determine the photon flux (ESI†). | ||
3.3 Gas diffusion through LCW wall
Oxygen gas plays an important role in many photodegradation reactions. The Teflon AF LCW comprises a gas permeable wall, which offers the possibility to influence the photodegradation process by adding or removing oxygen gas to or from the exposed solution. Oxygen diffusion was tested by using methylene blue (MB) as an indicator for oxygen. The LID cell was filled with an aqueous solution of MB, glucose and sodium hydroxide. In the presence of oxygen, MB is oxidized and the solution will turn blue, resulting in absorption of visible light by MB. When oxygen is removed or consumed, MB will be reduced by the sodium hydroxide and the solution turns colourless, resulting in a reduced visible absorbance. The decolourization reaction of MB by oxygen was recorded in real-time by measuring the transmitted light intensity of the LCW at the absorption maximum of MB (668 nm).Fig. 6 shows the LCW light transmittance in real-time (yellow curve) while the outer tubing of the LID cell, which initially contained air, was flushed with nitrogen gas. During the flushing, part of the LID cell was filmed using a camera and fragments of this footage are shown at the corresponding time points. After about 50 s, the nitrogen flow was turned on, and from 65 s onwards the transmitted light intensity increased, indicating decolourization, thus reduction of MB caused by oxygen depletion in the LCW. After about 200 s the nitrogen flow was turned off, and subsequently a reduction of the transmittance was observed, implying oxidation of MB due to presence of oxygen in the LCW. The latter means that spontaneous diffusion of oxygen occurs from air back into the solution. Since the solution inside the LCW is not stirred, we assume an oxygen gradient will exist from the wall to the middle of the LCW. As expected, the video recording of the colour changes showed no sign of inhomogeneity along the length dimension of the LCW. This experiment showed that the Teflon AF wall is gas permeable even at ambient pressure, which is in line with previous findings.41 The experiment also demonstrated that oxygen levels in the LCW can be controlled using the tube-in-tube design, and that the reaction process can be followed in real-time by absorbance spectroscopy. This is a clear advantage of the type I LCW over the type II, as it allows the study of photodegradation under oxygen-saturated conditions and evaluation of the effect of oxygen on degradation, providing more detailed mechanistic insight.
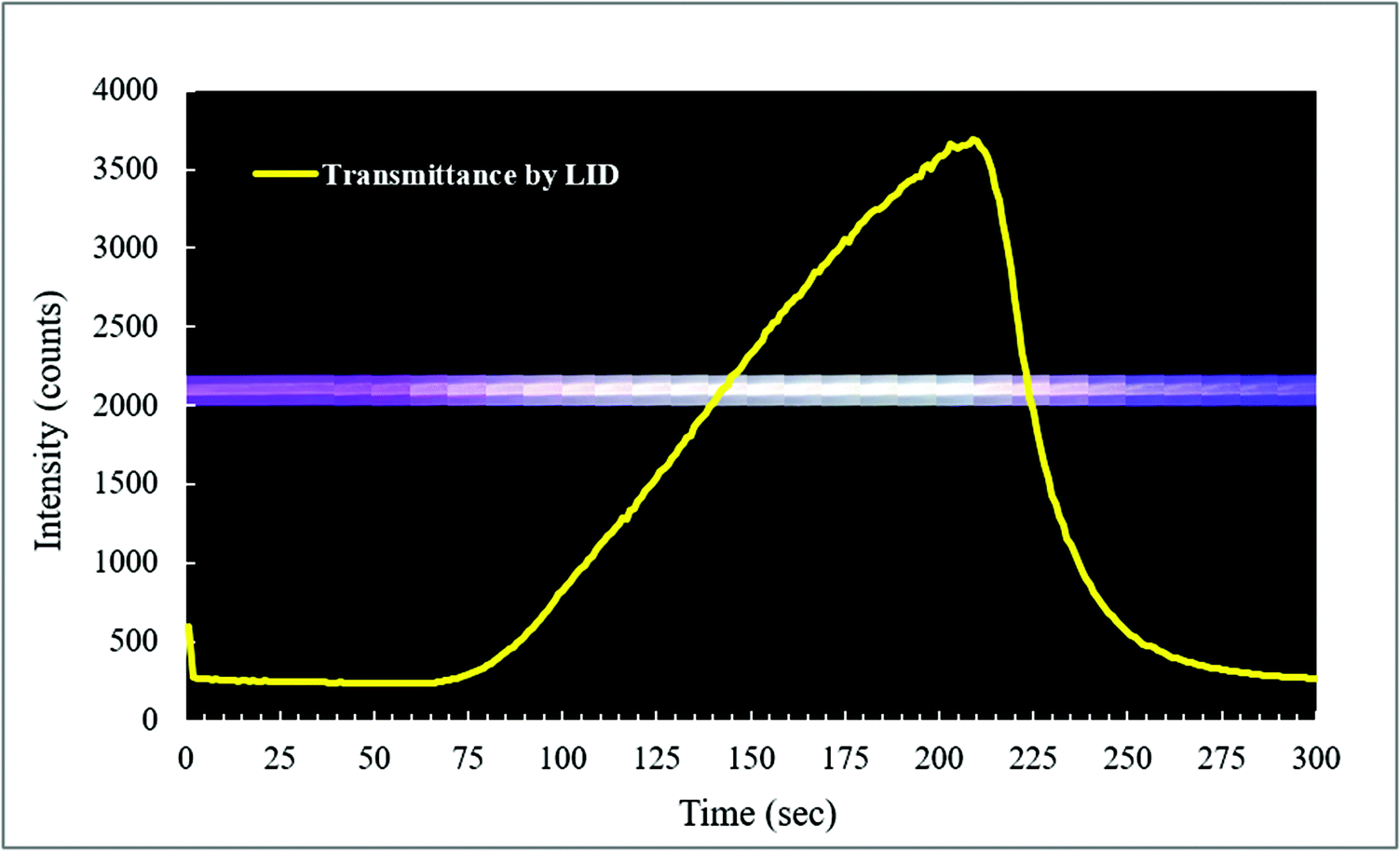 | ||
| Fig. 6 Monitoring of oxygen in the LCW. The LID cell was filled with a solution of MB, glucose and NaOH which was used as reagents to observe the presence of dissolved oxygen in the LCW. The yellow curve shows the measured transmittance of the LCW at 668 nm. The blueish trace shows the actual colour of the LCW at the corresponding time points. When the oxygen concentration decreases by flushing nitrogen gas through the outer tubing (50–200 s), MB is reduced and turns colourless. | ||
3.4 Monitoring the LID of EY and CV
Two proof-of-concept experiments were performed by monitoring the photodegradation of the dyes EY (Fig. D.1†) and CV (Fig. D.2†). The photodegradation processes of these dyes are well known, allowing thorough evaluation of the results obtained with the LCW setup and LC analyses. A 12 cm LCW was filled with a 5 × 10−7 M solution of EY (ε at 518 nm, 95![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cm−1 M−1), and subsequently exposed to light from the Xenon lamp for 3 hrs while recording absorption spectra in real time every 10 min. An aliquot of the starting (unexposed) EY solution was also analysed by LC with PDA detection. After 3 hrs the exposed EY solution was collected off-line by flushing the LID cell with 60 μL of MeOH. The collected sample was immediately analysed by LC-PDA in order to measure potential degradation products. The same procedure was followed for a 2.5 × 10−6 M solution of CV (ε at 590 nm, 87000 cm−1 M−1), but then using a 5 hr light exposure time.
000 cm−1 M−1), and subsequently exposed to light from the Xenon lamp for 3 hrs while recording absorption spectra in real time every 10 min. An aliquot of the starting (unexposed) EY solution was also analysed by LC with PDA detection. After 3 hrs the exposed EY solution was collected off-line by flushing the LID cell with 60 μL of MeOH. The collected sample was immediately analysed by LC-PDA in order to measure potential degradation products. The same procedure was followed for a 2.5 × 10−6 M solution of CV (ε at 590 nm, 87000 cm−1 M−1), but then using a 5 hr light exposure time.
 | ||
| Fig. 7 (a) Real-time absorption spectra of a solution of EY (5 × 10−7 M in water) present in the LID-cell during light irradiation for 3 hrs; (b) LC chromatograms of the EY solution before (at t = 0 hr) and after exposure (at t = 3 hr) in the LID cell. The insert shows the PDA spectra obtained during LC analysis at t = 0 hr and t = 3 hr for the peak at a retention time of 18.48 min. | ||
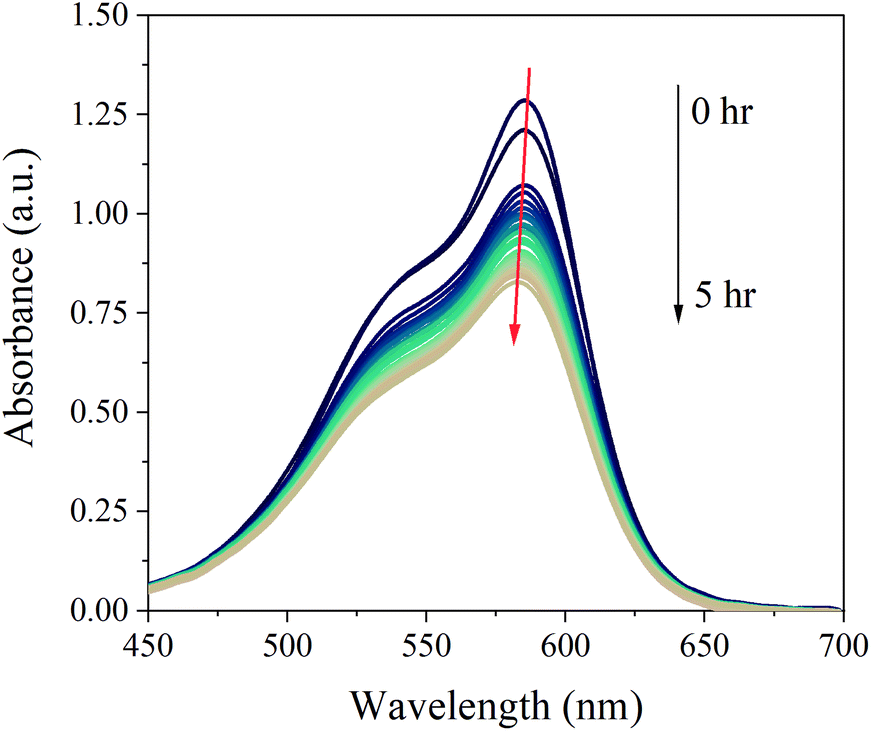 | ||
| Fig. 8 Real-time absorption spectra of a solution of CV (2.5 × 10−6 M) in water present in the LID cell during light irradiation for 5 hrs. | ||
A slight shift towards shorter wavelength of the absorption maximum was observed for the whole sample during the in situ absorption measurements in Fig. 8. Evaluating the absorption spectra of the degradation products measured by the LC-PDA (Fig. D.5†), a similar blueshift was noticed for each degradation product, which was also observed in degradation research studies of CV by Confortin et al., and others.1,45,46 This is most likely a consequence of the demethylation process of CV resulting in a 5 nm shift of the absorption maximum for each newly formed product. Demethylation also results in a decreased retention time, caused by an increased polarity of the degradation products, which can be seen in Fig. 9. An overview of the detected analytes and tentative assignments is given in Table D.1.† When comparing the obtained results with those by Confortin et al. peaks 2, 3, and 4 are most likely mono-, di-, and tri-demethylated CV, respectively. Peak 5 may be an isomer of tri-demethylated CV, and peaks 6 and 7 show similar absorption spectra as tetra-, and penta-demethylated CV. Three more peaks (8–10) were found of which peaks 8 and 9 show similar absorption maxima as that of diamond green B (Fig. D.6†). However, as Confortin also explains, these may also be assigned to N-oxide derivatives of CV. Peaks 11, 12 and 14 show absorbance in the UV, but remain unidentified as the concentration was too low to obtain well defined absorption spectra and are therefore not shown. Peak 13, however, showed an absorption maximum of 546 nm, which is similar to that of pararosaniline, which is completely demethylated CV. This was later confirmed by the analysis of a pararosaniline standard. The relative peak areas (derived from the LC-PDA signals) of CV and its degradation products, before and after irradiation, are shown in Fig. 10.
 | ||
| Fig. 9 Normalized LC-PDA chromatograms (at 595 nm) of the CV solution before (at t = 0 hr) and after exposure (at t = 5 hr) in the LID cell. The insert shows a zoom of the chromatogram obtained at t = 5 hr revealing the peaks of degradation products (2–13). | ||
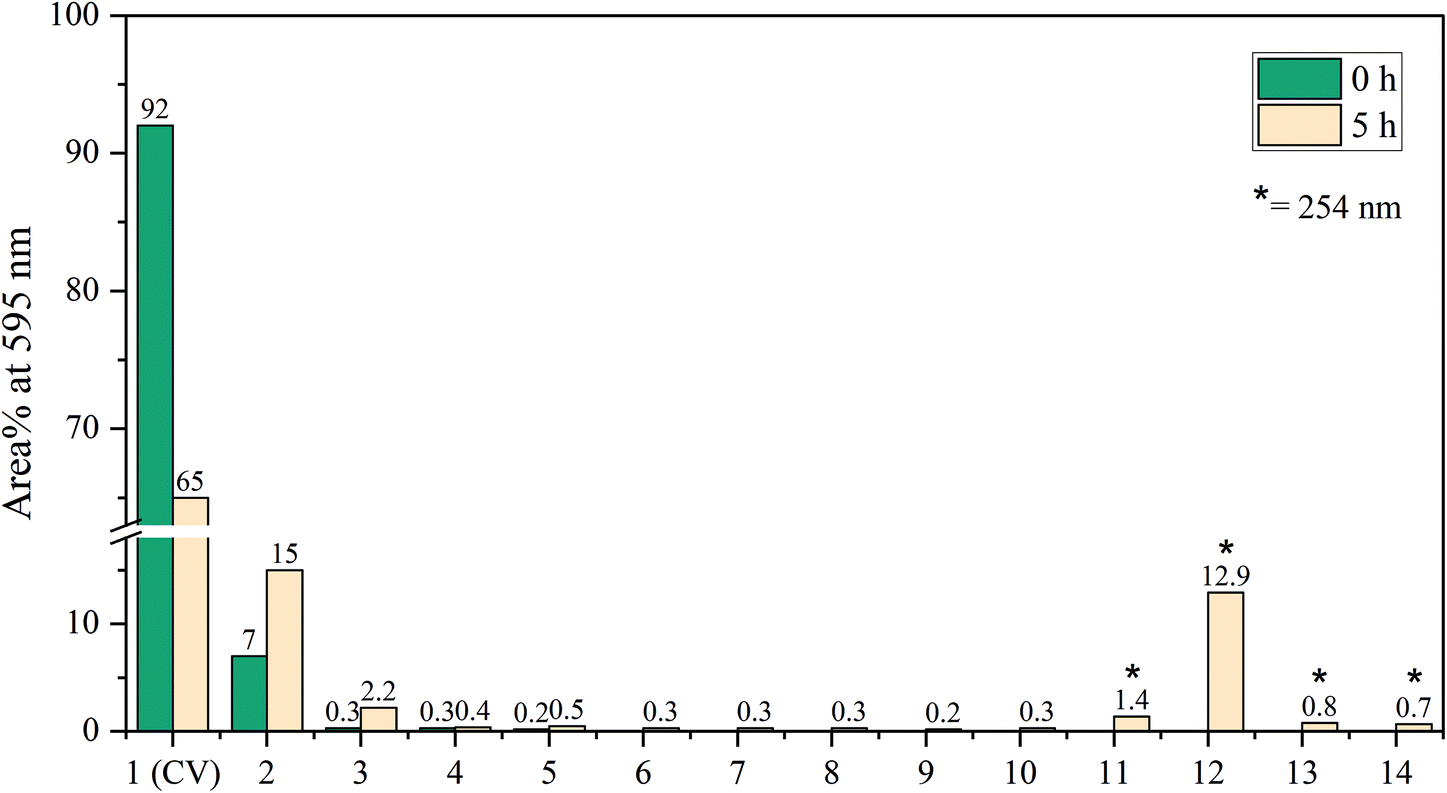 | ||
| Fig. 10 Relative peak areas of CV and degradation products obtained by LC-PDA before (0 hr) and after (5 hr) exposure in the LID-cell. Peak areas are extracted at 595 or 254 nm (*) if absorbance was in the UV. | ||
To make final conclusions about the identity of the abovementioned peaks, additional MS analyses should be performed. However, the most important result shown here is that a >30% reduction of the main component CV was obtained after 5 hrs of degradation. Weyermann and Confortin, who performed off-line degradation of CV in solution, reported near complete degradation after 25 and 115 hrs, respectively.1,45 For simple degradation processes, such as for EY, the decrease in absorbance is a straightforward measure for the degradation rate. For compounds like CV, however, for which the degradation products also absorb in the same wavelength range as the parent compound, it is slightly more complicated to use the absorbance for this purpose.
4. Conclusions
This study has shown that it is possible to initiate photodegradation of EY and CV while collecting real-time spectral data using an LID cell based on an LCW made of Teflon AF2400. The LCW is an efficient tool for illumination of the whole sample and is able to guide the light towards a detector. The results from the degradation experiments showed that real-time absorbance spectroscopy is useful for acquiring information on the stage of degradation. Changes in the shape of the absorption spectrum as well as the absorbance intensity are indicative for the extent of degradation. The results of the photodegradation of EY and CV are in line with previous studies, and the high photodegradation rate compared to conventional ‘in-beaker’ degradations looks promising.1,42,45,46 Clearly, the potential for on-line coupling of the cell to LC for further characterization of the degradants, comes as an advantage here.There is also room for improvement. The speed of photodegradation could be enhanced by improving the efficiency of light coupling into the cell, and automation of the injection system may result in more reproducible spectral data as variation in applied pressure is reduced. The long path length of the LCW is beneficial for (diluted) samples with low absorbances. The transmittance by the LCW, however, decreases with the length of the capillary, caused by pores and imperfections in the material. As a result, the intensity of the light inside the LCW also decreases with length. Therefore, for LID studies, medium-length LCW are preferred over very long ones to prevent the formation of a strong light-intensity gradient inside the LID cell.
Author contributions
I. Groeneveld: conceptualization, methodology, investigation, resources, writing – original draft and editing; Suzan E. Schoemaker: methodology, investigation; Govert W. Somsen: writing – review and editing, supervision; Freek Ariese: writing – review and editing, supervision; Maarten R. Van Bommel: writing – review and editing, funding acquisition.Conflicts of interest
The authors declare that there is no conflict of interests.Acknowledgements
We thank the Precision Mechanics and Engineering group at the Vrije Universiteit Amsterdam, and Dick van Iperen in particular, for the close collaboration towards designing and building the LID cells. This work is part of the TooCOLD project (Toolbox for studying the Chemistry of Light-induced Degradation; project number 15506) carried out in the TTW Open Technology Programme and is (partly) financed by the Netherlands Research Council (NWO).References
- D. Confortin, H. Neevel, M. Brustolon, L. Franco, A. J. Kettelarij, R. M. Williams and M. R. Van Bommel, J. Phys. Conf. Ser., 2010, 231, 1–9 CrossRef.
- M. J. Toohey and L. N. Bell, J. Food Process. Preserv., 2019, 43, 1–5 Search PubMed.
- P. I. Hora, P. J. Novak and W. A. Arnold, Environ. Sci. Water Res., 2019, 5, 897–909 RSC.
- M. Shamsipur, B. Hemmateenejad, N. J. Jahani and K. M. Majd, J. Photochem. Photobiol., A, 2015, 299, 62–68 CrossRef.
- V. G. Bório, A. U. Fernandes and L. Silveira, Instrum. Sci. Technol., 2016, 44, 189–198 CrossRef.
- R. Salgado, V. J. Pereira, G. Carvalho, R. Soeiro, V. Gaffney, C. Almeida, V. V. Cardoso, E. Ferreira, M. J. Benoliel, T. A. Ternes, A. Oehmen, M. A. M. Reis and J. P. Noronha, J. Hazard. Mater., 2013, 244–245, 516–527 CrossRef CAS PubMed.
- A. Y. C. Lin and M. Reinhard, Environ. Toxicol. Chem., 2005, 24, 1303–1309 CrossRef CAS PubMed.
- A. Y. C. Lin, X. H. Wang and W. N. Lee, Environ. Sci. Technol., 2013, 47, 4104–4112 CrossRef CAS PubMed.
- F. Gosetti, M. Bottaro, V. Gianotti, E. Mazzucco, P. Frascarolo, D. Zampieri, C. Oliveri, A. Viarengo and M. C. Gennaro, Environ. Pollut., 2010, 158, 592–598 CrossRef CAS PubMed.
- A. Kiss, S. Rapi and C. Csutorás, Microchem. J., 2007, 85, 13–20 CrossRef CAS.
- K. Sivagami, R. R. Krishna and T. Swaminathan, Sol. Energy, 2014, 103, 488–493 CrossRef CAS.
- V. T. Trang, Y. Kurogi, S. Katsuno, T. Shimamura and H. Ukeda, Int. Dairy J., 2008, 18, 344–348 CrossRef.
- G. J. M. Fechine, P. A. Christensen, T. A. Egerton and J. R. White, Polym. Degrad. Stab., 2009, 94, 234–239 CrossRef CAS.
- E. Yousif, N. Salih and J. Salimon, J. Appl. Polym. Sci., 2011, 120, 2207–2214 CrossRef CAS.
- D. A. Volmer, J. Chromatogr. A, 1998, 794, 129–146 CrossRef CAS PubMed.
- P. J. Harman, G. L. Blackman and G. Phillipou, J. Chromatogr. B: Biomed. Sci. Appl., 1981, 225, 131–138 CrossRef CAS.
- S. X. Peng and S. M. Dansereau, J. Chromatogr. A, 2001, 914, 105–110 CrossRef CAS PubMed.
- T. Pérez-Ruiz, C. Martínez-Lozano and M. D. García-Martínez, J. Pharm. Biomed. Anal., 2009, 50, 315–319 CrossRef PubMed.
- A. F. Kadjo, P. K. Dasgupta and C. P. Shelor, Anal. Chem., 2020, 92, 6391–6400 CrossRef CAS PubMed.
- L. Ganranoo, S. K. Mishra, A. K. Azad, A. Shigihara, P. K. Dasgupta, Z. S. Breitbach, D. W. Armstrong, K. Grudpan and B. Rappenglueck, Anal. Chem., 2010, 82, 5838–5843 CrossRef CAS PubMed.
- D. Kottke, B. B. Burckhardt, J. Breitkreutz and B. Fischer, Talanta, 2021, 226 DOI:10.1016/j.talanta.2021.122145.
- K. Bescherer, J. A. Barnes and H. P. Loock, Anal. Chem., 2013, 85, 4328–4334 CrossRef CAS PubMed.
- T. Rubles, D. Paige and C. Anastasio, Rev. Sci. Instrum., 2006, 77, 1–4 Search PubMed.
- Z. Liu and J. Pawliszyn, Anal. Chem., 2003, 75, 4887–4894 CrossRef CAS PubMed.
- A. Datta, I. Y. Eom, A. Dhar, P. Kuban, R. Manor, I. Ahmad, S. Gangopadhyay, T. Dallas, M. Holtz, H. Temkin and P. K. Dasgupta, IEEE Sens. J., 2003, 3, 788–795 CrossRef CAS.
- K. Flavin, H. Hughes and P. McLoughlin, Int. J. Environ. Anal. Chem., 2007, 87, 29–42 CrossRef CAS.
- B. J. Marquardt, P. G. Vahey, R. E. Synovec and L. W. Urgess, Anal. Chem., 1999, 71, 4808–4814 CrossRef CAS PubMed.
- S. Tanikkul, J. Jakmunee, M. Rayanakorn, K. Grudpan, B. J. Marquardt, G. M. Gross, B. J. Prazen, L. W. Burgess, G. D. Christian and R. E. Synovec, Talanta, 2003, 59, 809–816 CrossRef CAS PubMed.
- R. J. Dijkstra, A. N. Bader, G. P. Hoornweg, U. A. T. Brinkman and C. Gooijer, Anal. Chem., 1999, 71, 4575–4579 CrossRef CAS.
- Y. Tian, L. Zhang, J. Zuo, Z. Li, S. Gao and G. Lu, Anal. Chim. Acta, 2007, 581, 154–158 CrossRef CAS PubMed.
- R. Altkorn, I. Koev and M. J. Pelletier, Appl. Spectrosc., 1999, 53, 1169–1176 CrossRef CAS.
- R. Altkorn, I. Koev, R. P. Van Duyne and M. Litorja, Appl. Opt., 1997, 36, 8992–8998 CrossRef CAS PubMed.
- M. Brzozowski, M. O'Brien, S. V. Ley and A. Polyzos, Acc. Chem. Res., 2015, 48, 349–362 CrossRef CAS PubMed.
- S. Ponce, H. Christians, A. Drochner and B. J. M. Etzold, Chem. Ing. Tech., 2018, 90, 1855–1863 CrossRef CAS.
- C. Sambiagio and T. Noël, Trends Chem., 2020, 2, 92–106 CrossRef CAS.
- WPI, 4000 Series Liquid Waveguide Capillary Cell, Instruction Manual, 2014.
- G. Dlubek, J. Pionteck, K. Rätzke, J. Kruse and F. Faupel, Macromolecules, 2008, 41, 6125–6133 CrossRef CAS.
- A. Roibu, S. Fransen, M. E. Leblebici, G. Meir, T. Van Gerven and S. Kuhn, Sci. Rep., 2018, 8, 1–10 CAS.
- M. Irie, K. Sakemura, M. Okinaka and K. Uchida, J. Org. Chem., 1995, 60, 8305–8309 CrossRef CAS.
- T. Sumi, Y. Takagi, A. Yagi, M. Morimoto and M. Irie, Chem. Commun., 2014, 50, 3928–3930 RSC.
- M. O'Brien, I. Baxendale and S. Ley, Synfacts, 2010, 2010, 1199–1199 CrossRef.
- A. Alvarez-Martin, S. Trashin, M. Cuykx, A. Covaci, K. De Wael and K. Janssens, Dyes Pigm., 2017, 145, 376–384 CrossRef CAS.
- B. W. J. Pirok, G. Moro, N. Meekel, S. V. J. Berbers, P. J. Schoenmakers and M. R. van Bommel, J. Cult. Herit., 2019, 38, 29–36 CrossRef.
- N. Kuramoto and T. Kitao, Dyes Pigm., 1982, 3, 49–58 CrossRef CAS.
- C. Weyermann, D. Kirsch, C. Costa-Vera and B. Spengler, J. Forensic Sci., 2009, 54, 339–345 CrossRef CAS PubMed.
- G. Favaro, D. Confortin, P. Pastore and M. Brustolon, J. Mass Spectrom., 2012, 47, 1660–1670 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1an00272d |
| This journal is © The Royal Society of Chemistry 2021 |