
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Nuclear hyperpolarization of (1-13C)-pyruvate in aqueous solution by proton-relayed side-arm hydrogenation†
Laurynas
Dagys
 a,
Anil P.
Jagtap
bc,
Sergey
Korchak
bc,
Salvatore
Mamone
bc,
Philip
Saul
bc,
Malcolm H.
Levitt
*a and
Stefan
Glöggler
*bc
a,
Anil P.
Jagtap
bc,
Sergey
Korchak
bc,
Salvatore
Mamone
bc,
Philip
Saul
bc,
Malcolm H.
Levitt
*a and
Stefan
Glöggler
*bc
aSchool of chemistry, Highfield Campus, Southampton, SO171BJ, UK. E-mail: mhl@soton.ac.uk
bMax Planck Inst. Biophys. Chem., NMR Signal Enhancement Grp., Am Fassberg 11, D-37077 Göttingen, Germany. E-mail: stefan.gloeggler@mpibpc.mpg.de
cCenter for Biostructural Imaging of Neurodegeneration of UMG, Von-Siebold-Str. 3A, D-37075 Göttingen, Germany
First published on 15th January 2021
Abstract
We employ Parahydrogen Induced Polarization with Side-Arm Hydrogenation (PHIP-SAH) to polarize (1-13C)-pyruvate. We introduce a new method called proton-relayed side-arm hydrogenation (PR-SAH) in which an intermediate proton is used to transfer polarization from the side-arm to the 13C-labelled site of the pyruvate before hydrolysis. This significantly reduces the cost and effort needed to prepare the precursor for radio-frequency transfer experiments while still maintaining acceptable polarization transfer efficiency. Experimentally we have attained on average 4.33% 13C polarization in an aqueous solution of (1-13C)-pyruvate after about 10 seconds of cleavage and extraction. PR-SAH is a promising pulsed NMR method for hyperpolarizing 13C-labelled metabolites in solution, conducted entirely in high magnetic field.
1. Introduction
Hyperpolarization methods are a group of techniques that address the limited sensitivity of Nuclear Magnetic Resonance (NMR).1–9 These methods have led to promising progress towards highly polarized molecules for use in the chemical and biological sciences as molecular imaging agents8,10,11 some of which are already on their way to clinical diagnostics.12–14 Among the several metabolic substrates that have been hyperpolarized, (1-13C)-pyruvate is, by far, the most widely investigated.15,16 Pyruvate participates in several important metabolic processes, such as glycolysis, which makes it a compelling probe of energy metabolism in biological systems.12–17One of the fastest hyperpolarization methods is parahydrogen-induced polarization (PHIP).2,3,7,17–31 It has a number of advantages, such as its low-cost, compact and easy application without the need of dedicated cryo-cooled high-field magnets, and the ability to produce large quantities of polarized material on demand. The method uses spin order from the para-spin isomer of molecular hydrogen which is provided by chemical interactions between a substrate of interest, solvent and a catalyst.3,32
One of the most effective ways to hyperpolarize pyruvate with parahydrogen is the side-arm hydrogenation (SAH) procedure (Fig. 1).23–25 In PHIP-SAH, a 13C-labeled metabolite is attached to a side-arm with an unsaturated bond. This side-arm is subsequently hydrogenated in an organic solvent with para-enriched hydrogen resulting in high nuclear spin order. The order is transferred from the side-arm protons to the 13C of the target molecule by a variety of methods, as discussed below. After achieving strong 13C magnetization, the side-arm is cleaved off with an aqueous basic solution, thereby extracting the desired hyperpolarized metabolite into the aqueous phase.23 For biological uses, the pH of the obtained solution is adjusted as the last step.
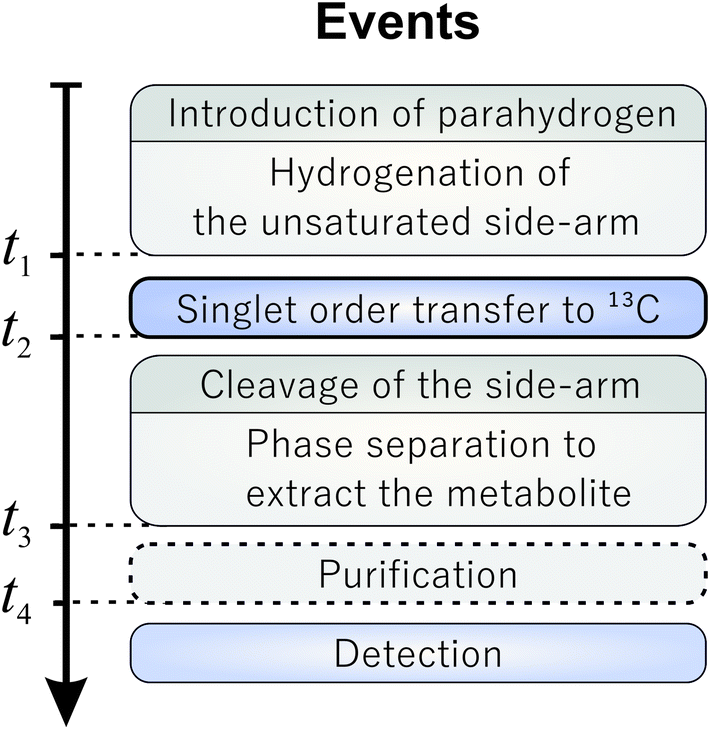 | ||
| Fig. 1 Generalized PHIP-SAH timing scheme. The indicated time points are related to the procedure depicted in Fig. 2. | ||
The achievable 13C magnetization yield is dictated by the achievable proton spin order, the polarization transfer step, and relaxation losses during the chemical reactions and pulse sequence. The most common way to perform the transfer step is to use field cycling techniques achieving about 5% 13C polarization of pyruvate enriched in the 1-13C position.16,33–39 This involves changing the magnetic field from earth to near zero field which converts the initial spin-order into 13C magnetization. However, the use of the low-field regime normally requires 1 m or more of separation between the site of spin order conversion and the site of the NMR or MRI experiment for metabolic studies (normally inside the bore of a high-field magnet).
Methods have been proposed in which the spin order transformation step is conducted entirely in high magnetic fields using radiofrequency pulse sequences.40–44 This is particularly useful for in vivo applications since it allows the polarization to be conducted in close proximity to the organism under study. Hence, polarization losses are minimized. Such a realization was recently demonstrated via the SAMBADENA (Synthesis Amid the Magnet Bore Allows Dramatically Enhanced Nuclear Alignment) approach and was so far shown for the xenobiotic hydroxyethyl propionate only.42,43 A first study to hyperpolarize pyruvate using pulsed PHIP-SAH required the use of two 13C sites, one located in the sidearm and one located in the target metabolite.40 After hydrogenation of the molecule the spin order is relayed via the sidearm-13C to the 13C of interest and cleavage yields the free metabolite. For free (1-13C)pyruvate 3.4% polarization was so far achieved. However, the cleavage experiments were only performed in organic solvents and no extraction into the aqueous phase was demonstrated.40
We introduce a new method called proton-relayed sidearm hydrogenation (PR-SAH), in which a bridging hydrogen nucleus is used instead of an additional 13C-labelled site, reducing the cost and effort of isotopic labelling with respect to rf-transfer methods. We also demonstrate that extraction of the hyperpolarized metabolite into an aqueous phase is possible without unacceptable loss of polarization.
The proposed molecular system for side-arm hydrogenation experiments is shown in Fig. 2(a). The new side arm (3-butyn-2-ol-d5 ) consists of a terminal triple bond, one proton for the relay and is otherwise deuterated. The sidearm is attached to the 1-13C-pyruvate moiety by a cleavable ester linkage. Addition of the para-enriched hydrogen (parahydrogen) to the unsaturated side-arm of the molecule I as shown in Fig. 2(a), deposits the singlet order of the parahydrogen between two, now vicinal, protons.
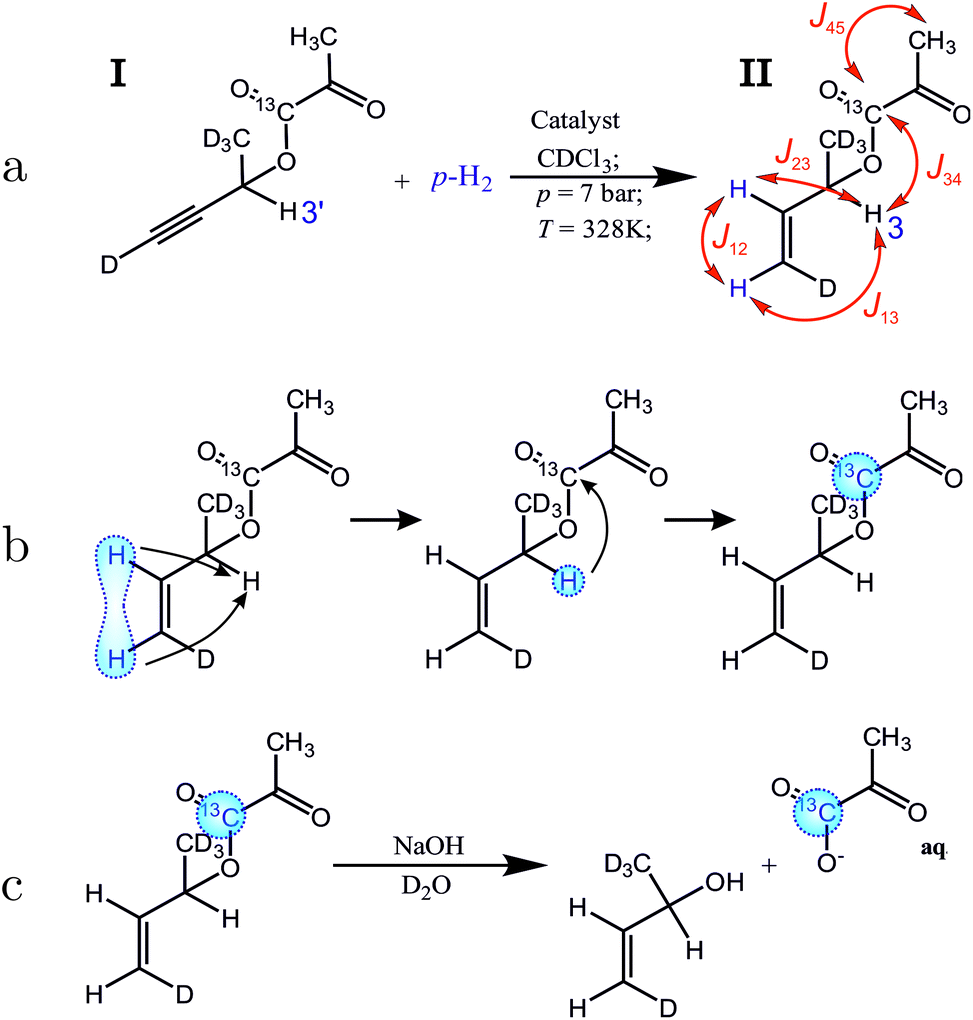 | ||
| Fig. 2 Proton-relayed side-arm hydrogenation (PR-SAH) scheme. (a) Hydrogenation of the precursor molecule I with parahydrogen to form the hyperpolarized molecule II. Red symbols and arrows represent J couplings between spins of interest: |J12| = 10.6 Hz, |J23| = 6.2 Hz, |J13| = ∼1 Hz, |J34| = 2.9 Hz, |J45| = 1.5 Hz. The chemical shifts of the protons of interest are as follows: (1) 5.20 ppm, (2) 5.88 ppm, (3) 5.44 ppm, (3′) 5.50 ppm. (b) Spin-order transfer from the former parahydrogen protons to the 13C of the pyruvate, via the relaying proton. (c) Cleavage reaction to detach the highly polarized pyruvate from the side-arm using a basic solution. | ||
The singlet nuclear spin order from the para-enriched hydrogen is first transferred to the “relay” proton, and then on to the pyruvate-13C, as shown in Fig. 2(b). These steps are performed by the radiofrequency pulse sequence discussed in this article. The polarized metabolite is released from the side-arm by a rapid injection of sodium hydroxide (Fig. 2(c)). The free pyruvate is dissolved into deuterated water whereas the catalyst stays in the organic phase. This also acts as an extraction step since the organic and aqueous phases separate, leaving highly polarized pyruvate dissolved in water. This finalizes all steps up to t3 marked in the general scheme (Fig. 1) and takes around 19 s in total. Our experiments show that after the polarization transfer (but before cleavage) 7.1% 13C polarization is obtained at the site of interest. After cleavage and phase extraction we achieved 4.3% 13C polarization of 1 mM pyruvate in the aqueous phase. Thus, PR-SAH could be a potentially useful cost-effective approach to perform hyperpolarized 13C experiments entirely in high field.
2. Methods
2.1. Materials
The precursor molecule I was synthesised in three steps including esterification of the side-arm 3-butyn-2-ol-d5 and (1-13C)pyruvic acid. The detailed synthesis and validation of the precursor compound can be found in ESI.† The catalyst [Rh(dppb)(COD)]BF4 (CAS Number 79255-71-3) was purchased from Sigma Aldrich. Other materials such as choloform-d1, ethanol-d1 and D2O were also provided by the same vendor.para-Enriched hydrogen was continuously produced by a Bruker parahydrogen generator (BPHG90), with a specified parahydrogen content of 82%. The hydrogen gas was pressurized to 10 bar.
The cleavage solution consisted of 150 mM sodium hydroxide dissolved in D2O.
2.2. Equipment
The hyperpolarization setup consists of a 5 mm NMR tube inside a cryomagnet to which 7 bar gauge pressure of parahydrogen is delivered. A plastic tubing and a thin glass capillary reaching to the bottom of the tube supplied the para-enriched hydrogen to the reaction mixture. The flow of the gas was controlled by magnetic valves located in close vicinity to the cryomagnet. The valves are driven by amplified electronic pulses from the NMR spectrometer triggered via the pulse program. The setup facilitated all of the following PHIP experiments.Experiments were conducted on a Bruker Avance AVANCE III HD spectrometer coupled with a 300 MHz Bruker Ascend magnet equipped with a 5 mm BBO probe. The excitation pulses were set to ∼26.5 kHz and ∼33.3 kHz nutation frequency for 1H and 13C respectively. All 1H pulses used an irradiation frequency corresponding to the chemical shift of 5.90 ppm, except for one selective pulse, which used a 1H irradiation frequency corresponding to 5.44 ppm, in order to match an individual alkene 1H resonance (see below). The shape of the selective pulse consisted of 1000 points approximating a 14.7 ms long Gaussian pulse with a truncation at 1% of the total height. The 13C spectra were collected with 256k real-data point density for 250 ppm width. Resonant 2H irradiation was applied throughout all experiments to decouple 2H from the rest of the spin system.
3. Proton-relayed side-arm hydrogenation
3.1. Sample preparation and hydrogenation
The synthesized precursor but-3-yn-2-yl-1,1,1,4-d4 2-oxopropanoate-1-13C (molecule I) and the catalyst were added into a small glass vial. The chloroform-d1 solvent was added and the mixture was sonicated for several minutes. The default 2 mL stock solution contained 5 mM of the catalyst and ∼2.5 mM of the precursor molecule. The cleavage experiments required the solvent to be a 9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mix of chloroform-d1 and ethanol-d1.
:1 mix of chloroform-d1 and ethanol-d1.
Prior to NMR experiments, the NMR tube was filled with a 150 μL aliquot of the stock solution. The solution was deoxygenated with nitrogen gas for 1 minute, inserted into the magnet and the temperature equilibrated at 55 °C.
The sample was pressurized with parahydrogen for 2 seconds and bubbled for 5 seconds. A 1 second delay was allowed for the bubbles to dissipate before the application of resonant rf pulses.
The side-arm hydrogenation experiments were repeated three times in order to assess the reproducibility of the procedure and to assess the standard deviation of the resulting polarization.
3.2. Polarization transfer
The polarization transfer sequence is shown in Fig. 3 and is a modified version of the ESOTHERIC (Efficient Spin Order Transfer to HEteronuclei with Relayed INEPT Chains) scheme containing 3 consecutive INEPT blocks.40,41,44 The pulse sequence operates under weak coupling conditions, which is fulfilled due to the large chemical shift differences between all of the coupled 1H spins with respect to their J-coupling. The exact timings of the sequence seen in Fig. 3 are τ1 = 74.62 ms, τ2 = 22.6 ms, τ3 = 24.6 ms, τ4 = 350 ms unless stated otherwise.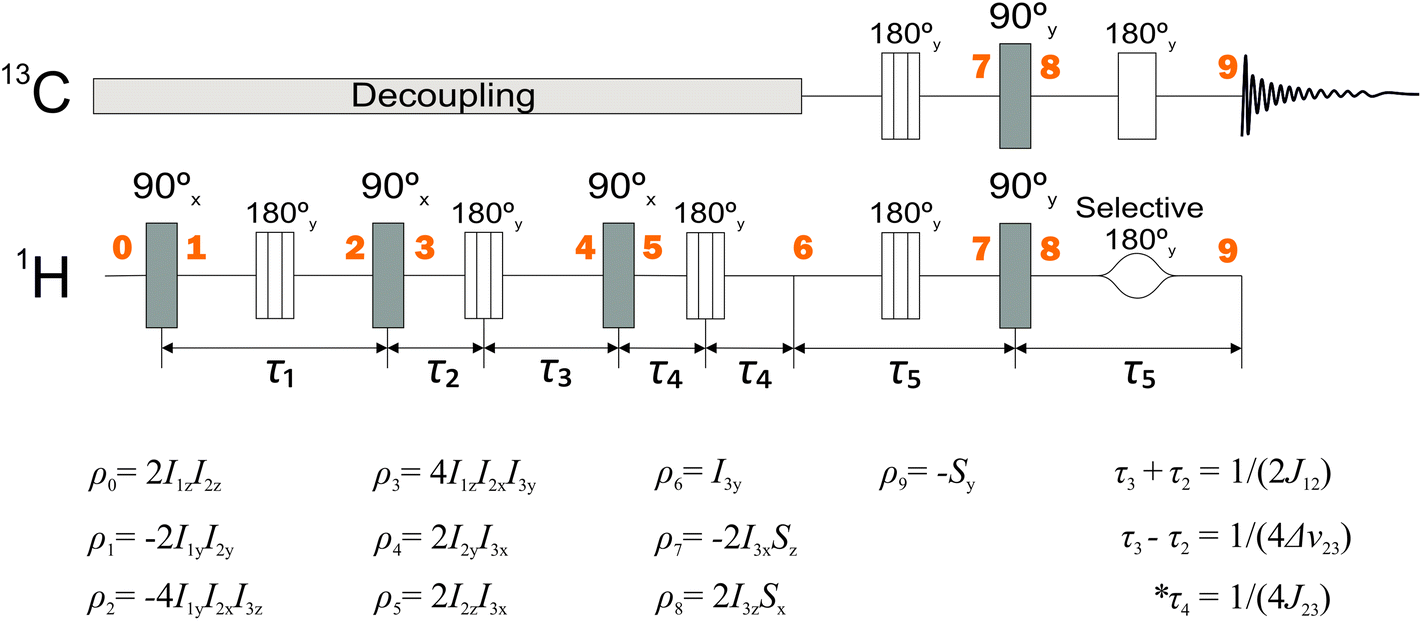 | ||
| Fig. 3 NMR pulse sequence used to perform the relayed polarization transfer depicted in Fig. 2(b). Spin density operators represent each point in time marked by bold numbers assuming J13 = 0 and ignoring any relaxation or deviations from the weak-coupling condition. 2H decoupling was used throughout the whole sequence and is not shown. The intervals τ1 and τ5 are given by τ1 = 1/(2J23) and τ5 = 1/(2J34). The values of the other delays are given in the figure. *In practice, the time interval τ4 is optimized for the best polarization yield. | ||
We treat molecule II as 4-spin system throughout the article ignoring the deuterons and the remote methyl protons of the pyruvate. The J-coupling values among the spins were determined from the multiplicity of the spectral lines and are given in the Fig. 2. No special attention was made to the sign of the couplings as they only determine the sign of the spin operator carried over during the sequence resulting in positive or negative polarization at the end of the sequence. With only one 13C to be polarized in the final solution only absolute spin order is considered. The 1H nuclei of the molecules I and II display chemical shifts that are well separated in the 1H NMR spectrum (Fig. 4). This means all protons fall within the weak coupling regime and as a result the parahydrogen singlet order is effectively projected onto the 2I1zI2z spin operator, as in the standard PASADENA experiment.45,46
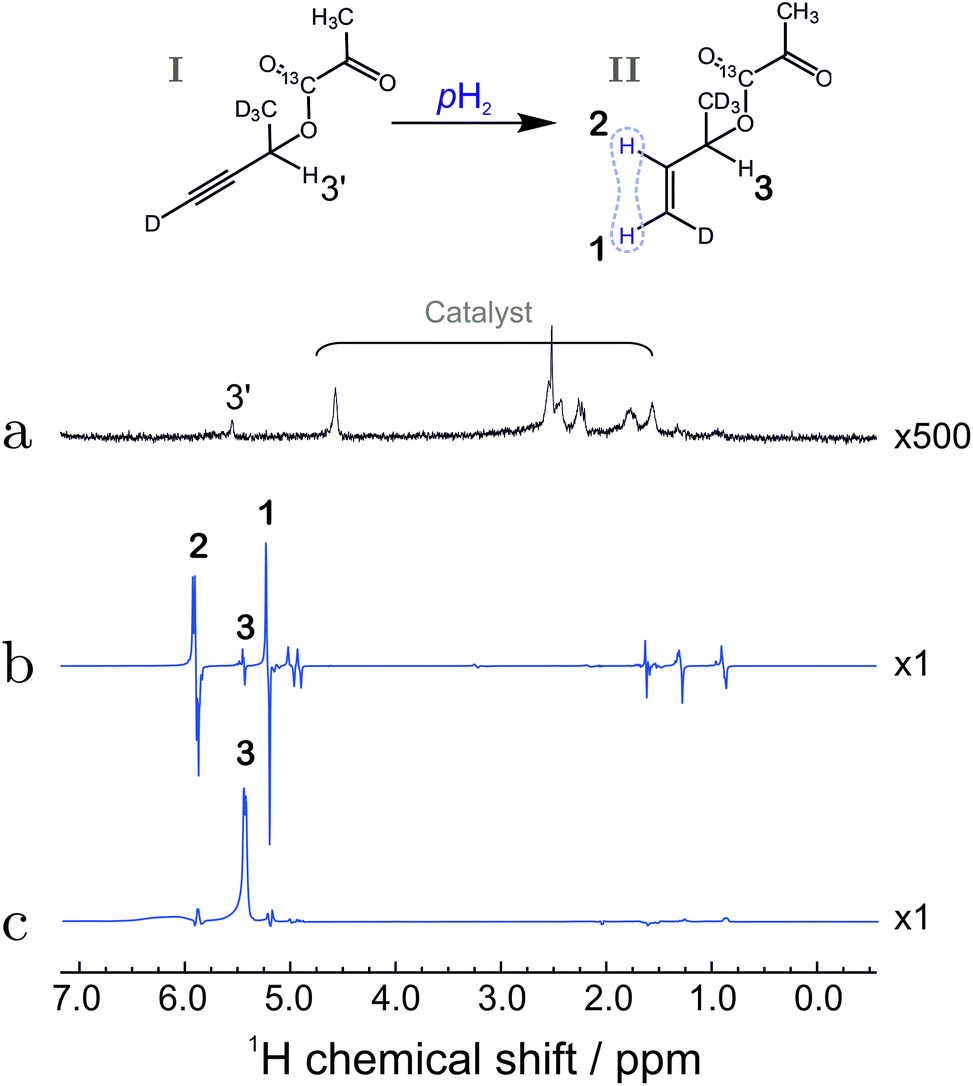 | ||
| Fig. 4 (a) Spectrum of compound I before hydrogenation, recorded by applying a π/2 pulse to a sample in thermal equilibrium (expanded vertically by a factor of 500). (b) Hyperpolarized 1H spectrum of compound II, recorded by applying a π/4 pulse to the sample after reaction with para-enriched hydrogen; (c) 1H spectrum after reaction of substance I with para-enriched hydrogen, and application of the pulse sequence in Fig. 3 up to time point 6. The spectrum displays the strongly enhanced signal of the relay proton (labelled 3). The bold numbers indicate the spectral lines associated with the protons of interest. For the chemical shifts, see the caption to Fig. 2. The other anti-phase features in the spectrum (b) originate from the fully hydrogenated form of the precursor (signals at 1.3 ppm and 0.9 ppm) as well as a hydrogenated 1,5-cyclooctadiene (signal at 1.6 ppm). The origin of the line at 5 ppm is not fully understood but may be associated with trans-hydrogenation product or impurities. | ||
The zz-order in the sequence (Fig. 3) is converted by a (π/2)x pulse into a 2I1yI2y state, which is not directly observable. The J23 coupling causes the state to evolve. At the time equal to 1/(2J23) a pure 4I1yI2xI3z is attained if chemical shifts and J13 couplings are neglected. The chemical shift, however, is refocused by placement of πy pulses. Following the same logic, this state would be transformed into a 2I2yI3y state using the J12 coupling instead.
Here, a slight shift of the π pulse (between point ‘3’ and point ‘4’ in the sequence) plays an important role. Note that the time τ2 and τ3 are different. This small difference allows evolution under the chemical shift of the third spin. This leads to a 2I2yI3x state at point ‘4’ which allows for the subsequent (π/2)x pulse to generate 2I2zI3x spin state.
The sequence efficiency here relies on the obtained spectral resolution and all peaks need to be separated. One reason is that a selective pulse on proton 3 needs to be applied during the transfer. The other is that one part of the sequence relies on the chemical shift difference Δν23 according to (1/(4Δν23)). Overlapping peaks would hence diminish the polarization transfer result.
Having acquired the 2I2zI3x state, it gets refocused to the I3y state during the third period (from point ‘5’ to ‘6’) that employs J12 coupling the second time. This means that neglecting the remote J13 coupling and after three consecutive INEPT blocks (up to point ‘6’) one attains the state which corresponds to observable transverse magnetization.
To test the polarization transfer efficiency up to this point ‘6’, the partial sequence in Fig. 3 was performed after which the 1H NMR signal was collected and Fourier transformed to obtain the NMR spectrum. The spectrum shows that only the relay proton site is significantly polarized (Fig. 4(c)). The calculated enhancement factor of 6405 corresponds to a polarization level of 13.9 ± 1.3%. This corresponds to ∼90% of the initial spin order deposited by the hydrogenation reaction. The small loss in polarization is attributed to relaxation losses and to the non-zero J13 coupling value which is equal to 1.1 Hz.
The result of the complete sequence is shown in Fig. 5 and shows the enhanced 13C peak of the 13C pyruvate moiety. The polarization level was estimated to be 7.1 ± 0.4% by comparing the hyperpolarized spectrum with the 13C spectrum recorded before the hydrogenation.
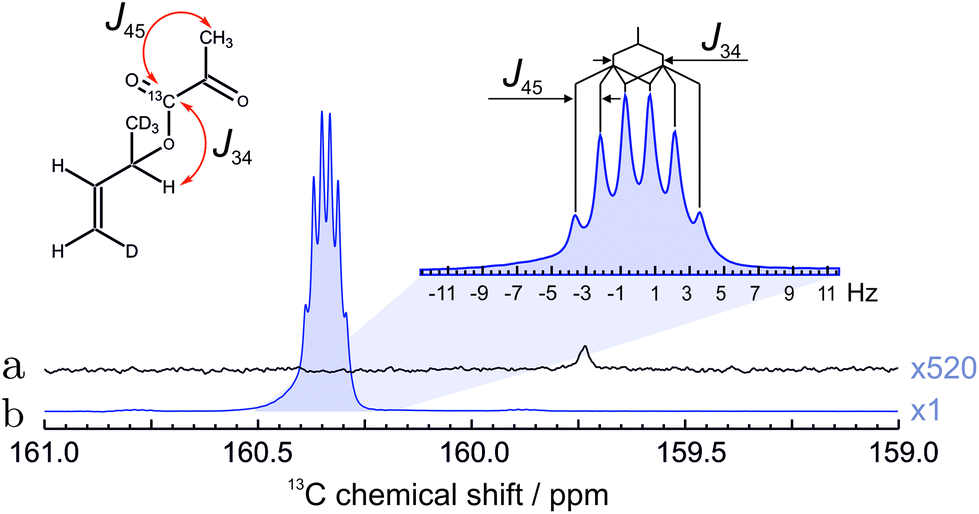 | ||
| Fig. 5 (a) The spectrum of thermally polarized pyruvate precursor before hydrogenation of the unsaturated side-arm. The spectrum was recorded under 1H decoupling and is multiplied 520 times for clarity. (b) 13C NMR spectrum of the hyperpolarized hydrogenation product collected using the optimized sequence, acquired without 1H decoupling during data acquisition. The expansion shows the multiplet structure due to the indicated 13C–1H J-couplings. The polarization level in (b) is estimated to be 7.1%, as determined from the ratio of the spectral intensities in (a) and (b). | ||
3.3. Cleavage
Before hydrolysis, 400 μL of sodium hydroxide solution was loaded into a syringe which was connected to the NMR tube by a Teflon capillary. After the 13C of the pyruvate derivative was polarized using the PR-SAH routine, the pressure inside the test tube was released and the cleaving solution was injected. The sample was mixed vigorously by nitrogen bubbling. After phases started to separate, the NMR tube was shifted such that only the aqueous phase was positioned within the excitation/detection coil. This was followed by a hard read-out pulse on the 13C channel.4. Results and discussion
The 13C spectrum of hyperpolarized pyruvate obtained by the total routine (in Fig. 2) is shown in Fig. 6. The increased linewidth of the hyperpolarized spectrum is attributed to magnetic susceptibility changes during injection of the cleavage solution.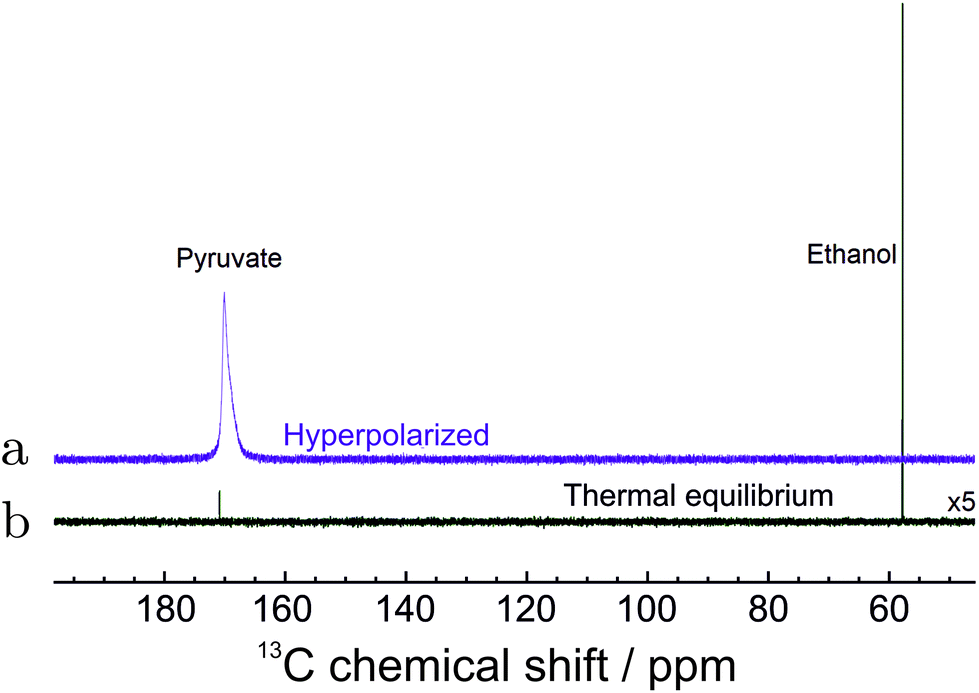 | ||
| Fig. 6 13C NMR spectra of hyperpolarized pyruvate in water. (a) The spectrum of hyperpolarized pyruvate dissolved in aqueous solution immediately after all steps in Fig. 2 were concluded. (b) The spectrum recorded with 320 transients after polarization has relaxed to thermal equilibrium at 55 °C. The spectrum is enlarged 5 times for clarity. The 13C polarization level in 1 mM solution is estimated to be 4.33% (see text). | ||
The final polarization level was assessed by the following procedure. After the PR-SAH routine was concluded, the sample was allowed to settle at 55 °C temperature. A thermally polarized spectrum was then recorded and compared to the enhanced one. The 13C reference spectrum of the cleaved pyruvate solution was accumulated with 320 repetitions and 2 min relaxation delay using 1H decoupling. Note: the reference spectra of the precursor molecule in Fig. 5 was, however, acquired with 52 transients and 20.3 times increased receiver sensitivity for quicker acquisition. The enhancement factor in Fig. 6 was estimated to be ∼8000 which in the corresponding magnetic field of 7.05 T at a temperature of 55 °C translates to 4.33 ± 0.18% polarization.
The concentration of the hydrolysed pyruvate in water was determined to be ∼1 mM which is consistent with the dilution caused by addition of water. This value was estimated by comparing the spectral intensity of the thermally polarized sample (Fig. 6(a)) with a reference sample of a well defined concentration using the same experimental conditions.
We would like to underline a few areas for improvement. First, the initial spin order created by the hydrogenation reaction is only a fraction of the full spin order stored in the para-enriched gases. The fine adjustments of optimal reaction conditions for rapid and selective hydrogenation could lead to a significantly higher 13C polarization. Separate effort would be also required to yield higher concentration of the (1-13C)pyruvate for future in vivo or in vitro studies. Second, the hydrolysis step could be automated to reduce the time for extraction and for the overall procedure to respect the relaxation of the hyperpolarized pyruvate. Third, new spin systems with a different set of scalar couplings could be designed to achieve even more effective proton-relay not only limited to pyruvate.
5. Conclusions
We have introduced a side-arm hydrogenation scheme that uses a relaying proton to hyperpolarize a metabolite with a pulsed NMR method. The routine successfully transferred spin-order from the side-arm to (1-13C)pyruvate with 46% efficiency which led to 7.6% of 13C polarization. The demonstrated hydrolysis and extraction of the hyperpolarized free pyruvate concluded in 4.3% of 13C polarization. This proves that the proton-relayed SAH method is effective enough to be applied for fast hyperpolarized metabolite delivery.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Marie Skłodowska-Curie program of the European Union (grant number 766402). We acknowledge funding by the Max Planck Society and the German Research Foundation (DFG) grant number DFG418416679. Authors also would like to thank Francesca Reineri and Andreas Trabesinger for their valuable input during the writing process. Open Access funding provided by the Max Planck Society.References
- T. Maly, G. T. Debelouchina, V. S. Bajaj, K.-N. Hu, C.-G. Joo, M. L. Mak-Jurkauskas, J. R. Sirigiri, P. C. A. van der Wel, J. Herzfeld, R. J. Temkin and R. G. Griffin, J. Chem. Phys., 2008, 128, 052211 CrossRef.
- F. Hill-Casey, A. Sakho, A. Mohammed, M. Rossetto, F. Ahwal, S. B. Duckett, R. O. John, P. M. Richardson, R. Virgo and M. E. Halse, Molecules, 2019, 24, 4126 CrossRef CAS.
- C. R. Bowers and D. P. Weitekamp, Phys. Rev. Lett., 1986, 57, 2645–2648 CrossRef CAS.
- F. D. Colegrove and P. A. Franken, Phys. Rev., 1960, 119, 680–690 CrossRef CAS.
- M. Ebert, T. Grossmann, W. Heil, E. Otten, R. Surkau, M. Thelen, M. Leduc, P. Bachert, M. Knopp and L. Schad, Lancet, 1996, 347, 1297–1299 CrossRef CAS.
- R. W. Adams, J. A. Aguilar, K. D. Atkinson, M. J. Cowley, P. I. Elliott, S. B. Duckett, G. G. Green, I. G. Khazal, J. López-Serrano and D. C. Williamson, Science, 2009, 323, 1708–1711 CrossRef CAS.
- J. Natterer and J. Bargon, Prog. Nucl. Magn. Reson. Spectrosc., 1997, 31, 293–315 CrossRef.
- S. Nelson, D. Vigneron, J. Kurhanewicz, A. Chen, R. Bok and R. Hurd, Appl. Magn. Reson., 2008, 34, 533–544 CrossRef CAS.
- K. Münnemann and H. W. Spiess, Nat. Phys., 2011, 7, 522 Search PubMed.
- R. Weissleder, Science, 2006, 312, 1168–1171 CrossRef CAS.
- R. Weissleder and U. Mahmood, Radiology, 2001, 219, 316–333 CrossRef CAS.
- J. W. Gordon, H.-Y. Chen, A. Autry, I. Park, M. Van Criekinge, D. Mammoli, E. Milshteyn, R. Bok, D. Xu, Y. Li, R. Aggarwal, S. Chang, J. B. Slater, M. Ferrone, S. Nelson, J. Kurhanewicz, P. E. Z. Larson and D. B. Vigneron, Magn. Reson. Med., 2019, 81, 2702–2709 CrossRef CAS.
- T. Harris, H. Degani and L. Frydman, NMR Biomed., 2013, 26, 1831–1843 CrossRef CAS.
- S. J. Nelson, J. Kurhanewicz, D. B. Vigneron, P. E. Z. Larson, A. L. Harzstark, M. Ferrone, M. van Criekinge, J. W. Chang, R. Bok, I. Park, G. Reed, L. Carvajal, E. J. Small, P. Munster, V. K. Weinberg, J. H. Ardenkjaer-Larsen, A. P. Chen, R. E. Hurd, L.-I. Odegardstuen, F. J. Robb, J. Tropp and J. A. Murray, Sci. Transl. Med., 2013, 5, 1946–6234 Search PubMed.
- K. M. Brindle, J. Am. Chem. Soc., 2015, 137, 6418–6427 CrossRef CAS.
- E. Cavallari, C. Carrera, S. Aime and F. Reineri, Chem. – Eur. J., 2017, 23, 1200–1204 CrossRef CAS.
- W. Iali, S. S. Roy, B. J. Tickner, F. Ahwal, A. J. Kennerley and S. B. Duckett, Angew. Chem., Int. Ed., 2019, 58, 10271–10275 CrossRef CAS.
- M. E. Gemeinhardt, M. N. Limbach, T. R. Gebhardt, C. W. Eriksson, S. L. Eriksson, J. R. Lindale, E. A. Goodson, W. S. Warren, E. Y. Chekmenev and B. M. Goodson, Angew. Chem., 2020, 59, 418–423 CrossRef CAS.
- S. S. Roy, K. M. Appleby, E. J. Fear and S. B. Duckett, J. Phys. Chem. Lett., 2018, 9, 1112–1117 CrossRef CAS.
- P. J. Rayner, B. J. Tickner, W. Iali, M. Fekete, A. D. Robinson and S. B. Duckett, Chem. Sci., 2019, 10, 7709–7717 RSC.
- J. Eills, E. Cavallari, C. Carrera, D. Budker, S. Aime and F. Reineri, J. Am. Chem. Soc., 2019, 141, 20209–20214 CrossRef CAS.
- B. Ripka, J. Eills, H. Kourilova, M. Leutzsch, M. H. Levitt and K. Muennemann, Chem. Commun., 2018, 54, 12246–12249 RSC.
- F. A. Gallagher, M. I. Kettunen, S. E. Day, D.-E. Hu, J. H. Ardenkjaer-Larsen, R. in't Zandt, P. R. Jensen, M. Karlsson, K. Golman, M. H. Lerche and K. M. Brindle, Nature, 2008, 453, 940–973 CrossRef CAS.
- M. Goldman, H. Johannesson, O. Axelsson and M. Karlsson, C. R. Chim., 2006, 9, 357–363 CrossRef CAS.
- K. Golman, R. in't Zandt and M. Thaning, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 11270–11275 CrossRef CAS.
- M. Itoda and Y. Naganawa, RSC Adv., 2019, 9, 18183–18190 RSC.
- K. V. Kovtunov, D. A. Barskiy, R. V. Shchepin, A. M. Co, K. W. Waddell, I. V. Koptyug and E. Y. Chekmenev, Anal. Chem., 2014, 86, 6192–6196 CrossRef CAS.
- A. Harthun, K. Woelk, J. Bargon and A. Weigt, Tetrahedron, 1995, 51, 11199–11206 CrossRef CAS.
- J. Bargon, J. Kandels, P. Kating, A. Thomas and K. Woelk, Tetrahedron Lett., 1990, 31, 5721–5724 CrossRef CAS.
- S. Aime, D. Canet, W. Dastru, R. Gobetto, F. Reineri, A. Viale, V. P. Giuria, L. De, H. Poincare, I. Nancy, B. P. Vandoeu and N. Cedex, J. Phys. Chem. A, 2001, 105, 6305–6310 CrossRef CAS.
- A. S. Kiryutin, K. L. Ivanov, A. V. Yurkovskaya, R. Kaptein and H. M. Vieth, Z. Phys. Chem., 2012, 226, 1343–1362 CrossRef CAS.
- R. W. Adams, J. A. Aguilar, K. D. Atkinson, M. J. Cowley, P. I. P. Elliott, S. B. Duckett, G. G. R. Green, I. G. Khazal, J. López-Serrano and D. C. Williamson, Science, 2009, 323, 1708–1711 CrossRef CAS.
- E. Cavallari, C. Carrera, T. Boi, S. Aime and F. Reineri, J. Phys. Chem. B, 2015, 119, 10035–10041 CrossRef CAS.
- E. Cavallari, C. Carrera, S. Aime and F. Reineri, J. Magn. Reson, 2018, 289, 12–17 CrossRef CAS.
- F. Reineri, B. Tommasco and S. Aime, Nat. Commun., 2015, 6, 2041–1723 Search PubMed.
- R. V. Shchepin, D. A. Barskiy, A. M. Coffey, I. V. Manzanera-Esteve and E. Y. Chekmenev, Angew. Chem., Int. Ed., 2016, 55, 6071–6074 CrossRef CAS.
- E. Cavallari, C. Carrera, M. Sorge, G. Bonne, A. Muchir, S. Aime and F. Reineri, Sci. Rep., 2018, 8, 2045–2322 CrossRef.
- R. V. Shchepin, D. A. Barskiy, A. M. Coffey, I. V. Manzanera Esteve and E. Y. Chekmenev, Angew. Chem., Int. Ed., 2016, 55, 6071–6074 CrossRef CAS.
- N. V. Chukanov, O. G. Salnikov, R. V. Shchepin, K. V. Kovtunov, I. V. Koptyug and E. Y. Chekmenev, ACS Omega, 2018, 3, 6673–6682 CrossRef CAS.
- S. Korchak, S. Yang, S. Mamone and S. Glöggler, ChemistryOpen, 2018, 7, 344–348 CrossRef CAS.
- S. Korchak, S. Mamone and S. Gloeggler, ChemistryOpen, 2018, 7, 672–676 CrossRef CAS.
- A. B. Schmidt, S. Berner, M. Braig, M. Zimmermann, J. Hennig, D. von Elverfeldt and J.-B. Hövener, PLoS One, 2018, 13, 1–15 Search PubMed.
- A. B. Schmidt, S. Berner, C. Müller, T. Lickert, N. Schwaderlapp, S. Knecht, J. G. Skinner, A. Dost, P. Rovedo, J. Hennig, D. von Elverfeldt and J. B. Hövener, Nat. Commun., 2017, 8, 14535 CrossRef CAS.
- S. Korchak, M. Emondts, S. Mamone, B. Blumich and S. Gloeggler, Phys. Chem. Chem. Phys., 2019, 21, 22849–22856 RSC.
- O. W. Sørensen, G. W. Eich, M. H. Levitt, G. Bodenhausen and R. R. Ernst, Prog. Nucl. Magn. Reson. Spectrosc., 1984, 16, 163–192 CrossRef.
- H. Sengstschmid, R. Freeman, J. Barkemeyer and J. Bargon, J. Magn. Reson., Ser. A, 1996, 120, 249–257 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/D0AN02389B |
| This journal is © The Royal Society of Chemistry 2021 |