
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Lithium-7 qNMR as a method to quantify lithium content in brines using benchtop NMR†
Juan F.
Araneda
 *a,
Paul
Hui
a,
Garett M.
Leskowitz
a,
Susanne D.
Riegel
a,
Rodrigo
Mercado
b and
Christopher
Green
b
*a,
Paul
Hui
a,
Garett M.
Leskowitz
a,
Susanne D.
Riegel
a,
Rodrigo
Mercado
b and
Christopher
Green
b
aNanalysis Corp., 1-4600 5 St NE, Calgary, AB T2E 7C3, Canada. E-mail: juan.araneda@nanalysis.com
bSociedad Química y Minera de Chile S.A (SQM), Laboratorio de Desarrollos Analíticos Av. Balmaceda 3228, Antofagasta, Chile
First published on 19th November 2020
Abstract
A novel 7Li quantitative NMR (qNMR) method to analyze lithium was developed to determine the lithium content in real brine samples using benchtop NMR instruments. The method was validated, and limits of detection and quantification of 40 and 100 ppm, respectively, were determined. Linearity, precision, and bias were also experimentally determined, and the results are presented herein. The results were compared to those obtained using atomic absorption (AA) spectroscopy, currently one of the few validated methods for the quantification of lithium. The method provides both accurate and precise results, as well as excellent correlation with AA. The absence of matrix effects, combined with no need for sample preparation or deuterated solvents, shows potential applicability in the mining industry.
Introduction
Lithium is used in many industrial manufacturing processes such as glass, ceramics, lubricating grease, and pharmaceuticals.1–4 However, its importance in the last two decades has increased due in part to the superior performance of lithium-ion batteries,5–7 which are widely used in a variety of electronic devices, such as notebooks, cellphones, cameras, etc.8 The rising demand for electric vehicles is projected to significantly increase the need for lithium in the coming years.9,10 This demand is expected to grow up to 900 ktons per year by 2025, which is approximately three times higher than in 2018.7Lithium is mainly sourced from either spodumene or brine, with the latter consisting of 59% of the market share.11,12 Lithium-containing brines are concentrated by solar evaporation in multiple stages and are monitored quantitatively at each step until the lithium content reaches approximately 6%.13–15 The most common techniques for this quantification are atomic absorption (AA) and inductively coupled plasma (ICP) spectroscopy. However, both methods require significant sample manipulation and are prone to interference caused by other ions present in solution, representing a significant matrix effect.16–19
Nuclear magnetic resonance (NMR) spectroscopy has become one of the most valuable tools a chemist has, partly due to its extraordinary ability for structural elucidation.20–24 By understanding the fundamental concepts of NMR spectroscopy (i.e., coupling constant, chemical shift, and integration), scientists can take advantage of this technique to gain insight into the different atoms present in a molecule, such as how they are connected and their relative spatial proximities.25,26 More recently, NMR spectroscopy has also been applied to quantitative applications (qNMR), particularly in the areas of natural products and medicinal chemistry, because it is non-destructive and inherently quantitative in nature.27–34 However, qNMR spectroscopy is not as widely used in industry as other techniques such as UV-Vis, GC-MS, ICP, AA and IR spectroscopy due to the cost and size associated with traditional high-field instruments.
Oftentimes, qNMR applications make use of nuclei with spin I = ½ (e.g., 1H, 19F, 31P) rather than quadrupolar nuclei, which have spin I > ½ (e.g., 7Li, 11B, 23Na, 27Al). The quadrupole moment associated with these nuclei represents an asymmetry in charge distribution, and its interaction with local fluctuating electric field gradients, provided by electrons and other moving charges, represents a relaxation mechanism. This interaction helps to explain the fact that quadrupolar nuclei usually give rise to broader signals than spin-1/2 nuclei. These broad signals can be problematic to qNMR applications, since the signal will be spread out over a larger frequency range, effectively reducing the signal intensity and consequently the signal-to-noise ratio (SNR).35 There are, however, a few reported applications that use quadrupolar nuclei in qNMR.36–38 For example, in 2018 Fernandez and coworkers reported the quantitative analysis of boric acid in biocides using 11B qNMR.39 However, to the best of our knowledge there are no reports of lithium NMR in quantitative applications.
In the last few years, the emergence of benchtop NMR spectrometers that are affordable, portable, and require minimal maintenance has accelerated the introduction of this technique in industrial settings.40–44 This is reflected by the increasing number of publications incorporating benchtop NMR instruments in a variety of industrial applications such as quality control, quality assurance, and process analytical technology (PAT).45–48 Due to the important role of lithium in the future global energy demand, we decided to explore the potential of benchtop NMR instruments in the quantification of this metal in brines. In this article, we present our findings in the direct quantification of lithium without any additional sample preparation, dilution, or need for deuterated solvents.
Experimental section
Brine samples
For this study, 16 samples taken from underground brine and evaporation ponds at SQM site at Salar de Atacama, near Antofagasta in northern Chile, were used. Samples were divided in two sets. One set from each brine was sent to Nanalysis Corp. in Calgary, Canada to be analyzed by NMR spectroscopy, and the other set remained at the SQM facilities in Chile to be analyzed by AA.Following collection, some samples with high lithium content presented crystallization of some of the salts after a few days, which happened during shipment from Chile to Canada. These samples were transferred to a flask, weighed, and heated to 60 °C for 1 hour. Distilled water was added until all the solids were dissolved. Samples were allowed to reach room temperature, and the weight was adjusted by evaporation of excess water or addition of distilled water until the initial weight was reached. Note that heating of the samples is not required for on-site analysis where shipment or storage of the samples is unnecessary, as the samples can be transferred to an NMR tube and analyzed directly, provided that this is done in the few days prior to the appearance of precipitate.
Instrumentation
All 7Li NMR spectra were obtained at 32 °C using a Nanalysis 60PRO benchtop NMR spectrometer at a 23.46 MHz lithium frequency (1.418 tesla). The qNMR experiments were performed with the following optimized parameters: spectral width, 50 ppm; spectral center, 0 ppm; number of complex-valued points, 4096; scans, 4; interscan delay, 115 seconds; pulse angle, 90° (as determined by nutation);49 acquisition time, 3.44 seconds. All NMR measurements were carried out with a fixed receiver gain of 35 dB to ensure consistency of quantitative results and to avoid receiver saturation. All experiments were conducted without any form of decoupling. NMR spectra were acquired without deuterated solvents, and samples were used as received without any additional preparation. Lithium chemical shift is provided relative to aqueous LiCl 3 M (0 ppm). A line-broadening factor of 1.0 Hz was applied to the free induction decays (FID) prior to Fourier transformation. 7Li NMR spectra were manually corrected for phase and baseline distortions using MestReNova software (v14.1.0). The region of integration was constant between samples (+3 to −3 ppm). Microsoft Excel was used for data analysis. To improve and monitor precision, each measurement was performed in triplicate.AA analysis was performed using a Thermo Elemental S4 system from Thermo Scientific running SOLAAR Data Station 11.0 software. The atomic line for Li at 670.8 nm was monitored with a 0.5 mm slit. The air/acetylene flow rate was maintained at 1.0 mL per minute. The instrument is equipped with a 50 mm burner at ∼45°. A sample aspiration rate of 7 mL per minute was used. The AA methodology used is in agreement with the Chilean Standard Method NCh3349/2020 for the analysis of brines and ASTM D3561-16. The method used five standards for the calibration curve with relative standard deviations below 2% and residuals below 2%. Microsoft Excel was used for data analysis.
Results and discussion
Optimization of qNMR method
Lithium has two NMR active nuclei, 6Li and 7Li, with natural abundances of 7.6 and 92.4% and gyromagnetic ratios of 3.9371 × 107 and 10.398 × 107 rad s−1 T−1, respectively. The spin quantum numbers are 1 for 6Li and 3/2 for 7Li. Due to its greater sensitivity, lithium-7 is the most used isotope in lithium NMR spectroscopy.50–52NMR is inherently quantitative and in most qNMR applications a standard (or calibrant) of known concentration is added to compare its relative integration with that of the compound of interest.31 In aqueous solution, the lithium ions are solvated with labile H2O ligands, which form a hydration shell.53,54 The 7Li NMR signal is therefore in a dynamic exchange regime that is fast on the NMR timescale, yielding a single resonance. The lithium content is accordingly determined as a total lithium composition, independent of the nature of the species (e.g., Li2CO3, LiCl, Li2SO4, etc.). This makes the use of an internal calibrant impractical and as a result, a calibration curve correlating the known concentration of the analyte and its signal response was used. The calibration curve was recorded by preparing several solutions of LiCl in H2O at concentrations of 0.25, 0.50, 1, 3, 7.5, 15 and 30% (w/w%).
The first step in developing a 7Li-NMR method to quantify lithium content involved the determination of the optimal acquisition parameters. Most of these can be extracted from literature (e.g., number of points, digital resolution, and pulse angle). However, the most crucial parameter for accurate quantification is the recycle time (i.e., interscan delay + acquisition time) between scans. It is generally agreed upon that for qNMR, an interscan delay of 5 to 7 times the longitudinal relaxation time T1 should be used to ensure that more than 99% of the equilibrium magnetization has been recovered before the next scan is acquired.26 Table S1† shows the T1 values of several LiCl solutions at different concentrations along with the T1 of four randomly selected brine samples. The T1 values of the solutions prepared in the lab range from 3.7 to 23.2 seconds and the values for the brine samples are on the order of 13 seconds.
Method validation
To ensure the proposed method was effective for the intended application, it was validated using samples prepared in the lab, reference standard materials, and lithium samples from brine pools. Linearity, intra-day and inter-day precision, limit of detection (LOD), limit of quantification (LOQ), and bias were evaluated.55–58The linear response of the instrument was determined by preparing LiCl solutions in H2O at concentrations of 0.25, 0.50, 1, 3, 7.5, 15 and 30% (w/w) and plotting the absolute integration area versus concentration. Fig. 1 shows that the instrument's response is linear over this range of percent composition, with a coefficient of determination (R2) of 0.9993.
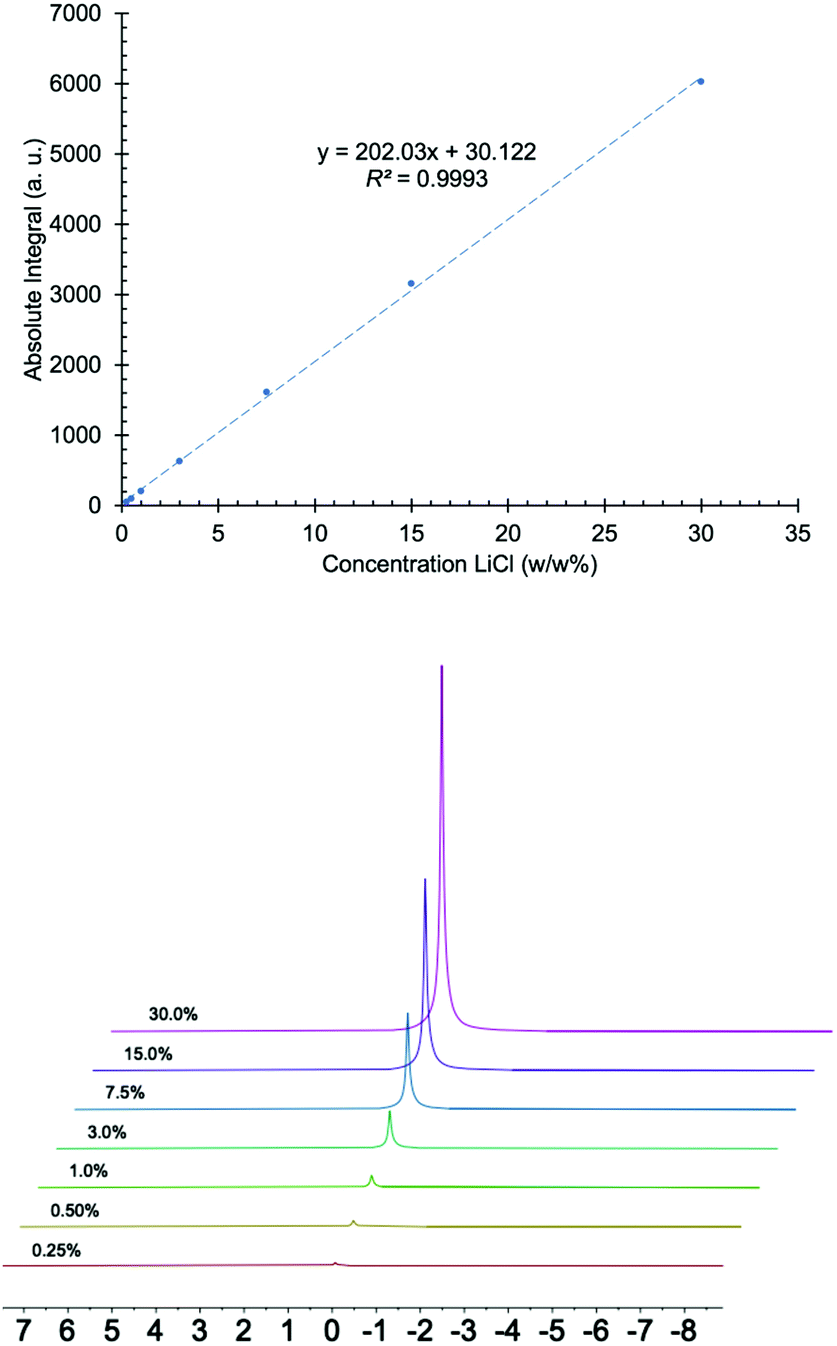 | ||
| Fig. 1 Top: graph of absolute integral versus concentration of LiCl made with 0.25, 0.50, 1, 3, 7.5, 15 and 30 (w/w%) solutions. Bottom: 7Li NMR spectra of LiCl solutions used to produce the graph. | ||
Brine samples contain several ions and elements in addition to lithium. Some of the most common species are sodium, potassium, boron, magnesium, calcium, chloride, and sulfate (Table 1). In some cases, the amount of such species in solution can be significantly higher than lithium itself. While such species in principle exhibit no absorption peaks in the 7Li NMR spectrum, in practice, small auxiliary effects attributable to these species may introduce complications in the instrument's response. Most notably, it's known that ionic strength (i.e., salt concentration of the samples) can affect T1 relaxation values, and variable salt concentrations can affect the tuning and matching of probes in high-field instruments.59,60 For these reasons, the matrix effect was investigated by comparing the slope of a calibration curve prepared using pure water with the slopes of two brine samples prepared using the standard addition method. The slope ratio (slope of standard addition/slope of pure sample) of 0.964 and 0.956 for the two brine samples suggests that, over the broad range of concentrations used, matrix effects should induce less than a few percent error in linear estimates of concentration (Fig. 2).
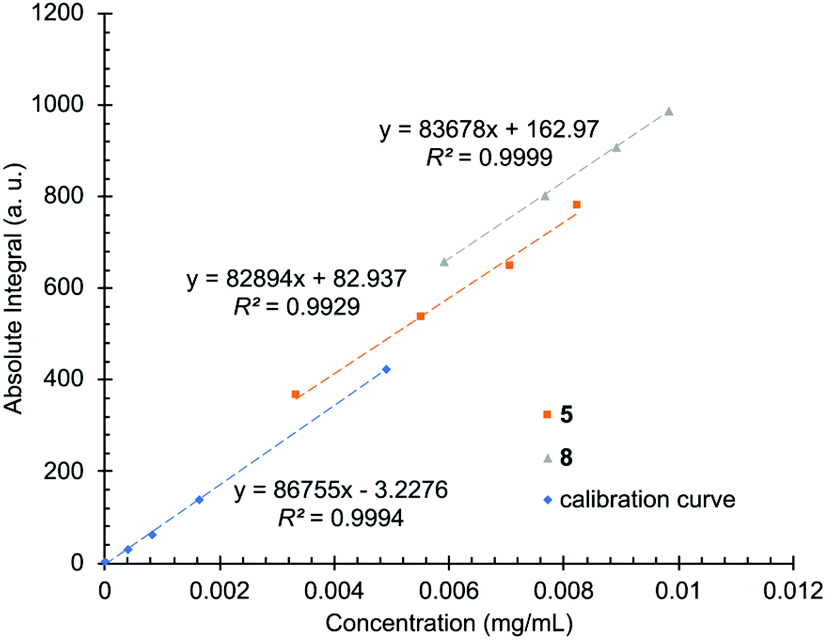 | ||
| Fig. 2 Calibration curves recorded with solutions of lithium chloride and distilled water (blue) and by spiking two brine samples (5 in orange and 8 in grey) with known amounts of lithium. | ||
| Sample | Lia (w/w%) | Kb (w/w%) | Nab (w/w%) | SO4b w/w%) | Clc (w/w%) | Bb (w/w%) | TDSd (g L−1) |
|---|---|---|---|---|---|---|---|
| a Determined by AA. b Determined by ICP-OES. c Determined by titration (Mohr's method). d Total dissolved solids (TDS) is a measure of the total ionic concentration of dissolved minerals in water (salinity). | |||||||
| 1 | 0.163 | 2.25 | 6.76 | 0.23 | 16.14 | 0.04 | 268 |
| 2 | 0.197 | 2.54 | 6.39 | 1.35 | 15.74 | 0.06 | 276 |
| 3 | 0.226 | 2.36 | 5.53 | 0.12 | 16.68 | 0.05 | 269 |
| 4 | 0.289 | 3.34 | 4.63 | 0.36 | 16.89 | 0.07 | 276 |
| 5 | 0.339 | 3.13 | 4.1 | 0.66 | 17 | 0.09 | 277 |
| 6 | 0.439 | 3.16 | 4.47 | 5.83 | 17.39 | 0.11 | 347 |
| 7 | 0.471 | 2.02 | 4.52 | 1.75 | 15.86 | 0.08 | 269 |
| 8 | 0.598 | 2.22 | 2.22 | 2.5 | 17.1 | 0.18 | 284 |
| 9 | 0.654 | 1.79 | 1.82 | 0.19 | 18.45 | 0.06 | 268 |
| 10 | 0.755 | 1.9 | 1.31 | 4.89 | 17.75 | 0.26 | 317 |
| 11 | 0.802 | 1.55 | 0.75 | 1.5 | 20.98 | 0.22 | 311 |
| 12 | 1.060 | 0.45 | 0.44 | 1.04 | 21.66 | 0.27 | 306 |
| 13 | 1.969 | 0.08 | 0.2 | 0.22 | 26.36 | 0.27 | 345 |
| 14 | 5.032 | 0.05 | 0.06 | 0.04 | 32.12 | 0.75 | 408 |
| 15 | 6.243 | 0.17 | 0.07 | 0.04 | 34.91 | 0.75 | 434 |
| 16 | 6.144 | 0.06 | 0.07 | 0.04 | 35.09 | 0.91 | 436 |
Nutation experiments are commonly used in NMR spectroscopy to calibrate the 90° pulse angle.49 If samples of high ionic strength affected the instrument's response, it would be evidenced by different results in the nutation experiments. A potential effect might be a decrease in the Q factor of the radio-frequency resonant detection circuit due to dielectric dissipation in the aqueous sample when the ionic strength of the solution is high.61 This could be expected to reduce the effective signal strength at high lithium concentrations or high concentrations of other ions. A significantly reduced Q factor will also cause the nutation rate to decrease, and so saltier samples should exhibit longer 90° nutation times. Table 2 shows very similar nutation values for samples containing different lithium chloride content (1 to 30 w/w%) and more ions in solution (samples 11 and 14). There is a monotonic trend in nutation times, but the nutation rate varies by only about 1.1% over the whole range of concentrations that was studied. While this is small compared to the level of precision observed in the concentration estimates herein (see below), an analysis with higher-precision measurements and sample-preparation methods should take this into account. The small variation in nutation rates (and the attendant signal amplification factors) should have a minimal effect on the accuracy and precision of the results presented herein.
| Sample | 90° Nutation (μs) |
|---|---|
| 11 | 24.17 |
| 14 | 24.07 |
| 30% | 24.25 |
| 15% | 24.22 |
| 3% | 24.03 |
| 1% | 23.98 |
The precision (repeatability) of the instrument was evaluated by performing intra- and inter-day experiments. Intra-day precision was evaluated by running a sample of LiCl (15 w/w%) in triplicate, and inter-day precision was studied by analyzing the same sample in triplicate for six consecutive days. In both cases, the absolute integration area was monitored (see ESI, Table S2†) and the relative standard deviations ranged from 0.03 to 0.30%. Precision and bias were also evaluated following the American Public Health Association (APHA) 1040 B guide for method development and evaluation. A certified standard solution of lithium carbonate for ICP containing 1002 mg L−1 ± 8 mg L−1 lithium was analyzed in 10 replicates. The standard deviation was determined to be 11 mg L−1 and the bias was −1 mg L−1.
Limit of detection (LOD) and limit of quantification (LOQ) are important validation parameters relevant for identifying and quantifying an analyte. For the lithium NMR studies the detection limit was established as the lowest concentration of Li+ that gave rise to a signal intensity for which the SNR equals 3 and the LOQ as the lowest concentration that gives a signal with an SNR of 10. In this method, the LOD was determined to be 40 ppm Li+, and the LOQ was determined to be 100 ppm Li+. It should be noted that LOQ is the concentration of analyte for which the signal of interest generates the minimal required SNR for the given method precision and different applications may require a higher SNR for LOQ.
Sample analysis
The linearity study showed in Fig. 1 displays an excellent linear correlation with an R2 value of 0.9993. However, to use a specific calibration curve, the validity of the linear regression model should also be evaluated.58,62 One of the simplest ways to evaluate a regression analysis is to examine the residual errors. If the regression model is valid, then the residual errors should be randomly distributed about an average residual error of zero. The calibration curve ranging from 0.25 to 30 (w/w%) LiCl showed excellent linear regression, but the residual error was larger for samples at low concentrations. For that reason, the calibration curve was split into two smaller ones, one ranging from 0.25 to 3 and the other from 3 to 30 (w/w%) LiCl. Samples were initially analyzed with the full calibration curve to estimate the lithium content and subsequently with the corresponding linear regression model to accurately determine the lithium content.The method was used to analyze real samples taken from brines and evaporation ponds and the results are shown in Table 3. The samples were analyzed by NMR directly from the brine pools, no sample preparation or dilution was performed. Samples were analyzed in triplicate, and in all cases, the relative standard deviation (RSD) values were below 2%. To evaluate the reliability of the NMR results, the samples were also analyzed by AA. The results show excellent correlation, with most of the results showing differences below 5%. The fact that the inter-day precision studies show excellent stability suggests that multiple recalibrations are not necessary with the NMR method, unlike AA.
| Sample | Li content by NMR | RSDa | Li content by AA | Diff.b (%) |
|---|---|---|---|---|
| a Relative standard deviation of NMR measurements. b ((Li content by NMR – Li content by AA)/(Li content by AA)) × 100. | ||||
| 1 | 0.166 | 1.86 | 0.163 | 1.8 |
| 2 | 0.201 | 1.06 | 0.197 | 2.0 |
| 3 | 0.222 | 0.83 | 0.226 | −1.8 |
| 4 | 0.292 | 1.56 | 0.289 | 1.0 |
| 5 | 0.349 | 0.70 | 0.339 | 2.9 |
| 6 | 0.467 | 0.68 | 0.439 | 6.4 |
| 7 | 0.478 | 0.34 | 0.471 | 1.5 |
| 8 | 0.617 | 0.23 | 0.599 | 3.0 |
| 9 | 0.684 | 0.46 | 0.654 | 4.6 |
| 10 | 0.811 | 0.51 | 0.755 | 7.4 |
| 11 | 0.850 | 0.55 | 0.802 | 6.0 |
| 12 | 1.136 | 0.27 | 1.060 | 7.2 |
| 13 | 2.110 | 0.13 | 1.969 | 7.2 |
| 14 | 5.271 | 0.06 | 5.032 | 4.7 |
| 15 | 6.380 | 0.11 | 6.243 | 2.2 |
| 16 | 6.480 | 0.04 | 6.144 | 5.5 |
One common approach to compare two analytical methods is the use of a regression line. In this approach, the results of the two methods are plotted on the two axes of a regression graph, with each point on the graph representing a single sample measured by the two analytical techniques being compared. In an ideal situation, if both techniques yield identical results, the regression line will have a slope of 1 and 0 intercept. However, in practice this almost never occurs and some deviation from the ideal results is inevitable.58 The NMR results were plotted on the y-axis and the AA data on the x-axis of Fig. 3. The intercept obtained is 0.0065, with the upper and lower confidence limits of 0.0366 and −0.0236. The slope of the graph is 1.0405, with the 95% confidence interval of 1.029–1.052 and the coefficient of determination (R2) is equal to 0.9996 (see ESI† for a detailed explanation). The results show that the confidence interval of the intercept contains the 0 value and the intercept is only slightly off the unit slope, demonstrating the high correlation between both techniques. NMR slightly overestimated the concentration of lithium at high concentrations, as reflected in the slope of Fig. 3, but the results are within the accepted values for the intended application.
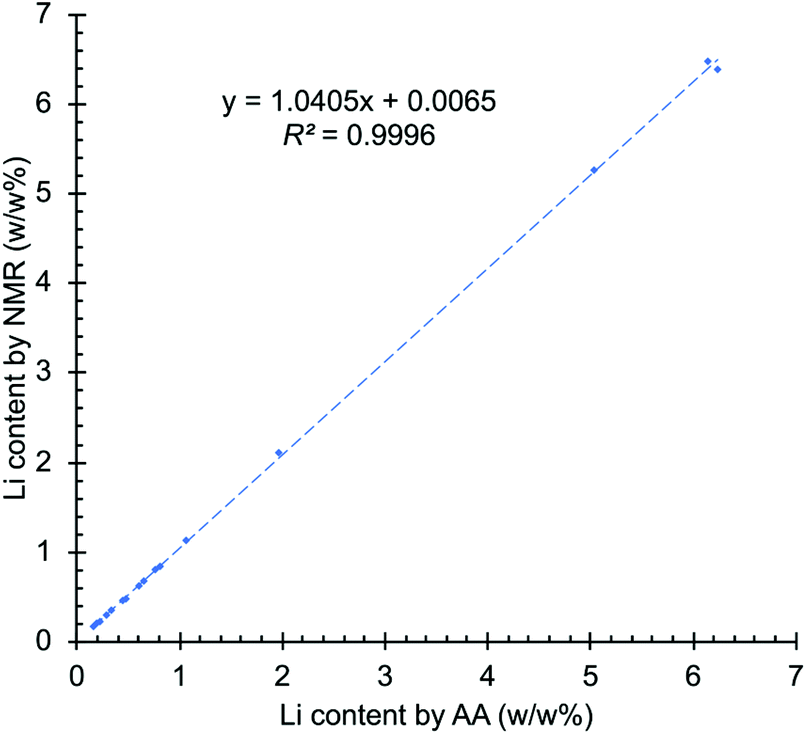 | ||
| Fig. 3 Graph of regression line analysis comparing results from NMR and AA. | ||
One of the major drawbacks of NMR spectroscopy as an analytical technique to quantify components is the lower sensitivity compared to other techniques such as ICP and AA.63,64 For example, in this particular application the LOD and LOQ experimentally determined by AA for lithium are 0.06 and 0.17 ppm, respectively. These values are significantly lower than the ones determined by low-field NMR (LOD was determined to be 40 ppm and the LOQ 100 ppm Li+). However, the method reported herein provides more than sufficient sensitivity to quantify the lithium content in real brine pool samples. The superior sensitivity of AA does not offer any particular benefit for this application.
Conclusions
A new method to determine lithium content in brine pools has been developed. Although traditional methods used in the lithium mining industry (e.g., ICP and AA) provide lower LOQ and LOD than NMR spectroscopy, the method reported herein provides more than sufficient sensitivity to analyze real brine pool samples. Unlike ICP and AA, this approach does not require sample preparation or dilution, and samples can be analyzed directly from the brine pools during different stages of the purification process. Moreover, the NMR concentration measurements were shown to be stable over many days, which promises increased reliability and reduced expense and inconvenience associated with day-to-day recalibration. It was also revealed that there is minimal matrix effect with the NMR spectroscopic method and excellent correlation with AA results, with most of the values showing differences below 5%. The reported NMR method does not require the use of deuterated solvents and the only consumables needed are the NMR tubes. This makes the NMR method very inexpensive compare to ICP and AA, which require regular maintenance and gases such as acetylene and argon. All these combined factors provide an attractive new method for the quantification of lithium in the mining industry. We are currently exploring additional applications of benchtop qNMR in various industrial settings.Conflicts of interest
J. F. A., P. H., G. M. L. and S. D. R. are employed by Nanalysis Corp.Acknowledgements
R. M. and C. G. would like to acknowledge SQM for financial support to explore new initiatives. J. F. A. would like to thank Felipe Verdugo for valuable discussions.Notes and references
- N. Linneen, R. Bhave and D. Woerner, Sep. Purif. Technol., 2019, 214, 168–173 CrossRef CAS.
- J. K. Rybakowski, ACS Chem. Neurosci., 2014, 5, 413–421 CrossRef CAS.
- A. Nassar and A. N. Azab, ACS Chem. Neurosci., 2014, 5, 451–458 CrossRef CAS.
- B. Swain, J. Chem. Technol. Biotechnol., 2016, 91, 2549–2562 CrossRef CAS.
- O. Gröger, H. A. Gasteiger and J.-P. Suchsland, J. Electrochem. Soc., 2015, 162, A2605–A2622 CrossRef.
- B. Dunn, H. Kamath and J. M. Tarascon, Science, 2011, 334, 928–935 CrossRef CAS.
- A. Battistel, M. S. Palagonia, D. Brogioli, F. La Mantia and R. Trócoli, Adv. Mater., 2020, 32, 1905440 CrossRef CAS.
- A. M. Haregewoin, A. S. Wotango and B. J. Hwang, Energy Environ. Sci., 2016, 9, 1955–1988 RSC.
- P. K. Choubey, M. S. Kim, R. R. Srivastava, J. C. Lee and J. Y. Lee, Miner. Eng., 2016, 89, 119–137 CrossRef CAS.
- J. Wang, M. Chen, H. Chen, T. Luo and Z. Xu, Procedia Environ. Sci., 2012, 16, 443–450 CrossRef CAS.
- J. Kogel, N. Trivedi, J. Barker and S. Krukowski, Industrial Minerals and Rocks: Commodities, Markets and Uses. Society for Mining, Metallurgy and Exploration, Society for Mining, Metallurgy, and Exploration, 7th edn, 2006 Search PubMed.
- P. Meshram, B. D. Pandey and T. R. Mankhand, Hydrometallurgy, 2014, 150, 192–208 CrossRef CAS.
- V. K. Silvana, F. R. Horacio and O. M. Agustina, World J. Res. Rev., 2016, 3, 66–70 Search PubMed.
- J. W. An, D. J. Kang, K. T. Tran, M. J. Kim, T. Lim and T. Tran, Hydrometallurgy, 2012, 117–118, 64–70 CrossRef CAS.
- B. Swain, Sep. Purif. Technol., 2017, 172, 388–403 CrossRef CAS.
- Y. Morishige and A. Kimura, SEI Tech. Rev., 2008, 106–111 Search PubMed.
- X. Wen, P. Ma, G. Zhu and Z. Wu, Rare Met., 2006, 25, 309–315 CrossRef CAS.
- A. Sapkota, M. Krachler, C. Scholz, A. K. Cheburkin and W. Shotyk, Anal. Chim. Acta, 2005, 540, 247–256 CrossRef CAS.
- B. Baraj, L. F. H. Niencheski, R. D. Trapaga, R. G. França, V. Cocoli and D. Robinson, Fresenius’ J. Anal. Chem., 1999, 364, 678–681 CrossRef CAS.
- D. Quaglio, S. Corradi, S. Erazo, V. Vergine, S. Berardozzi, F. Sciubba, F. Cappiello, M. E. Crestoni, F. Ascenzioni, F. Imperi, F. Delle Monache, M. Mori, M. R. Loffredo, F. Ghirga, B. Casciaro, B. Botta and M. L. Mangoni, ACS Med. Chem. Lett., 2020, 11, 760–765 CrossRef CAS.
- M. O. Marcarino, M. M. Zanardi, S. Cicetti and A. M. Sarotti, Acc. Chem. Res., 2020, 53, 1922–1932 CrossRef CAS.
- J. J. J. Van Der Hooft, R. C. H. De Vos, V. Mihaleva, R. J. Bino, L. Ridder, N. De Roo, D. M. Jacobs, J. P. M. Van Duynhoven and J. Vervoort, Anal. Chem., 2012, 84, 7263–7271 CrossRef CAS.
- A. V. Buevich and M. E. Elyashberg, J. Nat. Prod., 2016, 79, 3105–3116 CrossRef CAS.
- D. K. Menyhárd, G. Pálfy, Z. Orgován, I. Vida, G. M. Keserű and A. Perczel, Chem. Sci., 2020, 11, 9272–9289 RSC.
- V.-T. Vu, M.-T. Nguyen, W.-L. Wang, B.-N. Nguyen, G.-N. Pham, L.-Y. Kong and J.-G. Luo, Org. Biomol. Chem., 2020, 2, 6607–6611 RSC.
- N. E. Jacobsen, NMR Spectroscopy Explained: Simplified Theory, Applications and Examples for Organic Chemistry and Structural Biology, Wiley-Interscience, 2007 Search PubMed.
- G. F. Pauli, S. N. Chen, C. Simmler, D. C. Lankin, T. Gödecke, B. U. Jaki, J. B. Friesen, J. B. McAlpine and J. G. Napolitano, J. Med. Chem., 2014, 57, 9220–9231 CrossRef CAS.
- L. A. C. Pieters and A. J. Vlietinck, J. Pharm. Biomed. Anal., 1989, 7, 1405–1417 CrossRef CAS.
- U. Holzgrabe, R. Deubner, C. Schollmayer and B. Waibel, J. Pharm. Biomed. Anal., 2005, 38, 806–812 CrossRef CAS.
- T. Schoenberger, S. Menges, M. A. Bernstein, M. Pérez, F. Seoane, S. Sýkora and C. Cobas, Anal. Chem., 2016, 88, 3836–3843 CrossRef CAS.
- G. F. Pauli, B. U. Jaki and D. C. Lankin, J. Nat. Prod., 2007, 70, 589–595 CrossRef CAS.
- P. Giraudeau, Magn. Reson. Chem., 2017, 55, 61–69 CrossRef CAS.
- E. Hafer, U. Holzgrabe, K. Kraus, K. Adams, J. M. Hook and B. Diehl, Magn. Reson. Chem., 2020, 58, 653–665 CrossRef CAS.
- W. von Philipsborn, Pure Appl. Chem., 1986, 58, 513–528 CAS.
- J. T. Urban, Nuclear Magnetic Resonance Studies of Quadrupolar Nuclei and Dipolar Field Effects, University of California, Berkeley, 1997 Search PubMed.
- C. Gerardin, M. Haouas, C. Lorentz and F. Taulelle, Magn. Reson. Chem., 2000, 38, 429–435 CrossRef CAS.
- H. Maki, G. Sakata and M. Mizuhata, Analyst, 2017, 142, 1790–1799 RSC.
- L. M. Ravaglia, D. dos S. Freitas, T. G. Ricci, C. E. D. Nazario and G. B. Alcantara, Magn. Reson. Chem., 2020, 58, 186–190 CrossRef CAS.
- L. M. Aguilera-Sáez, J. R. Belmonte-Sánchez, R. Romero-González, J. L. Martínez Vidal, F. J. Arrebola, A. Garrido Frenich and I. Fernández, Analyst, 2018, 143, 4707–4714 RSC.
- L. Sassu, S. Puligheddu, C. Puligheddu and S. Palomba, Magn. Reson. Chem., 2020, 58, 1222–1233 CAS.
- J. C. Edwards, Magn. Reson. Chem., 2016, 54, 492–493 CrossRef CAS.
- P. Sagmeister, J. Poms, J. D. Williams and C. O. Kappe, React. Chem. Eng., 2020, 5, 677–684 RSC.
- W. G. Lee, M. T. Zell, T. Ouchi and M. J. Milton, Magn. Reson. Chem., 2020, 58, 1193–1202 CAS.
- S. Yamada, K. Ito, A. Kurotani, Y. Yamada, E. Chikayama and J. Kikuchi, ACS Omega, 2019, 4, 3361–3369 CrossRef CAS.
- K. Meyer, S. Kern, N. Zientek, G. Guthausen and M. Maiwald, Trends Anal. Chem., 2016, 83, 39–52 CrossRef CAS.
- F. Dalitz, M. Cudaj, M. Maiwald and G. Guthausen, Prog. Nucl. Magn. Reson. Spectrosc., 2012, 60, 52–70 CrossRef CAS.
- M. H. M. Killner, E. Danieli, F. Casanova, J. J. R. Rohwedder and B. Blümich, Fuel, 2017, 203, 171–178 CrossRef CAS.
- M. Grootveld, B. Percival, M. Gibson, Y. Osman, M. Edgar, M. Molinari, M. L. Mather, F. Casanova and P. B. Wilson, Anal. Chim. Acta, 2019, 1067, 11–30 CrossRef CAS.
- S. Berger and S. Braun, 200 and more NMR experiments: a practical course, Wiley-VCH, 2004 Search PubMed.
- H. J. Reich, D. P. Green and N. H. Phillips, J. Am. Chem. Soc., 1989, 111, 3444–3445 CrossRef CAS.
- H. J. Reich and K. J. Kulicke, J. Am. Chem. Soc., 1996, 118, 273–274 CrossRef CAS.
- L. M. Jackman, L. M. Scarmoutzos and C. W. Debrosse, J. Am. Chem. Soc., 1987, 109, 5355–5361 CrossRef CAS.
- J. C. Handman, J. Chem. Phys., 1962, 36, 1000–1015 CrossRef.
- G. V. Karpov, Chem. Phys. Lett., 2005, 402, 300–305 CrossRef CAS.
- G. Maniara, K. Rajamoorthi, S. Rajan and G. W. Stockton, Anal. Chem., 1998, 70, 4921–4928 CrossRef CAS.
- T. Schonberger, Y. B. Monakhova, D. W. Lachenmeier and T. Kuballa, EUROLABS Technical Report No. 01/2014 May 2014, 2014.
- J. C. Miller and J. N. Miller, Analyst, 1988, 113, 1351–1356 RSC.
- J. N. Miller, Analyst, 1991, 116, 3–14 RSC.
- S. K. Bharti and R. Roy, TrAC, Trends Anal. Chem., 2012, 35, 5–26 CrossRef CAS.
- O. Beckonert, H. C. Keun, T. M. D. Ebbels, J. Bundy, E. Holmes, J. C. Lindon and J. K. Nicholson, Nat. Protoc., 2007, 2, 2692–2703 CrossRef CAS.
- J. Mispelter, M. Lupu and A. Briguet, NMR Probeheads for Biophysical and Biomedical Experiments, Imperial College Press, 2nd edn, 2015 Search PubMed.
- D. Harvey , Analytical Chemisty 2.1, Open Education Resource (OER) LibreTexts Project.
- D. L. Olson, T. L. Peck, R. L. Magin, J. V. Sweedler and A. G. Webb, Science, 1995, 270, 1967–1970 CrossRef CAS.
- J. H. Lee, Y. Okuno and S. Cavagnero, J. Magn. Reson., 2014, 241, 18–31 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Table S1, T1 measurements; Table S2, intra- and inter-day precision; Table S3, precision and bias by NMR; Table S4, calibration curves; Table S5, residual outputs; regression analysis comparing results from NMR and AA (PDF). See DOI: 10.1039/d0an02088e |
| This journal is © The Royal Society of Chemistry 2021 |