
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCdSe nanocrystal sensitized photon upconverting film†
Emily M. Rigsby a,
Tsumugi Miyashitab,
Dmitry A. Fishmanc,
Sean T. Robertsd and
Ming L. Tang*a
a,
Tsumugi Miyashitab,
Dmitry A. Fishmanc,
Sean T. Robertsd and
Ming L. Tang*a
aDepartment of Chemistry, University of California Riverside, Riverside, CA 92521, USA. E-mail: mltang@ucr.edu
bDepartment of Bioengineering, University of California Riverside, Riverside, CA 92521, USA
cDepartment of Chemistry, University of California, Irvine, California 92697, USA
dDepartment of Chemistry, University of Texas at Austin, Austin, TX 78712, USA
First published on 20th September 2021
Abstract
Here, films using CdSe nanocrystal (NC) triplet photosensitizers in conjunction with diphenylanthracene (DPA) emitters were assembled to address several challenges to practical applications for solution-based photon upconversion. By using poly(9-vinylcarbazole) as a phosphorescent host in this film, volatile organic solvents are eliminated, the spontaneous crystallization of the emitter is significantly retarded, and ∼1.5% photon upconversion quantum yield (out of a maximum of 50%) is obtained. Transient absorption spectroscopy on nanosecond-to-microsecond time scales reveals this efficiency is enabled by an exceptionally long triplet lifetime of 3.4 ± 0.3 ms. Ultimately, we find the upconversion efficiency is limited by incomplete triplet–triplet annihilation, which occurs with a rate 3–4 orders of magnitude slower than in solution-phase upconversion systems.
Photon upconversion1–3 has promising photovoltaic and sensing applications2–5 if it can occur efficiently in thin films with low excitation densities. In this work, we address outstanding challenges using drop-cast films of CdSe nanocrystals (NCs) for light absorption in combination with anthracene emitters for emission. This hybrid system capitalizes on the synthetically tunable nature of inorganic NC absorption and emission. By transferring spin-triplet excitons from an inorganic NC to an organic acceptor, the benefits of strong semiconductor absorption and small exchange splitting can be coupled with triplet–triplet annihilation (TTA) and subsequent fluorescence from organic emitters. This is important because NCs can be made to absorb near-infrared (NIR) light much more strongly compared to their organic or lanthanide counterparts, and thus have great potential for efficiently converting NIR to visible light.
Much work has been done in the past to translate these solution-based upconversion systems to the solid phase.6–14 Previous work on DPA-containing supramolecular gels has focused on forming an annihilator-decorated polymer matrix doped with sensitizer to preserve efficient triplet energy transfer.7,9 Other groups have tried to dope a palladium porphyrin sensitizer directly into an emissive co-polymer host capable of TTA for upconversion, which resulted in efficiencies of up to 6% out of a possible 50%.8 Ogawa et al.15 have improved efficiencies by increasing the photoluminescence QY of the emitter in thin films by including singlet traps as dopants. However, the resulting reports have upconversion efficiencies that are typically much lower than those in solution. This is often due to decreased triplet mobility in rigid systems and increased aggregation of the sensitizer and molecular emitter.
Here, we report the first solution processed TTA-based photon upconversion film using semiconductor NCs to replace noble metal-containing porphyrin light absorbers in comparable all-organic TTA-based systems. We examine films of a CdSe NC photosensitizer/DPA emitter hybrid system that can convert green to violet light with QYs of over 10%16–18 (out of a maximum of 50%) in solution. Although the photon upconversion QY for the film here is an order of magnitude lower compared to solution, ∼1.5% (out of 50%), the QYs are comparable to all-organic systems,6 albeit at relatively high excitation densities exceeding 10 W cm−2.
This film consists of CdSe NCs and the DPA small molecule embedded in a wide-bandgap polymer poly(9-vinylcarbazole) (PVK) host material commonly used in organic light emitting diodes (LEDs). Importantly, PVK has been shown to enable efficient TTA for upconversion-based LEDs.19 The energy transfer processes that enable the function of this nanocrystal-based upconversion system are illustrated in Fig. 1a. 532 nm photons are absorbed by the CdSe NC sensitizer (green arrow), fuelling triplet energy transfer (TET, orange arrows) to the bound transmitter ligand, followed by TET to diphenylanthracene (DPA). DPA can then undergo triplet–triplet annihilation (TTA) to create an emissive singlet, producing violet 430 nm light. This TTA process occurs when two excited DPA molecules annihilate to form one DPA in a singlet excited state and one in the ground state. Fig. 1b shows the absorption (solid lines) and emission (dashed lines) of both the CdSe absorber with bound 9-anthracene carboxylic acid (9ACA) transmitter ligand and DPA emitter for this system. We focus on a blended film with all components mixed together in the same layer.
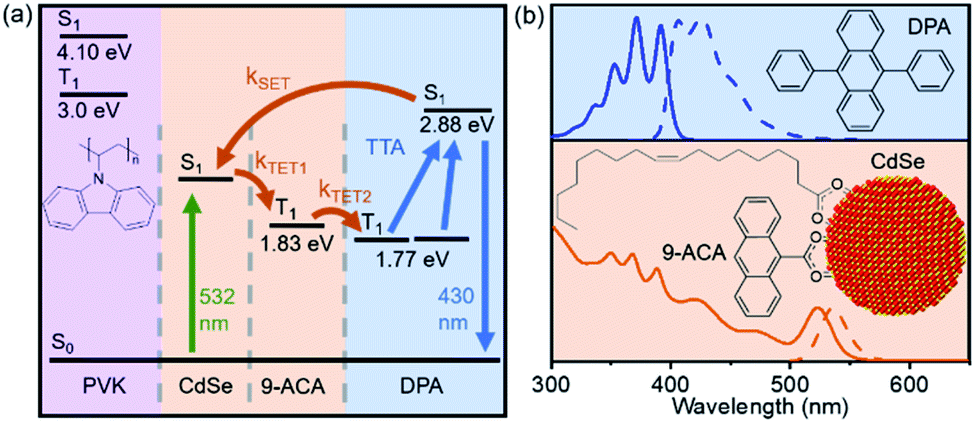 | ||
| Fig. 1 (a) Energy diagram depicting steps involved in photon upconversion: NC photoexcitation (green arrow), triplet energy transfer from CdSe to 9ACA (TET1), and triplet energy from 9ACA to DPA emitters (TET2) that emit via triplet–triplet annihilation (TTA, blue arrow). (b) Normalized absorbance (solid lines) and emission spectra (dashed lines) of DPA and 9ACA-functionalized CdSe nanocrystals dispersed in toluene. | ||
Air-free conditions were used for thin film fabrication with the goal of dispersing both CdSe sensitizers and DPA emitters evenly. To that end, PVK and DPA were dissolved in 1,2-dichlorobenzene (o-DCB), followed by the addition of 532 nm absorbing CdSe nanocrystals functionalized with 9ACA transmitter ligands (see SI for ligand exchange details, Fig. S1†). Once fully dissolved, this PVK solution was drop-cast onto glass coverslips. After annealing at 60 °C for ten minutes, the films were left to dry overnight in a glovebox. The films were then sealed using UV-cured epoxy. Profilometry measurements reveal that each drop-cast layer is approximately 6 microns thick (Fig. S2†).
Photon upconversion was performed using a 532 nm CW laser in a front-face fluorescence geometry. The upconversion signal varied based on the spatial location at which each film is illuminated, indicating these drop-cast thin films are not homogenous (Fig. 2a). The average upconversion signal detected by illuminating points near the center of the film was uniform, with large increases in upconversion signal being found at the film's edges where its thickness was greatest and it came into contact with the epoxy. PL mapping experiments confirm that the CdSe is concentrated at the edges of the film and that clusters of emissive CdSe NCs are non-uniformly distributed in the middle of the film (Fig. S3A†). These spots appear to correspond to the areas of highest upconversion. These best upconverting spots at the edges are not included in our analysis due to the film's increased thickness and variability of absorption in these regions. DPA is more uniformly distributed within the films, as PL mapping exciting with 405 nm shows uniform DPA emission throughout the film (Fig. S3B†).
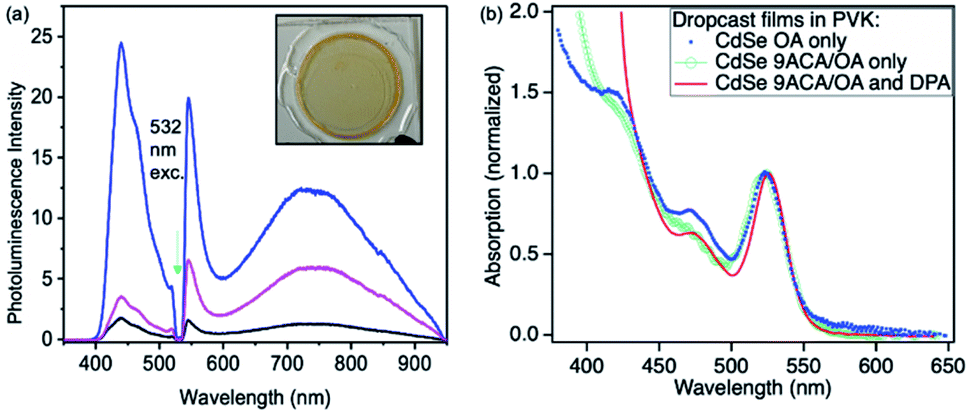 | ||
| Fig. 2 (a) Front face photon upconversion on the film consisting of CdSe 9ACA/OA and DPA in PVK (inset) with excitation using 532 nm CW light (green arrow), resulting in photoluminescence (PL) from DPA. Band edge and trap state emission from CdSe NCs is also observed. PL spectra from three different regions are shown to highlight the film's spatial inhomogeneity. The film's corresponding absorption spectrum is shown in (b) in red. For comparison, absorption spectra of films containing only CdSe OA NCs and CdSe 9ACA/OA are shown in blue squares and open green circles, respectively. | ||
The absorption spectra corresponding to the middle of the film is shown in Fig. 2b (red line). We measured the film's relative emission quantum yield by employing a rhodamine 6G (R6G) thin film as the fluorescent standard in the same PVK polymer host. The fluorescence QY of the R6G standard was measured in an integrating sphere to be 24% (as described in the ESI†). This R6G reference film was then placed in the same front-face detection geometry and used to quantify the upconversion signal for all films. To verify the accuracy of this approach, the CdSe containing thin film was measured in both the integrating sphere and front-face geometry. Similar upconversion QYs of ∼1.5% were obtained in both relative and absolute upconversion QY measurements. In general, the average photon upconversion QY is about 1.5% (out of a 50% maximum) for the film using a 532 nm CW laser with power densities exceeding 10 W cm−2 (Fig. S5†). At the edges, the upconversion signals were approximately 4-fold higher. In comparison, in solution, these CdSe 9ACA/OA NCs in 3 mM of DPA in toluene have a photon upconversion QY of 10.0% (out of 50% maximum).18 We note that without the 9ACA transmitter on the CdSe nanocrystals, no photon upconversion was observed in either thin films or solution.
Using the Beer–Lambert law, we calculated the concentration of CdSe 9ACA/OA and DPA in the film assuming the extinction coefficients of DPA and CdSe are the same in toluene and PVK, as there are minimal lineshape changes (Table S1†). To further explore the energy transfer processes within this film, we made several control samples to study with transient absorption spectroscopy using nanosecond pulsed excitation. The first control sample was a PVK film containing 20 wt% of DPA that was prepared and sealed in an air-free nitrogen glovebox, which allowed for characterization of DPA in the matrix. Two additional control samples of CdSe embedded in PVK without DPA were made, the first with CdSe capped with its native oleic acid (OA) ligands (CdSe OA, blue squares, Fig. 2b); and the second containing CdSe capped with 9ACA and OA transmitter ligands (CdSe 9ACA/OA) (green open circles, Fig. 2b).
Eqn (1) relates the overall photon upconversion efficiency to the efficiencies of each energy transfer step shown in Fig. 1a:
 | (1) |
To quantify ϕDPA, the DPA fluorescence QY, we used the integrating sphere to characterize the 20 wt% DPA in PVK thin film, excited at 365 nm. The resulting ϕDPA was 66%, which is lower than its value in toluene20 (ϕDPA = 90%). This decrease in ϕDPA could be due to aggregation of DPA in the polymer matrix, or an inner filter effect from the high concentration of DPA in PVK (0.574 M). Evidence for the former is seen in the red-shifted PL of DPA while evidence for the latter stems from a truncation of the blue shoulder of DPA's emission spectrum for the dropcast film when compared to DPA in toluene (Fig. S6†).
Transient absorption (TA) spectroscopy can directly probe the rates of energy transfer in this hybrid system as the excited state absorption (ESA) of the lowest triplet state of anthracene can be clearly measured. Fig. 3a–c show ns-TA spectra corresponding to the CdSe OA, CdSe 9ACA/OA, and CdSe 9ACA/OA + DPA in PVK films, respectively (Fig. S7† shows the corresponding near-infrared region). In Fig. 3a, in the CdSe OA in PVK film, a negative signal corresponding to the ground-state bleach (GSB) of CdSe is observed where the CdSe NC absorbs maximally. CdSe/OA also has an ESA centered at 450 nm.
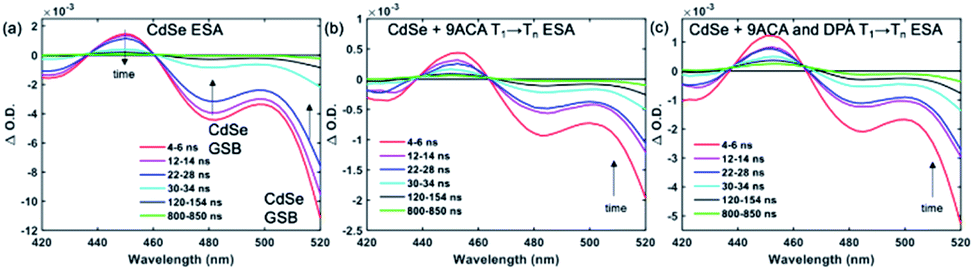 | ||
| Fig. 3 (a) Nanosecond transient absorption (TA) spectra for the (a) CdSe OA, (b) CdSe 9ACA/OA and (c) CdSe 9ACA/OA + DPA in PVK films respectively. | ||
To visualize the anthracene triplet clearly, double-difference (dd) TA spectra were obtained by subtracting the CdSe contribution at each time trace, normalizing at the 2nd GSB minimum (482.5 nm), where there is no laser scatter nor contribution from the anthracene. Fig. 4a shows dd spectra of the isolated 9ACA ESA from the CdSe 9ACA/OA sample (ns-TA spectra in Fig. 3b). We note that the anthracene ESA corresponding to its T1 → Tn transition is red-shifted in PVK compared to free DPA in toluene.21 Focusing on the anthracene triplet kinetics centered from 440–455 nm obtained from the dd spectra of the CdSe 9ACA/OA in PVK sample (Fig. 4a), we were able to fit an exponential triplet rise of 29.2 ± 2.1 ns and a 9ACA lifetime of 46.1 ± 0.5 μs after subtracting the CdSe component (Fig. 4c, for fitting details see Fig. S8†). In toluene, the corresponding TET1 rise time is 113 ± 1 ns.18 From biexponential fits to the CdSe/OA in PVK sample at the same wavelength (Fig. S9†), the amplitude-averaged lifetime of CdSe/OA was obtained according to eqn S4,† τCdSe-OA = 35.8 ns. The 9ACA rise time includes contributions from the CdSe lifetime and TET1 rate. Using τTET1 = 158 ns, we calculate the efficiency of TET1 using eqn S5† to obtain ϕTET1 = 0.18.
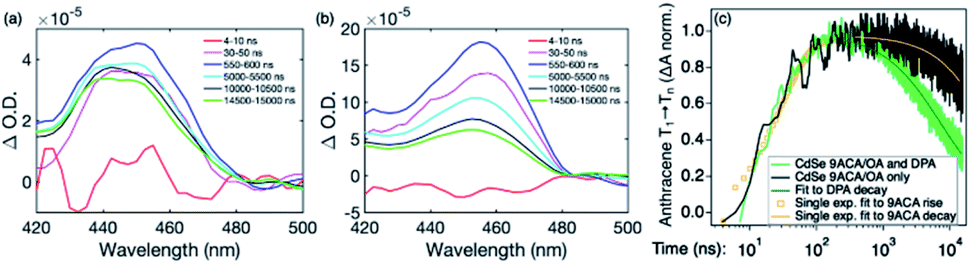 | ||
| Fig. 4 Double difference TA spectra for (a) CdSe 9ACA/OA and (b) CdSe 9ACA/OA + DPA in PVK films with the CdSe contribution subtracted. An excited state absorption corresponding to the anthracene spin-triplet exciton is clearly observed (c) Normalized dd TA kinetics spectrally integrated from 440–455 nm highlighting the growth and decay of anthracene triplets (both 9ACA and DPA). Triplet kinetics for the CdSe 9ACA/OA film are shown in black while kinetics for the CdSe 9ACA/OA + DPA film appear in green. All TA data was collected following 532 nm excitation. | ||
We next sought to understand the TTA efficiency using TA spectroscopy. Fig. 4b shows the dd spectra for the anthracene triplet ESA in the CdSe 9ACA/OA + DPA sample (ns-TA spectra in Fig. 3c). Since DPA is the major component of the thin film, the TA spectra and its evolution are dominated by DPA because the molar ratio of DPA to CdSe is 546![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (Table S1†). Since we are unable to distinguish between contributions from the 9ACA and DPA triplet to this dataset, we ignore the growth of this ESA that is dominated by kTET1 and focus only on its decay. Triplet decay kinetics of the dropcast film, CdSe 9ACA/OA + DPA in PVK are described by concurrent first- and second-order processes:
:1 (Table S1†). Since we are unable to distinguish between contributions from the 9ACA and DPA triplet to this dataset, we ignore the growth of this ESA that is dominated by kTET1 and focus only on its decay. Triplet decay kinetics of the dropcast film, CdSe 9ACA/OA + DPA in PVK are described by concurrent first- and second-order processes:
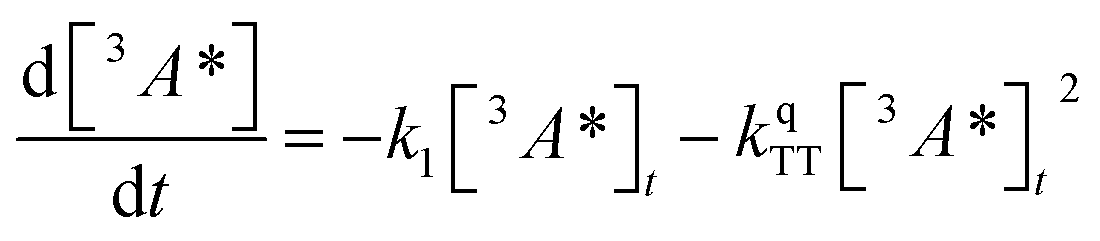 | (2) |
where
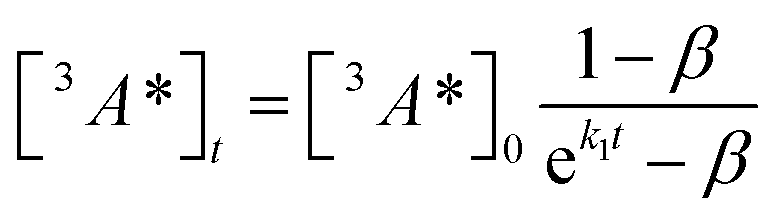
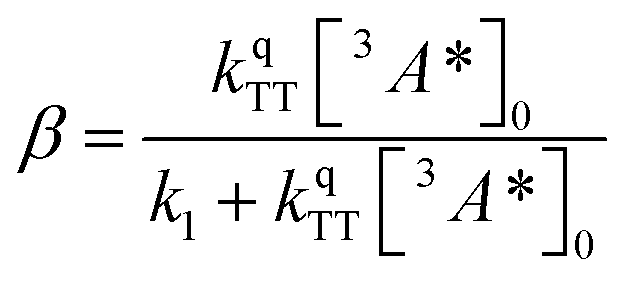 | (3) |
The DPA decay (neon green, Fig. 4c) of the CdSe 9ACA/OA + DPA in PVK film can be fit with eqn (3) (olive green line, Fig. 4c and S10†) where 1/k1 = 3.4 ± 0.3 ms and 1/kqTT ∼7.15 ± 0.02 μs M in the films. In comparison, DPA in its triplet excited state has a comparable lifetime in toluene of 8.61 ms,23 and kTTA is 3.14 × 109 M−1 s−1 (1/kTTA = 3.18 × 104 μs M).24 Considering the high concentration of DPA in PVK (0.574 M), we expect that kqTT ∼108 s−1 M−1 if TTA dominates the 2nd order decay and is diffusion limited like in solution. The fact that kqTT is ∼4 orders of magnitude lower in the PVK film suggests that the rigid nature of PVK limits TTA, perhaps by limiting the orbital overlap between two DPA molecules necessary for TTA. The diffusion of triplets is also inhibited in the solid state.25–27 We note that the DPA lifetime here is an under-estimate because of FRET-based back transfer from DPA to the CdSe (kSET, Fig. 1a). Lin et al. reported that using a layered morphology, back transfer in their molecular films decreased to 8% from 60.3% compared to the blended structure used here.6 A layered film is a good way of minimizing the parasitic reabsorption by the CdSe NC light absorbers.
To conclude, we have successfully transformed our nanocrystal sensitized TTA-based upconversion system from volatile organic solvents to a micron-thick film with upconversion quantum yields on par with the best organic TTA-based thin films. The use of PVK allows a high concentration of DPA to be incorporated while preventing phase separation of the photosensitizer and annihilator. The key bottleneck in these thin films is the inefficient annihilation of triplet pairs to form singlets, which may be addressed with transmitter ligands that facilitate triplet energy transfer via a hopping mechanism28 or with shorter solubilizing ligands on the NCs.29 Our current upconversion efficiency of 0.015 out of a maximum of 0.5 can be significantly improved by increasing the DPA fluorescence QY and modifying our fabrication techniques to produce more uniform thin films. Better control over the distribution of CdSe 9ACA/OA within the films, e.g. by spin-coating, will enhance TET2 and therefore the overall upconversion efficiencies of these films. With future improvements and the viability of environmentally friendly, non-toxic InP or silicon based light absorbers,30 these films have great potential to be used in practical solar or biomedical applications.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
MLT acknowledges Air Force Office of Scientific Research (AFOSR) Award FA9550-19-1-0092 for equipment and the National Science Foundation grant (OISE-1827087) for salary support. STR acknowledges support from the National Science Foundation (CHE-2003735), Robert A. Welch Foundation (Grant F-1885), and a Sloan Fellowship. DF acknowledges NSF grant 1532125.Notes and references.
- Z. Huang, X. Li, M. Mahboub, K. M. Hanson, V. M. Nichols, H. Le, M. L. Tang and C. J. Bardeen, Nano Lett., 2015, 15, 5552–5557 CrossRef CAS PubMed.
- C. Mongin, S. Garakyaraghi, N. Razgoniaeva, M. Zamkov and F. N. Castellano, Science, 2016, 351, 369–372 CrossRef CAS PubMed.
- M. Wu, D. N. Congreve, M. W. B. Wilson, J. Jean, N. Geva, M. Welborn, T. Van Voorhis, V. Bulovic, M. G. Bawendi and M. A. Baldo, Nat. Photonics, 2016, 10, 31–34 CrossRef CAS.
- R. S. Khnayzer, J. Blumhoff, J. A. Harrington, A. Haefele, F. Deng and F. N. Castellano, Chem. Commun., 2012, 48, 209–211 RSC.
- B. D. Ravetz, A. B. Pun, E. M. Churchill, D. N. Congreve, T. Rovis and L. M. Campos, Nature, 2019, 565, 343–346 CrossRef CAS PubMed.
- T.-A. Lin, C. F. Perkinson and M. A. Baldo, Adv. Mater., 2020, 32, 1908175 CrossRef CAS PubMed.
- V. Gray, B. Küçüköz, F. Edhborg, M. Abrahamsson, K. Moth-Poulsen and B. Albinsson, Phys. Chem. Chem. Phys., 2018, 20, 7549–7558 RSC.
- V. Jankus, E. W. Snedden, D. W. Bright, V. L. Whittle, J. A. G. Williams and A. Monkman, Adv. Funct. Mater., 2013, 23, 384–393 CrossRef CAS.
- D. F. B. d. Mattos, A. Dreos, M. D. Johnstone, A. Runemark, C. Sauvée, V. Gray, K. Moth-Poulsen, H. Sundén and M. Abrahamsson, J. Chem. Phys., 2020, 153, 214705 CrossRef PubMed.
- P. F. Duan, N. Yanai, Y. Kurashige and N. Kimizuka, Angew. Chem., Int. Ed., 2015, 54, 7544–7549 CrossRef CAS PubMed.
- P. F. Duan, N. Yanai, H. Nagatomi and N. Kimizuka, J. Am. Chem. Soc., 2015, 137, 1887–1894 CrossRef CAS PubMed.
- S. Hisamitsu, N. Yanai and N. Kimizuka, Angew. Chem., Int. Ed., 2015, 54, 11550–11554 CrossRef CAS PubMed.
- N. Kimizuka, N. Yanai and M. Morikawat, Langmuir, 2016, 32, 12304–12322 CrossRef CAS PubMed.
- T. Ogawa, N. Yanai, A. Monguzzi and N. Kimizuka, Sci. Rep., 2015, 5, 10882 CrossRef CAS PubMed.
- T. Ogawa, M. Hosoyamada, B. Yurash, T.-Q. Nguyen, N. Yanai and N. Kimizuka, J. Am. Chem. Soc., 2018, 140, 8788–8796 CrossRef CAS PubMed.
- X. Li, A. Fast, Z. Y. Huang, D. A. Fishman and M. L. Tang, Angew. Chem., Int. Ed., 2017, 56, 5598–5602 CrossRef CAS PubMed.
- X. Li, Z. Huang, R. Zavala and M. L. Tang, J. Phys. Chem. Lett., 2016, 7, 1955–1959 CrossRef CAS PubMed.
- E. M. Rigsby, T. Miyashita, P. Jaimes, D. A. Fishman and M. L. Tang, J. Chem. Phys., 2020, 153, 114702 CrossRef CAS PubMed.
- D. Di, L. Yang, J. M. Richter, L. Meraldi, R. M. Altamimi, A. Y. Alyamani, D. Credgington, K. P. Musselman, J. L. MacManus-Driscoll and R. H. Friend, Adv. Mater., 2017, 29, 1605987 CrossRef PubMed.
- S. L. Murov, I. Carmichael and G. L. Hug, Handbook of photochemistry, CRC Press, 1993 Search PubMed.
- J. De Roo, Z. Huang, N. J. Schuster, L. S. Hamachi, D. N. Congreve, Z. Xu, P. Xia, D. A. Fishman, T. Lian, J. S. Owen and M. L. Tang, Chem. Mater., 2020, 32, 1461–1466 CrossRef CAS.
- S. M. Bachilo and R. B. Weisman, J. Phys. Chem. A, 2000, 104, 7711–7714 CrossRef CAS.
- V. Gray, D. Dzebo, A. Lundin, J. Alborzpour, M. Abrahamsson, B. Albinsson and K. Moth-Poulsen, J. Mater. Chem. C, 2015, 3, 11111–11121 RSC.
- V. Gray, A. Dreos, P. Erhart, B. Albinsson, K. Moth-Poulsen and M. Abrahamsson, Phys. Chem. Chem. Phys., 2017, 19, 10931–10939 RSC.
- V. Gray, K. Moth-Poulsen, B. Albinsson and M. Abrahamsson, Coord. Chem. Rev., 2018, 362, 54–71 CrossRef CAS.
- B. Joarder, N. Yanai and N. Kimizuka, J. Phys. Chem. Lett., 2018, 9, 4613–4624 CrossRef CAS PubMed.
- Y. Murakami and K. Kamada, Phys. Chem. Chem. Phys., 2021, 23, 18268–18282 RSC.
- Z. Huang, Z. Xu, T. Huang, V. Gray, K. Moth-Poulsen, T. Lian and M. L. Tang, J. Am. Chem. Soc., 2020, 142, 17581–17588 CrossRef CAS PubMed.
- L. Nienhaus, M. Wu, N. Geva, J. J. Shepherd, M. W. B. Wilson, V. Bulovic, T. Van Voorhis, M. A. Baldo and M. G. Bawendi, ACS Nano, 2017, 11, 7848–7857 CrossRef CAS PubMed.
- P. Xia, E. K. Raulerson, D. Coleman, C. S. Gerke, L. Mangolini, M. L. Tang and S. T. Roberts, Nat. Chem., 2020, 12, 137–144 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Complete description of experimental methods, synthesis, and data fitting procedures. See DOI: 10.1039/d1ra06562a |
| This journal is © The Royal Society of Chemistry 2021 |