
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe impact of molecular orientation on carrier transfer characteristics at a phthalocyanine and halide perovskite interface
Saqib Javaidab and
Geunsik Lee *a
*a
aCenter for Superfunctional Materials, Center for Wave Energy Materials, Department of Chemistry, Ulsan National Institute of Science and Technology (UNIST), Ulsan 44919, Korea
bMMSG, Theoretical Physics Division, PINSTECH, P.O. Nilore, Islamabad, Pakistan. E-mail: gslee@unist.ac.kr
First published on 27th September 2021
Abstract
We have studied the interface properties of metal phthalocyanine (MPc, M = Zn, Cu) molecules at a methylammonium lead iodide (MAPbI3) surface using density functional theory (DFT) based simulations. From the adsorption energies, the face-on orientation is found to have an order of magnitude stronger binding energy than the edge-on orientation, where CuPc binds a little stronger than ZnPc with its closer interfacial distance. Our detailed analysis of interface electronic structure suggests that the edge-on configuration possesses a large energy barrier for the hole transfer from MAPbI3 to MPc molecules. In contrast, the face-on configuration has no such barrier, facilitating the hole transfer, while at the same time the desirable alignment of the conduction band suppresses the electron–hole recombination. Therefore, the face-on configuration is clearly found to be more suitable for the photovoltaic process, in line with the experimental reports. Our work emphasizes the impact of MPc orientation upon perovskite solar cell efficiency besides other factors such as Pc thin film's mobility and morphology, and provides insightful guidance to efficient and stable hole transport layers.
1. Introduction
With efficiency exceeding 25%,1,2 perovskite halide based solar cells are the most promising candidates to replace silicon based solar technology.3–5 Perovskite halide materials are represented by the formula APbX3, where A is the organic cation while X is the halide anion.6 In general, a perovskite halide photovoltaic device comprises electron and hole transport layers (ETL and HTL, respectively) that are placed adjacent to the perovskite layer to extract photo-excited charge carriers toward their respective electrodes.7 Thus, the efficiency of the device depends critically upon the perovskite/charge transport layer interface where the charge separation process takes place.8,9Until recently, Spiro-OMeTAD has been employed widely as the HTL.10 However, Spiro-OMeTAD suffers from a number of issues that inhibit its commercial utilization.11–13 For example, it is well known that Spiro-OMeTAD is not stable in hot and humid conditions. Moreover, it is amongst the most expensive parts of the device due to the multistep synthesis and purification. Consequently, the replacement of Spiro-OMeTAD is the key towards the development of stable and cheap photovoltaic devices. In this regard, metal-phthalocyanine (MPc; M = 3d metal, Pc = N8C32H16) molecules are amongst the most promising materials. These molecules are generally less expensive than Spiro, thermally stable and can be grown into structurally ordered thins films.14–16 Thus far, various studies have successfully employed phthalocyanine (Pc) molecules based HTL within perovskite halide solar cells.11,12
An interesting aspect of Pc materials is their orientation flexibility which can be exploited to tune photovoltaic properties. In general, Pc molecules are adsorbed in edge-on orientation (molecule standing up on the surface) on perovskite halide surfaces; however, a transformation from edge-on to face-on orientation (molecule lying flat on the surface) leads to significant improvement in photovoltaic efficiency.17,18 This increase in efficiency is attributed in part to higher mobility in the denser face-on film, though the impact of molecular orientation on the charge transfer process at perovskite/HTL interface remains unexplored. A previous DFT study investigated the face-on ZnPc/MAPbI3 interface which provided useful insights regarding the nature of the heterostructures.19 However, a direct comparison between the electronic structures of face-on and edge-on perovskite/Pc interfaces is still lacking. To bridge this gaps, we have studied the ZnPc/MAPbI3 and CuPc/MAPbI3 interfaces in both edge-on and face-on configurations by employing DFT simulations. It should be pointed out that CuPc is widely used material in opto-electronic devices due to its superior transport properties.20 Through this comparative study, we show that the hole transfer from MAPbI3 to MPc in edge-on configuration is impeded due to the presence of a large interface energy barrier. In contrast, face-on orientation facilitates hole transfer and at the same time suppresses electron–hole recombination, which are partly responsible for its superior performance.
2. Methods
All DFT calculations have been performed using planewave pseudopotential approach as implemented in VASP suite.21 Generalized gradient approximation (GGA) in its PBE parameterization is used as exchange correlation functional.22 It is well known that GGA fails to incorporate the van der Waals (vdW) interactions which is necessary to study the molecular adsorption on the surfaces. Therefore, vdW interactions are included by adding Tkatchenko and Scheffler (TS) dispersion correction to the total energy and forces.23 Moreover, the estimation of electronic structure and band gaps are further improved by employing HSE06 hybrid functional including spin–orbit coupling.24Previous experimental work has shown that the average Pc-surface orientation is ∼75° for the edge-on configuration.25 Here, we have adopted this value for the supercell formation of edge-on configuration. In contrast, Pc molecules were kept parallel to the MAPbI3 surface in the case of face-on orientation. A vacuum layer of ∼15 Å was provided to avoid spurious interactions between periodic images of MAPbI3/MPc system. Brillion zone sampling has been performed on a 2 × 2 × 1 k-mesh. However, no significant change in the electronic structure was observed as k-mesh was reduced to a single Γ-point. Thus, for computationally expensive hybrid HSE06 plus the spin–orbit coupling (HSE06 + SOC) calculations, only Γ-point is employed. Spin polarized calculations were performed for MAPbI3/CuPc owing to the local magnetic moment of Cu, though spin polarization was not required for MAPbI3/ZnPc. Finally, we also checked the validity of our results by employing dipolar correction which provided essentially similar results.
For MAPbI3, we have chosen room temperature tetragonal phase (lattice parameters; a = 8.85, c = 12.65). Specifically, MAPbI3 surface is modelled by taking experimentally prevalent (001) surface,26 corresponding to 2 × 2 MAPbI3 unit cell. Furthermore, PbI2 termination is considered for MAPbI3 (001) surface which is known to be energetically optimum.27 The band gap obtained by employing HSE06 + SOC approach was 1.66 eV which is in excellent agreement with the experimental values.28 As for ZnPc and CuPc, the optimized structural parameters and HUMO–LUMO gaps were found to be in good agreement with the previous DFT calculations.29,30 Structural optimization was performed for whole MAPbI3/MPc system. The convergence criteria for energy and forces during the electronic and structural optimizations were set to 10−5 eV and 0.015 eV Å−1, respectively. To analyze charge transfer at interface, Bader charges were calculated.31
3. Results and discussion
Fig. 1 depicts different possible adsorption sites of MPc molecules on MAPbI3 (001) surface that are studied in this work, namely on the top of Pb, I and hollow sites. For face-on configuration, these adsorption sites correspond to the position of center 3d metal of MPc molecules above the substrate. In the case of edge-on configuration, the main interaction between MAPbI3 surface and MPc molecules takes place through two peripheral hydrogen atoms of MPc, present at the interface (see Fig. 2). Thus, the adsorption site of MPc for edge-on geometry above MAPbI3 surface corresponds to the mean position of these two hydrogen atoms.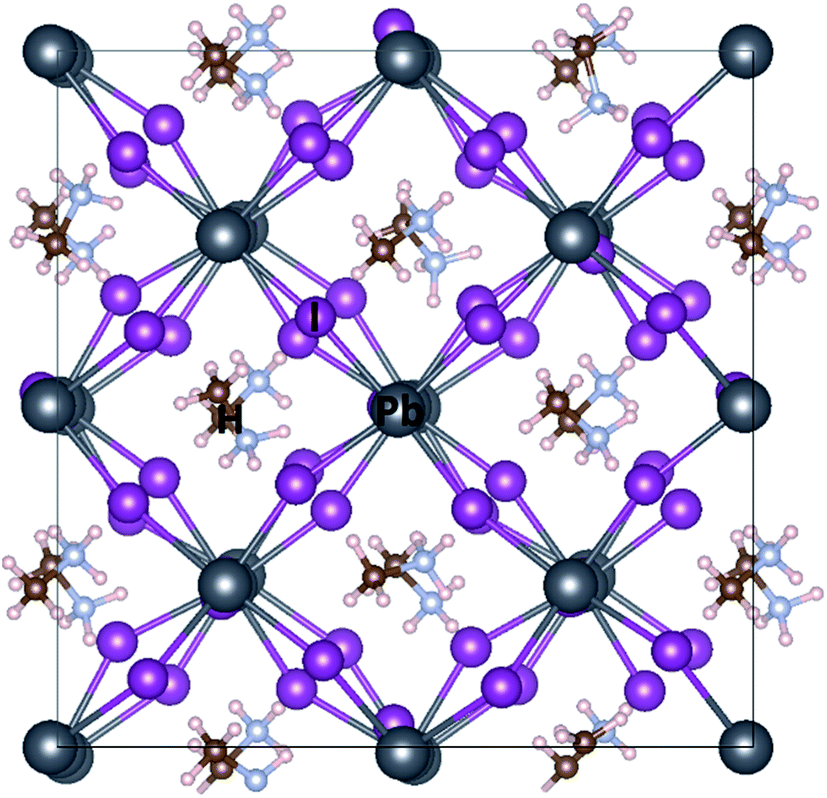 | ||
| Fig. 1 Various adsorption positions of ZnPc or CuPc molecule on MAPbI3(001) surface studied in this work, namely Pb, I and hollow (H) sites. Atomic color scheme: Pb (grey), I (purple), N (light blue), C (brown) and H (white). The dark solid lines represent the lateral supercell size. | ||
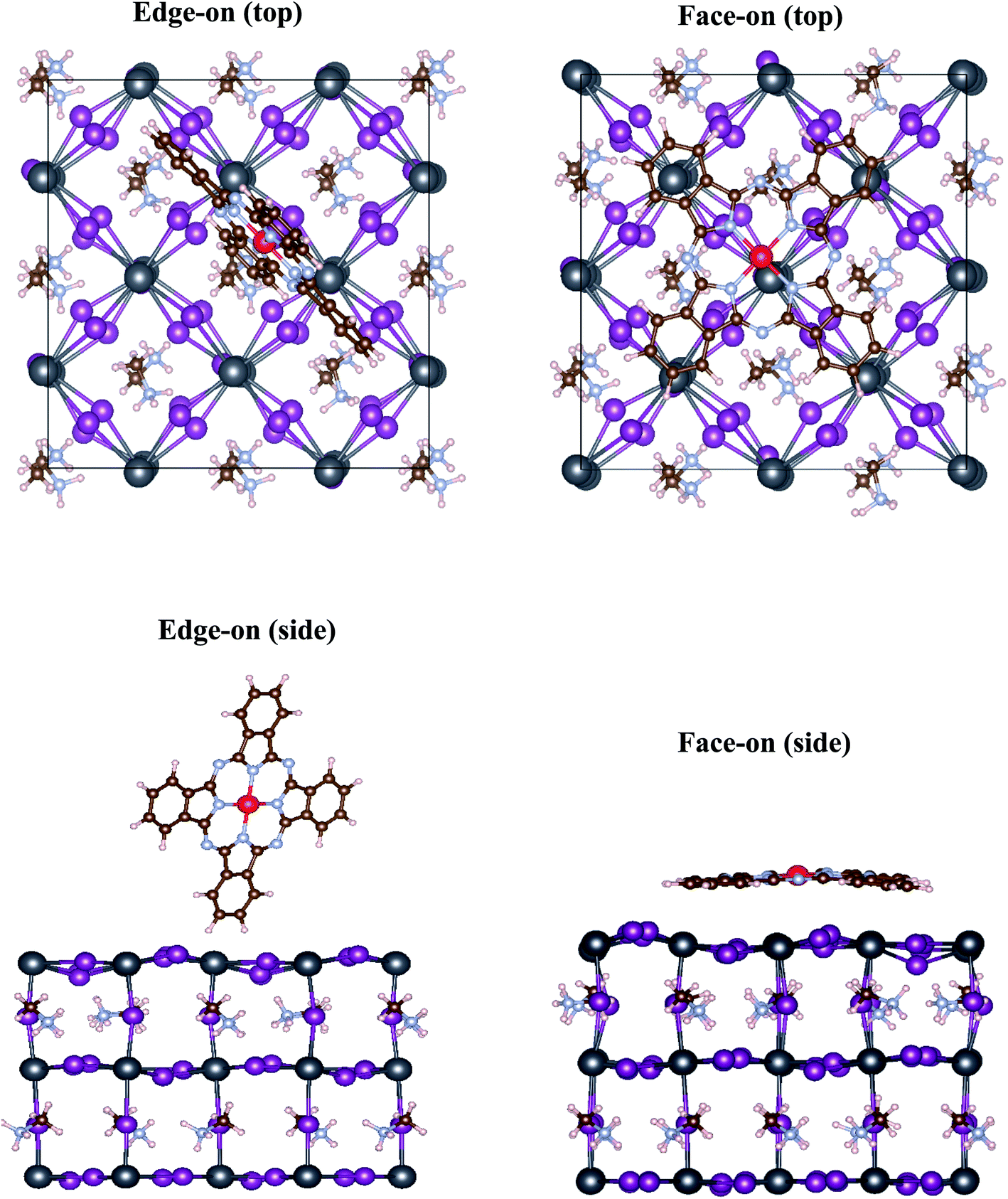 | ||
| Fig. 2 Top and side views of the optimized MAPbI3/phthalocyanine interfaces for edge-on and face-on configurations. The most stable adsorption locations of MPc for edge-on and face-on orientations are hollow and Pb sites, respectively. Atomic color scheme: Pb (grey), I (purple), N (light blue), C (brown), H (white), red (phthalocyanine's center Zn or Cu atom). Note that the MAPbI3 slab is shifted so that phthalocyanine molecule is at the center of the cell. | ||
Table 1 further compares the adsorption and structural properties of MAPbI3/MPc interface within face-on and edge-on configurations. We first discuss the results of face-on orientation. The I top site is found to be energetically most stable for ZnPc. However, the energy difference between Pb and I top sites is negligibly small (∼0.03 eV per cell) so the adsorption on both Pb and I top sites should be feasible for MAPbI3/ZnPc. On the other hand, the energy difference between hollow and Pb/I top sites is rather large (∼0.20 eV per cell) highlighting the unsuitability of hollow site for MPc adsorption in face-on geometry. For CuPc, Pb top site becomes more favorable than I, though the energy difference between the two sites still remains rather small (<0.10 eV per cell). For a proper comparison of adsorption properties, we will mainly focus on the Pb top site for both ZnPc and CuPc in the following. The MAPbI3–MPc distances (dint) are rather similar (∼3.15–3.20 Å) for MAPbI3/ZnPc and MAPbI3/CuPc interfaces. It should be pointed that this distance corresponds to the minimum vertical separation between MPc molecules and MAPbI3, therefore, different atoms within MPc molecule exhibit varying distance from MAPbI3 surface whose heights may be larger than the value reported in Table 1. Overall, CuPc is adsorbed relatively closer to MAPbI3 as compared to ZnPc as highlighted in Table 1. In particular, Cu within CuPc seems to interact more strongly with the substrate than Zn in ZnPc since Cu–Pb bond distance is ∼0.20 Å shorter than that of Zn–Pb.
| Face-on | Edge-on | |||||
|---|---|---|---|---|---|---|
| dint (Å) | Eads (eV) | Site | dint (Å) | Eads (eV) | Site | |
| MAPbI3/ZnPc | 3.21 | −2.54 | Pb/I | 3.03 | −0.36 | Hollow |
| MAPbI3/CuPc | 3.16 | −2.59 | Pb | 3.02 | −0.34 | Hollow |
In order to ascertain the strength of interfacial interactions, the adsorption energy (Eads) is calculated as:
| Eads = E(MAPbI3–MPc) − E(MAPbI3) − E(MPc) | (1) |
As for the edge-on configuration, the optimum adsorption site is hollow in contrast to the case of face-on configuration, as shown in Table 1. The absence of atoms in the top layer of MAPbI3 at the hollow site means that MPc molecule can come much closer to the substrate and maximize the interfacial interactions. The MAPbI3–MPc separation is 3.03 Å for both ZnPc and CuPc which corresponds to the distance between peripheral hydrogen atoms of MPc and the nearest neighbor I. Owing to the interface geometry in edge-on configuration, other atoms of MPc are at a large distance from MAPbI3 substrate so they do not interact with underlying MAPbI3 substrate. Most crucially, Eads for edge-on orientation is roughly an order of magnitude lower than that of face-on orientation. Nevertheless, despite much weaker binding, an energetically stable interface is formed for edge-on configuration, in line with the experimental findings.32 For comprehensive description of relative stability of two molecular orientations, realistic factors need to be considered such as the adsorbate–adsorbate interaction (e.g. the intermolecular binding energy of −4.9 eV per molecule as calculated for MPc crystal) and/or uneven surface of MAPbI3 (e.g. edge-on orientation on a flat surface caused by a slanted one nearby as illustrated in ref. 18).
Fig. 2 depicts the optimized interface geometry for face-on and edge-on orientations. As discussed earlier, only hydrogen atoms of MPc can interact with the underlying MAPbI3 surface for the edge-on configuration. Thus, the interaction is weak and ZnPc/CuPc retain their planar conformation. On the other hand, impact of interfacial interaction is much more evident for the face-on orientation. As a consequence of structural optimization, the center metal (Cu/Zn) of MPc moves slightly away from Pb site such that N atom of MPc is placed at the top on I atom of MAPbI3 surface (top view). Crucially, a noticeable deviation from square planar geometry of MPc is observed in face-on case; outer edges of MPc molecule move towards MAPbI3 substrate (side view) and as a consequence, MPc molecule is distorted. Specifically, the vertical distance between center metal and peripheral hydrogen is calculated to be 0.45 Å for ZnPc and 0.40 Å for CuPc while its value is zero for the pristine MPc with planar D4h symmetry. The presence of this bending in MPc molecules underscores the presence of more attractive interaction of peripheral atoms with the substrate than inner atoms, albeit rather weak overall due to large adsorption distances.
Fig. 3 elaborates the impact of MPc molecular orientation on the interfacial electronic structure as calculated from HSE06 + SOC approach. Here, we mainly focus on the interface atomic layers, i.e., the atoms that are directly involved in the charge transfer process within a photovoltaic device. Thus, only peripheral hydrogen atoms are considered for edge-on configuration since only these atoms interact with the substrate, where the highest occupied molecular orbital (HOMO) driven by carbon atoms does not couple directly with surface PbI2 layer. Conversely, for the face-on case, all atoms of MPc are included owing to the flat geometry of MPc at the interface. Similarly, for MAPbI3 substrate the top PbI2 layer is considered. Density of states (DOS) for edge-on orientation is qualitatively similar for MAPbI3/ZnPc (Fig. 3a) and MAPbI3/CuPc (Fig. 3b). The valence band maxima (VBM) and conduction band minima (CBM) are dominated by MAPbI3 surface Pb and I atoms. Importantly, the main contribution MPc H atoms comes from deep inside the valence band, i.e., ∼1.0 eV below VBM. Besides, the contribution from MPc H in conduction band lies at least ∼1.6–1.7 eV above the CBM. In contrast, a noticeable difference is observed for face-on configuration (Fig. 3c and d). The VBM is dominated by MPc C atoms, and it lies ∼0.4–0.5 eV above the peak mainly contributed by top PbI2 layer of MAPbI3. Therefore, an appreciable band offset (difference between VBM of MAPbI3 and HOMO of MPc) is observed for the valence band. The main contribution for the CBM comes from MAPbI3 while MPc (C, N) states appear ∼1.0 eV higher. Similar to the case of edge-on, electronic structure of face-on configuration is not significantly affected by the change of 3d metal of MPc molecule.
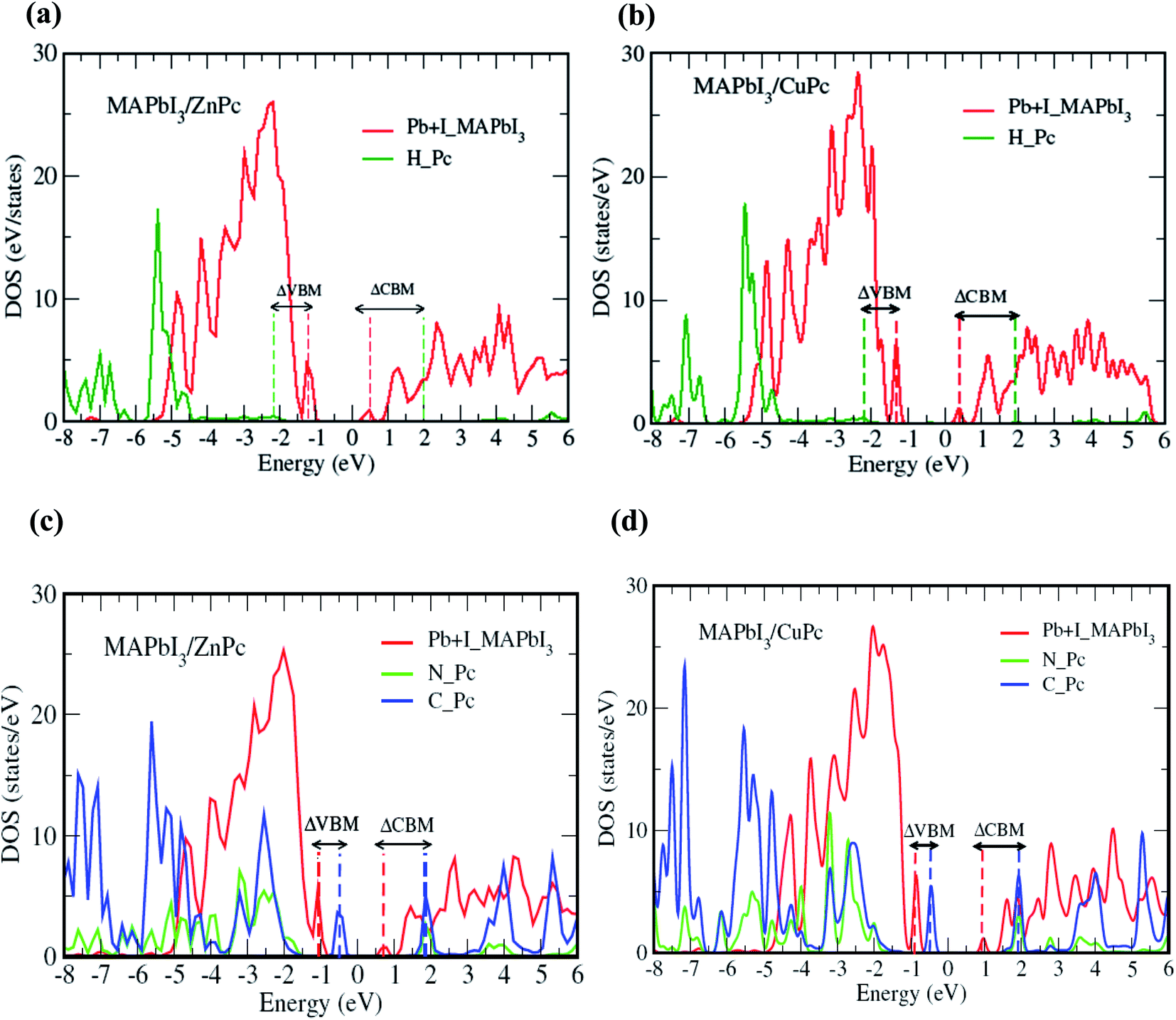 | ||
| Fig. 3 MAPbI3/MPc interface (Pb + I for MAPbI3, H or C + N for MPc) DOS with (a) M = Zn, (b) M = Cu for edge-on configuration, and (c) M = Zn, (d) M = Cu for face-on orientation. Dashed vertical lines indicate the position of band minima/maxima while double arrow horizontal lines present the band offsets. Zero of energy corresponds to the Fermi level (EF). It is noted that Zn or Cu contribution is not appreciable for HOMO or LUMO in (c) and (d). | ||
In light of DOS results presented above, Fig. 4 depicts the interface energy level diagram which is crucial for the understanding of charge transfer process within photovoltaic devices. For brevity, we focus on MAPbI3/CuPc case as the results for MAPbI3/ZnPc are qualitatively similar. For edge-on orientation, the intermediate H state of CuPc lies deep inside the valence band. Thus, a hole created by photoexcitation of MAPbI3 cannot be easily transferred to HOMO of CuPc due to the presence of large energy barrier (∼0.80 eV) through peripheral H. In contrast, no such energy barrier exists for face-on orientation, thus hole transfer across the interface is facilitated. Moreover, the lowest unoccupied molecular orbital (LUMO) of CuPc is situated at a much higher energy than the conduction band of MAPbI3 for both orientations. This means that photoexcited electrons in the conduction band of MAPbI3 cannot be transferred to the LUMO of CuPc. This electron blockage is helpful in reducing the deleterious recombination process.
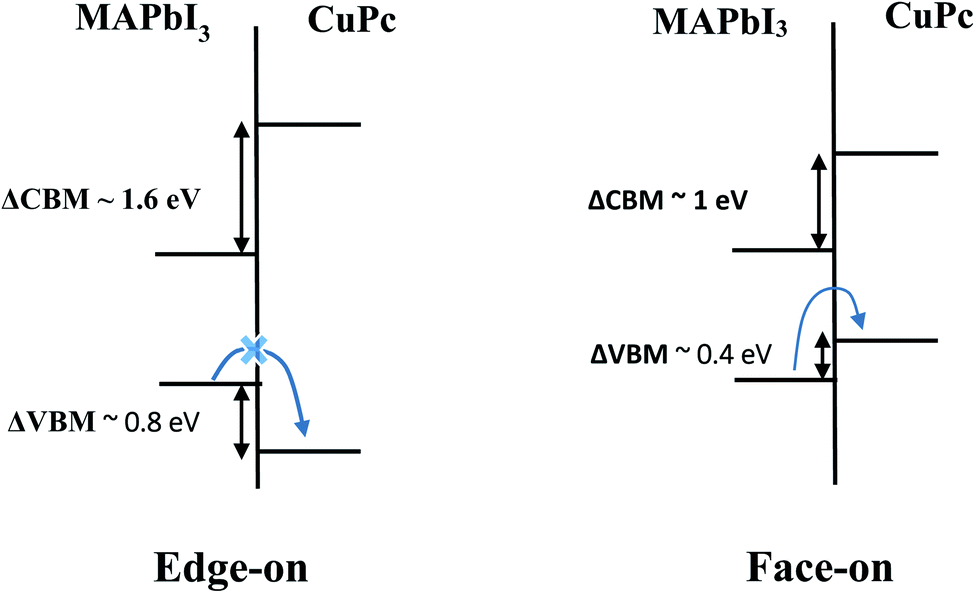 | ||
| Fig. 4 Schematic depiction of the energy level alignment at MAPbI3/CuPc interface for (left) edge-on and (right) face-on orientations. For the edge-on orientation, hole transfer is impeded due to presence of large barrier (∼0.8 eV). In contrast, the face-on orientation facilitates hole transfer due to favorable valence band offset of ∼0.4 eV. | ||
Additional impacts of MPc molecular orientation on the perovskite solar cell performance is described. As we checked the muffin-tin charges of a Pb and an I surface atom with the muffin-tin radii of 1.7 and 1.5 Å, respectively, before/after face-on molecule adsorption, they amount 2.81/2.79 and 4.30/4.31, respectively, for the face-on orientation, 2.82/2.81 and 4.32/4.32, respectively, for the edge-on. It indicates negligible charge transfer at the interface or no interface dipole, which is confirmed with our data in Table 2. In addition, as also listed in Table 2, our ionization energies of all four heterostructures by HSE06 + SOC are almost equal to that (6.10 eV) of clean MAPbI3. This indicates that the orbital hybridization at the interface is negligible, and that the electronic structures of individual components remain almost intact before and after adsorption. Nonetheless, two peripheral hydrogen atoms for the edge-on configuration causes the substantial amount of hole injection barrier. In fact, from Fig. 4, the hole injection barrier corresponds to the potential energy height (Vb ∼ 1.2 eV) for an electron at HOMO of MPc to be transmitted to VBM of MAPbI3. To estimate roughly the suppressed magnitude of hole injection rate for the edge-on compared to face-on, the tunneling probability formula exp[−2(2meVb/ℏ2)1/2L] is adopted with L ∼ 1.0 Å (corresponding to C–H bond length), which gives ∼0.3.
| Charge transfer (e) | Ionization energy (eV) | |||
|---|---|---|---|---|
| Face-on | Edge-on | Face-on | Edge-on | |
| MAPbI3/ZnPc | −0.010 | +0.034 | 6.09 | 6.07 |
| MAPbI3/CuPc | −0.013 | +0.033 | 6.10 | 6.09 |
Controlling the molecular orientation to tune photovoltaic properties is already well established in organic solar cells.33,34 Current findings provide helpful insight about detailed interface properties of perovskite and MPc. However, our result of Fig. 4 suggests a great enhancement of hole transfer rate when the face-on configuration is stabilized as usually done in the experiments by proper peripheral functionalization. Nevertheless, commonly observed multiple facets or domains of perovskite surface results in non-uniform stacking of MPc molecules around the boundaries, which would impede the carrier mobility. Thus attempts to have sufficient face-on contacts with proper functionalization might not be satisfactory for highly improving the efficiency, furthermore MPc's capability of surface defect passivation is unknown. Instead, one might introduce a proper capping interlayer35,36 for not only passivating surface defects, but regulating a rough surface for better MPc contact.
4. Conclusions
We have studied the MAPbI3/Pc interface in both the edge-on and face-on configurations by employing DFT based simulations. These results show that interfacial interaction are much stronger for the face-on orientation since the adsorption energy is an order of magnitude higher for face-on configuration as compared to edge-on case. As a consequence of stronger coupling with underlying MAPbI3, Pc molecular geometry is distorted for face-on configuration while it retains it square planar geometry within edge-on orientation. Beside much weaker interfacial coupling, electronic structure calculations indicate that edge-on orientation does not support the hole transfer process due to unfavorable valence band offset. In contrast, face-on interface facilitates hole transfer while at the same time suppressing the electron–hole recombination process. Overall, our results underscore the impact of molecular orientation on interface energy alignment and charge transfer process.Author contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
We acknowledge the support of MIS at PINSTECH, and PIEAS, Islamabad, and KISTI (KSC-2021-CRE-0188) and UNIST Supercomputing Center with regards to providing the computational resources. This work has been supported by Pakistan Atomic Energy Commission, Basic Science Research Program (2021R1A2C1006039, 2019R1A4A1029237) through National Research Foundation of Korea.References
- M. A. Green, E. D. Dunlop, J. Hohl-Ebinger, M. Yoshita, N. Kopidakis and A. W. Ho-Baillie, Solar cell efficiency tables (Version 55), Prog. Photovoltaics, 2020, 28(1), 3–15 CrossRef PubMed.
- S. I. Seok, T.-F. Guo, S. Lee, K. Choi, C. H. Min, M. Y. Woo and J. H. Noh, Photon recycling in halide perovskite solar cells for higher efficiencies, MRS Bull., 2020, 45(6), 439–448 CrossRef CAS.
- D. W. Ferdani, S. R. Pering, D. Ghosh, P. Kubiak, A. B. Walker, S. E. Lewis, A. L. Johnson, P. J. Baker, M. S. Islam and P. J. Cameron, Partial cation substitution reduces iodide ion transport in lead iodide perovskite solar cells, Energy Environ. Sci., 2019, 12(7), 2264–2272 RSC.
- H. Fu, Review of lead-free halide perovskites as light-absorbers for photovoltaic applications: from materials to solar cells, Sol. Energy Mater. Sol. Cells, 2019, 193, 107–132 CrossRef CAS.
- D. A. Egger, A. Bera, D. Cahen, G. Hodes, T. Kirchartz, L. Kronik, R. Lovrincic, A. M. Rappe, D. R. Reichman and O. Yaffe, What remains unexplained about the properties of halide perovskites?, Adv. Mater., 2018, 30(20), 1800691 CrossRef PubMed.
- A. Senocrate, I. Spanopoulos, N. Zibouche, J. Maier, M. S. Islam and M. G. Kanatzidis, Tuning Ionic and Electronic Conductivities in the “Hollow” Perovskite {en} MAPbI3, Chem. Mater., 2021, 33(2), 719–726 CrossRef CAS.
- S. Javaid, C. W. Myung, J. Yun, G. Lee and K. S. Kim, Organic cation steered interfacial electron transfer within organic–inorganic perovskite solar cells, J. Mater. Chem. A, 2018, 6(10), 4305–4312 RSC.
- D. Meggiolaro, E. Mosconi, A. H. Proppe, R. Quintero-Bermudez, S. O. Kelley, E. H. Sargent and F. De Angelis, Energy Level Tuning at the MAPbI3 Perovskite/Contact Interface Using Chemical Treatment, ACS Energy Lett., 2019, 4(9), 2181–2184 CrossRef CAS.
- L. K. Ono and Y. Qi, Surface and interface aspects of organometal halide perovskite materials and solar cells, J. Phys. Chem. Lett., 2016, 7(22), 4764–4794 CrossRef CAS PubMed.
- Y. Li, H. Li, C. Zhong, G. Sini and J.-L. Brédas, Characterization of intrinsic hole transport in single-crystal spiro-OMeTAD, npj Flexible Electron., 2017, 1(1), 1–8 CrossRef CAS.
- M. Urbani, G. de la Torre, M. K. Nazeeruddin and T. Torres, Phthalocyanines and porphyrinoid analogues as hole-and electron-transporting materials for perovskite solar cells, Chem. Soc. Rev., 2019, 48(10), 2738–2766 RSC.
- Y. Matsuo, K. Ogumi, I. Jeon, H. Wang and T. Nakagawa, Recent progress in porphyrin-and phthalocyanine-containing perovskite solar cells, RSC Adv., 2020, 10(54), 32678–32689 RSC.
- N. J. Jeon, H. Na, E. H. Jung, T.-Y. Yang, Y. G. Lee, G. Kim, H.-W. Shin, S. I. Seok, J. Lee and J. Seo, A fluorene-terminated hole-transporting material for highly efficient and stable perovskite solar cells, Nat. Energy, 2018, 3(8), 682–689 CrossRef CAS.
- H. Yamada, T. Shimada and A. Koma, Preparation and magnetic properties of manganese(II) phthalocyanine thin films, J. Chem. Phys., 1998, 108(24), 10256–10261 CrossRef CAS.
- G. Mattioli, F. Filippone, P. Alippi, P. Giannozzi and A. A. Bonapasta, A hybrid zinc phthalocyanine/zinc oxide system for photovoltaic devices: a DFT and TDDFPT theoretical investigation, J. Mater. Chem., 2012, 22(2), 440–446 RSC.
- M. Grobosch, C. Schmidt, R. Kraus and M. Knupfer, Electronic properties of transition metal phthalocyanines: The impact of the central metal atom (d5–d10), Org. Electron., 2010, 11(9), 1483–1488 CrossRef CAS.
- G. Yang, Y.-L. Wang, J.-J. Xu, H.-W. Lei, C. Chen, H.-Q. Shan, X.-Y. Liu, Z.-X. Xu and G.-J. Fang, A facile molecularly engineered copper(II) phthalocyanine as hole transport material for planar perovskite solar cells with enhanced performance and stability, Nano Energy, 2017, 31, 322–330 CrossRef CAS.
- Y. Kim, T.-Y. Yang, N. Jeon, J. Im, S. Jang, T. Shin, H.-W. Shin, S. Kim, E. Lee, J. H. Noh, S. I. Seok and J. Seo, Engineering interface structures between lead halide perovskite and copper phthalocyanine for efficient and stable perovskite solar cells, Energy Environ. Sci., 2017, 10(10), 2109–2116 RSC.
- H. Qin, L. Xu and D. Zhong, First-Principles Study of Zinc Phthalocyanine Molecules Adsorbed on Methylammonium Lead Iodide Surfaces, J. Phys. Chem. C, 2020, 124(9), 5167–5173 CrossRef CAS.
- N. Marom, X. Ren, J. E. Moussa, J. R. Chelikowsky and L. Kronik, Electronic structure of copper phthalocyanine from G 0 W 0 calculations, Phys. Rev. B: Condens. Matter Mater. Phys., 2011, 84(19), 195143 CrossRef.
- G. Kresse and J. Furthmüller, Efficiency of ab initio total energy calculations for metals and semiconductors using a plane-wave basis set, Comput. Mater. Sci., 1996, 6(1), 15–50 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Generalized gradient approximation made simple, Phys. Rev. Lett., 1996, 77(18), 3865 CrossRef CAS PubMed.
- A. Tkatchenko and M. Scheffler, Accurate molecular van der Waals interactions from ground-state electron density and free-atom reference data, Phys. Rev. Lett., 2009, 102(7), 073005 CrossRef PubMed.
- A. V. Krukau, O. A. Vydrov, A. F. Izmaylov and G. E. Scuseria, Influence of the exchange screening parameter on the performance of screened hybrid functionals, J. Chem. Phys., 2006, 125(22), 224106 CrossRef PubMed.
- D. Qi, J. Sun, X. Gao, L. Wang, S. Chen, K. P. Loh and A. T. Wee, Copper Phthalocyanine on Hydrogenated and Bare Diamond (001)-2 × 1: Influence of Interfacial Interactions on Molecular Orientations, Langmuir, 2010, 26(1), 165–172 CrossRef CAS PubMed.
- J. H. Heo, S. H. Im, J. H. Noh, T. N. Mandal, C.-S. Lim, J. A. Chang, Y. H. Lee, H.-j. Kim, A. Sarkar and M. K. Nazeeruddin, Efficient inorganic–organic hybrid heterojunction solar cells containing perovskite compound and polymeric hole conductors, Nat. Photonics, 2013, 7(6), 486–491 CrossRef CAS.
- G. Volonakis and F. Giustino, Ferroelectric graphene–perovskite interfaces, J. Phys. Chem. Lett., 2015, 6(13), 2496–2502 CrossRef CAS PubMed.
- M. A. Green, Y. Jiang, A. M. Soufiani and A. Ho-Baillie, Optical properties of photovoltaic organic–inorganic lead halide perovskites, J. Phys. Chem. Lett., 2015, 6(23), 4774–4785 CrossRef CAS PubMed.
- O. Arillo-Flores, M. M. Fadlallah, C. Schuster, U. Eckern and A. Romero, Magnetic, electronic, and vibrational properties of metal and fluorinated metal phthalocyanines, Phys. Rev. B: Condens. Matter Mater. Phys., 2013, 87(16), 165115 CrossRef.
- I. E. Brumboiu, S. Haldar, J. Luder, O. Eriksson, H. C. Herper, B. Brena and B. Sanyal, Influence of electron correlation on the electronic structure and magnetism of transition-metal phthalocyanines, J. Chem. Theory Comput., 2016, 12(4), 1772–1785 CrossRef CAS PubMed.
- W. Tang, E. Sanville and G. Henkelman, A grid-based Bader analysis algorithm without lattice bias, J. Phys.: Condens. Matter, 2009, 21, 084204 CrossRef CAS PubMed.
- T. Duong, J. Peng, D. Walter, J. Xiang, H. Shen, D. Chugh, M. Lockrey, D. Zhong, J. Li and K. Weber, Perovskite solar cells employing copper phthalocyanine hole-transport material with an efficiency over 20% and excellent thermal stability, ACS Energy Lett., 2018, 3(10), 2441–2448 CrossRef CAS.
- B. P. Rand, D. Cheyns, K. Vasseur, N. C. Giebink, S. Mothy, Y. Yi, V. Coropceanu, D. Beljonne, J. Cornil and J. L. Brédas, The impact of molecular orientation on the photovoltaic properties of a phthalocyanine/fullerene heterojunction, Adv. Funct. Mater., 2012, 22(14), 2987–2995 CrossRef CAS.
- I. Avilov, V. Geskin and J. Cornil, Quantum-Chemical Characterization of the Origin of Dipole Formation at Molecular Organic/Organic Interfaces, Adv. Funct. Mater., 2009, 19(4), 624–633 CrossRef CAS.
- E. M. Kim, S. Javaid, J. H. Park and G. Lee, Edge Functionalized Graphene Nanoribbons with tunable band alignments for Carrier Transport Interlayer in Organic-Inorganic Perovskite Solar Cells, Phys. Chem. Chem. Phys., 2020, 22, 2955–2962 RSC.
- S. Javaid, M. J. Akhtar, E. M. Kim and G. Lee, Band gap tuning and excitonic effect in chloro-fluorinated graphene, Surf. Sci., 2019, 686, 39 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2021 |