
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceImpact of tensile and compressive forces on the hydrolysis of cellulose and chitin†
Hirokazu
Kobayashi
 *a,
Yusuke
Suzuki
ab,
Takuya
Sagawa
ac,
Kyoichi
Kuroki
ab,
Jun-ya
Hasegawa
a and
Atsushi
Fukuoka
*a
*a,
Yusuke
Suzuki
ab,
Takuya
Sagawa
ac,
Kyoichi
Kuroki
ab,
Jun-ya
Hasegawa
a and
Atsushi
Fukuoka
*a
aInstitute for Catalysis, Hokkaido University, Kita 21 Nishi 10, Kita-ku, Sapporo, Hokkaido 001-0021, Japan. E-mail: kobayashi.hi@cat.hokudai.ac.jp; fukuoka@cat.hokudai.ac.jp
bGraduate School of Chemical Sciences and Engineering, Hokkaido University, Kita 13 Nishi 8, Kita-ku, Sapporo, Hokkaido 060-8628, Japan
cDepartment of Industrial Chemistry, Faculty of Engineering, Tokyo University of Science, 12-1 Ichigayafunagawara-machi, Shinjuku-ku, Tokyo, 162-0826, Japan
First published on 5th June 2021
Abstract
Mechanochemistry enables unique reaction pathways in comparison to conventional thermal reactions. Notably, it can achieve selective hydrolysis of cellulose and chitin, a set of abundant and recalcitrant biomass, by solvent-free ball-milling in the presence of acid catalysts. Although the merits of mechanochemistry for this reaction are known, the reaction mechanism is still unclear. Here, we show how the mechanical forces produced by ball-milling activate the glycosidic bonds of carbohydrate molecules towards hydrolysis. This work uses experimental and theoretical evaluations to clarify the mechanism. The experimental results reveal that the ball-mill accelerates the hydrolysis by mechanical forces rather than local heat. Meanwhile, the classical and quantum mechanics calculations indicate the subnano to nano Newton order of tensile and compressive forces that activate polysaccharide molecules in the ball-milling process. Although previous studies have taken into account only the stretching of the molecules, our results show that compressive forces are stronger and effective for the activation of glycosidic bonds. Accordingly, in addition to stretching, compression is crucial for the mechanocatalytic reaction. Our work connects the classical physics of ball-milling on a macro scale with molecular activation at a quantum level, which would help to understand and control mechanochemical reactions.
Introduction
The applications of mechanochemical reactions have expanded to a wide range of research fields,1 including alloying,2 organic synthesis,3,4 polymer engineering,5,6 sensing7,8 and imaging.9 The synergistic effects of physical and chemical factors also play key roles in forming asymmetric organs in living matter.10–12 The mechanochemical approach provides characteristic results that are rarely achieved using conventional thermal reactions.Recently, the use of mechanical forces of ball-milling has attracted interest in the hydrolysis of carbohydrate polymers, specifically cellulose and chitin (Fig. 1).13,14 Cleavage of their β-1,4-glycosidic bonds by hydrolysis produces glucose and N-acetylglucosamine (NAG), respectively. These monomers are promising feedstock for sustaining our demand for energy and chemical products. However, low reactivity of the polymers has largely blocked this biorefinery pathway. Hydrolysis of chitin is especially difficult because the process currently needs, for example, excess concentrated hydrochloric acid and enzymes.15,16 The resulting high price of NAG has limited its use in cosmetic and health care applications. Nonetheless, the ball-milling method can selectively hydrolyse cellulose and chitin in the presence of catalytic amounts of acids, thus being attractive.17–25
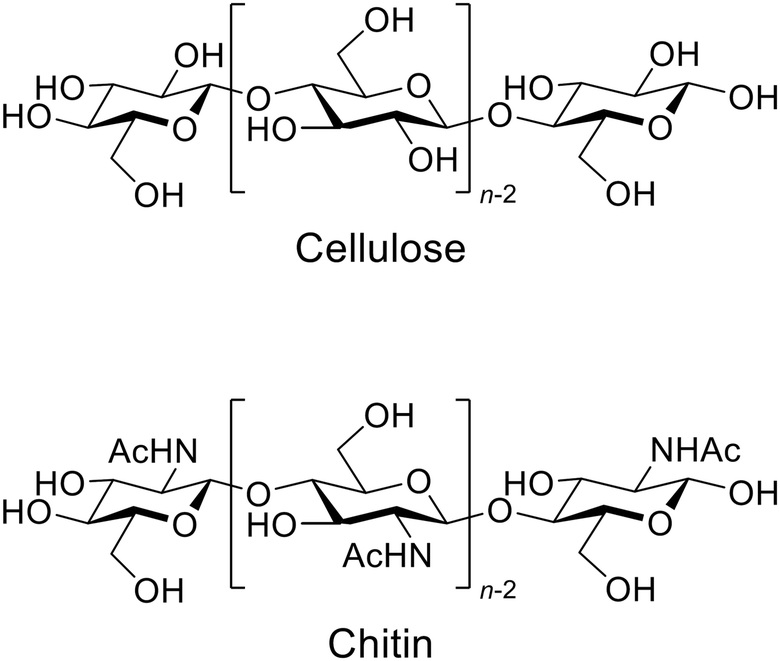 | ||
| Fig. 1 Structure of cellulose and chitin. Chitin contains amino groups instead of acetamide in part. | ||
In the development of mechanically-promoted hydrolysis, Blair et al. reported that the ball-milling of cellulose with a delaminated kaolinite produced soluble products such as glucose.17 Mechanical forces possibly assist the acid-catalysed hydrolysis reactions, which is called mechanocatalysis. Subsequent studies have shown that sulphuric acid catalyst further accelerates the conversion of cellulose in the milling and identified that the main products are glucans with β-1,4- and α-1,6-glycosidic bonds (Fig. 2, a2).18–21 These products are different from that of the conventional thermal reactions in water, namely β-1,4-glucans (h2). For the formation of α-1,6-linkage, instead of a water molecule (h1), a hydroxy group in the C6 position in another oligomer likely attacks an oxocarbenium ion intermediate (a1), which produces the unusual bond. The water-soluble properties of the products19 allow easy hydrolysis to glucose and further chemical conversion including the use of continuous flow systems.26–28 The mechanical treatment is applicable to the hydrolysis of chitin to oligomers.22,24,25 It is noteworthy that the acetamido groups in chitin are preserved during the mechanocatalytic process. This result is in contrast to that of liquid-phase thermal hydrolysis reactions using acid catalysts, where the hydrolysis of glycosidic bonds and amide bonds occurs in parallel.29 The oligomer product can be converted to NAG, its methyl glycoside and 2-acetamido-2-deoxysorbitol in good yields.22,30,31 Accordingly, mechanocatalytic hydrolysis is a useful method with different characteristics from thermal reactions.
 | ||
| Fig. 2 Scission of glycosidic bonds by ball-milling in the presence of acid catalysts. | ||
Clarification of the reaction mechanism is essential to understand the uniqueness of mechanocatalytic hydrolysis reactions. For a broader view of mechanochemical reactions, atomic force microscope (AFM) has been used to examine the effect of mechanical forces.32 Organic molecules attached to the probe of AFM were pulled by moving a cantilever with measuring force, which showed that tensile forces of a nano Newton order cleave a variety of covalent bonds. Another example is that a polymer containing spiropyran rings opens the rings by stretching the polymer.7 These insights motivated us to speculate that tensile stress produced by ball-milling selectively activates the glycosidic bonds of chitin existing within the long chain of the polymer.22 By contrast, amide bonds in side-chains might not receive strong forces. Very recently, a computational study clarified that tensile forces reduce the activation energy of the hydrolysis of glycosidic bonds.33 Furthermore, Hasegawa et al. theoretically demonstrated that tensile forces in the polymer chain direction change the conformation of protonated chitin molecules.34 This deformation facilitates the cleavage of glycosidic bonds to produce oxocarbenium ion intermediates for hydrolysis. By contrast, the amide bond is not activated even in the presence of tensile forces stretching the C–N bond. Under acidic conditions, the slowest elemental step in the hydrolysis of amide is water addition, which receives little effect from the force. This feature is different from that under basic conditions, where the C–N bond cleavage is slow.35 These studies provide an understanding of the effect of mechanical forces in part for the selective hydrolysis at a molecular level. However, no report has shown the relationship between the macroscale physical actions of ball-milling and the eventual cleavage of glycosidic bonds. Moreover, these studies have considered only the stretching of molecules.
In this work, we conceptually link the classical physics of ball collision and hydrolysis at the quantum chemistry level. Initially, our work demonstrates the difference between mechanocatalytic hydrolysis using a planetary ball-mill and thermal hydrolysis. The experimental results clarify that mechanical forces are essential for the acid-catalysed hydrolysis in the ball-milling processes. To understand the mechanism, our work employs a physical simulation that estimates the forces applied to the polysaccharide particles during ball-milling. Density functional theory (DFT) calculations reproduce the force-induced molecular deformation including both stretching and compression to understand the relationship between the macro-level ball collision and the nano-scale quantum chemistry. The accuracy of DFT in energy for such unusual chemical structures is proved by comparing with the molecular orbital methods including multi reference systems. Finally, we discuss the correlation between the deformation and energy profiles for hydrolysis, which reveals that compressive forces may play important roles in hydrolysis, in addition to tensile forces.
Results and discussion
Comparison of mechano- and thermo-catalytic hydrolysis
Ball-mill produces mechanical forces and local heat on the substrate by the collision of balls, at the same time. To clarify which predominantly drives the hydrolysis, we compared the ball-milling and thermal reactions. In these tests, cellulose and chitin were impregnated with H3PO4 catalyst. Physisorbed water on the samples (ca. 5 wt%) hydrolysed the substrates. No solvent was used because the milling was carried out under dry conditions. During ball-milling at room temperature, the milling energy elevated the temperature of the sample to 40 to 50 °C. Due to the local heat formation by ball collision, the temperature instantly further increases. Therefore, the reaction temperature for thermal hydrolysis was set at 60–120 °C. Additionally, the ball-mill commonly amorphises cellulose and chitin, which motivated us to use amorphous substrates in the thermal system for better comparison (Fig. S1, ESI†).First, we conducted the hydrolysis of cellulose using ball-mill or heat, and compared the oligomer products of the two systems. Ball-milling H3PO4-impregnated cellulose gave an off-white solid powder having 65% solubility in water. As cellulose is insoluble in water even in amorphous form, the solubility indicates the formation of water-soluble products in 65% yield. The soluble fraction contained glucose and its oligomers, corresponding to 63% yield (Fig. 3A), viz., a good material balance. Cellulose contained only β-1,4-glycosidic bonds to connect glucose units, but the nuclear magnetic resonance (NMR) analysis showed that the produced oligomers had a small number of α-1,6-bonds in addition to them (Fig. S2, ESI†). Furthermore, β-1,6-anhydride structures were found at the reducing ends in a small ratio. These structures are produced by inter- and intra-molecular attack of the C6–OH groups to the oxocarbenium ion, the active intermediate given by the cleavage of glycosidic bonds (α-1,6 formation: Fig. 2 right side; anhydride: Fig. S3, ESI†). In contrast to the ball-milling system, thermal hydrolysis of amorphous cellulose using the same acid produced a blackish brown solid material at 120 °C (Fig. 3A), thus suggesting the formation of carbonaceous materials and humins. The solid contained glucose oligomers linked with various kinds of glycosidic bonds as indicated by NMR analysis (Fig. S2, ESI†). This is different from the observation that thermal hydrolysis in water commonly produces oligomers with β-1,4-glycosidic bonds and glucose.36,37 Due to lack of solvent water, the thermal reaction is reversible under the reaction conditions, which eventually gives a variety of glycosidic bonds based on thermodynamic equilibrium (Fig. S3, ESI†).38,39 At the optimised reaction time (6 h, see Table S1, ESI† for details), the solubility of the sample was high (78%), but the total yield of the oligomers and glucose was only 25% (Fig. 3A). Thermal hydrolysis was significantly less selective than ball-milling hydrolysis.
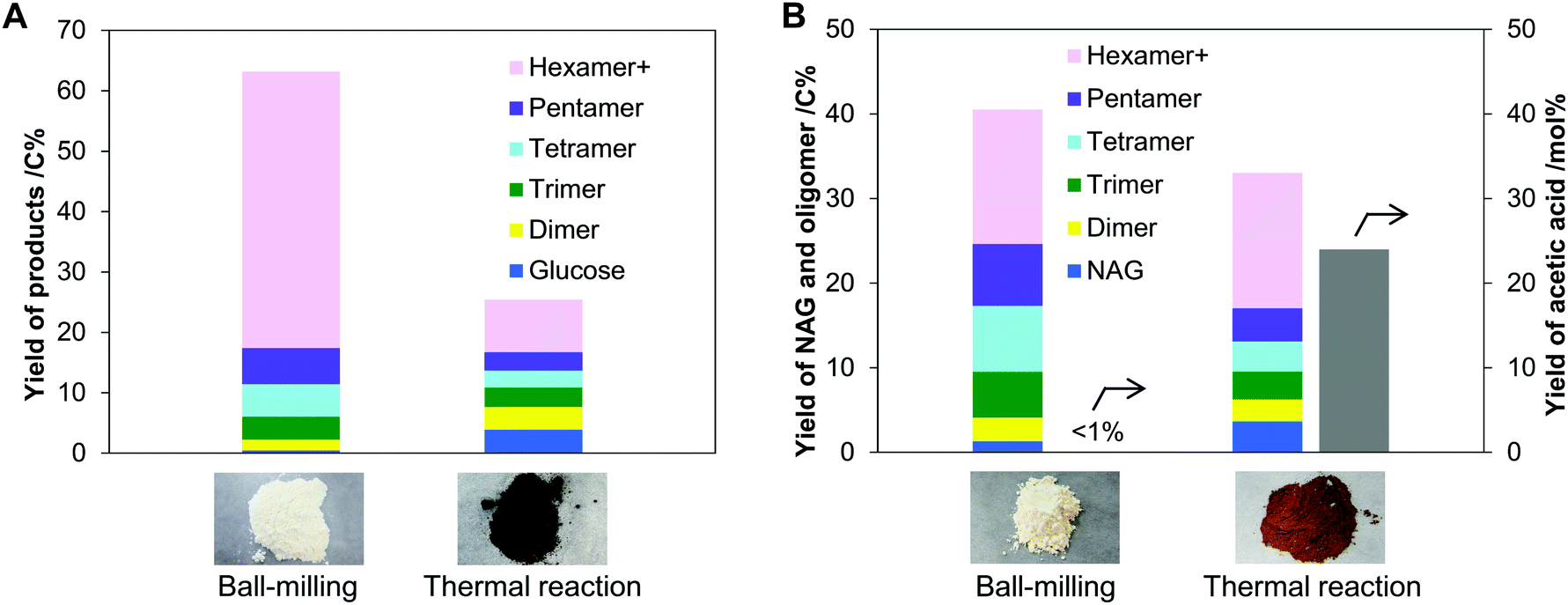 | ||
| Fig. 3 Hydrolysis with H3PO4 catalyst by the ball-milling and thermal methods: (A) cellulose and (B) chitin. Reaction conditions (common): substrate/catalyst ratio (S/C) = 5.0 mol-monomer unit per mol, no solvent, adsorbed water content ca. 5 wt%. Ball-milling: sample 5.0 g, 500 rpm, ø5 Al2O3 balls 100 g, 25 °C, and 12 h. Thermal reaction: sample 1.0 g, 120 °C, 6 h for cellulose, 3 h for chitin (the optimum times). The pictures show samples after the reactions. “Hexamer+” indicates hexamer and larger oligomers. | ||
Hydrolysis of chitin was performed to show an essential difference in selectivity between ball-milling and thermal reactions. The ball-milling process produced NAG in 1.3% yield and oligomers in 39% yield (Fig. 3B). The NAG oligomers contained β-1,4-glycosidic bonds with a small fraction of 1,6-bonds (Fig. S4, ESI†), similar to glucose oligomers produced by ball-milling. We emphasise no formation of acetic acid (AcOH) by the hydrolysis of amide groups in chitin. In a sharp contrast, a thermal reaction at 120 °C gave a large amount of AcOH (24 mol% yield) in addition to NAG (3.7%) and oligomers (29%). Testing under varied reaction temperatures (60–120 °C) and time intervals (0.5–12 h) has shown that the deacetylation side reaction is unavoidable in the thermal hydrolysis system (Table S2, ESI†).
Consequently, the selectivity difference between ball-milling and thermal reactions is obvious. We conclude that the mechanical treatment accelerates the hydrolysis reaction mainly by mechanical forces.
Classical mechanics for ball collision
A simple model of planetary milling with one ball was employed to estimate a representative collision velocity of the ball to polysaccharide samples on the pot wall (details: Fig. S5, ESI†). Fig. 4A shows the resulting trajectory of the ball, which was determined with particular parameters shown in Methods. The calculation reveals that the ball departs from the pot wall (named detachment), flies in the pot, and eventually hits the pot wall (denoted collision) at a velocity of 4.08 m s−1 from almost the vertical direction.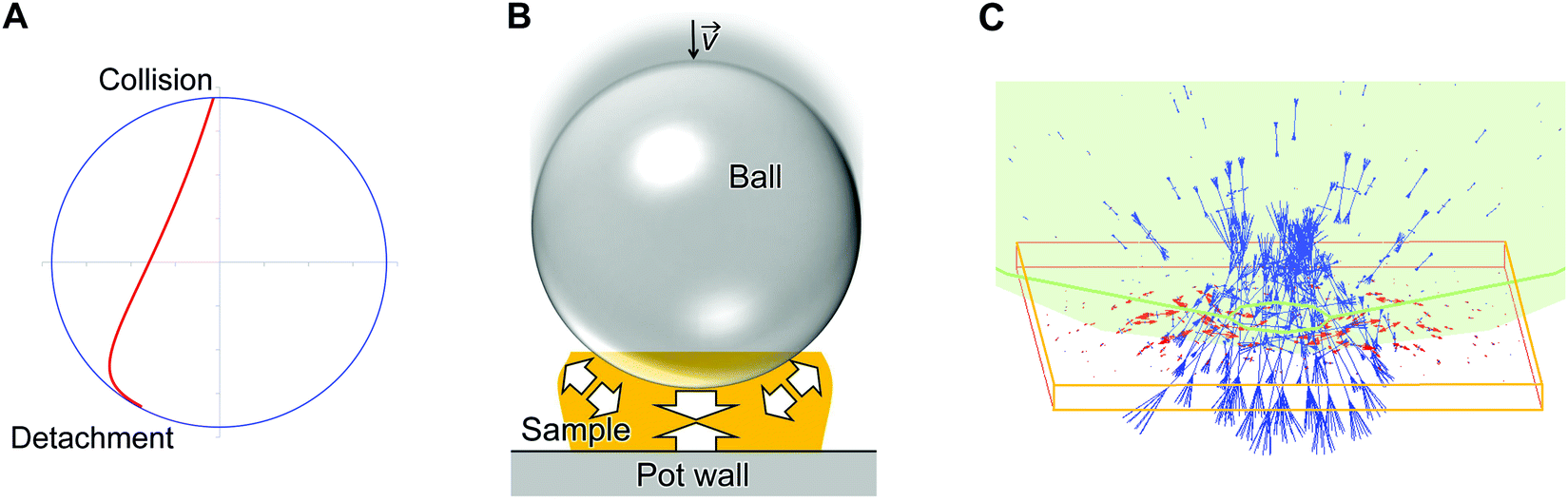 | ||
| Fig. 4 Collision of ball to polysaccharide samples in planetary milling. (A) Trajectory of the flying ball shown in an internal coordinate of the pot until the collision. Blue circle indicates inner wall of the pot, and red line indicates the trajectory. (B) Schematic for the collision of the ball to a cellulose or chitin particle on the pot wall. The transparent orange rectangle indicates the sample. The white arrows schematically represent the forces produced by collision. (C) A typical result of the collision simulation. The transparent green object indicates the ball. Blue and red lines show compressive and tensile stresses, respectively. | ||
We estimated forces applied to cellulose and chitin particles by the ball collision with a finite element method (denoted FEM; see Methods). This simulation supposed that a ball with a diameter of 5 mm caused an elastic collision to a flat plane of the samples from the vertical direction (Fig. 4B and C). The ball was made of α-Al2O3 and had an initial ball velocity of 4.08 m s−1, to be consistent with the actual experiments and the aforementioned calculation. The thickness of the cellulose and chitin samples was changed from 0.08 to 0.4 mm as plausible values based on the experimental observation (Fig. S6, ESI†).
In the initial calculation, we supposed the collision of the ball to a cellulose sample with 0.4 mm thickness having an isotropic Young's modulus of 137 GPa, the value observed for polymer chain direction in crystalline cellulose.40 As a result, the collision made the maximum compressive and tensile stresses of 4.9 GPa and 0.66 GPa, respectively, at 2.6 μs after starting the collision (Table 1, entry 1). The stress deformed the sample, and the degree was 2.0% around the centre point of collision. To verify the accuracy of the FEM, the simplified condition allowed us to analytically approximate the collision equations estimating compressive forces as shown in the ESI† (Fig. S7 and eqn (S3)–(S5)). The calculation has shown that the maximum compressive force is 4.6 GPa, which is similar to that determined by the FEM (4.9 GPa).
| Entry | Sample | Thickness/mm | Collision timef/μs | Deformationf/% | Maximum stress/GPa | |
|---|---|---|---|---|---|---|
| Compression | Tension | |||||
| a Using isotropic Young's modulus of 137 GPa. b Collision in the direction of polymer chain. c Collision in the transverse direction. d Amorphous cellulose. e Using isotropic Young's modulus of 59 GPa. f Time and deformation degree at the largest stress found. g Actual stress is larger because the elastic modulus of amorphous cellulose increases by deformation, and moreover, stress concentration happens due to the disordered molecular arrangement. | ||||||
| 1 | Cellulosea | 0.4 | 2.6 | 2.0 | 4.9 | 0.66 |
| 2 | Celluloseb | 0.08 | 2.3 | 7.5 | 7.4 | 1.0 |
| 3 | Celluloseb | 0.4 | 3.3 | 2.3 | 4.2 | 0.65 |
| 4 | Cellulosec | 0.08 | 5.3 | 17 | 4.7 | 1.5 |
| 5 | Cellulosec | 0.4 | 7.3 | 6.4 | 2.7 | 0.73 |
| 6 | Cellulosed | 0.08 | 6.8 | 23 | (2.7)g | (0.35)g |
| 7 | Cellulosed | 0.4 | 12 | 8.1 | (1.2)g | (0.17)g |
| 8 | Chitine | 0.08 | 2.5 | 8.6 | 6.3 | 0.80 |
| 9 | Chitine | 0.4 | 4.0 | 2.9 | 3.6 | 0.48 |
The FEM was extended to various conditions for the collision to cellulose. First, we took into account the fact that cellulose has a anisotropic elastic moduli of 137 GPa for the direction of polymer chain and ca. 10 GPa41 for the transverse direction. The collision from the polymer chain direction gave compressive stresses in the range of 4.2–7.4 GPa and tensile stresses in the range of 0.65–1.0 GPa (Table 1, entries 2 and 3), and that from the axial face provided their intensities in the range of 2.7–4.7 GPa and 0.73–1.5 GPa, respectively (entries 4 and 5). Subsequently, the collision of the ball to amorphous cellulose (Young's modulus 5.6 GPa)42 was calculated. This condition gave 1.2 to 2.7 GPa of compressive stresses (entries 6 and 7), but the actual strength should be larger because the elastic modulus of amorphous cellulose gradually increases with the compression.43 Further examinations with changing the collision angle and including friction coefficients indicated compressive (1.3–4.9 GPa) and tensile stresses (0.56–1.5 GPa) (Table S3, ESI†) similar to those described above.
Finally, chitin samples with 0.08 to 0.4 mm thickness were hit by the Al2O3 ball, where Young's modulus of 59 GPa44 was used for the polymer chain direction. The elastic modulus lower than that of cellulose (137 GPa) is mainly due to the slightly bent conformation of chitin molecules in the crystal, so that the deformation is easier than that of cellulose.44 The calculations provided similar results (compressive stress 3.6–6.3 GPa, tensile stress 0.48–0.80 GPa; Table 1, entries 8 and 9) to those for cellulose.
Based on the aforementioned results, we conclude that cellulose and chitin are deformed by a gigapascal order of stresses. Compressive stresses are stronger than tensile ones during the collision event. Multiplying the stress intensity by the cross-sectional area of one carbohydrate molecule in their polymer chain direction (0.32 nm2 for cellulose, 0.44 nm2 for chitin)45,46 gives roughly a subnano to nano Newton order of forces. For example, the maximum compressive and tensile stresses found in our calculations (7.4 GPa and 1.5 GPa) correspond to 2.4 nN and 0.48 nN, respectively, for a cellulose molecule. This magnitude of forces would be applied to pull or push the molecular chains.
Molecular-level effect of the mechanical forces
We calculated the energy and force required for the deformation of cellulose and chitin molecules, for which cellobiose and N,N’-diacetylchitobiose were employed as their minimal models. Among deformation types, previous studies have shown that elongation of molecular chain is impactful for activation of polymers.33,34,47–49 Meanwhile, our calculations based on classical mechanics have suggested that compressive force is stronger than tension in planetary milling. Therefore, this work considers the effect of compression in addition to that of stretching. The B3LYP/6-31+G(d,p) level of theory50–52 was utilised for this purpose owing to the good accuracy as evaluated in our benchmark tests (see Methods and Fig. S8, S9 and Table S4, ESI†).To clarify the effect of tensile forces, after optimising the conformation of cellobiose, two oxygen atoms at the tips of the molecule were separated up to 13% away from the original distance (Fig. 5A). As described below, this elongation degree was large enough to consider the effect of tensile forces produced during the ball-milling process. Subsequently, at each elongation degree, the geometry of atoms other than the two oxygen atoms was optimised to minimise the electronic energy. This process, called the COGEF method, accurately estimates the effect of bond elongation.53,54 Stretching the molecule by 13% required 207 kJ mol−1, which was still smaller than the bonding energies of C–C and C–O (>300 kJ mol−1). Covalent bonds are unlikely cleaved in this range of stretching. The tensile force given as the derivative of the potential curve almost linearly increased to 4.9 nN with increasing distance of the two oxygen atoms by 12% (Fig. 5A, blue line), namely, harmonic oscillator-like behaviour in this range. The force–elongation relationship corresponds to a molecular Young's modulus of 130 GPa (4.9 nN/0.12/0.32 nm2 = 1.3 × 1011 N m−2), which is close to the experimental value for crystalline cellulose (137 GPa). N,N′-Diacetylchitobiose provided a similar result to cellobiose; the force was almost proportional to the stretching amount and reached 5.0 nN at 12% elongation (Fig. 5B, blue line).
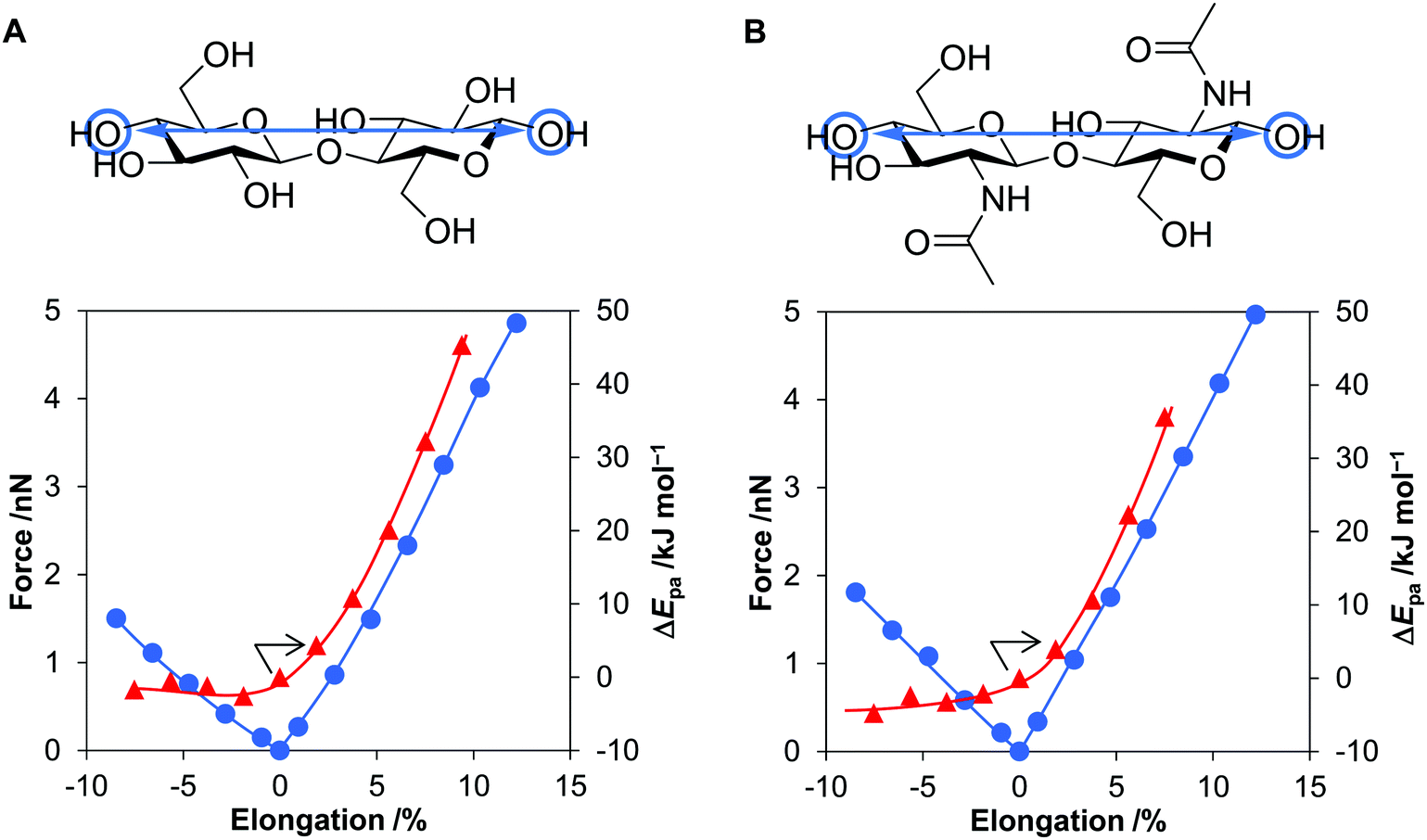 | ||
| Fig. 5 Effects of elongation or compression of sugars: (A) cellobiose and (B) N,N’-diacetylchitobiose. Blue circles show forces and red triangles indicate changes in proton affinity (ΔEpa) by deformation. | ||
As the second type of force, cellobiose and N,N′-diacetylchitobiose molecules were compressed in the direction of the polymer chain (Fig. 5, minus direction in the x-axis). Due to bending of the molecules, the O–O length was shortened with 45–55% of the forces required for stretching. Therefore, compression is easier than stretching.
We also focused on the changes in proton affinity (Epa) at the deformed states. The stretching-induced increase in Epa was proposed by Beyer.55 Indeed, previous work suggested the selective increase in basicity of the O atom in a glycosidic bond under the conditions.33 Furthermore, a natural bond orbital (NBO) analysis showed increased electron density on the O atom in glycosidic bonds at the stretched states.34 Therefore, our DFT calculations quantitatively evaluated ΔEpa by the deformation of cellobiose and N,N′-diacetylchitobiose (Fig. 5, red lines). The value increased in a convex downward curve with increasing elongation degree for both molecules. For instance, a 5.6% elongation of cellobiose elevated the energy by 20 kJ mol−1, corresponding to an increase in pKa value by 3.5 at 298 K. Accordingly, the stretching facilitates the protonation process in terms of thermodynamic equilibrium. In contrast, compression gave almost no influence on the value. The increase in Epa is specific to the elongation.
We calculated energy diagrams for the acid-catalysed scission of a glycosidic bond of stretched cellobiose (Fig. 6A, C1–O4′ elongation). H3PO4 coordinated to the glycosidic bond to reproduce our real experimental conditions. The obtained potential curves are slightly different from actual ones under force fields, as the COGEF method does not give a constant force during the reaction. For the conformation in the absence of forces, namely, no stretching of the molecule, the potential curve was continuously in an uphill nature along the C1–O4′ bond length (Fig. 6C, black line). Eventually, the elongation produced an oxocarbenium ion and a glucose molecule at 121 kJ mol−1, which was similar to typical activation energies found in the thermocatalytic hydrolysis of cellobiose by acids.56 In the transient state (C1–O4’ = 0.18 nm), an E3 conformation57 was observed (Fig. 6B, “Original”; atomic coordinate Table S5, ESI†). This structure is similar to the 4E structure found in a general mechanism for cellulose hydrolysis.58 In both conformations, C1–O4′ bond stands perpendicular to the ring, and therefore the glycosidic bond is cleaved in an anti-periplanar conformation against a lone pair of O5 (Fig. S10, ESI†). This is an analogue of typical E2 reactions.59
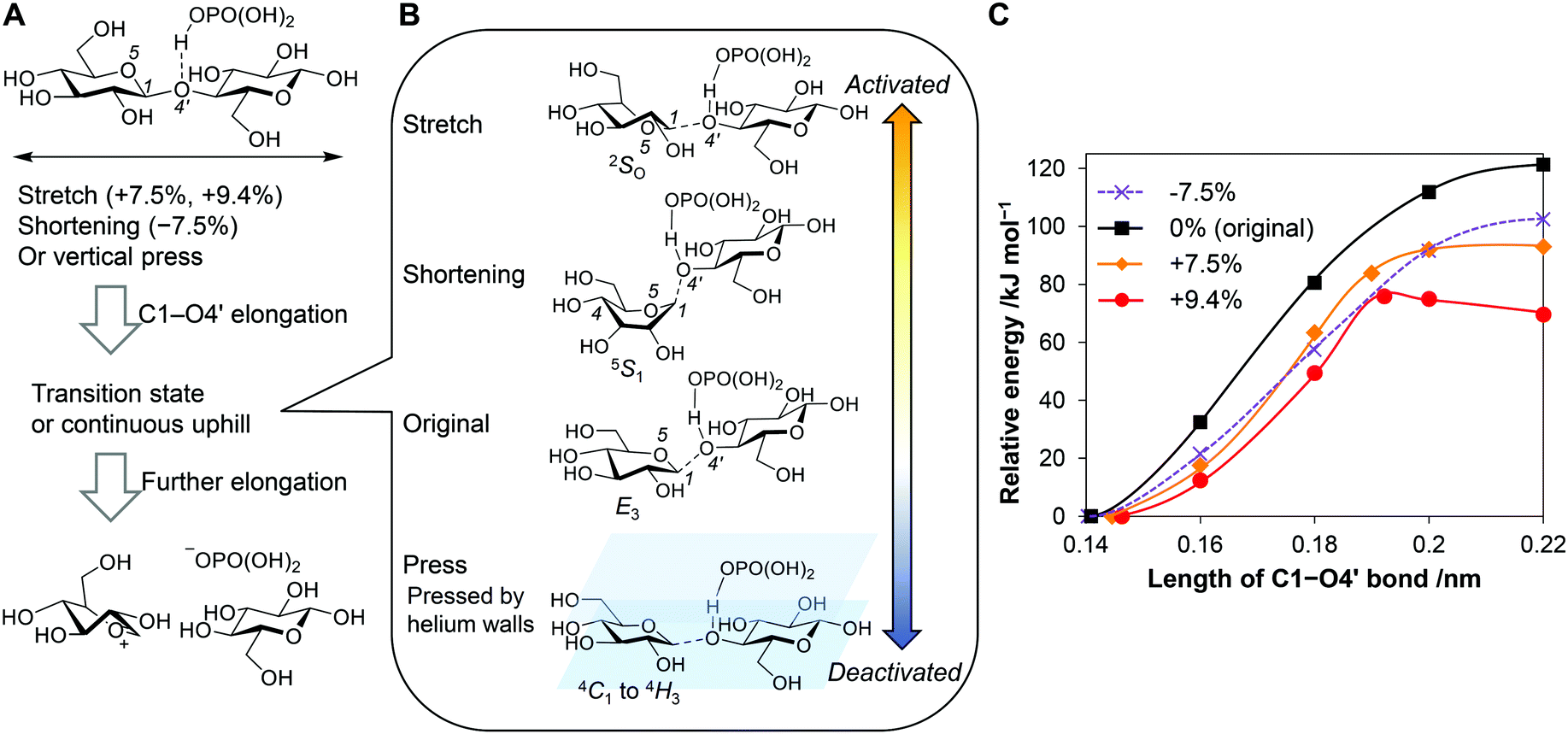 | ||
| Fig. 6 Scission of glycosidic bond of cellobiose in the presence of forces. (A) Schematic of the reaction. (B) Structures at transition or the transient states. (C) Potential curves as a function of C1–O4′ bond length for the stretched or shortened molecule. H3PO4 coordinates on O4′ of cellobiose as an acid catalyst. | ||
Stretching of the molecule facilitated the cleavage of glycosidic bonds. The deformation decreased the reaction barrier to 93 kJ mol−1 at 7.5% elongation and to 76 kJ mol−1 at 9.4% (Fig. 6C, orange and red lines). The carbohydrate molecule receives forces for microseconds during the ball collision, whereas chemical reactions occur in femto- to pico-seconds.60 Due to the different time scales, the molecule has a large chance to overcome the lowered potential barrier in the presence of tensile forces. However, the forces required to stretch the molecules by 7.5% and 9.4% (>2 nN; Fig. 5A) are larger than the estimated tensile forces in the classical mechanics calculations (up to 0.48 nN). A cellulose sample only infrequently receives tensile forces strong enough to be activated, for example, when stress concentration happened by a disordered arrangement of molecules or lever-arm effects.61,62
For the system at 9.4% elongation, a maxima was found in the reaction coordinate (Fig. 6B, “Stretch”; atomic coordinate Table S6, ESI†). Notably, the structure is 2SO, which is suitable to keep the long length of the molecule. This structure gives a syn-periplanar conformation between the C1–O4′ bond and a lone-pair of O5 for the dissociation (Fig. S10, ESI†). Thermal reactions rarely experience elimination reactions in this conformation.59 Tensile forces not only decrease the barrier of the reaction but also can change the reaction mechanism.
Shortening the molecular length of cellobiose by 7.5% decreased the barrier height to 102 kJ mol−1 (Fig. 6C, purple line; atomic coordinate Table S7, ESI†). The force offered a twisted boat conformation (5S1) having a shorter distance between C1 and C4 than that in the armchair form (Fig. 6B, “Shortening”). The conformation enables an anti-periplanar type elimination, which is similar to the 4E and E3 structures. Accordingly, the compression also facilitates the hydrolysis reaction. The activation degree is lower and the reaction mechanism is different, compared to the stretching. It is noteworthy that the 7.5% shortening requires a 1.6 nN of compressive force, while our simulation of ball collision has indicated that forces up to 2.4 nN are available. Therefore, even though compressive forces activate glycosidic bonds less strongly than tensile ones, the better availability may lead to similar or rather higher contributions in the mechanocatalytic hydrolysis.
As an additional discussion, we evaluated the possibility that the tensile force directly breaks the glycosidic bonds. DFT calculations have suggested that this is possible when a naked proton activated the glycosidic bond (Fig. S11, ESI†). However, under such conditions, the thermal hydrolysis reaction takes place very rapidly (activation energy 7.4 kJ mol−1) before the force cleaves the bond. Moreover, the reaction could produce an unusual bicyclic compound (Fig. S12, ESI†) that has not been observed in actual experiments. Thus, the direct cleavage of glycosidic bonds by forces is likely a minor route in the ball-milling reactions. The details are discussed in the ESI.†
Besides, we pressed cellobiose from its axial face by sandwiching the molecule within inert walls made of helium (Fig. 6B, “Press”). A number of He atoms were placed under and over a cellobiose molecule, and then the He walls compressed the cellobiose molecule by exchange interactions (see Fig. S13, ESI† for details). However, the activation energy increased (127 kJ mol−1 at a wall distance of 7 Å and 173 kJ mol−1 at 5 Å, Fig. S13 and Table S8, ESI†). Thus, this force deactivates glycosidic bonds.
Conclusions
Planetary milling accelerates the hydrolysis of cellulose and chitin impregnated with an acid catalyst. Rather than local heat, mechanical forces play predominant roles in the enhancement. Thus, the reaction is categorised as mechanocatalysis. Our physical modelling of the collision of an Al2O3 ball to the polysaccharide particles during the milling process estimates that the collision gives subnano to nano Newton order of forces to the molecules. Compressive forces are larger than tensile ones. DFT calculations indicate that the tension in the direction of the polymer chain activates the glycosidic bonds of the polysaccharide molecules. However, the force required is larger than the estimated available strength. Therefore, the tensile force-driven activation may not be very frequent during the milling process. By contrast, compressive forces in the same direction less strongly activate the chemical bonds, but the mechanical treatment provides forces large enough to do so. Accordingly, these results suggest that compressive forces are as important as or more important than tensile forces. As our DFT calculations used dimer models, to clarify the applicability of this finding to actual large polymers within solids is a potential subject in the future.In a different viewpoint, our work indicates the contradiction that ball-milling primarily gives compressive forces, whereas tensile ones are more effective in activating polysaccharide molecules. If a new mechanical method is developed to apply tensile forces to samples selectively and efficiently, it will probably improve the reaction efficiency. These insights show the importance of an integrated view of macro-scale mechanics and nano-scale chemistry and would assist in understanding and controlling various mechanochemical reactions.
Methods
Materials
Microcrystalline cellulose was obtained from Aldrich (Avicel PH-101). Chitin, 85% H3PO4 and diethyl ether were received from Wako Pure Chemical Industry.Hydrolysis reactions
For the mechanocatalytic hydrolysis, a biomass sample (5.0 g) was added to a diethyl ether solution of H3PO4 (15 mL) and dried using a rotary evaporator at 50 kPa at 303 K. The substrate/catalyst ratio was 5.0 mol-monomer unit per mol. The resulting sample was milled with α-Al2O3 balls (∅ 5, 100 g) in an alumina pot (Fritsch; 250 mL, inner diameter 74.8 mm) in a planetary mill (Fritsch, P-6; revolution diameter 105 mm, revolution–rotation ratio = 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :−1.82) at 500 rpm for a designated time. 100 mg of the solid product was added to 10 mL of water and sonicated for 10 min at 25 °C. After filtrating the solution using a hydrophilic polytetrafluoroethylene membrane (pore 0.1 μm), the solution was analysed by a high-performance liquid chromatograph (HPLC; Shimadzu, Prominence) equipped with three SB802.5HQ columns (Shodex; ∅ 8, 300 mm length) connected in series and a refractive index detector (RID). The mobile phase was pure water. Yields of sugar compounds were calculated based on the number of carbon atoms (C%). Acetic acid was quantified with an Aminex HPX-87H column (Bio-rad; ∅ 7.8, 300 mm length) using 5 mM H2SO4 aqueous solution as the mobile phase. Yield of acetic acid was defined as mole of acetic acid/mole of NAG unit. The solid residue on the membrane filter was weighed after drying in an oven at 110 °C to calculate the solubility of the product.
:−1.82) at 500 rpm for a designated time. 100 mg of the solid product was added to 10 mL of water and sonicated for 10 min at 25 °C. After filtrating the solution using a hydrophilic polytetrafluoroethylene membrane (pore 0.1 μm), the solution was analysed by a high-performance liquid chromatograph (HPLC; Shimadzu, Prominence) equipped with three SB802.5HQ columns (Shodex; ∅ 8, 300 mm length) connected in series and a refractive index detector (RID). The mobile phase was pure water. Yields of sugar compounds were calculated based on the number of carbon atoms (C%). Acetic acid was quantified with an Aminex HPX-87H column (Bio-rad; ∅ 7.8, 300 mm length) using 5 mM H2SO4 aqueous solution as the mobile phase. Yield of acetic acid was defined as mole of acetic acid/mole of NAG unit. The solid residue on the membrane filter was weighed after drying in an oven at 110 °C to calculate the solubility of the product.
For thermal hydrolysis, a biomass sample was milled for 2 h to make it amorphous, and afterward, the sample was impregnated with H3PO4 in the same way. Initial amorphisation was performed so that structures of the substrates were similar in mechanocatalytic and thermal reactions. The sample (1.0 g) was put into a closed pressure-resistant glass tube (20 mL) in an oil bath to keep water in the system. The thermal reaction was carried out at a certain temperature (60–120 °C) for a designated time (0.5–12 h). Products were analysed in the same manner as above.
Nuclear magnetic resonance (NMR) analysis of the products was performed using an ECX-600 (JEOL; 1H 600 MHz) after dissolving the sample in D2O. 2,2-Dimethyl-2-silapentane-5-sulphonic acid sodium salt (DSS) was used as an internal standard.
Classical mechanics calculations
We estimated the velocity of a ball in planetary milling using a mathematical model.63 In the calculations, slipping of the ball on a pot wall, gravity and air resistance were not included because their effect on ball velocity was small as shown in the literature.63 Details for the derivation of equations are shown in the ESI† (Fig. S4, eqn (S1) and (S2)).A finite element method was used to estimate the force generated by ball collision on a rectangular shape of cellulose or chitin sample on the physical simulator named Femtet (version 2020.1; Murata Software). The program divided matter into a set of tetrahedrons and optimised the position of vertexes to balance forces using an iterative solver. The parameters used were as follows: α-Al2O3 ball with 5 mm diameter; mesh size 0.7 mm for the ball, 0.2 mm for the sample; Young's modulus 400 GPa for α-Al2O3, designated values for cellulose and chitin; and Poisson's ratio 0.30 for cellulose and chitin.64 A flat surface on the polygonised ball hit the sample not to overestimate compressive forces. Non-linear deformation factors were included in the calculations so that a large deformation gave an accurate force of repulsion.
Quantum calculations
The calculations were performed using the Gaussian 16 (rev. A03) software. The solvent effect was included by the self-consistent reaction field method with an integral equation formalism polarised continuum model (IEFPCM).65 A previous related work also utilised a similar polarised continuum model to include the solvent effect.49 Diethyl ether was used to reproduce the solvation effect of solid cellulose and chitin due to similar dielectric constants.66 Based on the Born–Oppenheimer approximation,67 the potential energy curve for the bond elongation or shortening was obtained from the electronic energies with fixing nucleus at each position. We did not include the kinetic energy of nucleus in the total energy of the system, as the inclusion naturally distorted the harmonic oscillator-like energy curve.As the calculation level, we employed the Hartree–Fock/DFT hybrid functional named B3LYP with the Pople's double-ζ basis set 6-31+G(d,p)50–52 owing to good accuracy in energy as follows. To evaluate the usability of DFT for the unusual chemical structures in the presence of forces, we compared B3LYP with the full configuration interaction (full-CI) in bond elongation of H2 and with the multi-configurational second-order perturbation theory (CASPT2)68 in that of CO (Fig. S7 and S8, ESI†). B3LYP provided potential curves similar to those of the references in both cases, showing good reliability in terms of energy. For the benchmark of DFT functionals and basis sets, we calculated energies of bond elongation and protonation of dimethyl ether as the minimal model of a glycosidic bond, using B3LYP, CAM-B3LYP,69 M06-2X,70 MN1571 and ωB97XD72 with aug-PC-173 and 6-31+G(d,p). The results were compared with reference data obtained by means of the coupled cluster with single, double and perturbative triple excitations [CCSD(T)].74 This benchmark test showed that B3LYP/6-31+G(d,p) was the best of the combinations tested (Table S3, ESI†).
Author contributions
H. K. and A. F. wrote the manuscript. Y. S. and T. S. performed experiments. K. K. provided preliminary analysis to distinguish mechanochemical and thermal reactions. H. K. and Y. S. carried out classical mechanics calculations. H.K. performed quantum calculations by discussing with J. H. A. F. supervised this project. All the authors approved the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by Grant-in-Aid for Scientific Research (B) (KAKENHI, 18H01781; 20H02685; 21H01973) from Japan Society for the Promotion of Science (JSPS). We utilised the NMR instrument in the Open Facility of Institute for Catalysis (ICAT), Hokkaido University. Computations were partially performed using the computation system in Research Cluster for Sustainable Catalyst, ICAT.References
- J.-L. Do and T. Friščić, ACS Cent. Sci., 2017, 3, 13–19 CrossRef CAS.
- M. Vaidya, G. M. Muralikrishna and B. S. Murty, J. Mater. Res., 2019, 34, 664–686 CrossRef CAS.
- J. Andersen and J. Mack, Green Chem., 2018, 20, 1435–1443 RSC.
- G.-W. Wang, Chem. Soc. Rev., 2013, 42, 7668–7700 RSC.
- M. A. Ghanem, A. Basu, R. Behrou, N. Boechler, A. J. Boydston, S. L. Craig, Y. Lin, B. E. Lynde, A. Nelson, H. Shen and D. W. Storti, Nat. Rev. Mater., 2021, 6, 84–98 CrossRef CAS.
- T. Matsuda, R. Kawakami, R. Namba, T. Nakajima and J. P. Gong, Science, 2019, 363, 504–508 CrossRef CAS PubMed.
- D. A. Davis, A. Hamilton, J. Yang, L. D. Cremar, D. Van Gough, S. L. Potisek, M. T. Ong, P. V. Braun, T. J. Martinez, S. R. White, J. S. Moore and N. R. Sottos, Nature, 2009, 459, 68–72 CrossRef CAS PubMed.
- D. Yildiz, C. Baumann, A. Mikosch, A. J. C. Kuehne, A. Herrmann and R. Göstl, Angew. Chem., Int. Ed., 2019, 58, 12919–12923 CrossRef CAS PubMed.
- T. Seki, N. Tokodai, S. Omagari, T. Nakanishi, Y. Hasegawa, T. Iwasa, T. Taketsugu and H. Ito, J. Am. Chem. Soc., 2017, 139, 6514–6517 CrossRef CAS.
- T. Mammoto and D. E. Ingber, Development, 2010, 137, 1407–1420 CrossRef CAS.
- M. Hashimoto, K. Shinohara, J. Wang, S. Ikeuchi, S. Yoshiba, C. Meno, S. Nonaka, S. Takada, K. Hatta, A. Wynshaw-Boris and H. Hamada, Nat. Cell Biol., 2010, 12, 170–176 CrossRef CAS.
- A. Erzberger, A. Jacobo, A. Dasgupta and A. J. Hudspeth, Nat. Phys., 2020, 16, 949–957 Search PubMed.
- Q. Zhang and F. Jérôme, ChemSusChem, 2013, 6, 2042–2044 CrossRef CAS PubMed.
- E. Calcio Gaudino, G. Cravotto, M. Manzoli and S. Tabasso, Chem. Soc. Rev., 2021, 50, 1785–1812 RSC.
- M. Ishikawa, F. Nanjo and K. Sakai, Pat. JP1822027, 1994.
- N. Yan and X. Chen, Nature, 2015, 524, 155–157 CrossRef CAS.
- S. M. Hick, C. Griebel, D. T. Restrepo, J. H. Truitt, E. J. Buker, C. Bylda and R. G. Blair, Green Chem., 2010, 12, 468 RSC.
- N. Meine, R. Rinaldi and F. Schüth, ChemSusChem, 2012, 5, 1449–1454 CrossRef CAS PubMed.
- A. Shrotri, L. K. Lambert, A. Tanksale and J. Beltramini, Green Chem., 2013, 15, 2761–2768 RSC.
- S. Furusato, A. Takagaki, S. Hayashi, A. Miyazato, R. Kikuchi and S. T. Oyama, ChemSusChem, 2018, 11, 888–896 CrossRef CAS PubMed.
- P. Dornath, H. J. Cho, A. Paulsen, P. Dauenhauer and W. Fan, Green Chem., 2015, 17, 769–775 RSC.
- M. Yabushita, H. Kobayashi, K. Kuroki, S. Ito and A. Fukuoka, ChemSusChem, 2015, 8, 3760–3763 CrossRef CAS.
- X. Chen, H. Yang, Z. Zhong and N. Yan, Green Chem., 2017, 19, 2783–2792 RSC.
- G. Margoutidis, V. H. Parsons, C. S. Bottaro, N. Yan and F. M. Kerton, ACS Sustainable Chem. Eng., 2018, 6, 1662–1669 CrossRef CAS.
- X. Ma, G. Gözaydın, H. Yang, W. Ning, X. Han, N. Y. Poon, H. Liang, N. Yan and K. Zhou, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 7719–7728 CrossRef CAS.
- J. Hilgert, N. Meine, R. Rinaldi and F. Schüth, Energy Environ. Sci., 2013, 6, 92–96 RSC.
- A. Shrotri, H. Kobayashi, A. Tanksale, A. Fukuoka and J. Beltramini, ChemCatChem, 2014, 6, 1349–1356 CAS.
- S. Liu, Y. Okuyama, M. Tamura, Y. Nakagawa, A. Imai and K. Tomishige, ChemSusChem, 2015, 8, 628–635 CrossRef CAS.
- A. Einbu and K. M. Vårum, Biomacromolecules, 2007, 8, 309–314 CrossRef CAS PubMed.
- F. D. Bobbink, J. Zhang, Y. Pierson, X. Chen and N. Yan, Green Chem., 2015, 17, 1024–1031 RSC.
- H. Kobayashi, K. Techikawara and A. Fukuoka, Green Chem., 2017, 19, 3350–3356 RSC.
- M. K. Beyer and H. Clausen-Schaumann, Chem. Rev., 2005, 105, 2921–2948 CrossRef CAS PubMed.
- S. Amirjalayer, H. Fuchs and D. Marx, Angew. Chem., Int. Ed., 2019, 58, 5232–5235 CrossRef CAS PubMed.
- D. De Chavez, H. Kobayashi, A. Fukuoka and J. Hasegawa, J. Phys. Chem. A, 2021, 125, 187–197 CrossRef CAS.
- M. F. Pill, A. L. L. East, D. Marx, M. K. Beyer and H. Clausen-Schaumann, Angew. Chem., Int. Ed., 2019, 58, 9787–9790 CrossRef CAS PubMed.
- R. Rinaldi and F. Schüth, ChemSusChem, 2009, 2, 1096–1107 CrossRef CAS.
- P. Chen, A. Shrotri and A. Fukuoka, ChemSusChem, 2019, 12, 2576–2580 CrossRef CAS.
- J. Hirayama, H. Kobayashi and A. Fukuoka, Bull. Chem. Soc. Jpn., 2020, 93, 273–278 CrossRef CAS.
- N. Hamaguchi, H. Hirai, K. Aizawa and M. Takada, J. Appl. Glycosci., 2015, 62, 7–13 CrossRef CAS.
- I. Sakurada, Y. Nukushina and T. Ito, J. Polym. Sci., 1962, 57, 651–660 CrossRef CAS.
- R. Wagner, R. Moon, J. Pratt, G. Shaw and A. Raman, Nanotechnology, 2011, 22, 455703 CrossRef PubMed.
- K. Kulasinski, S. Keten, S. V. Churakov, D. Derome and J. Carmeliet, Cellulose, 2014, 21, 1103–1116 CrossRef CAS.
- F. R. P. Pienaar, N. J. Eaton and A. Pizzi, J. Macromol. Sci., Part B: Phys., 1989, 28, 115–129 CrossRef.
- Y. Ogawa, R. Hori, U.-J. Kim and M. Wada, Carbohydr. Polym., 2011, 83, 1213–1217 CrossRef CAS.
- Y. Nishiyama, P. Langan and H. Chanzy, J. Am. Chem. Soc., 2002, 124, 9074–9082 CrossRef CAS.
- P. Sikorski, R. Hori and M. Wada, Biomacromolecules, 2009, 10, 1100–1105 CrossRef CAS PubMed.
- S. W. Schmidt, A. Kersch, M. K. Beyer and H. Clausen-Schaumann, Phys. Chem. Chem. Phys., 2011, 13, 5994–5999 RSC.
- M. B. Larsen and A. J. Boydston, J. Am. Chem. Soc., 2013, 135, 8189–8192 CrossRef CAS PubMed.
- M. F. Pill, S. W. Schmidt, M. K. Beyer, H. Clausen-Schaumann and A. Kersch, J. Chem. Phys., 2014, 140, 044321 CrossRef PubMed.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS PubMed.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- W. J. Hehre, R. Ditchfield and J. A. Pople, J. Chem. Phys., 1972, 56, 2257–2261 CrossRef CAS.
- M. K. Beyer, J. Chem. Phys., 2000, 112, 7307–7312 CrossRef CAS.
- I. M. Klein, C. C. Husic, D. P. Kovács, N. J. Choquette and M. J. Robb, J. Am. Chem. Soc., 2020, 142, 16364–16381 CrossRef CAS PubMed.
- M. K. Beyer, Angew. Chem., Int. Ed., 2003, 42, 4913–4915 CrossRef CAS PubMed.
- O. Bobleter, Prog. Polym. Sci., 1994, 19, 797–841 CrossRef CAS.
- H. Satoh and S. Manabe, Chem. Soc. Rev., 2013, 42, 4297–4309 RSC.
- C. Loerbroks, R. Rinaldi and W. Thiel, Chem. – Eur. J., 2013, 19, 16282–16294 CrossRef CAS PubMed.
- F. M. Bickelhaupt, J. Comput. Chem., 1999, 20, 114–128 CrossRef CAS.
- A. H. Zewail, J. Phys. Chem. A, 2000, 104, 5660–5694 CrossRef CAS.
- P. Dopieralski, P. Anjukandi, M. Rückert, M. Shiga, J. Ribas–Arino and D. Marx, J. Mater. Chem., 2011, 21, 8309–8316 RSC.
- H. M. Klukovich, T. B. Kouznetsova, Z. S. Kean, J. M. Lenhardt and S. L. Craig, Nat. Chem., 2013, 5, 110–114 CrossRef CAS PubMed.
- M. Abdellaoui and E. Gaffet, Acta Metall. Mater., 1995, 43, 1087–1098 CrossRef CAS.
- R. J. Roberts, R. C. Rowe and P. York, Int. J. Pharm., 1994, 105, 177–180 CrossRef CAS.
- J. Tomasi, B. Mennucci and R. Cammi, Chem. Rev., 2005, 105, 2999–3094 CrossRef CAS PubMed.
- Y. Ishida, M. Yōshino, M. Takayanagi and F. Irie, J. Appl. Polym. Sci., 1959, 1, 227–235 CrossRef CAS.
- M. Born and R. Oppenheimer, Ann. Phys., 1927, 389, 457–484 CrossRef.
- J. J. W. McDouall, K. Peasley and M. A. Robb, Chem. Phys. Lett., 1988, 148, 183–189 CrossRef CAS.
- T. Yanai, D. P. Tew and N. C. Handy, Chem. Phys. Lett., 2004, 393, 51–57 CrossRef CAS.
- Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2008, 120, 215–241 Search PubMed.
- H. S. Yu, X. He, S. L. Li and D. G. Truhlar, Chem. Sci., 2016, 7, 5032–5051 RSC.
- J.-D. Chai and M. Head-Gordon, Phys. Chem. Chem. Phys., 2008, 10, 6615–6620 RSC.
- F. Jensen, J. Chem. Phys., 2001, 115, 9113–9125 CrossRef CAS.
- G. E. Scuseria, C. L. Janssen and H. F. Schaefer III, J. Chem. Phys., 1988, 89, 7382–7387 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Supplementary experimental data, details for theoretical calculations, validation and additional results of DFT calculations. See DOI: 10.1039/d1cp01650d |
| This journal is © the Owner Societies 2021 |