
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIdentifying cysteine residues susceptible to oxidation by photoactivatable atomic oxygen precursors using a proteome-wide analysis†
Ankita
Isor
a,
Benjamin V.
Chartier
b,
Masahiro
Abo
b,
Emily R.
Currens
a,
Eranthie
Weerapana
b and
Ryan D.
McCulla
 *a
*a
aDepartment of Chemistry, Saint Louis University, 3501 Laclede Ave, Saint Louis, MO 63103, USA. E-mail: ryan.mcculla@slu.edu
bDepartment of Chemistry, Boston College, Chestnut Hill, MA 02467, USA
First published on 19th January 2021
Abstract
The reactivity profile of atomic oxygen [O(3P)] in the condensed phase has shown a preference for the thiol group of cysteines. In this work, water-soluble O(3P)-precursors were synthesized by adding aromatic burdens and water-soluble sulphonic acid groups to the core structure of dibenzothiophene-S-oxide (DBTO) to study O(3P) reactivity in cell lysates and live cells. The photodeoxygenation of these compounds was investigated using common intermediates, which revealed that an increase in aromatic burdens to the DBTO core structure decreases the total oxidation yield due to competitive photodeoxygenation mechanisms. These derivatives were then tested in cell lysates and live cells to profile changes in cysteine reactivity using the isoTOP-ABPP chemoproteomics platform. The results from this analysis indicated that O(3P) significantly affects cysteine reactivity in the cell. Additionally, O(3P) was found to oxidize cysteines within peptide sequences with leucine and serine conserved at the sites surrounding the oxidized cysteine. O(3P) was also found to least likely oxidize cysteines among membrane proteins.
Introduction
Reactive oxygen species (ROS), including superoxide (O2˙−), hydroxyl radical (˙OH), and peroxide (H2O2), have been investigated for their role in regulating cell signalling in diseases such as cancer.1,2 Excessive ROS levels have shown to cause apoptosis or necrosis;1 however, low levels have been implicated in inducing signalling pathways that lead to cell survival and proliferation.2 Within proteomes, ROS often modify proteins by oxidizing nucleophilic cysteine residues that function as redox signalling intermediates.3,4 Among the different types of ROS, atomic oxygen [O(3P)] has not been studied within the cell due to the lack of “clean” sources in condensed phase, and its transient nature that makes it difficult to detect.5–8 A search for clean O(3P) sources has been a challenge because of the high energy photons associated with generating the oxidant.9 In 1997, Dibenzothiophene-S-oxide (DBTO), an aromatic heterocycle, was found to photo-deoxygenate upon exposure to UV-A irradiation, producing O(3P) and the corresponding sulphide in condensed phase through an S–O bond cleavage.10 Since then, DBTO and its derivatives have been used to explore O(3P) reactivity in solution.11,12 DBTO has also been derivatized to study the effect of O(3P) on lipids, regulatory proteins, and DNA.13–16 However, availability of DBTO derivatives to explore the effect of O(3P) oxidation within a biological setting still remains a challenge due to limited water solubility and the need for UV-A light closer to ∼320 nm to drive photo-deoxygenation. Numerous efforts have been made to derivatize DBTO to include functional groups in its structure that increase water solubility, thus improving its compatibility for biological studies,17,18 and benzannulation of DBTO to red-shift the absorbance.19 Benzonaphthothiophene-S-oxide (1) has been previously synthesized to allow the use of longer wavelength UV-A light sources as short wavelength UV-A irradiation may have deleterious effects on biomolecules.19–22 These strategies have driven studies to explore the effect of O(3P) on lipids, regulatory proteins, and DNA (Fig. 1).13–16 | ||
| Fig. 1 Benzannulated DBTO derivative: benzonaphthothiophene-S-oxide. | ||
The gas phase reactivity of O(3P) has illustrated its swift and selective nature. The rates for oxidation of alkenes, thiols, and sulphides are 100 times faster than other functional groups like alkanes, alcohols, and aldehydes.23,24 Recently, O(3P) reactivity was profiled using DBTO as an O(3P)-precursor. A preference for primary thiols, conjugated aromatic alkenes, and sulphides was observed.12 The study determined that there were two oxidation processes: one through freely diffusing O(3P) from the photodeoxygenation of DBTO, and the other involved thiol-oxidation prior to escape from solvent cage around DBTO (Fig. 2).12 Solvent cage oxidation was significant at high thiol concentrations, and moderate concentrations of alcohol-containing bifunctional thiols, due to hydrogen bonding between DBTO and the hydroxyl proton of the bifunctional thiols.12 The reaction rate for freely diffusing O(3P) produced from photodeoxygenation of DBTO was higher for primary thiols when compared to benzylic, secondary, and tertiary thiols.12
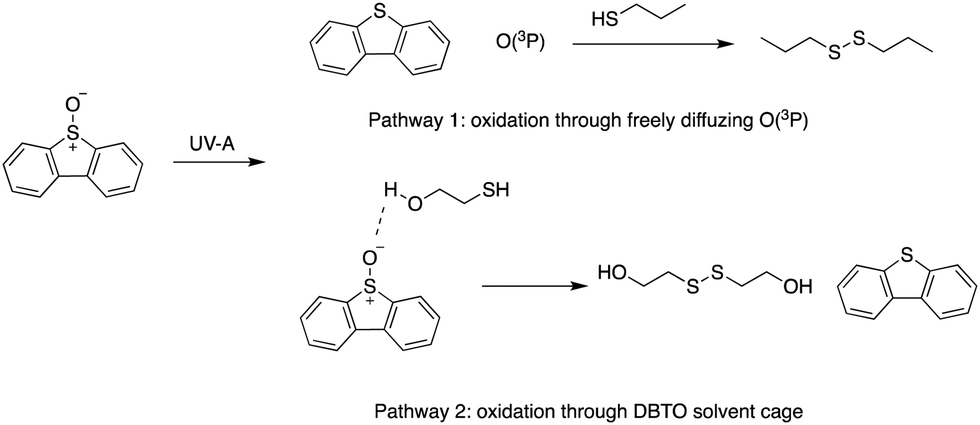 | ||
| Fig. 2 Oxidation pathways for thiols using DBTO as an O(3P) pre-cursor outlined in Omlid et al., 2017. | ||
This selectivity towards thiols was also demonstrated for the kinase Adenosine-5′-phosphosulfate kinase (APSK), where UV-A irradiation of a water-soluble DBTO derivative selectively oxidized regulatory cysteines to cystine.15 This study presented the possibility of using photodeoxygenation of biologically-compatible DBTO derivatives to selectively oxidize regulatory cysteines in enzymes. Photoactivable O(3P) precursors can further be used to explore the effect of O(3P)-induced oxidative stress in redox-mediated cell signalling. To facilitate these studies, it is important to understand the inherent proteome-wide selectivity of O(3P). Here, live cells were treated with biologically compatible photoactivable O(3P) precursor and the extent of oxidation across hundreds of reactive cysteines in the proteome was monitored. The O(3P) precursors used in our studies (2 and 3; Fig. 3) are water-soluble and display red-shifted absorbance, based on previous strategies to improve biocompatibility.17–19
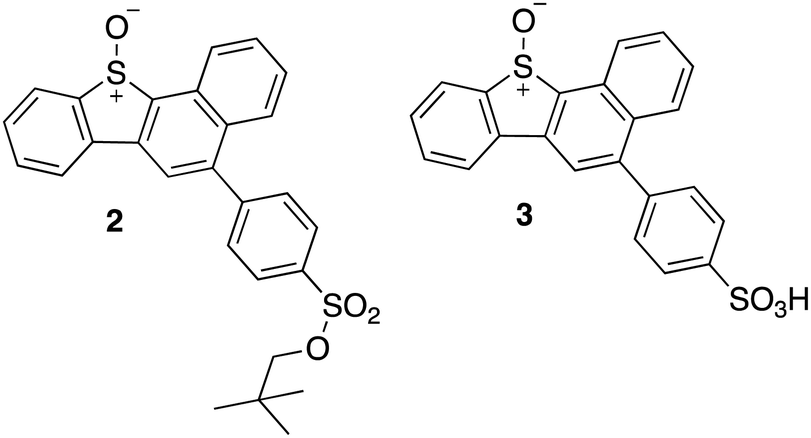 | ||
| Fig. 3 Synthesized water-soluble and red-shifted DBTO derivatives to study cysteine oxidation in cell lysates. | ||
Results and discussion
Synthesis of red-shifted water-soluble DBTO derivatives
Compound 1 was synthesized using previously described methods outlined in Scheme 1.19 Compounds 2 and 3 were synthesized using a modular synthetic approach outlined previously for water-soluble sulphonic acid DBTO derivatives.17 Sulphide 1S was brominated to give 5S. A 2D 1H–1H COSY was performed to determine the position of bromine substitution on 5S (ESI,† Section S1). The presence of a singlet in 1H-NMR ruled out the possibility of the electrophilic substitution at C-10 or C-4. Furthermore, the 2D 1H–1H COSY spectra revealed that the peaks at 7.89–7.80 ppm (H-2 and H-3, 2H) and 7.61–7.57 ppm (H-8 and H-9, 2H) were each coupled to two other peaks with single integrations [H-8 and H-9 were coupled to H-10 (8.19–8.17 ppm) and H-7 (8.56–8.53 ppm); H-2 and H-3 were coupled to H-1 (8.35–8.32 ppm) and H-4 (8.24–8.20 ppm)]. Additionally, the singlet peak with 1H integration was not coupled to any other peak. This observation indicated that there were two sets of four protons on two aromatic rings. Based on this evidence, it was concluded that bromine was at position 5 in 5S. The benzannulated sulphide 5S, was then oxidized using mCPBA to synthesize 5. Suzuki coupling was then used to add a neopentyloxysulphonyl phenyl group to 5, yielding 2 in 86.9% yield (Scheme 2). A previously published deprotection protocol for the neopentyl group was then performed to synthesize the sulphonic acid derivative 3 (Scheme 2).25 | ||
| Scheme 1 Synthetic outline for 1 (Zheng et al., 2016).19 | ||
Synthesis of a hydrophobic DBTO derivative representing the core structure of 2 and 3
O(3P) produced during the photodeoxygenation of aromatic sulphoxides has been traditionally investigated through a set of common intermediate experiments with toluene.19,26–28 This is due to the transient nature of O(3P), which makes it difficult to detect through conventional spectroscopic methods.8 One of the limitations of these common intermediate experiments is its incompatibility with aromatic sulphoxides with a sulphonic ester or a sulphonic acid group due to their poor solubility in toluene. In order to circumvent this limitation, a derivative 6, was synthesized that represented the core motif of 2 and 3. This derivative, 6, was then used in further studies to analyze the photochemistry of UV-A induced photodeoxygenation of 2 and 3 (Scheme 3). | ||
| Scheme 2 Synthetic outline for 2 and 3. | ||
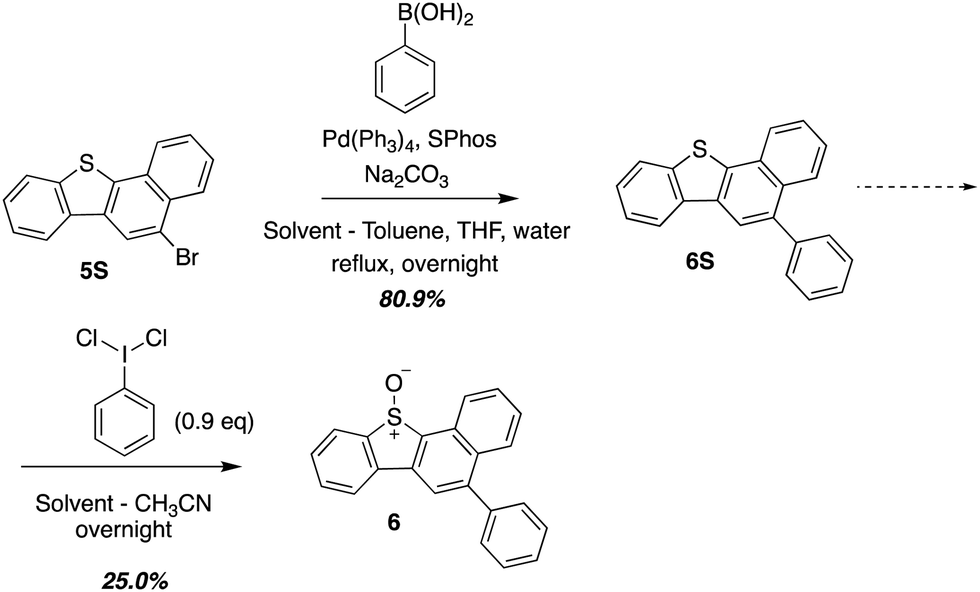 | ||
| Scheme 3 Synthesis of 6 which represents the core structure of 2 and 3. | ||
UV-vis absorbance of 1–3
A limitation that restricts the use of DBTO in biological settings is that it absorbs poorly at wavelengths higher than 340 nm. To minimize background absorption by biomolecules and to lessen the effects of high frequency UV light-induced damage on DNA, it is desirable to use light sources above 340 nm. The ground state absorption spectra for DBTO, 1–3, and 6 are included in Fig. 4. For DBTO, the λmax for the second and third absorption bands were observed to be at 280 nm and 320 nm. The λmax for the longest wavelength absorption band for 1, 2, 3, and 6 were at 340 nm, 346 nm, 349 nm, and 346 nm, respectively. This red-shifted absorption was observed due to the benzo group in 1 along with the phenyl group in 2, 3 and 6. The molar absorptivity falls below 2000 M−1 cm−1 at 362 nm, 360 nm, and 364 nm for 2, 3 and 6 respectively. These parameters in absorption spectra indicate that 1–3 can be used in biological settings with light sources above 340 nm.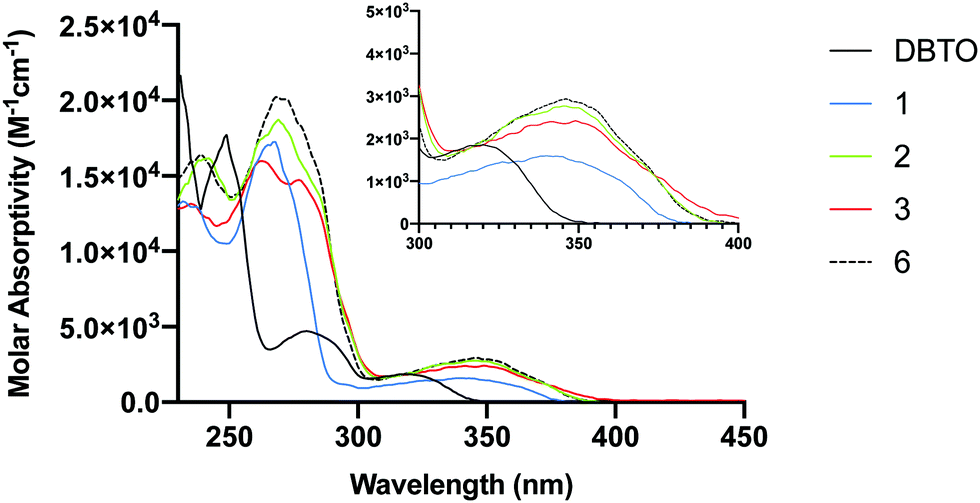 | ||
| Fig. 4 UV-Vis absorption spectra for DBTO, 1–3, and 6. | ||
Quantum yield
Quantum yield is an indicator of photodeoxygenation efficiency. Quantum yield measurements for the formation of the corresponding sulphide (ϕ+sulphide) upon UV-A irradiation of 1 and 6 in acetonitrile were performed. The quantum yield for 6 was found to be 0.0039 ± 0.0000 at 330 ± 5 nm which is comparable to the previously reported ϕ+sulphide of 0.0047 for 1 at 330 ± 3 nm in acetonitrile.19 It is almost twice the ϕ+sulphide of 0.0026 for DBTO at 320 ± 14 nm in acetonitrile.10 The ϕ+sulphide for 1 and 6 at 350 ± 5 nm were experimentally found to be 0.0050 ± 0.0016 and 0.0045 ± 0.0012, respectively. Both 1 and 6 have a higher quantum yield at 350 nm when compared to 330 nm which is supported by their longest wavelength absorption bands in Fig. 4. This also supports the hypothesis that the series of compounds (1–3) with extended chromophores are promising O(3P)-precursors to be used in biological settings with higher wavelength UV sources to minimize the deleterious effect of high frequency UV radiation.Photodeoxygenation of 1 and 6
In this study, photodeoxygenation of 1 and 6 was explored using common intermediate experiments with toluene (Fig. 5). In these common intermediate experiments, the aromatic sulphoxide was dissolved in toluene and then irradiated using broadly emitting UV bulbs centered at 350 nm (fwhm 325–375 nm). The possible toluene oxidation products, namely benzaldehyde, benzyl alcohol, and cresols, were quantified and compared to the toluene oxidation profile generated on photodeoxygenation of DBTO under similar conditions (Fig. 5). If the toluene oxidation profile of the aromatic sulphoxide is analogous to the toluene oxidation profile of a known O(3P)-precursor like DBTO,10 then it is suggested that derivatives 2 and 3 generate O(3P) upon UV-A irradiation.19,26–28 This approach is not very robust since previous studies have found that these common intermediate experiments are sensitive to irradiation periods, headspaces in the cuvette, and dissolved molecular oxygen.26,27 Larger headspaces and longer duration of UV-A exposure have shown to increase benzylic oxidation products.27 Although steps are taken to minimize the levels of dissolved oxygen through argon sparging, the benzylic oxidation products tend to increase in presence of dissolved residual molecular oxygen.11,28 This increase in benzylic oxidation is due to O(3P) reacting with dissolved molecular oxygen to form ozone that favours oxidization at the benzylic position. 11 Because of these reasons, cresol formation and the ratio of o-cresol and m/p-cresols produced through ring oxidation is a more reliable indicator of O(3P) generation on photodeoxygenation. | ||
| Fig. 5 Common intermediate experiment with toluene. | ||
Table 1 summarizes the percent yields of toluene oxidation products for DBTO, 1, and 6. When product ratios for o-cresol and m- & p-cresols were compared between DBTO and derivatives 1 and 6, it was observed that the ratios generated from this work were comparable to each other and ranged from 1.4–1.5. The yields for benzylic oxidation were lower compared to ring oxidation for DBTO which was not observed for the common intermediate experiments for 1 and 6 from this work. The oxidation profile for 1 has been reported previously where benzylic oxidation was slightly lower compared to ring oxidation.19 The ratios of products were compared instead of specific yields from previous experimental data because of the sensitive nature of these common intermediate experiments.27 Since the ratios of cresols are comparable to DBTO, we can conclude that O(3P) is a product in the photodeoxygenation of 1 and 6. The toluene oxidation profile generated from 6 is similar to 1 with low total oxidation percent yields of 10.4% and 7.5%, respectively, when compared to DBTO which had a total toluene oxidation percent yield of 57%. The low percent yield of oxidized toluene is suggestive of other competitive photodeoxygenation mechanisms that did not produce O(3P).
| Sulphoxide | Product yields (%) | Ratio (o-cresol and m/p-cresols) | Total toluene oxidation % yield | |||
|---|---|---|---|---|---|---|
| Benzaldehyde | Benzyl alcohol | o-Cresol | m & p-Cresolc | |||
| Yields of toluene oxidation products were calculated relative to sulphide formation (DBT, 1S, and 6S) on photodeoxygenation of DBTO, 1, and 6. Error was reported at 95% confidence intervals.a Results from ref. 19.19b Results from this study.c m-Cresol and p-cresol were quantified as single peak. | ||||||
| DBTOa | 3 ± 1 | 5 ± 1 | 20 ± 1 | 18 ± 1 | 1.1 | 46 ± 2 |
| DBTOb | 8.4 ± 0.7 | 7.4 ± 5.1 | 24 ± 2 | 17 ± 1 | 1.4 | 57 ± 6 |
| 1a | 1.7 ± 0.4 | 3.4 ± 0.6 | 5.5 ± 0.3 | 2.7 ± 0.2 | 2 | 13.3 ± 0.8 |
| 1b | 1.6 ± 1.4 | 2.4 ± 0.8 | 2.1 ± 0.2 | 1.4 ± 0.2 | 1.5 | 7.5 ± 1.6 |
| 6b | 2.3 ± 0.3 | 2.9 ± 0.1 | 3.1 ± 0.6 | 2.1 ± 0.1 | 1.5 | 10.4 ± 0.7 |
The low toluene oxidation yields have been observed in common intermediate experiments with DBTO derivatives that have aromatic burdens or expanded chromophores in previous studies.19,27 The release of O(3P) from DBTO has been hypothesized to be result of T2 state dissociation of DBTO* using CASSSCF and MRMP2 calculations through direct irradiation.29 These calculations have shown that the T2 state of DBTO is lower in energy than the optimized S1 state, which allows for intersystem crossing from S1 to T2, as shown in Fig. 6. The calculations further revealed that dissociation from T2 state results in the release of thiophene and O(3P) and a barrier to S–O cleavage was found around T1 surface.29 However, experimental results through sensitization experiments showed photodeoxygenation of DBTO through triplet sensitization30 and that deoxygenation can also be initiated through bimolecular photoreduction of DBTO.18,31 The low toluene yields from Table 1 suggest that there are competing mechanisms in play leading to photodeoxygenation of 6 that doesn’t release O(3P). Presumably, non-atomic oxygen release mechanism proceeds through the T1 state. This is supported by computational investigation of energy states in a 2016 study, which found that the excited singlet and triplet energies decreased with increasing aromatic burdens on DBTO chromophores with a concurrent increase in ΔEST.19 The decrease in excited energy states and increase in ΔEST was hypothesized to lead to increased partitioning from the S1 state and T2 into the in the T1 state. Thus, more exothermic processes leading to T1 were considered to disfavour O(3P) release and resulting in photodeoxygenation through mechanisms from triplet sensitized DBTO and bimolecular photoreduction.
 | ||
| Fig. 6 Modified Jablonski diagram for photodeoxygenation of DBTO. | ||
To verify if this theory could be expanded to the DBTO derivatives discussed in this work, T1 excited state energies were calculated for DBTO, 1–3, and 6 using HSEH1PBE method and 6-311G(d,p) as basis set.32–34 The optimized geometries for DBTO, 1–3, and 6 and their triplet excited state energies based on computational calculations are shown in ESI,† 2A–E and Table 2, respectively. The triplet excited state energies of 1–3 and 6 are comparable to each other but lower than DBTO by almost 10 kcal mol−1. This trend indicates that the intersystem crossing from S1 to T1 and the non-radiative relaxation from T2 to T1 may be more efficient than release of O(3P). As described above, increased T1 population is expected to result in deoxygenation that does not produce O(3P) simultaneously resulting in a decrease in total toluene oxidation due to the decrease in the amount of O(3P) production for 1 and 6.
| Compound | T 1 (kcal mol−1) |
|---|---|
| DBTO | 60.5 |
| 1 | 50.2 |
| 2 | 47.9 |
| 3 | 48.1 |
| 6 | 48.6 |
Cysteine oxidation profile in cells
To determine the proteome-wide selectivity of the O(3P) generated by the water-soluble and red-shifted DBTO derivatives 1–3, we used the isotopic tandem orthogonal proteolysis–activity-based protein profiling (isoTOP-ABPP) platform (Fig. S1, ESI†). This platform monitors changes in cysteine reactivity across different biological samples. Cysteine reactivity is monitored through the use of an iodoacetamide (IA)-alkyne probe. Oxidation of a cysteine residue by the generated O(3P) will result in formation of oxidized species, including disulphides and sulphenic acids, which will not react with the IA-alkyne probe. Initially, we assessed how the DBTO derivatives affected cysteine reactivity in cell lysates, using a gel-based analysis. Compounds were added to HeLa cell lysates at a concentration of 10 μM, followed by UV-A irradiation to initiate photodeoxygenation and generate O(3P). Control samples were treated with 1–3 but maintained in the dark to minimize O(3P) generation. The cell lysates were treated with IA-alkyne and IA-alkyne-labelled proteins were conjugated to TAMRA-azide using copper-catalysed azide–alkyne cycloaddition (CuAAC). Gel electrophoresis and imaging by in-gel fluorescence reveals the degree to which each O(3P) affected cysteine reactivity across the proteome. Oxidation of a cysteine by the O(3P) generated from 1–3 would result in a loss of fluorescence signal due to a loss in cysteine nucleophilicity. As anticipated, lysates incubated with 1–3 displayed reduced fluorescence upon UV-irradiation compared to lysates that were incubated with 1–3 in the dark (Fig. 7A). The most significant loss of labelling was observed with lysate incubated with 1, and due to this high potency, 1 was then chosen for subsequent live-cell treatments.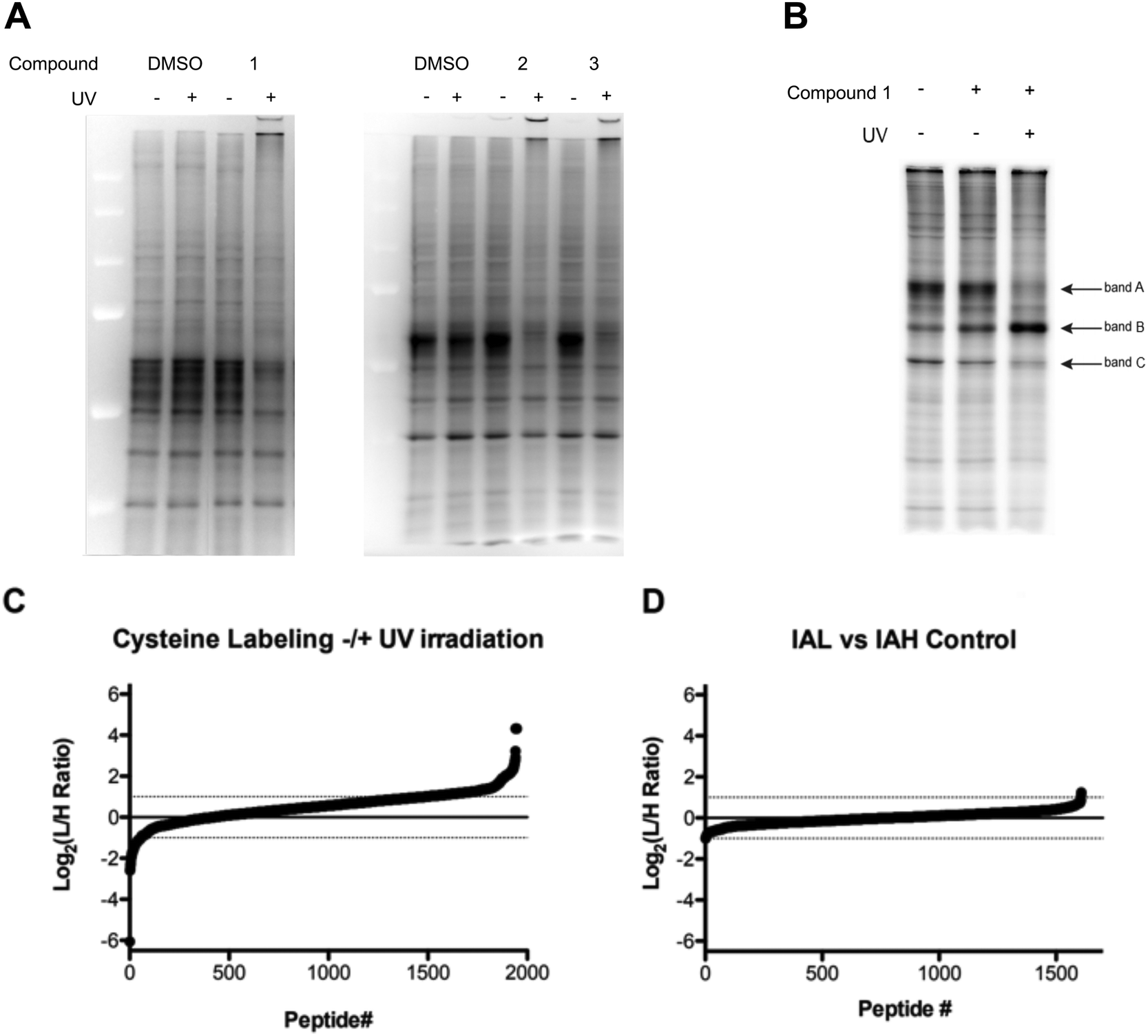 | ||
| Fig. 7 Cysteine reactivity surveyed by Iodoacetamide labeling (A) iodoacetamide labeling in lysate treated with DBTO derivatives 1–3 ± UV irradiation; (B) iodoacetamide labeling in lysates from live HeLa cells ± treatment with 1 ± UV irradiation; (C) cysteine reactivity from HeLa cells treated with 1 ± UV irradiation; (D) cysteine reactivity from HeLa cells treated with 1 – UV irradiation. | ||
For treatment of live cells, HeLa cells were incubated with 10 μM of the DBTO derivative 1 in culture media and subjected to UV irradiation or incubation in the dark. Minimal differences in iodoacetamide labelling were observed between cells treated with, or without 1 in the absence of UV exposure (Fig. 7B). Upon treatment with 1 and UV irradiation the majority of protein bands showed a decrease in fluorescence signal, indicating a loss in cysteine reactivity. The loss of labelling can be most clearly seen in bands A and C, however there was one notable exception as band B displayed increased labelling when treated with 1 and irradiation. This observed increase in labelling could be due to process initiated by the irradiation of 1 leading to structural changes or unfolding that could expose cysteines that are typically buried. It should be noted that UV-irradiation alone had no significant effect on labelling for cell lysates as shown in Fig. 7A. Normalization for both lysate and live cell experiments was confirmed by Coomassie staining (Fig. S2, ESI†).
To identify and quantify sites of cysteine oxidation by O(3P) generated from 1, isoTOP-ABPP analysis was performed. The isoTOP-ABPP analysis utilizes isotopically tagged IA-alkyne probes, IA-light and IA-heavy, to modify reactive cysteine residues in two different samples. Lysates from HeLa cells treated with 1 without UV irradiation were labelled with IA-light, while lysates from cells treated with 1 and irradiation were labelled with IA-heavy.35 IA-labelled proteins were then conjugated to a photo-cleavable biotin-azide, enriched on streptavidin beads and subjected to on-bead trypsin digestion. IA-labelled peptides were then released by photo-cleavage for mass-spectrometry (MS) analysis (Fig. S1, ESI†). Differences in cysteine reactivity across the two samples were quantified by comparing signal intensities between light and heavy labelled variants of each peptide. Oxidation by 1 in the irradiated sample would result in a loss in cysteine reactivity, which would be reflected by light:heavy ratios (RL:H) greater than 1 (plotted as log2 values, Fig. 7C).
Relative cysteine reactivity in irradiated and non-irradiated samples were quantified for ∼2000 cysteine residues within the HeLa-cell proteome. In general, there was a global decrease in cysteine reactivity in the irradiated sample, resulting in a median log2(RL:H) of 0.54, suggesting oxidation of a large number of cysteines upon treatment with O(3P). This reduction in cysteine reactivity was further illustrated by the fact that most cysteines (1528, 78.5%) exhibited a reduction in labelling (log2(RL:H > 0)) upon treatment with 1 and irradiation, with 461 of these cysteines displaying at least a two-fold reduction (log2(RL:H ≥ 1), 23.6%). This decrease in labelling indicated oxidation of the cysteines by O(3P) generated as a result of photodeoxygenation of 1 in UV. This overall increase in cysteine oxidation with 1 and UV suggests that O(3P) oxidation is not particularly specific in attacking certain cysteines. This could be due to indirect activation of other redox enzymes by O(3P) in addition to direct O(3P) oxidation of cysteines in proteins. Redox enzymes have shown to engage in oxidative behaviour like metalloenzymes through a metal–oxygen complex. Metalloenzymes like P450 catalyse oxidation by forming such a complex,36–38 and O(3P) may have a similar reactivity as these metalloenzymes.39
Consistent with the in-gel fluorescence results, a small subset of peptides (2.9% of peptides) were shown to have a significant increase (log2(RL:H ≤ −1)) in cysteine labelling following treatment with 1 and irradiation.
To demonstrate the quantitative accuracy of this isoTOP-ABPP analysis, two un-irradiated samples were compared to demonstrate that there is minimal variation in cysteine reactivity across two identical samples. In this control sample, two identical aliquots of lysate from cells treated with 1 and without irradiation were labelled with light and heavy probes. Mass-spectrometry analysis quantified reactivity for 1608 cysteines. The median log2(RL:H) was 0.01 and only 3 peptides displayed a two-fold reduction (1) or increase (2) in labelling. Together, these data provide significant evidence of cysteine oxidation by O(3P) generated by photodeoxygenation of 1 in live cells.
The peptide sequences with cysteines that displayed a two-fold reduction in labelling were segmented based on their cellular localization using R and dplyr package (Fig. 8).40,41 The peptides were distributed mostly in the cytoplasm (38.6%), nucleus (20.2%), or localized to the cytoplasm and/or nucleus (8%); while about 5.6% of the peptides were localized to the mitochondria and cytoplasm/nucleus. This was slightly altered compared to proteome mapping data in HeLa cell, which in a 2011 study reported a distribution of ∼35% of the proteome to the nucleus, ∼30% to the membrane, and ∼10% to mitochondria.42
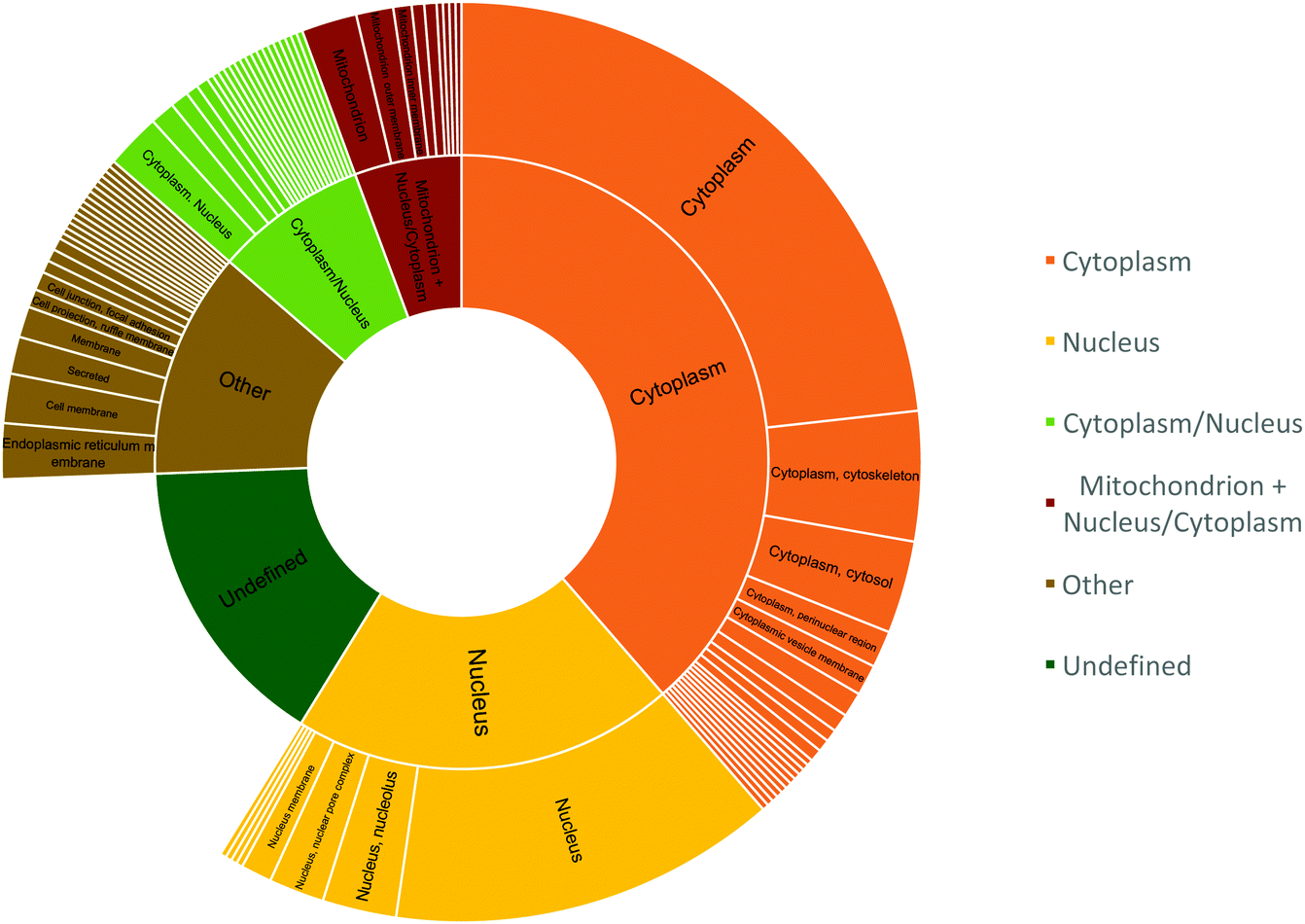 | ||
| Fig. 8 Cellular location of peptides that showed two-fold reduction in labelling. The inner circle represents the peptide count for cellular components and the outer circle represents the specific location within those components. | ||
Since cytosol accounts for 70% of the cell by volume and is ∼80% composed of water, the matrix provides a favourable hydrophilic platform for diffusing O(3P) to oxidize proteins that constitute ∼20–30% of the cytosol by volume.43–46 The identification of mitochondrial proteins, albeit with reduced efficiency, indicates the ability of compound 1 to diffuse into the mitochondria of living cells.
Sequence conservation analysis in the peptide sequences
To determine if there were any sequence or structural motifs that dictated increased reactivity with O(3P), various parameters were bioinformatically explored. Since three-dimensional structural information is lacking for many of the identified hits, the sequence conservation at the amino acids to the left and right of the oxidized cysteines were used as proxy. This analysis was only performed for those peptides identified to contain a single cysteine residue as a site of oxidation. The sequences were then further separated by extracting characters from the sequence string in Microsoft Excel to generate fragments to the left and right of the modified cysteines. The fragments were then filtered based on length and the fragments that did not have four amino acids after separation in excel were excluded from any further analysis (Table S1, ESI†). Fragment A accounted for the four amino acids to the left of the modified cysteine and Fragment B accounted for the four amino acids to the right of the modified cysteine. The fragments were then analysed using WebLogo where the height of the stack at a position indicated the sequence conservation at that site and the height of the letter code for the amino acid indicated the amino acid's frequency at that site.47,48 This sequence separation and conservation analysis was performed for the 461 peptides that showed two-fold reduction in labelling (log2(RL:H ≥ 1)), 80 peptides that had the least difference in labelling (−0.05 < log2(RL:H) < 0.05), and the 200 peptide sequences that had an increase in labelling (log2(RL:H ≤ 0)). These analyses were then used to compare and identify trends in sequence conservation between the peptides with the highest and lowest cysteine reactivity with O(3P). Additionally, in the sequence logos, the colour of the letter code for the amino acid is indicative of the hydrophilicity of the amino acid. The colour scheme is a reasonable gauge to examine the effect of hydrophilicity on the selective cysteine reactivity of O(3P). The analysis for both sets of peptide sequences are presented in Fig. 9. The sequence logos for Fragment A on peptides with highest reduction and increase in labelling show that the second amino acid position is conserved more as compared to the other sites. | ||
| Fig. 9 (Generated using WebLogo):47,48 (A) sequence Logos for peptides with a single modified cysteine exhibiting two-fold reduction in labeling segmented by “C” in the sequence (B) sequence Logos for peptides with a single modified cysteine that showed an increase in labeling segmented by “C” in the sequen. | ||
On analysing the specific amino acids, it was observed leucine (L) had a higher probability of being in position 1 for the peptides that show more cysteine oxidation, glycine (G) had the highest probability of being in position 3 for the peptides with no significant reactivity towards O(3P), and Alanine (A) was conserved at position 2 for the peptides that revealed highest increase in labelling. Serine (S) was also conserved in positions 1, 2 and 3 for the cysteines with the highest reactivity towards O(3P) oxidation in Fragment A.
Similarly, for Fragment B, position 2 and 3 exhibited more sequence conservation for all peptides. For the peptides with at least two-fold reduction in labelling, these positions had leucine (L) and serine (S) conserved. Interestingly, Fragment B for the peptide sequences with an increase in labelling showed the highest sequence conservation with leucine (L), alanine (A), valine (V), and glutamic acid (E) at 2 and 3 with high frequencies. For peptide sequences with no O(3P) reactivity, leucine (L) was conserved at position 1 and 3, and glutamic acid (E) was conserved at position 2 with the highest probability. Most of the amino acids that are included in the stacks are neutral and hydrophilic in nature with less frequency of hydrophobic amino acids for both sets of peptides.
On comparative analysis, the frequency of leucine (L) and serine (S) in Fragment A and B for Fig. 9A indicate the likelihood of these amino acids surrounding a O(3P)-sensitive cysteine, whereas aspartic acid (D), glycine (G) and alanine (A) are more likely to be present to the left of and leucine (L) with glutamic acid (E) are more likely to be present to the right of the cysteines in the peptide sequences with least O(3P) sensitivity. Alanine (A) is more likely to be present to the left and along with leucine (L) to the right of a cysteine with O(3P)-sensitivity possibly leading to cystine reduction instead of O(3P)-mediated oxidation.
Trends in top 10 cysteine residues most sensitive to oxidation
The 10 peptides that had cysteines with highest reduction, no difference, and increase in labelling are included in Tables S2–S4 (ESI†), respectively. The peptide sequences were further analysed on a structural level to evaluate if solvent accessibility of sulphur atoms of the cysteines within proteins is a key driver of O(3P)-oxidation. This hypothesis was based on O(3P)'s transient and diffusive nature which would facilitate oxidation because of its diffusive and transient nature.8,45The crystal structure of the proteins listed in Tables S2–S4 (ESI†) were examined to see if cysteines were solvent accessible or buried in the hydrophobic core of a protein. Crystal structures of proteins were found in PDB using Swiss Model Repository.49–53 Some of these proteins did not have the crystal structure of the regions with sequences containing the modified cysteines (ANKHD1, LRPRRC, DCAF7, LRRC40, HSPA4, ILVBL, IARS, ANLN, and LRRC47) while those that had resolved crystal structures in RCSB-PDB (rcsb.org) were further analysed Tables S5–S7 (ESI†).50 For the cysteines in proteins that have multiple chains with the same sequence and UniProt reference sequence, we explored the possibility for the modified cysteine to be included in every monomer chain as they have the same sequence. Additionally, homology models 4ATB, 6ZP4, and 2MQP were used as representative structures for the three proteins ILF3, EIF3CL, and HNRNPL, respectively because of their high sequence overlap.54–56
The solvent accessible surface area was then calculated for the sulphur atom in the cysteine using Prime in Maestro Version 12.0.012, Schrödinger, LLC.57–60 The accessible surface areas of sulphur atoms in cysteines for each peptide are shown in Tables S5–S7 (ESI†) and the average area is plotted on Fig. 10A–C. The colour of the column cell titled “Solvent accessibility for Sulphur (Å2)” in Tables S5–S7 (ESI†) are based on the total solvent accessibility label generated through Maestro Prime Energy Visualization (Structure analysis) software; red indicates solvent accessibility and green indicates solvent inaccessibility.57–59
 | ||
| Fig. 10 Solvent accessible surface area of sulphur atoms in cysteines with highest reduction in labeling of peptides with resolved crystal structures. | ||
From Table S5 (ESI†), there was no definitive trend indicating that the solvent accessibility of the sulphur atoms in oxidized cysteines of the peptides was a determining factor for oxidation by O(3P) (Fig. 10A). On closer analysis of the data, we observed that five out of the eight cysteines were not solvent accessible. For the cysteines that were not solvent accessible or buried in the hydrophobic core, we observed bound ligands to the sulphur atom of the cysteine or bound to amino acids around the cysteine. Zinc binds to C185 in TK1 (1XBT) and the cysteine in each chain is in the thiolate form that binds to Zn2+ in the structure.61 Cysteine C326 of PKM is solvent inaccessible and is completely buried in the tetrameric pyruvate kinase assembly.62 However, C326 is exposed to oxidizing conditions because of constant association and dissociation of its monomers.63 Oxidation of C326 decreases the activity of the enzyme because it doesn’t allow for the monomers to associate and form the active tetramer.63 For TUBB (5N5N), the cysteines C203 is slightly solvent accessible but C213 and C241 in all chains are solvent inaccessible. The sulphur atom in C241 in chain A is also deprotonated (thiolate form) whereas the C241 in other chains are not. The structure 5N5N had G2P as a ligand bound to Asn206, Tyr224 and Asn228.64 For RRP8 (2ZFU), C332 is not very solvent accessible but the sulphur is deprotonated (thiolate form) and it is close to the binding site of the ligand S-adenosylhomocysteine in the crystal structure.65 The ligand had binding sites at Asp334 and Leu335, and C332 was close to the binding pocket.65 On applying the weighted average to calculate the average solvent accessible surface area of the sulphur atoms in Table S5 (ESI†), a value of 2.51 Å2 was obtained.
Similar analysis was performed for proteins identified in Table S6 (ESI†) and the solvent accessibility of the sulphur atoms of the cysteines with the no difference in labelling are plotted on Fig. 10B. There were no conclusive trends from the solvent accessibility of the sulphur atoms because four out of the six cysteines were not solvent accessible. However, two of the sequences, SF3A1 (6FF7) and HNRNPL (2MQP), had extremely high areas exposed to the solvent as shown in Fig. 10B. Because of these high solvent accessible areas, the weighted average for sequences with least O(3P)-sensitivity was calculated to be 18.4 Å2. The chains that were used to analyze the solvent accessibility in these set of peptides had no ligands associated.
For the peptide sequences that had the highest increase in labelling indicating reduction as opposed to oxidation, it was observed that six out of the ten cysteines identified in the crystal structures were not solvent accessible (Table S7, ESI,† and Fig. 10C). In the crystal structure for 4HDO, C118 is solvent accessible and is surrounded by binding sites for GNP at positions 116, 117, 119, and 120.66 In 6RIR, C123 in both chains are surrounded by GTP ligand binding sites at 124, 125, 127, 128.67 Similarly, for 5LPN, the cysteine of interest is the binding site of GNP.68 These ligand binding sites can be a consequence of the experimental protein structure determination in the presence of a ligand. The average accessible surface area using weighted average for peptides with cysteines of interest on multiple chains was found to be 3.44 Å2.
As shown in Fig. 10D, the average solvent accessible surface area of the sulphur atom in cysteines is higher for peptides with no difference or increased labelling, which indicates no oxidation by O(3P). In contrast, peptides with a reduction in labelling due to the presumed oxidation by O(3P) have the lowest average solvent accessibility. Therefore, solvent accessibility cannot be identified as a driver to higher cysteine reactivity for O(3P) oxidation based on quantitative analysis. On assessing the location of these proteins, it was observed that all the peptides in Table S7 (ESI†) are also located in the membrane except ECHS1, LRRC47 and MDH2, which indicates that O(3P) has least preference for with cysteines within the membrane proteins. Out of the (200 peptides that showed increase in labelling), 19.5% are located in the membrane as opposed to 15.4% proteins found among the 461 peptides with the highest reduction in labelling. However, when the 2.9% peptides with the highest increase in labelling (log2(RL:H ≤ −1)) were segmented by sub-cellular location, 34% of the peptides were located in the membrane.
From the data on surrounding amino acids and the crystal structure analysis of the peptides with the highest levels of cysteine oxidation and peptides that showed a decrease in cysteine oxidation, we observed that the oxidized cysteines within the peptides were less solvent accessible than the cysteines that were not reactive to O(3P). Further sequence conservation analysis revealed that there was a preference for leucine (L) and serine (S) amino acids surrounding the oxidized cysteines that showed the highest reactivity of O(3P) contrary to the conservation of alanine (A), glycine (G), and leucine (L) in the peptides with no cysteine reactivity to O(3P). We also found that membrane proteins are more likely to undergo cystine reduction when treated with 1 and UV. This may be due to lower accessibility of those cysteines to O(3P) as compared to accessibility to cysteines among proteins in the cytoplasm and nucleus.
Conclusion
Derivatives of DBTO, 2 and 3, were synthesized that had improved water solubility and an extended chromophore to red-shift the absorption spectra of these compounds to increase their biological compatibility. The photodeoxygenation of these derivatives were explored by synthesizing derivative 6 that represented the heterocyclic aromatic core structure of 2 and 3. The common intermediate experiments with toluene revealed that the oxidation profile of 6 was different when compared to DBTO. The oxidation profile for 6 had a higher benzylic oxidation and an overall lower oxidation yield. This was attributed to aromatic burdens in the structure of 6 compared to DBTO. Previous studies correlated a decrease in T1 energy state to an increase in aromatic burdens on the core DBTO structure. As O(3P) release has been posited to result from the dissociative T2 state, a lower T1 energy state favours exothermic processes like ISC from S1 to T1 and non-radiative relaxation from T2 to T1 state, which decreases T2 energy state population producing O(3P). Computational energy calculations revealed that the T1 energy for 6 was 11.9 kcal mol−1 lower than the T1 energy state of DBTO supporting the hypothesis. Therefore, the higher benzylic oxidation and lower oxidation yields observed for 6 was attributed to competitive photodeoxygenation mechanisms other than the release of O(3P) from the dissociative T2 energy state.Derivatives 2 and 3 along with a previously synthesized DBTO derivative, 1, were then tested in cell lysates to observe changes in cysteine reactivity on O(3P) release from photodeoxygenation of the derivatives. A significant decrease in free cysteines was observed in samples that were treated with the three different derivatives and UV-A irradiation. Derivative 1 was found to be the most potent and therefore tested in live cells to quantitatively profile the difference in cysteine reactivity due to oxidation by O(3P) using isoTOP-ABPP analysis. Among the ∼2000 cysteines quantified, 78.5% of the cysteines exhibited a reduction in labelling indicating that O(3P) oxidizes a large proportion of cysteines in the cell. The peptides with the highest proportion of cysteines in the cell. The peptides with the highest reduction in labelling were then explored to determine if the solvent accessibility of these cysteines made it susceptible to O(3P) oxidation. Using the sequence information and computational protein modelling software, it was found that these cysteines are likely to be not solvent accessible when compared to the proteins with the lowest levels of cysteine reactivity towards O(3P). However, O(3P) disfavours cysteines among membrane proteins as membrane proteins constitute the majority of peptides that showed an increase in labelling post compound treatment with UV.
Experimental
Starting materials, reagents, solvents and catalysts were purchased from Sigma-Aldrich or Fisher Scientific unless stated otherwise. 4-(Neopentyloxysulphonyl)phenylboronic acid was purchased form Combi-Blocks, and Pd2(dba)3 was purchased from TCI Chemicals. All solvents used were ACS grade except Acetonitrile which was LCMS/HPLC grade. GCMS was performed on Shimadzu GCMS-QP2010S. NMR was performed on Bruker NMR 400 MHz Avance III, and HRMS was obtained on Orbitrap Q-Exactive (Thermo Scientific). UV-Vis spectra were acquired using Thermo Scientific Nanodrop 2000c. GC injections and calibration curves were analysed using Shimadzu GC-2010 Plus with an auto-sampler. HPLC traces and calibration curve injections were performed on Agilent 1200 series (Quad pump, DAD, autosampler) equipped with a C18, 5 μm CLIPEUS column (150 × 4.6 mm). All graphs were generated on GraphPad Prism in the manuscript and Microsoft excel in the ESI.†Synthesis of 1–3
Yield: 1427.5 mg (71.1%).
1H-NMR (400 MHz): δ 8.03 (d, 1H, J = 8.8 Hz), 7.90 (d, 1H, J = 7.6 Hz), 7.58–7.57 (m, 3H), 7.46 (td, 1H, J = 7.9 Hz, 1.0 Hz), 7.42–7.38 (m, 4H), 7.31 (d, 1H, J = 7.3 Hz), 7.20 (d, 1H, J = 16.3 Hz) (chemical shifts are consistent with previously published results in the literature).69
Yield: 248.5 mg (71.3%) – white crystalline solid.
1H-NMR (400 MHz): δ 8.25–8.22 (m, 1H), 8.19 (d, 1H, J = 8.6 Hz), 8.16 (d, 1H, J = 8.1 Hz), 7.99 (t, 2H, J = 7.7 Hz), 7.89 (d, 1H, J = 8.6 Hz), 7.64 (t, 1H, J = 7.2 Hz), 7.58 (t, 1H, J = 6.8 Hz), 7.55–7.48 (m, 2H) (chemical shifts are consistent with previously published results in the literature).19
Yield: 67.0 mg (24.2%) – white powdery solid.
1H-NMR (400 MHz): δ 8.55 (d, 1H, J = 8.3 Hz), 8.11–8.06 (m, 2H), 7.96 (d, 1H, J = 8.2 Hz), 7.90 (dd, 2H, J = 8.0 Hz, 5.4 Hz), 7.73–7.69 (m, 1H), 7.66–7.58 (m, 2H), 7.56–7.52 (m, 1H) (chemical shifts are consistent with previously published results in the literature).19
Yield: 285.3 mg (85.9%) – white crystalline solid.
1H-NMR (400 MHz): δ 8.91 (s, 1H), 8.56–8.53 (m, 1H), 8.35–8.32 (m, 1H), 8.24–8.20 (m, 1H), 8.19–8.17 (m, 1H), 7.84–7.80 (m, 2H), 7.61–7.57 (m, 2H).
13C-NMR (100 MHz): δ 139.01, 137.16, 135.55, 132.90, 130.46, 129.64, 128.47, 127.57, 127.49, 126.64, 124.89, 124.83, 123.69, 122.97, 121.59, 120.06.
GCMS (EI): calcd 311.96 found 312 (100%), 311 (97%) [note: unable to ionize in ESI+ LCMS].
Yield: 52.4 mg (44.7%) – white powdery solid.
1H-NMR (400 MHz): δ 8.56 (d, 1H, J = 8.0 Hz), 8.36 (d, 1H, J = 8.5 Hz), 8.23 (s, 1H), 8.07 (d, 1H, J = 7.4 Hz), 7.86 (d, 1H, J = 7.6 Hz), 7.79–7.69 (m, 2H), 7.66 (td, 1H, J = 7.5 Hz, 1.1 Hz), 7.75 (td, 1H, J = 7.6 Hz, 1.1 Hz).
13C-NMR (100 MHz): δ 145.46, 140.28, 136.41, 135.92, 132.76, 132.34, 131.51, 130.00, 129.76, 129.25, 128.75, 128.67, 127.64, 124.61, 123.22, 122.19.
HRMS (ESI+): m/z [M + H] calcd [C16H10OBrS]+ 328. 9635 found 328.9622.
Yield: 65.9 mg (86.9%) – light pale yellow-white powdery solid.
1H-NMR (400 MHz): δ 8.65 (d, 1H, J = 8.3 Hz), 8.12–8.09 (m, 3H), 7.88 (d, 1H, J = 7.6 Hz), 7.83–7.80 (m, 2H), 7.77–7.72 (m, 3H), 7.65 (t, 1H, J = 7.6 Hz), 7.60–7.55 (m, 2H), 3.85 (s, 2H), 1.00 (s, 9H).
13C-NMR (100 MHz): δ 145.65, 145.38, 144.19, 141.00, 137.18, 136.31, 135.20, 132.82, 132.00, 131.31, 130.89, 129.96, 129.27, 128.32, 128.14, 127.74, 126.98, 124.79, 122.25, 120.17, 80.15, 32.04, 26.29.
HRMS (ESI+): m/z [M + H] calcd [C27H25O4S2]+ 477.1194 found 477.1177.
Yield: 15.4 mg (86.3%) – sticky yellow-white solid.
1H-NMR (400 MHz): 8.52 (d, 1H, J = 8.3 Hz), 8.17–8.06 (m, 5H), 7.96 (d, 1H, J = 8.5 Hz), 7.81–7.75 (m, 2H), 7.68–7.62 (m, 4H).
13C-NMR (100 MHz): δ 146.05, 144.30, 141.25, 138.52, 137.24, 135.53, 133.04, 132.13, 130.62, 129.73, 129.58, 128.68, 127.49, 127.14, 126.96, 125.97, 123.46, 122.70, 120.09.
HRMS (ESI−): m/z [M-H] calcd [C22H13O4S2]+ 405.0261 found 405.0264.
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 522 mg, 12.75 mmol) were dissolved in 50 mL THF, 15 mL toluene, and 5 mL water in a two-neck round bottom flask. The flask was then sparged with nitrogen for 30 minutes. Pd(PPh3)4 (109 mg, 0.0943 mmol) and 2-dicyclohexylphosphino-2′,6′-dimethoxybiphenyl (19 mg, 0.047 mmol) was added to the flask and then the reaction was refluxed overnight. The reaction flask was then cooled and 30 mL CH2Cl2 was added. The resulting solution was then washed with Millipore water twice and the organic layer was dried using anhydrous MgSO4. The solvent was then removed under reduced pressure and the crude product was purified using high pressure flash chromatography (silica, hexanes, Rf = 0.23).
522 mg, 12.75 mmol) were dissolved in 50 mL THF, 15 mL toluene, and 5 mL water in a two-neck round bottom flask. The flask was then sparged with nitrogen for 30 minutes. Pd(PPh3)4 (109 mg, 0.0943 mmol) and 2-dicyclohexylphosphino-2′,6′-dimethoxybiphenyl (19 mg, 0.047 mmol) was added to the flask and then the reaction was refluxed overnight. The reaction flask was then cooled and 30 mL CH2Cl2 was added. The resulting solution was then washed with Millipore water twice and the organic layer was dried using anhydrous MgSO4. The solvent was then removed under reduced pressure and the crude product was purified using high pressure flash chromatography (silica, hexanes, Rf = 0.23).
Yield: 234.8 mg (80.9%) – white powdery solid.
1H-NMR (400 MHz): δ 8.23–8.20 (m, 2H), 8.13 (s, 1H), 8.01–7.99 (m, 2H), 7.65 (t, 1H, J = 7.0 Hz), 7.61–7.48 (m, 8H).
13C-NMR (100 MHz): 141.01, 139.48, 138.25, 137.10, 136.92, 132.38, 131.21, 130.54, 129.40, 128.60, 127.65, 126.89, 126.57, 126.51, 125.06, 124.84, 123.21, 121.84, 120.83.
HRMS (ESI+): m/z calcd [C22H15S]+ 311.0894 found 311.0881.
:1 EtAc in Hexanes)).
Yield: 13.8 mg (25.0%) – white powdery solid.
1H-NMR (400 MHz): 8.61 (d, 1H, J = 8.3 Hz), 8.08 (d, 1H, J = 7.5 Hz), 7.94 (d, 1H, J = 8.5 Hz), 7.86 (d, 1H, J = 7.6 Hz), 7.83 (s, 1H), 7.71 (t, 1H, J = 7.7 Hz), 7.63 (t, 1H, J = 7.5 Hz), 7.59–7.52 (m, 7H).
13C-NMR (100 MHz): 146.62, 145.72, 139.83, 137.59, 135.25, 132.70, 132.62, 131.29, 130.04, 129.67, 128.87, 128.77, 128.40, 127.78, 127.62, 127.53, 124.48, 122.20, 120.03.
HRMS (ESI+): m/z calcd [C22H15SO]+ 327.0843 found 327.0829.
Quantum yield
Quantum yield experiments were carried out by dissolving 6 in acetonitrile (1.3 mM) and optical density of the resulting solution was recorded to be >2 at 330 and 350 nm. The solution was then transferred to a quartz cuvette and sparged with argon for 20–30 min. The solution was then allowed to photolyze in a monochromator (Photon Technologies International) for 1 h 30 min at 330 ± 5 nm. The experiment was carried out to low sulphoxide conversion to sulphide (<30%). The concentration of sulphide formed as a result of photodeoxygenation was then determined using HPLC injections and calibration curves. Two trials were performed, and error was calculated at 95% confidence interval. Rearrangement of azoxybenzene to 2-hydroxyazobenzene was used as a chemical actinometer to calculate flux.70Common intermediate test
Common intermediate experiments were carried out by dissolving DBTO, 1, and 6 in toluene with a final concentration of 8–10 mM. 6 was quantified to be 88% pure by HPLC with unknown impurities which did not undergo any increase in concentration when monitored before and after UV irradiation. The solution was then transferred to a quartz cuvette and argon-sparged for 25 minutes. The solution was then placed in a Luzchem photoreactor with 8 LZC-UVA bulbs for 1 h 15 min to 1 h 37 min. These experiments were carried out to low sulphoxide conversion to sulphide (<20%). Toluene oxidation products were identified and quantified using GC injections and calibration curves with Dodecane as standard. For m/p-cresol calibration curves, separate calibration curves were made for m-cresol and p-cresol or a 1.96:1 solution of m-cresol and p-cresol was made, and the areas were divided using that ratio to generate the curve. The areas quantified from GC injections of the common intermediate experiment solution were quantified as m-cresol and p-cresol and then the concentrations were averaged. Sulphide formation as a result of photodeoxygenation was quantified using HPLC injections and calibration curves. Two trials were performed, and error was calculated at 95% confidence interval.
Calculation of T1 energy state
Energies for DBTO, derivatives 1–3 and 6 in T1 state was determined by optimizing geometries using HSEH1PBE/6-311G(d,p) level of theory. The T1 energy state was then determined by calculating the difference between the sum of electronic and thermal free energies for two optimized geometry calculations with charge = 0, multiplicity = 1 and charge = 0, multiplicity = 3, respectively for the derivative. These geometry optimizations were performed with Gaussian 09 suite of programs.32Evaluation of cysteine oxidation in cell lysates
HeLa cells were grown to ∼90% confluency at 37 °C under a 5% CO2 atmosphere in culture media consisting of DMEM supplemented with 10% FBS and 1% PSA. Cells were harvested by scraping, and pellets were washed ×3 with DPBS (300 g × 5 min). Pellets were suspended in DPBS and lysed by sonication to yield cell lysates. Lysates were centrifuged at 14000 rpm (FA-45-18-11 rotor) for 15 minutes. Protein concentration of the supernatant was determined using a Bradford Assay and lysates were normalized to 2.0 mg mL−1. Lysates were then treated with 10 μM of DBTO derivatives (1–3) for one hour either under UV exposure (360 nm) or in the dark on ice.
Evaluation of cysteine oxidation in live cells
HeLa cells were grown to ∼90% confluency at 37 °C under a 5% CO2 atmosphere in culture media consisting of DMEM supplemented with 10% FBS and 1% PSA. Media was removed and cells were washed twice with DPBS and treated with 10 μM of DBTO derivative 1 in clear RPMI for 10 minutes at room temperature in the dark. Cells were then placed on ice either in the dark or under UV exposure (360 nm) for 10 minutes, before being incubated at 37 °C for 10 minutes. Treatment media was removed and cells were washed twice with DPBS and then harvested by scraping. Pellets were washed with DPBS, and lysed by sonication. Lysate protein concentrations were determined by Bradford assay and normalized to a concentration of 2.0 mg mL−1.In-gel fluorescence analysis
0.1 mg of lysates were labelled by iodoacetamide-alkyne for one hour and then rhodamine-azide was appended via copper catalysed azide–alkyne cycloaddition (CuAAC) with tris(2-carboxyethyl)phosphine hydrochloride (TCEP), tris[(1-benzyl-1H-1,2,3-triazol-4-yl)methyl]amine (TBTA ligand), and copper(II) sulphate. CuACC reactions were run for 1 h at 25 °C with vortexing every 15 minutes. Reactions were quenched with the addition of 50 μL of 2× SDS-PAGE loading buffer, before being heated at 85 °C for 10 min. Samples were analysed by SDS-PAGE (10% polyacrylamide resolving gel). In-gel rhodamine fluorescence was imaged using a Bio-Rad ChemiDoc MP System.Mass-spectrometry analysis
HeLa cells treated with DBTO derivative 1 (10 μM in clear RPMI media) ±10 min UV exposure were normalized to a protein concentration of 1 mg mL−1. 1 mL of samples were labelled with an isotopically encoded light or heavy iodoacetamide alkyne (final concentration 100 μM) for 1 hour. A photo-cleavable biotin-azide tag (from Click Chemistry Tools) was appended to the alkyne via CuAAC. Click reactions were run for an hour at 25 °C with vortexing every 15 min. Upon completion, click reactions were centrifuged for 4 minutes at 6500 g (FA-45-18-11 rotor) to pellet proteins. Pellets were sonicated and resuspended in cold methanol. The resuspended light and heavy labelled samples were combined and washed again with cold methanol. Samples were resuspended in 1 mL of 1.2% SDS in DPBS with sonication and heating (85 °C, 5 min).The labelled proteome samples in 1.2% SDS in DPBS were diluted with 5 mL of DPBS to a final concentration of 0.2% SDS. Streptavidin-agarose beads (100 μL) were added to the samples which were then incubated overnight at 4 °C. Samples were resolubilized at 25 °C for 2 hours before beads were washed with 5 mL 0.2% SDS/DPBS (×1), 5 mL DPBS (×3), 5 mL water (×3). Bead pellets were generated by centrifugation (1400 g, 3 min) between each of the washes. Washed beads were suspended in 500 μL of 6 M urea/DPBS and reduced with 10 mM dithiothreitol (DTT, 10 mM final concentration) and heated at 65 °C for 20 min. Following DTT reduction, iodoacetamide was added (20 mM final concentration) and samples were incubated at 37 °C for 30 min. After alkylation, reactions were diluted with the addition of 950 μL of DPBS and centrifuged (1400 g, 2 min) to generate bead pellets. Supernatant was removed and beads were resuspended in 200 μL of 2 M urea, with 1 mM CaCl2, and 2 μg trypsin (Promega gold). Trypsin digestion proceeded overnight at 37 °C. Once digestion was complete, beads were pelleted with centrifugation and washed with 500 μL DPBS (×3) and 500 μL water. Beads were resuspended in 250 μL water and placed under UV lamp (360 nm) with gentle agitation via stirring for 3 hours. After UV cleavage, beads were centrifuged, and the supernatant was washed twice with 75 μL; the original supernatant and those from the two washes were combined to yield ∼350 μL of labelled peptides in DPBS. Formic acid (17.5 μL) was added to the samples and then the samples were stored at −20 °C until mass spectrometry analysis.
LC/LC-MS/MS analysis was performed on an LTQ-Orbitrap Discovery mass spectrometer (ThermoFisher) coupled to an Agilent 1200 series HPLC. Peptides were pressure loaded onto a 250 μm fused silica column packed with 4 cm of Aqua C18 reverse phase resin (Phenomenex). Loaded peptides were eluted onto a 100 μm fused biphasic column packed with 10 cm C18 and 4 cm Partisphere strong cation exchange resin (SCX, Whatman) with a multidimensional LC-MS protocol (MudPIT). Peptides were eluted to the C18 from the SCX with five salt pushes (0%, 50%, 80, 100%, and 100% 500 mM ammonium acetate) followed a gradient of 5–100% Buffer B in Buffer A (Buffer A: 95% water, 5% acetonitrile, and 0.1% formic acid; Buffer B: 20% water, 80% acetonitrile and 0.1% formic acid) (Weerapana, Speers, & Cravatt, 2007). Flow rate through the column was set to ∼0.25 μL min−1, with a spray voltage of 2.75 kV. One full MS1 scan (400–1800 MW) was followed by 8 data dependent scans of the nth most intense ions.
Tandem MS data was analyzed using the SEQUEST algorithm. A static modification of +57.02146 was specified in order to account for alkylation by iodoacetamide while differential modifications of +228.1375 and +234.1576 were specified on cysteine to account for modifications by the light and heavy probe respectively. SEQUEST output files were filtered using DTASelect2.071 and light:heavy ratios quantified using CIMAGE as previously described.72
Determination of sequence conservation and frequency of amino acid in peptide sequences
The amino acid sequences were separated using different commands to extract characters from the text string on Microsoft Excel to account for four amino acids to the left and the right of the cysteines in the sequence. If the separation through string function led to fragments less than four amino acids, then the fragments were excluded (Table S1, ESI†). The amino acids to the left was treated as Fragment A and to the right was treated as Fragment B. In the instance that sequence had two cysteines were oxidized, then Fragment C and D were introduced to account for the amino acids to the left and right of the second oxidized cysteines (Fig. S3, ESI†). These sequences were then converted to .fasta files using and ran on WebLogo software to generate sequence logos.47,48Calculation of accessible surface area of sulphurs in oxidized cysteine in a peptide
The accessible solvent surface area for oxidized cysteine was calculated using Maestro software by Schrödinger, LLC.60 The .pdb files were imported and protein preparation wizard tool was used to optimize the structure (references for pdb files are included in ESI†). The protein structures were split by chains and solvent accessibility was determined using Prime energy visualization tool.57–59Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Science Foundation under CHE-1900417 and the National Institutes of Health under R35GM134964.References
- C. Fleury, B. Mignotte and J. L. Vayssière, Biochimie, 2002, 84, 131–141 CrossRef CAS.
- B. Kalyanaraman, G. Cheng, M. Hardy, O. Ouari, B. Bennett and J. Zielonka, Redox Biol., 2018, 15, 347–362 CrossRef CAS.
- M. Schieber and N. S. Chandel, Curr. Biol., 2014, 24, R453–R462 CrossRef CAS.
- T. Nietzel, J. Mostertz, F. Hochgräfe and M. Schwarzländer, Mitochondrion, 2017, 33, 72–83 CrossRef CAS.
- E. Zadok, D. Amar and Y. Mazur, J. Am. Chem. Soc., 1980, 102, 6369–6370 CrossRef CAS.
- E. Zadok, S. Rubinraut and Y. Mazur, J. Org. Chem., 1987, 52, 385–390 CrossRef CAS.
- U. K. Kläning, K. Sehested and T. Wolff, J. Chem. Soc., Faraday Trans. 1, 1984, 80, 2969 RSC.
- G. Bucher and J. C. Scaiano, J. Phys. Chem., 1994, 98, 12411–12413 CrossRef.
- E. Lucien and A. Greer, J. Org. Chem., 2001, 66, 4576–4579 CrossRef CAS.
- D. D. Gregory, Z. Wan and W. S. Jenks, J. Am. Chem. Soc., 1997, 119, 94–102 CrossRef CAS.
- K. B. Thomas and A. Greer, J. Org. Chem., 2003, 68, 1886–1891 CrossRef CAS.
- S. M. Omlid, M. Zhang, A. Isor and R. D. McCulla, J. Org. Chem., 2017, 82, 13333–13341 CrossRef CAS.
- J. Korang, I. Emahi, W. R. Grither, S. M. Baumann, D. A. Baum and R. D. McCulla, RSC Adv., 2013, 3, 12390–12397 RSC.
- M. T. Bourdillon, B. A. Ford, A. T. Knulty, C. N. Gray, M. Zhang, D. A. Ford and R. D. McCulla, Photochem. Photobiol., 2014, 90, 386–393 CrossRef CAS.
- M. Zhang, G. E. Ravilious, L. M. Hicks, J. M. Jez and R. D. McCulla, J. Am. Chem. Soc., 2012, 134, 16979–16982 CrossRef CAS.
- J. T. Petroff, A. Isor, S. M. Chintala, C. J. Albert, J. D. Franke, D. Weinstein, S. M. Omlid, C. K. Arnatt, D. A. Ford and R. D. McCulla, RSC Adv., 2020, 10, 26553–26565 RSC.
- S. Omlid, A. Isor, K. Sulkowski, S. Chintala, J. Petroff and R. McCulla, Synthesis, 2018, 2359–2366 CAS.
- J. Korang, W. R. Grither and R. D. McCulla, J. Am. Chem. Soc., 2010, 132, 4466–4476 CrossRef CAS.
- X. Zheng, S. M. Baumann, S. M. Chintala, K. D. Galloway, J. B. Slaughter and R. D. McCulla, Photochem. Photobiol. Sci., 2016, 15, 791–800 RSC.
- D. I. Pattison, A. S. Rahmanto and M. J. Davies, Photochem. Photobiol. Sci., 2012, 11, 38–53 RSC.
- R. P. Rastogi, Richa, A. Kumar, M. B. Tyagi and R. P. Sinha, J. Nucleic Acids, 2010, 2010, 592980 Search PubMed.
- S. Roy, Advances in Experimental Medicine and Biology, Springer, New York LLC, 2017, vol. 996, pp. 207–219 Search PubMed.
- J. T. Herron and R. E. Huie, J. Phys. Chem. Ref. Data, 1973, 2, 467–518 CrossRef CAS.
- W. S. Nip, D. L. Singleton and R. J. Cvetanovic, J. Am. Chem. Soc., 1981, 103, 3526–3530 CrossRef CAS.
- J. C. Roberts, G. Huai, A. Gopalsamy, A. Kongsjahju and R. J. Patch, Tetrahedron Lett., 1997, 38, 355–358 CrossRef CAS.
- S. M. Chintala, J. T. Petroff, A. Barnes and R. D. McCulla, J. Sulfur Chem., 2019, 40, 503–515 CrossRef CAS.
- J. T. Petroff, S. M. Omlid, S. M. Chintala and R. D. McCulla, J. Photochem. Photobiol., A, 2018, 358, 130–137 CrossRef CAS.
- R. D. McCulla and W. S. Jenks, J. Am. Chem. Soc., 2004, 126, 16058–16065 CrossRef CAS.
- S. A. Stoffregen, S. Y. Lee, P. Dickerson and W. S. Jenks, Photochem. Photobiol. Sci., 2014, 13, 431–438 RSC.
- E. M. Rockafellow, R. D. McCulla and W. S. Jenks, J. Photochem. Photobiol., A, 2008, 198, 45–51 CrossRef CAS.
- J. W. Cubbage, T. A. Tetzlaff, H. Groundwater, R. D. Mcculla, M. Nag and W. S. Jenks, J. Org. Chem., 2001, 66, 8621–8628 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revis. B.01, Gaussian, Inc., Wallingford CT, 2009 Search PubMed.
- D. Avcı, S. Altürk, F. Sönmez, Ö. Tamer, A. Başoğlu, Y. Atalay, B. Z. Kurt and N. Dege, Tetrahedron, 2018, 74, 7198–7208 CrossRef.
- D. Avcl, S. Bahçeli, O. Tamer and Y. Atalay, Can. J. Chem., 2015, 93, 1147–1156 CrossRef.
- M. Abo, C. Li and E. Weerapana, Mol. Pharm., 2018, 15, 743–749 CrossRef CAS.
- F. P. Guengerich, ACS Catal., 2018, 8, 10964–10976 CrossRef CAS.
- F. P. Guengerich, Chem. Res. Toxicol., 2001, 14, 611–650 Search PubMed.
- J. Que L. and Y. Watanabe, Science, 2001, 292, 651–653 CrossRef.
- A. T. Pudzianowski and G. H. Loew, Int. J. Quantum Chem., 1983, 23, 1257–1268 CrossRef CAS.
- R: A language and environment for statistical computing, R Foundation for Statistical Computing, Vienna, Austria, URL https://www.R-project.org/ Search PubMed.
- H. Wickham and R. Francois, R Packag. version 0.4.2., 2015 Search PubMed.
- N. Nagaraj, J. R. Wisniewski, T. Geiger, J. Cox, M. Kircher, J. Kelso, S. Pääbo and M. Mann, Mol. Syst. Biol., 2011, 7, 548 CrossRef.
- K. Luby-Phelps, Mol. Biol. Cell, 2013, 24, 2593–2596 CrossRef CAS.
- K. Luby-Phelps, Int. Rev. Cytol., 2000, 192, 189–221 CAS.
- S. M. Omlid, S. A. Dergunov, A. Isor, K. L. Sulkowski, J. T. Petroff, E. Pinkhassik and R. D. Mcculla, Chem. Commun., 2019, 55, 1706–1709 RSC.
- R. J. Ellis, Trends Biochem. Sci., 2001, 26(10), 597–604 CrossRef CAS.
- G. E. Crooks, G. Hon, J. M. Chandonia and S. E. Brenner, Genome Res., 2004, 14, 1188–1190 CrossRef CAS.
- T. D. Schneider and R. M. Stephens, Nucleic Acids Res., 1990, 18, 6097–6100 CrossRef CAS.
- A. Bateman, Nucleic Acids Res., 2019, 47, D506–D515 CrossRef.
- H. M. Berman, J. Westbrook, Z. Feng, G. Gilliland, T. N. Bhat, H. Weissig, I. N. Shindyalov and P. E. Bourne, Nucleic Acids Res., 2000, 28, 235–242 CrossRef CAS.
- A. Waterhouse, M. Bertoni, S. Bienert, G. Studer, G. Tauriello, R. Gumienny, F. T. Heer, T. A. P. De Beer, C. Rempfer, L. Bordoli, R. Lepore and T. Schwede, Nucleic Acids Res., 2018, 46, W296–W303 CrossRef CAS.
- S. Bienert, A. Waterhouse, T. A. P. De Beer, G. Tauriello, G. Studer, L. Bordoli and T. Schwede, Nucleic Acids Res., 2017, 45, D313–D319 CrossRef CAS.
- K. Arnold, L. Bordoli, J. Kopp and T. Schwede, Bioinformatics, 2006, 22, 195–201 CrossRef CAS.
- U. M. Wolkowicz and A. G. Cook, Nucleic Acids Res., 2012, 40, 9356–9368 CrossRef CAS.
- M. Thoms, R. Buschauer, M. Ameismeier, L. Koepke, T. Denk, M. Hirschenberger, H. Kratzat, M. Hayn, T. Mackens-Kiani, J. Cheng, J. H. Straub, C. M. Stürzel, T. Fröhlich, O. Berninghausen, T. Becker, F. Kirchhoff, K. M. J. Sparrer and R. Beckmann, Science, 2020, 369, 1249–1255 CrossRef CAS.
- M. Blatter and F. Allain, RCSB PDB – 2MQP: Structural Investigation of hnRNP L bound to RNA.
- M. P. Jacobson, D. L. Pincus, C. S. Rapp, T. J. F. Day, B. Honig, D. E. Shaw and R. A. Friesner, Proteins: Struct., Funct., Genet., 2004, 55, 351–367 CrossRef CAS.
- M. P. Jacobson, R. A. Friesner, Z. Xiang and B. Honig, J. Mol. Biol., 2002, 320, 597–608 CrossRef CAS.
- Schrödinger Release 2020-2: Prime, Schrödinger, LLC.
- Schrödinger Release 2020-2: Maestro, Schrödinger, LLC, New York, NY, 2020.
- M. Welin, U. Kosinska, N.-E. Mikkelsen, C. Carnrot, C. Zhu, L. Wang, S. Eriksson, B. Munch-Petersen and H. Eklund, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 17970–17975 CrossRef CAS.
- H. R. Christofk, M. G. Vander Heiden, N. Wu, J. M. Asara and L. C. Cantley, Nature, 2008, 452, 181–186 CrossRef CAS.
- A. R. Mitchell, M. Yuan, H. P. Morgan, I. W. McNae, E. A. Blackburn, T. Le Bihan, R. A. Homem, M. Yu, G. J. Loake, P. A. Michels, M. A. Wear and M. D. Walkinshaw, Biochem. J., 2018, 475, 3275–3291 CrossRef CAS.
- A. Vemu, J. Atherton, J. O. Spector, C. A. Moores and A. Roll-Mecak, Mol. Biol. Cell, 2017, 28, 3564–3572 CrossRef CAS.
- A. Murayama, K. Ohmori, A. Fujimura, H. Minami, K. Yasuzawa-Tanaka, T. Kuroda, S. Oie, H. Daitoku, M. Okuwaki, K. Nagata, A. Fukamizu, K. Kimura, T. Shimizu and J. Yanagisawa, Cell, 2008, 133, 627–639 CrossRef CAS.
- A. R. Gingras, W. Puzon-McLaughlin and M. H. Ginsberg, J. Biol. Chem., 2013, 288, 23639–23649 CrossRef CAS.
- D. Waschbüsch, E. Purlyte, P. Pal, E. McGrath, D. R. Alessi and A. R. Khan, Structure, 2020, 28, 406–417.e6 CrossRef.
- A. Rai, A. Oprisko, J. Campos, Y. Fu, T. Friese, A. Itzen, R. S. Goody, E. M. Gazdag and M. P. Müller, eLife DOI:10.7554/eLife.18675.
- Y. Tominaga, M. L. Lee and R. N. Castle, J. Heterocycl. Chem., 1981, 18, 977–979 CrossRef CAS.
- N. J. Bunce, J. Lamarre and S. P. Vaish, Photochem. Photobiol., 1984, 39, 531–533 CrossRef CAS.
- J. K. Eng, A. L. McCormack and J. R. Yates, J. Am. Soc. Mass Spectrom., 1994, 5, 976–989 CrossRef CAS.
- E. Weerapana, C. Wang, G. M. Simon, F. Richter, S. Khare, M. B. D. Dillon, D. A. Bachovchin, K. Mowen, D. Baker and B. F. Cravatt, Nature, 2010, 468, 790–795 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: 1. Supporting information, 2. Excel workbook with protein analysis. See DOI: 10.1039/d0cb00200c |
| This journal is © The Royal Society of Chemistry 2021 |