Energy-efficient electrochemical ammonia production from dilute nitrate solution†
Received
25th October 2022
, Accepted 9th January 2023
First published on 24th January 2023
Abstract
Highly efficient electrochemical nitrate reduction could become a key process for sustainable ammonia production overcoming many limitations of the Haber–Bosch process. Current state-of-the-art electrocatalysts have severe drawbacks regarding yield, selectivity and energy efficiency when dealing with dilute nitrate solutions. Herein, we report a layered double hydroxide (LDH)/Cu foam hybrid electrocatalyst that offers a potential solution to this challenge. The [Ni0.75Fe0.25(OH)2](CO3)0.125 (Ni3Fe–CO3 LDH) exhibits an appropriate kinetic energy barrier for the Volmer step generating hydrogen radicals as well as suppressing H–H bond formation by inhibition of the Heyrovsky step. The electrochemically generated hydrogen radicals transfer to a Cu surface enabling NO3− reduction to NH3. The Ni3Fe–CO3 LDH/Cu foam hybrid electrode exhibits an 8.5-fold higher NH3 yield compared to a pristine Cu surface, while exhibiting an NH3 selectivity of 95.8% at 98.5% NO3− conversion. The best half-cell energy efficiency (36.6%) was recorded while achieving 96.8% faradaic efficiency at −0.2 V in 5 mM NO3−(aq).
Broader context
Significant attention has recently been directed towards developing electrocatalysts as an alternative technology to the traditional Haber–Bosch process. An alternative approach could involve the electrocatalytic reduction of nitrate (NO3−) to ammonia (NH3). To provide a sustainable and energy-efficient technical solution will require the use of earth abundant elements as well as utilising natural sources of NO3− (below 10 mM). Herein, we report an energy-efficient hybrid electrocatalyst for the reduction dilute NO3− solution to ammonia. The electrocatalyst operates via a hydrogen radical strategy leading to ultrahigh performance for NH3 production at low NO3− concentrations, thus paving the way to realising a sustainable and energy-efficient NH3 economy.
|
1 Introduction
The high energy density of ammonia (4.32 kW h L−1) means it has become an chemical of high interest as we consider carbon free energy carries in addition to remaining a pivotal feedstock of the agricultural, plastic, pharmaceutical and textile industries.1–4 For over 100 years the Haber–Bosch process has monopolised NH3 production; utilising 1–2% of the world's energy supply and producing 1% of the total global energy related CO2 emissions in order to produce NH3.5,6 Furthermore, 80% of the energy demand derives from steam methane reforming (SMR) to supply H2 gas as a feedstock to the Haber–Bosch process. Electrochemical H2 production using electricity from renewable technologies and its subsequent utilization could be a source of ‘green NH3’. Although the efficiency and stability of polymer electrolyte membrane (PEM) electrolysers for green H2 production have developed significantly, it requires at least 30.3–35.3 GJ per tonammonia at an operating efficiency of even up to 60–70%. Additionally, N2 production requires additional 2.7 GJ per tonammonia using an air-separation unit and Haber–Bosch loop compressors to supply N2 to proceed the Haber–Bosch process with green H2. These costs are currently still higher than the conventional Haber–Bosch process (below 30 GJ per tonammonia).54,55 In this regard, electrochemical nitrogen reduction (NRR) has recently sparked global research interests to produce NH3 as an alternative to Haber–Bosch process. To date, it has suffered from low yield (below 3 × 10−10 mol s−1 cm−2) and faradaic efficiency (FE, lower than 10%), challenged by the N![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N bond strength (941 kJ mol−1), poor N2 solubility in aqueous solution (0.66 mmol L−1 under ambient conditions), and the competing hydrogen evolution reaction (HER).7,8
N bond strength (941 kJ mol−1), poor N2 solubility in aqueous solution (0.66 mmol L−1 under ambient conditions), and the competing hydrogen evolution reaction (HER).7,8
Electrochemical nitrate reduction (NitRR) has the potential to offer a sustainable route to solve the environmental issues of denitrification and the energy demands of NH3 production. Low N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bonding energy (204 kJ mol−1) and high NO3− solubility offer the potential for high electrocatalytic performance of NitRR compared to NRR.9 In recent years, some NitRR electrocatalysts have been developed that can surpass the NH3 yield of Haber–Bosch process (<0.2 mol gcatalyst−1 h−1)10,11 and exceed the current density of 2 A cm−2 for NH3 production.12 In spite of these achievements, high-yield NH3 production from NO3− presents two major obstacles that need to be improved if it is to deliver a scalable practical solution. Firstly, highly performing electrocatalysts typically operate in highly concentrated NO3− electrolyte (100–1000 mM),5,10,13–16 requiring additional and expensive costs associated with concentrating nitrate solutions. In reality, most nitrate sources such as textile industrial wastewater (<10 mM) and polluted ground water (<2 mM) have low nitrate concentrations.17 Therefore, electrochemical low concentrated nitrate reduction is demanding because the faradaic efficiency decreases dramatically when the nitrate concentration is below 10 mM due to nitrate diffusion and the kinetically competitive HER reaction.18 Secondly, a high operating voltage needs to be applied to the solution in order to achieve a sufficiently high catalytic NH3 production. Nitrate reduction is a complex 8-electron reaction involving several intermediates (e.g., NO2 and NO) and slow multi-electron mediated reaction rates. Decentralised remote production of green ammonia from diluted aqueous nitrate solutions could be realised by developing an advanced electrode system to deliver high energy efficiency, selectivity and NO3− conversion rate using renewal electricity.
O bonding energy (204 kJ mol−1) and high NO3− solubility offer the potential for high electrocatalytic performance of NitRR compared to NRR.9 In recent years, some NitRR electrocatalysts have been developed that can surpass the NH3 yield of Haber–Bosch process (<0.2 mol gcatalyst−1 h−1)10,11 and exceed the current density of 2 A cm−2 for NH3 production.12 In spite of these achievements, high-yield NH3 production from NO3− presents two major obstacles that need to be improved if it is to deliver a scalable practical solution. Firstly, highly performing electrocatalysts typically operate in highly concentrated NO3− electrolyte (100–1000 mM),5,10,13–16 requiring additional and expensive costs associated with concentrating nitrate solutions. In reality, most nitrate sources such as textile industrial wastewater (<10 mM) and polluted ground water (<2 mM) have low nitrate concentrations.17 Therefore, electrochemical low concentrated nitrate reduction is demanding because the faradaic efficiency decreases dramatically when the nitrate concentration is below 10 mM due to nitrate diffusion and the kinetically competitive HER reaction.18 Secondly, a high operating voltage needs to be applied to the solution in order to achieve a sufficiently high catalytic NH3 production. Nitrate reduction is a complex 8-electron reaction involving several intermediates (e.g., NO2 and NO) and slow multi-electron mediated reaction rates. Decentralised remote production of green ammonia from diluted aqueous nitrate solutions could be realised by developing an advanced electrode system to deliver high energy efficiency, selectivity and NO3− conversion rate using renewal electricity.
Advanced electrodes based on noble metals (e.g. Pd and Ru) have been explored as NitRR electrocatalysts,19,20 however, their high price and abundance limits their large-scale application. Recent electrocatalysts have focused on earth-abundant transition metal in order to overcome the inherent disadvantages of noble metal-based electrocatalysts. However, most of the transition metals have their own inherent problems because NO3− has typically a low binding affinity for transition metals in an aqueous solution.21,22 Moreover, the transition metals are more likely to form M–H bonds for competitive HER, leading to low selectivity and low catalytic efficiency for NitRR.23 Significantly, copper metal catalysts have favorable binding energy for NO3− due to favourable energy level match between Cu's d-orbitals and nitrate's LUMO π* molecular orbital.24 Furthermore, the surfaces of Cu metal catalysts suppress HER and promote electron transfer, catalysing NO3− to NO2− conversion which is a rate-determining step of electrochemical nitrate reduction.25 Nevertheless, Cu metal catalysts commonly undergo rapid deactivation owing to their strong adsorption of NitRR intermediates (*NO3, *NO2, *NO, *N, *NH and *NH2), thus suppressing the further electrochemical reduction to NH3.
In this context, hydrogen radical supplier mediated Cu metal electrocatalysts could be a novel solution to address the high NO3− concentration issues and the disadvantages of Cu metal by separating the sites of hydrogen radical formation and the sites of nitrate reduction because hydrogen radicals can reduce NitRR intermediates on selectively NO3−-adsorbed Cu surface. As an active NitRR site, the highly porous Cu foam electrode is utilised both as a reduction reaction site enabling the selective NO3− adsorption on its surface at low NO3− concentrations and as an electrical conductor decreasing the materials cost for large-scale application. This compares favourably to electrodes that utilise nickel foam or carbon based substrates (carbon paper, carbon cloth) that only act as electrical conductors.10,13–16,18,23,24,43–45,47,48,53 As a hydrogen radical supplier, layered double hydroxides (LDH; 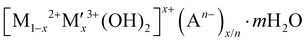 ) could be a unique catalyst platform because of its high overpotential for HER if the Heyrovsky step (Hads* + H2O + e− → H2 + OH) would be suppressed and if the Volmer step (H2O + e− → Hads* + OH−) would be activated. Very few articles have been reported on electrochemical NitRR using an LDH because high voltage should require to drive the cathodic reaction in order to overcome the low inherent electrical conductivity. To date, few LDHs have been used as active materials in electrochemical NitRR, in each case both a high voltage and high nitrate concentration were needed to achieve a high FE which decreases the overall energy efficiency (i.e. [NO3−] = 14 mM [NO3−] for Co1Fe–CO3 LDH;43 [NO3−] = 25 mM for Cu2CoAl–CO3 LDH;52 [NO3−] = 100 mM for Co3AlCO3 LDH53). In this work, we have explored a range of designer LDH decorated Cu foam electrodes (LDH/Cu foam) in order to investigate the role of mixed transition metal combinations to generate hydrogen radicals. We have prepared 12 LDH/Cu foam samples (LDHs = Ni3Al–CO3, Ni3V–CO3, Ni3Fe–CO3, Ni3Co–CO3, Co3Al–CO3, Co3V–CO3, Co3Fe–CO3, Zn3Al–CO3, Zn3V–CO3, Zn3Fe–CO3, Mg3Al–CO3 and Mg3Fe–CO3) which are representative active elements in electrocatalysts and evaluated them for electrochemical NitRR. The ultimate aim of the work is to increase the energy efficiency for selective nitrate reduction at low NO3− concentration. We found that the Ni3Fe–CO3 LDH/Cu foam electrode achieved the best electrochemical NitRR producing a 95.8% NH3 selectivity with a 98.5% NO3− conversion in 5 mM NO3−. Over 1 h the system exhibited a NH3 productivity of 1.261 mg cm−2 h−1 and a 96.8% FE at −0.2 V. This equates to a 36.6% half-cell energy efficiency. As far as we are aware, this is the highest energy efficient electrochemical NitRR to date.
) could be a unique catalyst platform because of its high overpotential for HER if the Heyrovsky step (Hads* + H2O + e− → H2 + OH) would be suppressed and if the Volmer step (H2O + e− → Hads* + OH−) would be activated. Very few articles have been reported on electrochemical NitRR using an LDH because high voltage should require to drive the cathodic reaction in order to overcome the low inherent electrical conductivity. To date, few LDHs have been used as active materials in electrochemical NitRR, in each case both a high voltage and high nitrate concentration were needed to achieve a high FE which decreases the overall energy efficiency (i.e. [NO3−] = 14 mM [NO3−] for Co1Fe–CO3 LDH;43 [NO3−] = 25 mM for Cu2CoAl–CO3 LDH;52 [NO3−] = 100 mM for Co3AlCO3 LDH53). In this work, we have explored a range of designer LDH decorated Cu foam electrodes (LDH/Cu foam) in order to investigate the role of mixed transition metal combinations to generate hydrogen radicals. We have prepared 12 LDH/Cu foam samples (LDHs = Ni3Al–CO3, Ni3V–CO3, Ni3Fe–CO3, Ni3Co–CO3, Co3Al–CO3, Co3V–CO3, Co3Fe–CO3, Zn3Al–CO3, Zn3V–CO3, Zn3Fe–CO3, Mg3Al–CO3 and Mg3Fe–CO3) which are representative active elements in electrocatalysts and evaluated them for electrochemical NitRR. The ultimate aim of the work is to increase the energy efficiency for selective nitrate reduction at low NO3− concentration. We found that the Ni3Fe–CO3 LDH/Cu foam electrode achieved the best electrochemical NitRR producing a 95.8% NH3 selectivity with a 98.5% NO3− conversion in 5 mM NO3−. Over 1 h the system exhibited a NH3 productivity of 1.261 mg cm−2 h−1 and a 96.8% FE at −0.2 V. This equates to a 36.6% half-cell energy efficiency. As far as we are aware, this is the highest energy efficient electrochemical NitRR to date.
2 Results and discussion
A hybrid LDH-Cu foam electrode for electrochemical nitrate reduction is illustrated in Fig. 1(a), where hydrogen radicals produced from water by a LDH are transferred to a Cu foam surface for electrochemical NitRR. LDHs adopt a brucite-like layered structure with cationic and anionic layers. The positively charged metal cationic layers (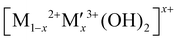 ) are balanced by solvated anionic layers (An−) as shown in Fig. S1 (ESI†).28 Powder X-ray Diffraction (PXRD) patterns for 12 LDHs with different combinations of metal cations (M2+: Ni, Co, Zn, Mg and M′(3+x)+: Al, Fe, Co, V) are shown in Fig. S2 (ESI†), the materials show the Bragg reflections at 2θ = 11° and 22° that can be indexed as (003) and (006) reflections for a prototypical LDH unit cell. In Table S1 (ESI†), calculated interlayer distance (d003) and lattice parameters correspond to a hydrotalcite-3R type LDH with the R3m symmetry.
) are balanced by solvated anionic layers (An−) as shown in Fig. S1 (ESI†).28 Powder X-ray Diffraction (PXRD) patterns for 12 LDHs with different combinations of metal cations (M2+: Ni, Co, Zn, Mg and M′(3+x)+: Al, Fe, Co, V) are shown in Fig. S2 (ESI†), the materials show the Bragg reflections at 2θ = 11° and 22° that can be indexed as (003) and (006) reflections for a prototypical LDH unit cell. In Table S1 (ESI†), calculated interlayer distance (d003) and lattice parameters correspond to a hydrotalcite-3R type LDH with the R3m symmetry.
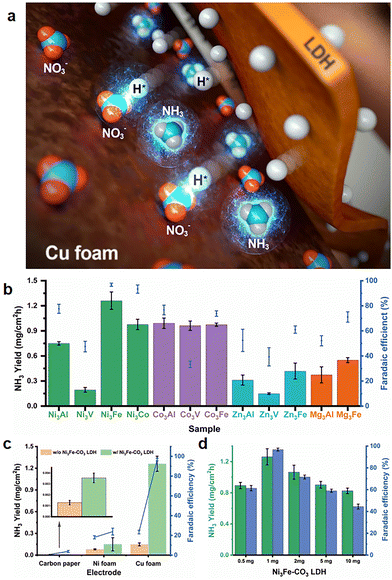 |
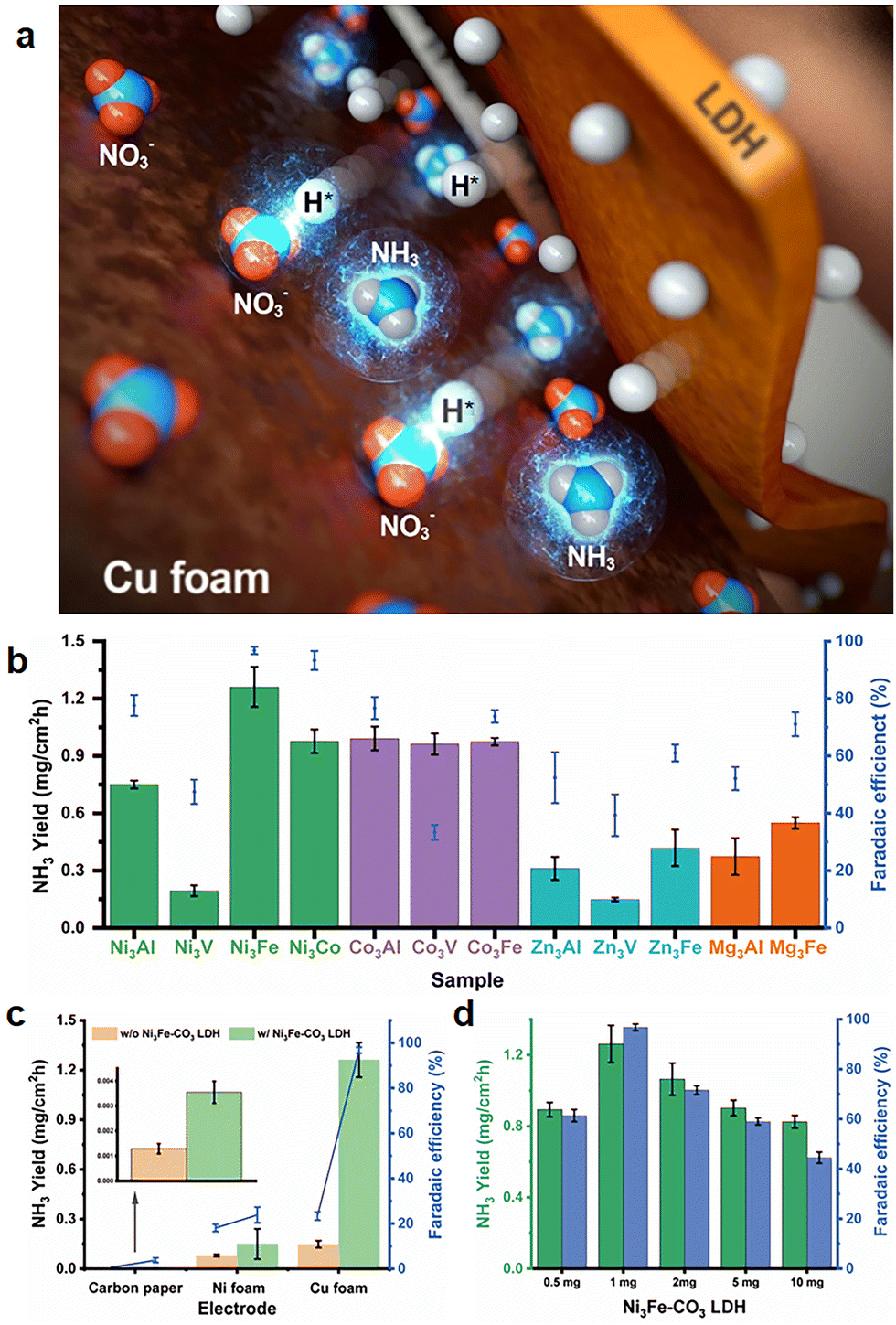
| | Fig. 1 Layered double hydroxide-Cu foam electrochemical nitrate reduction. (a) Schematic illustration of a hybrid multi-metallic LDH modified Cu foam electrode for electrochemical nitrate reduction, where hydrogen radicals produced and transferred to nitrate adsorbed on Cu foam surface by a LDH in an aqueous solution. (b) NH3 productivity (mg cm−2 h−1) and faradaic efficiency (%) using multi-metallic transition metal LDH/Cu foam electrodes. (c) NH3 productivity and faradaic efficiency (%) using the electrodes w/and w/o Ni3Fe–CO3 LDH. (d) The effect of Ni3Fe–CO3 LDH loading on NH3 productivity and faradaic efficiency. All electrocatalysis reactions was operated at 1 M KOH electrolyte with 5 mM of KNO3 at −0.2 V (vs. RHE). | |
Twelve LDHs were prepared to investigate the optimum transition metal combinations for the hydrogen radical supplier role, and electrochemical NitRR was conducted for an hour using a chronoamperometric technique with an LDH/Cu foam electrode as a working electrode, applying a −0.2 V potential in 1 M KOH electrolyte with 5 mM KNO3. As shown in Fig. 1(b), the Ni and Co based LDHs show relatively higher yield and FE than the Zn and Mg based LDHs except for the V containing LDHs because V ions in the LDH were active sites for HER suppressing NH3 production. In the case of Zn3Fe–CO3 and Zn3V–CO3 LDH, ZnO phase was mixed with LDH phase. To clarify the ZnO effect, we have measured electrochemical NitRR of pure ZnO (Fig. S3, ESI†). It was found out that pure ZnO has similar properties compared to Zn3Fe–CO3 and Zn3V–CO3 LDH, which is not proper candidate for electrochemical NitRR. Among the twelve types of LDH, the Ni3Fe–CO3 LDH/Cu foam electrode exhibits the highest catalytic activity with a yield rate of 1.261 mg cm−2 h−1 and 96.8% of a FE from NO3− to NH3, at −0.2 V. The hydrogen radical supplier role of the LDH was confirmed in Fig. 1(c), showing NH3 yield rate and FE depending on the electrode type and the presence of the Ni3Fe–CO3 LDH. When carbon paper (CP) is utilised as an electrode, the conversion and FE for NH3 production are neglectable regardless of the Ni3Fe–CO3 LDH because both CP and Ni3Fe–CO3 LDH are not the active sites for electrochemical NO3− reduction. In the case of the Ni foam electrode, a yield rate (and FE) before and after introducing the Ni3Fe–CO3 LDH on the surface of Ni foam electrode exhibit 0.080 mg cm−2 h−1 (18.1%) and 0.150 (23.8%) mg cm−2 h−1, respectively. The Ni foam electrode has an inherent activity for electrochemical NitRR, however, the formation of Ni–H bonds facilitates competitive HER, leading to a low yield and low FE. Introducing the Ni3Fe–CO3 LDH onto a Ni foam surface leads to a slight increase in NH3 production rate and FE. A Cu foam electrode displays a NH3 production rate of 0.149 mg cm−2 h−1 which is approximately 1.9 times higher than that of the Ni foam. This supports the hypothesis that Cu foam has more favorable active sites for electrochemical NitRR. Nonetheless, the FE of 23.4% for a Cu foam is very similar to that of Ni foam, implying that N-containing intermediate poisoning (*NO3, *NO2, *NO, *N, *NH and *NH2) during NitRR suppresses NH3 production because Cu has selective *NO3 adsorption followed by unfavourable ΔG for *NO2 adsorption.58,59 Remarkably, the NH3 production rate improved more than 8.5 times and the FE reached 96.8% by introducing Ni3Fe–CO3 LDH onto the Cu foam surface. As the Ni3Fe–CO3 LDH proved to be inactive for NitRR, it mainly acts as a hydrogen radical supplying role during the electrochemical reaction. Surface coverage effects with Ni3Fe–CO3 LDH were revealed in Fig. 1(d). The amount of Ni3Fe–CO3 LDH loading on the foam was varied from 0.5 mg to 10 mg, we observed a volcanic dependence for both yield rate and FE as a function of LDH loading. Maximum electrocatalytic performance was observed at a loading of 1 mg. We suggest this is due to the LDH particles blocking NO3− access to the active Cu surface.
To further understand the Ni3Fe–CO3 LDH/Cu foam electrode, the structure of Ni3Fe–CO3 LDH was analysed by PXRD and Fourier transform-infrared (FT-IR) spectroscopy. The PXRD (Fig. 2(a)) contains a series of basal and non-basal plane Bragg reflections which demonstrate the highly crystalline nature of the LDH phases.28 The interlayer anions can be identified as CO32− by the asymmetric stretching band for CO32− at around 1357 cm−1 in the FT-IR (Fig. S4, ESI†). Additionally, two absorbances found at around 3800–2600 cm−1 and at 1635 cm−1 can be ascribed to the stretching modes of the hydrogen-bonded HO⋯H2O interactions and the angular deformation of the interlayer H2O respectively.29 The thermogravimetric analysis (TGA) shows the typical two step weight loss profile for an LDH (Fig. S5, ESI†). The desorption of water in the interlayer region occurs below 200 °C, and the LDH structure collapses due to dehydration of hydroxyl ions on the cationic layers and the pyrolysis of anions in the interlayers between 200 °C and 400 °C.30 The X-ray absorption spectroscopy (XAS) were conducted to probe the local electronic structure around Ni and Fe cations in Ni3Fe–CO3 LDH. The valence state of Ni in Ni3Fe–CO3 LDH was revealed by comparing the Ni K-edge X-ray absorption near-edge spectroscopy (XANES) spectra of Ni(OH)2 and Ni metal (Fig. 2(b)). The Ni K-edge XANES spectra show a strong absorption edge at approximately 8350 eV, implying that Ni ions are divalent. Both Ni3Fe–CO3 LDH and Ni(OH)2 have similar shape spectra with absorption edges at around 8350 eV.31 The Fourier transformed extended X-ray absorption fine structure (FT-EXAFS) spectra were analysed to elucidate the coordination sphere of Ni. Ni3Fe–CO3 LDH and Ni(OH)2 exhibit the same radial Ni–O bonding at 1.56 Å (Fig. S6a, ESI†). However, the radial distance for Ni–metal bonding in Ni3Fe–CO3 LDH (2.70 Å) is slightly shorter than that of Ni(OH)2 (2.76 Å). Ni metal has only Ni–Ni bonds at 2.17 Å, matching well with a previous study.32 The Fe oxidation state and coordination environment were analysed using Fe K-edge XANES and FT-EXAFS. Ni3Fe–CO3 LDH contains trivalent Fe3+ (Fig. 2(c)), as evidenced by the rising edge position being right-shifted in the order of Fe metal (Fe0), Fe3O4 (Fe2+ and Fe3+), γ-Fe2O3 (Fe3+) and Ni3Fe–CO3 LDH. The right-shifted absorption edge between Fe3O4 and γ-Fe2O3 is derived from Fe2+ ions occupying the octahedral sites in the inverse spinel structure of γ-Fe2O3. Moreover, the Fe K-edge FT-EXAFS indicates that Ni3Fe–CO3 LDH has Fe–O and Fe-metal distances of 1.56 Å and 2.67 Å respectively. We note that γ-Fe2O3 and Fe3O4 have slightly shifted bond lengths as a result of their different crystal structures compared to Ni3Fe–CO3 LDH. (Fig. S6b, ESI†). Transmission electron microscopy (TEM) confirmed the morphology of Ni3Fe–CO3 LDH. As shown in Fig. S7a (ESI†), Ni3Fe–CO3 LDH has a nanoplatelet morphology with a platelet diameter below 50 nm. The high-resolution transmission electron microscopy (HR-TEM) image (Fig. 2(d)), exhibits lattice fringes of 0.78 nm corresponding to the (003) Bragg reflection of the Ni3Fe–CO3 LDH from a platelet with a 5–7 nm thickness (Fig. S7b, ESI†). The high angle annular dark field (HAADF)-scanning transmission electron microscopy (STEM) image (Fig. 2(e)) and STEM-energy dispersive spectrometer (EDS) analysis (Fig. 2(f) and (g)) provides clear evidence that Ni and Fe are well-dispersed. The Ni3Fe–CO3 LDH/Cu foam electrode was also characterised by HAADF-STEM image and STEM-EDS mapping, showing that Ni3Fe–CO3 LDH is well-attached to Cu foam surface (Fig. 2(h) and 2(i)). The N2 isotherm for Ni3Fe–CO3 LDH is categorised as type-4 with a BET specific surface area of 59.9 m2 g−1, the pore size distribution is presented in Fig. S8a and b (ESI†).32
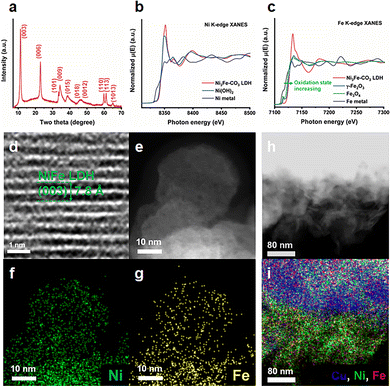 |
| | Fig. 2 Structural studies of electrocatalysts: (a) PXRD of Ni3Fe–CO3 LDH, (b) Ni K-edge XANES Ni3Fe–CO3 LDH with suitable reference materials, (c) Fe K-edge XANES of Ni3Fe–CO3 LDH with suitable reference materials, (d) HR-TEM image of Ni3Fe–CO3 LDH, (e) HAADF-STEM image of Ni3Fe–CO3 LDH, (f) STEM Ni mapping image of Ni3Fe–CO3 LDH, (g) STEM Fe mapping image for Ni3Fe–CO3 LDH, (h) a HAADF-STEM image, (i) STEM Cu, Ni, Fe mapping image for the Ni3Fe–CO3 LDH/Cu foam electrode. | |
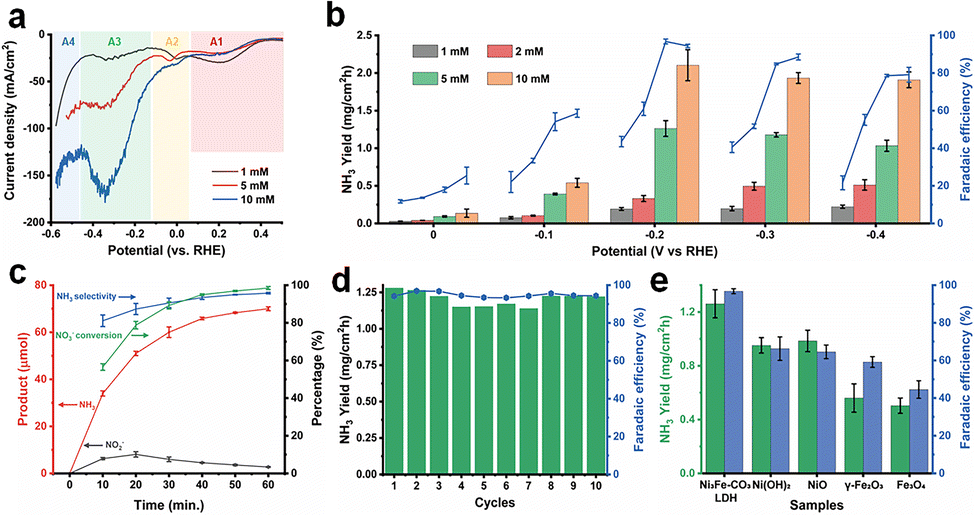
We investigated the electrochemical performance in NitRR and evaluated the synergistic effects between Ni3Fe–CO3 LDH and Cu foam at ambient conditions. The electrocatalysis was performed by a three-electrode system using Hg/HgO as the reference electrode, coiled platinum wire as the counter electrode, and Ni3Fe–CO3 LDH deposited on a Cu foam as the working electrode assembled with a H-type cell under 1 M KOH electrolyte with/without NO3− ions. High-purity argon gas was bubbled into the cathodic chamber during electro-reduction to prevent possible side-competitive reactions such as NRR. The NO2− and NH3 concentrations were quantified using a colourimetric method (Fig. S9a–d, ESI†). Firstly, linear weep voltammetry (LSV) experiments were conducted to assess NitRR activity of the Ni3Fe–CO3 LDH/Cu foam electrode at different nitrate concentrations at a scan rate of 5 mV s−1 (Fig. 3(a)). Electrocatalytic reduction on Cu surfaces involves four main reactions denoted A1–A4.33
| | | A1: NO3− + H2O +2e− → NO2− + 2OH− | (1) |
| | | A2: NO2− + 4H2O + 4e− → NH2OH + 5OH− | (2) |
| | | A3: NO2− + 5H2O + 6e− → NH3 + 7OH− | (3) |
| | | A4: HER from competing adsorption of H* | (4) |
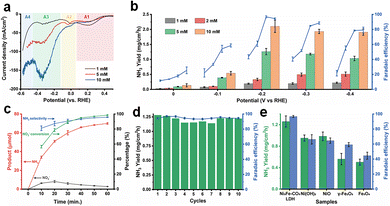 |
| | Fig. 3 Electrochemical performance of the Ni3Fe–CO3 LDH LDH/Cu foam electrode. (a) Linear sweep voltammetry (LSV) of the Ni3Fe–CO3 LDH LDH/Cu foam electrode depending on the different KNO3 concentration. The scan rate of LSV is 5 mV s−1. A1: NO3− + H2O +2e− → NO2− + 2OH−, A2: NO2− + 4H2O + 4e− → NH2OH + 5OH−, A3: NO2− + 5H2O + 6e− → NH3 + 7OH−, A4: HER from competing adsorption of H* (b) NH3 productivity and faradaic efficiency as a function of KNO3 concentration and applied potential. (c) Calculated NH3 selectivity, NO3− conversion and the amount of NH3 and NO2− production as a function of time (d) NH3 production rate and faradaic efficiency as a function of cycle number. (e) Comparison of NH3 productivity and faradaic efficiency with those of selected reference materials for an hour using chronoamperometric analysis. In (c)–(e), the potential was applied to −0.2 V (vs. RHE) at 1 M KOH with 5 mM of KNO3. All electrochemical reactions were conducted using the Ni3Fe–CO3 LDH/Cu foam electrode (1 mg cm−2) in 1 M KOH electrolyte. | |
The current density increased with the NO3− concentration, implying the Ni3Fe–CO3 LDH/Cu foam was electrocatalytically active for NitRR. Especially, current density is enhanced massively at A3 because the electrode specially facilitates the conversion of NO3− into NH3 compared to other reduction reactions. In Fig. S10 (ESI†), Cu foam also has the NitRR activity determined by the current density difference between the absence and presence of nitrate. However, the maximum current density (−42.8 mA cm−2) at A3 is significantly lower than that of −79.4 mA cm−2 after Ni3Fe–CO3 LDH incorporation with 5 mM NO3− condition. In accordance with LSV curves, NH3 productivity and FE for Ni3Fe–CO3 LDH/Cu foam electrode were measured as a function of NO3− concentrations (1 mM, 2 mM, 5 mM and 10 mM) at applied voltages from 0 V to −0.4 V as shown in Fig. 3(b). Each reaction was conducted using the chronoamperometric method for 1 h in 15 mL electrolyte containing 1 M KOH. The NH3 yield increases with the decreasing potential until −0.2 V for all nitrate concentrations. When a more negative potential than −0.2 V is applied, the NH3 productivity drops slightly because of competition with HER. Furthermore, NO3− conversion rates are very fast with most of the NO3− converted within an hour for all NO3− concentrations, up to a maximum of 98.5% in 5 mM [NO3−]. The rapid kinetics is illustrated by a decrease in current density within an hour (Fig. S11, ESI†). The FE was observed to vary as a function of NO3− concentrations and applied voltage (Fig. 3(b)). There was little change as a function of NO3− concentrations at 0 V which is attributed to lack of reduction energy. When the reduction voltage was between −0.1 V to −0.4 V, it was found that the FE was nearly linearly dependent on the NO3− concentration until it reached 5 mM and then saturated. This suggests first-order reaction kinetics at NO3− concentrations below 5 mM. The FE reached maximum values at −0.2 V and decreased at more negative voltages. This is a consequence of a competitive HER becoming active over −0.2 V at all NO3− concentrations. We found that the FE can reach 96.8% in 5 mM at −0.2 V, which represents a 36.6% energy-efficiency for conversion of NO3− to NH3. The evolved products were analysed every 10 min at 5 mM NO3− and −0.2 V. Both NH3 and NO2 evolved within 10 min (Fig. 3(c)) with 56.5% NO3− conversion. After 10 minutes, the quantity of NO2 decreased while NH3 increased gradually reaching 95.8% NH3 selectivity and 98.5% of NO3− conversion after 60 min. Neither H2 nor N2 was detected during the electrochemical reaction using a gas chromatography (GC) equipped with a capillary column and mass selective detector (MSD), indicating no HER or nitrification reactions are taking place. However, the concentration of the initial 74.2 μmol NO3− source rapidly reduced to zero after 60 min, only NO3−, NO2− and NH3 can be determined during electrochemical NitRR. (Fig. S12a, ESI†) Moreover, LSV curves (Fig. S12b, ESI†) shows that most of electrochemical reduction reactions finished after 50 min, which matched well with the time-dependent analysis of products in Fig. S12a (ESI†). The Ni3Fe–CO3 LDH/Cu foam electrode exhibited excellent stability as shown in Fig. 3(d), sustaining a NH3 productivity of >1.138 mg cm−2 h−1, and a FE of >93.3% after 10 cycles of operation in 5 mM NO3− at −0.2 V. The role of Ni and Fe in the Ni3Fe–CO3 LDH/Cu foam electrode during electrochemical NitRR was investigated by looking at other related reference materials including Ni(OH)2, NiO, γ-Fe2O3 and Fe3O4 on Cu foam. As a hydrogen radical supplier in electrochemical NitRR two processes need to be optimised, the kinetic energy barrier for the Volmer reaction should be matched with the potential range for NO3− reduction. If the energy barrier is too low, a chain reaction will occur leading to HER. Meanwhile, if the energy barrier is too high, water will not be dissociated into hydrogen radicals. In addition, the Heyrovsky reaction should be suppressed to prevent H–H bond formation during electrochemical NitRR. In this regard, Ni3Fe–CO3 LDH is an ideal candidate as a hydrogen radical supplier because of having a high kinetic energy barrier for the Volmer reaction (HER overpotential of >210 mV at 10 mA cm−2) and a rate-limiting Heyrovsky reaction which prevents H–H formation during electrochemical NitRR.26,27Fig. 3(e) shows that Ni(OH)2 and NiO result in high NH3 production rate of 0.952 mg cm−2 h−1 and 0.985 mg cm−2 h−1, respectively, which are comparable to that of Ni3Fe–CO3 LDH. However, their FE of 66.3% and 64.6% are significantly lower than that of the Ni3Fe–CO3 LDH. We postulate that Ni oxide-based materials could produce and transfer hydrogen radicals facilitating electrochemical NitRR on a Cu surface. However, it still provides an available surface for the Tafel reaction – H2 production via adsorbed hydrogen facilitated on a Ni–O–Ni surface results in a decreased FE.34,35 In the case of Fe oxide-based materials, both γ-Fe2O3 and Fe3O4 have lower NH3 productivity and FE, meaning the absence of active production and transfer abilities for hydrogen radicals due to low Fe–H binding energy compared to Ni–H bonding.36,37 Remarkably, the Ni3Fe–CO3 LDH had both Ni hydroxide layers facilitating the production of hydrogen radicals and Fe heteroatoms transferring hydrogen radicals to Cu sites by decreasing the bonding energy of metal–H bonding and inhibiting the Tafel step, thereby enabling the high yield rate and FE in electrochemical NitRR.
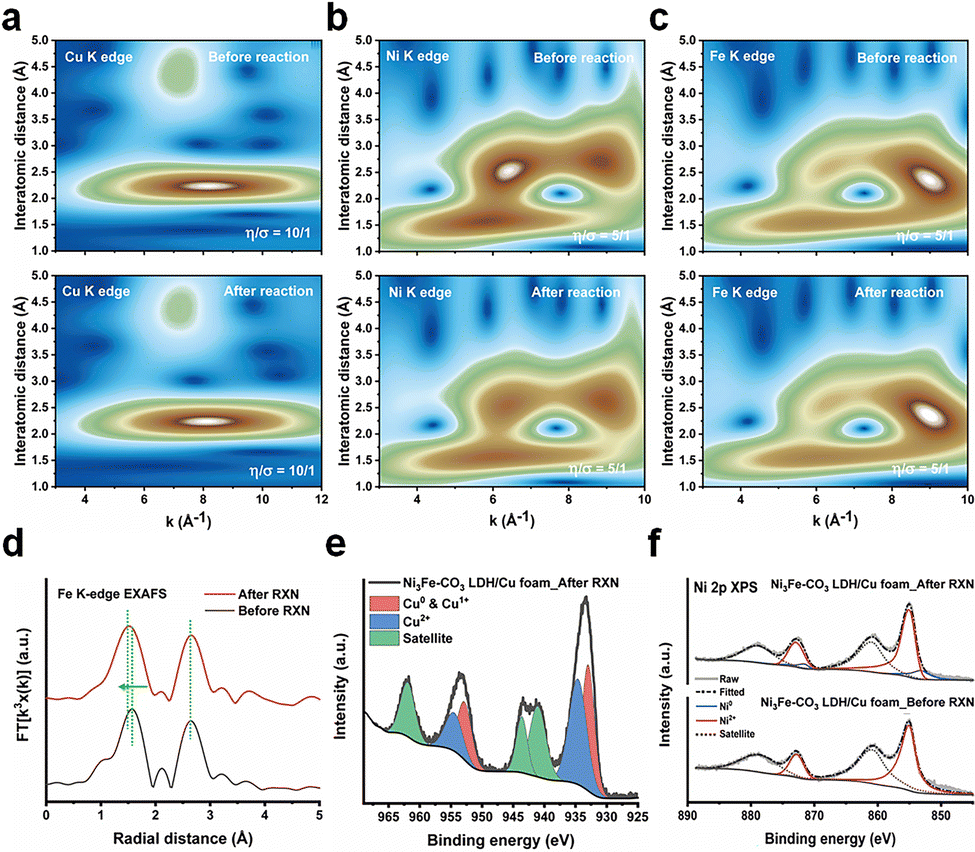
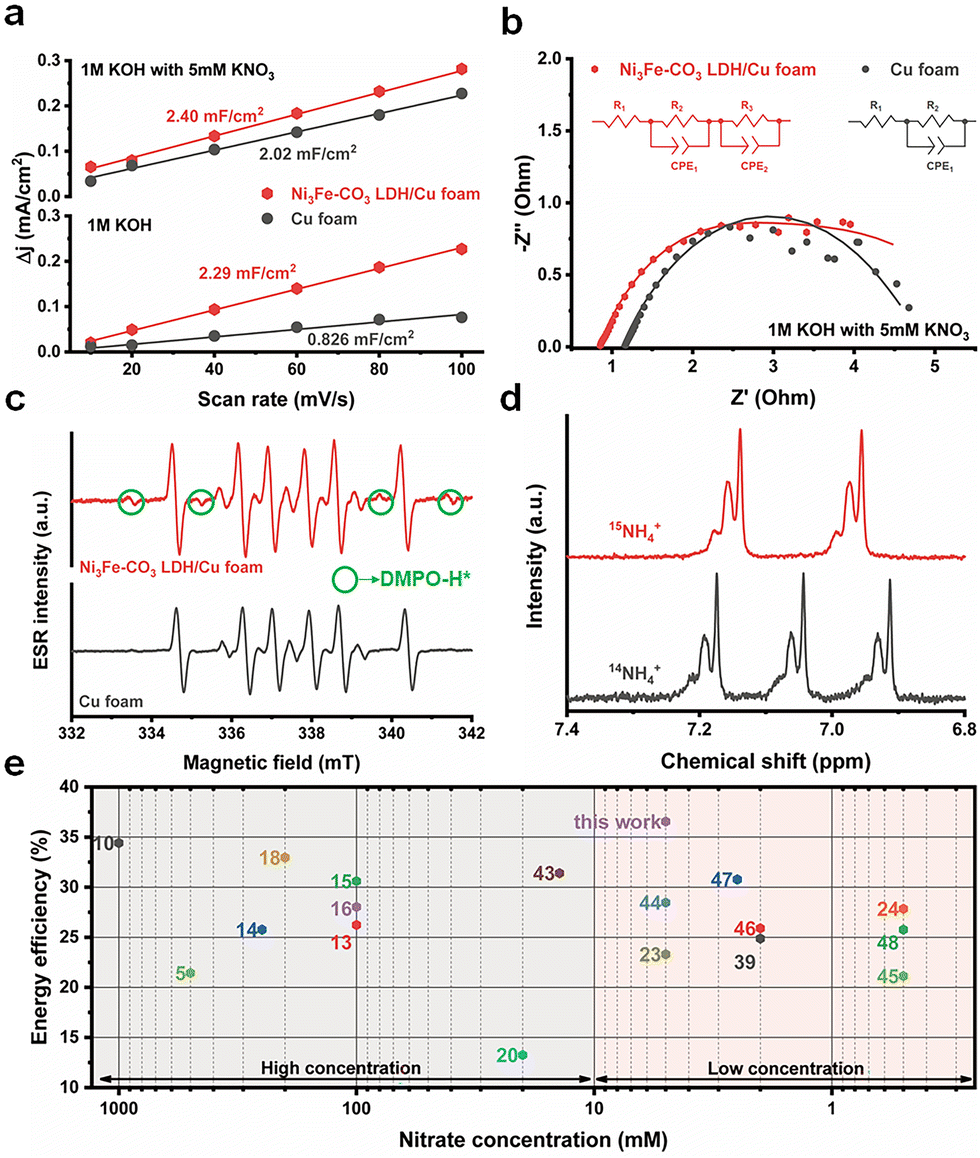
Ex situ XAS and X-ray photon spectroscopy (XPS) were performed to elucidate the bulk and surface structural changes of the Ni3Fe–CO3 LDH/Cu foam electrode before and after electrochemical NitRR. Wavelet transformed (WT)-EXAFS were analyzed to confirm the EXAFS structure both in k and r space simultaneously.38 The Cu K-edge WT-EXAFS shows only two main maxima identified by Cu–Cu scattering of the first shell at k ≈ 8.17 Å−1 and R ≈ 2.24 Å and by Cu–Cu scattering of the second shell at k ≈ 7.16 Å−1 and R ≈ 4.40 Å as shown in Fig. 4(a). Additionally, the Cu K-edge XANES (Fig. S13a, ESI†) and FT-EXAFS (Fig. S13b, ESI†) did not change, indicating the bulk structure of the Cu foam remains after the reaction. The Ni3Fe–CO3 LDH also retains its bulk structure after reaction as determined by Ni K-edge (WT-EXAFS in Fig. 4(b), XANES in Fig. S14a (ESI†) and FT-EXAFS in Fig. S14b, ESI†) and Fe K-edge (WT-EXAFS in Fig. 4(c) and XANES in Fig. S15, ESI†). However, the 1st Fe–O shell changed from 1.57 Å to 1.53 Å after electrochemical NitRR (Fig. 4(d)), suggesting that Fe affects the hydrogen radical transfer owing to lower binding strength of hydrogen to a Fe3+ centre.37 Using XPS analysis, a more significant change can be observed on the surface structure after NO3− reduction compared to that of the bulk structure. The Cu 2p XPS indicated Cu in the Ni3Fe–CO3 LDH/Cu foam is metallic before the reaction (Fig. S16, ESI†), however, after NO3− reduction both Cu2O and CuO phases were detected (Fig. 4(e)). This indicated that in situ electrochemical reconstruction occurs during the electrochemical reaction on its surface, by forming the Cu/Cu2O interface resulting in *NOH reaction, thus becoming an additional benefit for a high NH3 selectivity.39 Furthermore, the XPS data reveal that both Ni2+ (Fig. 4(f)) and Fe3+ (Fig. S17, ESI†) cations were reduced after the reaction. We observe the formation of metallic Ni on the Ni3Fe–CO3 LDH surface after NitRR. This suggests that Ni3Fe–CO3 LDH promote hydrogen radical transfer by hydrogen spillover mechanism.40 This surface reduction can also be observed by cyclic voltammetry (CV) of Ni3Fe–CO3/Cu foam and Cu foam. The curves show that new reduction and oxidation events take place depending on the direction of the applied potential upon introduction of Ni3Fe–CO3 LDH in the 1 M KOH. This indicates that the surface of LDH can be reduced during electrochemical NitRR (Fig. S18, ESI†). Nevertheless, we observe stable performance as the bulk structure of these materials is maintained despite surface changes during the electrochemical processes. Several electrochemical and spectroelectrochemical techniques were used in order to further elaborate the role of Ni3Fe–CO3 LDH in electrochemical NitRR (Fig. 5). Firstly, the electrochemically active surface area (ECSA) was obtained by determining the electrochemical double-layer capacitance (Cdl) from CV at various scan rates as shown in Fig. 5(a). In 1 M KOH without NO3−, the Ni3Fe–CO3 LDH/Cu foam electrode shows 2.29 mF cm−2 of Cdl which is 2.77 times higher than that of the Cu foam electrode. This indicated that both H2O and OH− could be adsorbed actively on Ni3Fe–CO3 LDH rather than on a Cu surface. Whereas in 1 M KOH with 5 mM KNO3, the Cdl of the Cu foam electrode increased 2.45 times implying that most NO3− adsorbs on the Cu surface because the Cdl of the Ni3Fe–CO3 LDH with or without NO3− is the same. Secondly, the charge transfer characteristics were studied using potentiostatic electrochemical impedance spectroscopy (pEIS) in 1 M KOH with 5 mM KNO3 electrolyte at −0.2 V, fitted by a proper equivalent circuit having the series resistance (R1), Cu foam/electrolyte resistance (R2), and the Ni3Fe–CO3 LDH/electrolyte interface resistance (R3) as shown in Fig. 5(b) and Table S2 (ESI†). It is found that the R2 value (0.277 Ω) for the Ni3Fe–CO3 LDH/Cu foam electrode is about 13.5 times lower than that for the Cu foam electrode (3.658 Ω), indicating that the charge transfer characteristics are significantly enhanced by hydrogen radical supply from Ni3Fe–CO3 LDH. Thirdly, electron spin resonance (ESR) spectroscopy was carried out to confirm the production of hydrogen radicals during elecectrochemical NitRR (Fig. 5(c)). To capture these unstable radical species, a chronoampherometric measurement was carried out using 50 mM 5,5-dimethyl-1-pyrroline-N-oxide (DMPO) in 1 M KOH electrolyte at –0.2 V. Regardless of the Ni3Fe–CO3 LDH, both spectra have superoxide (O2−) radicals generated from dissolved oxygen molecules in the KOH electrolyte. However, the ESR signal of hydrogen radicals was observed when the Ni3Fe–CO3 LDH is introduced onto the Cu surface, providing direct evidence for the hydrogen radical generating capability of Ni3Fe–CO3 LDH.41 To determine the kinetics regarding hydrogen radical generation and transfer to Ni3Fe–CO3 LDH/Cu foam electrode, the Tafel plots were analysed under HER conditions of 1 M KOH electrolyte (Fig. S19, ESI†). In principle, if the Volmer step is rate-determining for the overall reaction rate, adsorbed hydrogen radicals would be consumed quickly by the faster Heyrovsky and/or Tafel step on the surface of electrode. It means that the Tafel slope has single value because the reaction is limited by hydrogen coverage. On the other hand, if Heyrovsky step is the rate-determining step, the hydrogen radicals would fully cover the electrode surface resulting in the inflection of the Tafel slope.56 In Fig. S19a (ESI†), the Tafel plots of Cu foam has a singular slope, meaning the rate-determining step is Volmer step. After incorporating Ni3Fe–CO3 LDH (Fig. S19b, ESI†), the Tafel slope of electrode has an inflection depending on the negative overpotential. This means the overall rate cannot increase fast enough because the surface adsorption of hydrogen radicals has been fully saturated.57 From this kinetics analysis, Ni3Fe–CO3 LDH/Cu foam electrode can generate hydrogen radicals at negative potential, but Cu foam cannot because of rate-determining Volmer reaction. Furthermore, radical trapped DMPO mediated ESR spectra of Ni foam, Ni3Fe–CO3/Ni foam, C paper and Ni3Fe–CO3/C paper has carried out to study the synergistic effect between Ni3Fe–CO3 LDH and Cu foam for hydrogen radical generation and transfer. In the case of Ni3Fe–CO3/Ni foam and Ni foam (Fig. S20a, ESI†), only small amount of superoxide radicals was observed, but none was observed using C paper and Ni3Fe–CO3/C paper (Fig. S19b, ESI†).41 This is highly supportive of synergy between the Ni3Fe–CO3 and Cu foam when it comes to these hydrogen radicals mediated processes (Fig. S20c, ESI†). Lastly, a K15NO3 isotope experiment was performed to trace the origin of the NH3 generated by chronoamperometric analysis both in 5 mM K14NO3 and K15NO3 electrolyte. In Fig. 5(d), the 1H NMR spectrum exhibits a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:1 triplet with a spacing of 52.2 Hz ascribed to 14NH4+. In the case of the K15NO3 electrolyte solution, the 1H NMR spectrum shows clear doublet with a spacing of 73.1 Hz corresponding to 15NH4+, as a result of scalar interaction between 1H and 15N.42 These observations confirm that NH3 derives from NO3− dissolved
in the solution phase. Fig. 5(e) and Table S3 (ESI†) provide a comparison of the half-cell energy efficiency of recently reported electrocatalysts and this work. It supports the claim that the half-cell energy efficiency of the Ni3Fe–CO3 LDH/Cu foam is superior to those of other recent electrocatalysts both at high and low NO3− concentrations, achieving 36.6% and 36.2% at 5 mM and 10 mM NO3− concentration respectively.5,10,13–16,18,20,23,24,39,43–48
:1:1 triplet with a spacing of 52.2 Hz ascribed to 14NH4+. In the case of the K15NO3 electrolyte solution, the 1H NMR spectrum shows clear doublet with a spacing of 73.1 Hz corresponding to 15NH4+, as a result of scalar interaction between 1H and 15N.42 These observations confirm that NH3 derives from NO3− dissolved
in the solution phase. Fig. 5(e) and Table S3 (ESI†) provide a comparison of the half-cell energy efficiency of recently reported electrocatalysts and this work. It supports the claim that the half-cell energy efficiency of the Ni3Fe–CO3 LDH/Cu foam is superior to those of other recent electrocatalysts both at high and low NO3− concentrations, achieving 36.6% and 36.2% at 5 mM and 10 mM NO3− concentration respectively.5,10,13–16,18,20,23,24,39,43–48
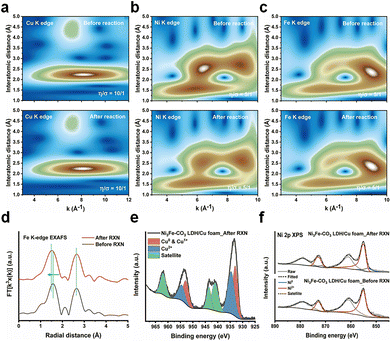 |
| | Fig. 4 X-Ray spectroscopic studies of electrocatalyst before and after electrochemical NO3− reduction. (a) Cu K-edge XAS, (b) Ni K-edge XAS, and (c) Fe K-edge WT-EXAFS before and after electrochemical NitRR for the Ni3Fe–CO3 LDH/Cu foam. (d) Fe K-edge EXAFS of the Ni3Fe–CO3 LDH/Cu foam electrode, and (e) Cu 2p XPS of the Ni3Fe–CO3 LDH/Cu foam electrode after electrochemical NitRR. (f) Ni 2p XPS of Ni3Fe–CO3 LDH/Cu foam electrodes before and after the electrochemical NitRR. RXN means electrochemical NitRR and after RXN sample has analysed after 1 cycle of the electrochemical reaction conducted by chronoamperometric technique for an hour at −0.2 V (vs. RHE) in 1 M KOH with 5 mM of KNO3. | |
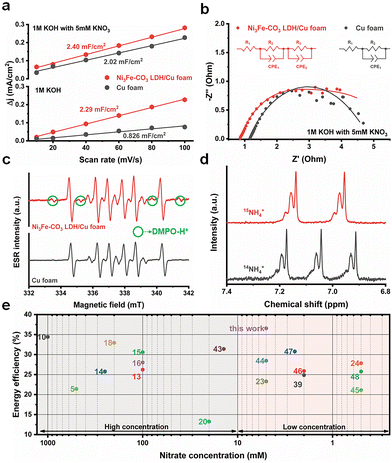 |
| | Fig. 5 (a) Double layer capacitance for the Ni3Fe–CO3 LDH/Cu foam and Cu foam electrodes with/without 5 mM KNO3 in 1 M KOH electrolyte based on the cyclic voltammetry profiles at various scan rates. (b) The Nyquist plots for the EIS spectra of the Ni3Fe–CO3 LDH/Cu foam and Cu foam electrodes at −0.2 V (vs. RHE) under 1 M KOH with 5 mM KNO3. (c) ESR spectra of the solutions obtained by the Ni3Fe–CO3 LDH/Cu foam and Cu foam electrodes in presence of 50 mM DMPO in 1 M KOH. The electrochemical reaction was conducted by chronoamperometric technique for an hour to trap the hydrogen radicals. (d) 1H NMR spectra of the electrolyte after electrochemical NitRR using the Ni3Fe–CO3 LDH/Cu foam electrocatalyst at −0.2 V (vs. RHE) using K15NO3 as a feedstock. The electrochemical reaction was conducted by chronoamperometric technique for an hour under 1 M KOH electrolyte. (e) Half-cell energy efficiencies of this work as a function of [NO3−] compared with the recently reported electrocatalysts (The number indicates cited reference). X-Axis (nitrate concentration) was log10-scaled. Data in Table S2 (ESI†). | |
3 Conclusions
In conclusion, we demonstrated robust and exceptional catalytic NitRR performance of a Ni3Fe–CO3 LDH/Cu foam electrode under low NO3− concentration. Ni3Fe–CO3 LDH/Cu foam exhibits 8.5-fold higher productivity compared to pristine Cu foam. Our electrocatalyst exhibits a 95.8% NH3 selectivity at 98.5% NO3− conversion within an hour. A 36.6% of half-cell energy efficiency was achieved, while yielding a production rate of 1.261 mg cm−2 h−1 and 96.8% of FE at –0.2 V in 5 mM NO3−. These results support that the hybrid electrode structure offers both a highly porous Cu foam allowing the selective binding for NO3− and also a Ni3Fe–CO3 LDH coating facilitating hydrogen radical production/transfer, thus providing a new strategy for surface engineering involving hydrogen radicals in catalysis.
4 Experimental details
4.1. Preparation
Chemicals.
The detailed chemicals are given in the ESI.†
Synthesis of layered double hydroxides.
Layered double hydroxides (LDH) were synthesised by either co-precipitation or hydrothermal methods. The ratio of M2+ to M′3+ cations was 3:1 and all metal sources were MCl2 and M′Cl3 salts. In the case of Ni3Al–CO3, Ni3Fe–CO3, Ni3Co–CO3, Co3Al–CO3, Co3Fe–CO3, Zn3Al–CO3, Mg3Al–CO3 and Mg3Fe–CO3 a co-precipitation method was employed. For the Ni3Al–CO3 LDH, an aqueous solution of sodium carbonate (1.0 M) with sodium hydroxide (1.0 M) was pre-heated in a round flask at 60 °C. And then, 0.64 M of the metal precursors (0.48 M of NiCl2 and 0.16 M of AlCl3) was added to a pre-heated solution in a drop-wise manner. For Ni3Fe–CO3, Ni3Co–CO3, Co3Al–CO3, Co3Fe–CO3, Zn3Al–CO3, and Mg3Al–CO3 after drop-wise addition, the reaction was heated for 24 h at 60 °C. The suspension produced was centrifugated several times at 6000 rpm for 10 min with water. Finally, the centrifugated powder was dried overnight at 30 °C in a vacuum oven. For the Co3V–CO3 LDH, an aqueous solution of sodium carbonate (1.0 M) with sodium hydroxide (1.0 M) was pre-heated in a round flask at 85 °C. And then, 0.64 M of the metal precursors (0.48 M of CoCl2 and 0.16 M of VCl3) were added to a pre-heated solution in a drop-wise manner. After drop-wise addition, the reaction was heated for 24 h at 85 °C. The suspension produced was centrifugated several times at 6000 rpm for 10 min with water. Finally, the Co3V–CO3 LDH powder was dried overnight at 30 °C in a vacuum oven. Ni3V–CO3, Zn3V–CO3 and Zn3Fe–CO3 LDHs were synthesised by a urea hydrolysis method. 0.04 M of metal precursor solution with 0.0625 M urea (80 mL of the total solution) was heated in the 125 mL Teflon-lined stainless-steel autoclave for 12 h at 120 °C. The suspension produced was centrifugated several times at 6000 rpm for 10 min with water. Finally, the pre-synthesised powder was dried overnight at 30 °C in a vacuum oven.
Preparation of the LDH/Cu foam electrode.
First, an electrocatalyst ink was prepared for the fabrication of the electrode. 10 mg of as-prepared powder was dissolved into 1 mL of ethanol with 50 μL of perfluorinated resin solution containing Nafion™ 1100W. Second, a 2 cm × 1 cm Cu foam was cut before ultrasonication pre-cleaning with acetone, 3 M HCl and DI water in sequence for 15 min to remove any contamination and possible surface oxide layers. Then, 100 μL of as-prepared electrocatalyst ink (1 mg of LDH) was slowly deposited on 1 cm × 1 cm size of the Cu foam. Finally, the LDH/Cu foam electrode was dried at room temperature for 12 hours.
4.2. Electrochemical characterisation
Electrochemical measurements.
All the electrochemical measurements were carried out using a Gamry (Reference 3000) workstation. The coiled Pt wire counter electrode and Hg/HgO reference electrode (filled with 1 M KOH) were used to measure electrochemical nitrate reduction properties in an H-type electrolytic cell separated by a Nafion-117 membrane. An aqueous electrolyte of 1 M KOH (pH 14) was used as an electrolyte, and KNO3 was added into the cathode compartment (15 mL) as the reactant and continuously purged with Ar gas (99.999%) during NO3− reaction. Unless otherwise specified, the concentration of NO3− was 5 mM and chronoamperometric experiments are carried out for 1 h at a constant potential. The scan rate of the current–voltage curve was 5 mV s−1 and electrochemical impedance spectroscopy analysis was performed from 0.1 Hz to 0.1M Hz. The measured potentials (vs. the Hg/HgO) were converted into the reversible hydrogen electrode (VRHE) scale by using the Nernst equation
where VHg/HgO is the experimental penitential value against the Hg/HgO reference electrode, pH is 14 at a 1 M KOH aqueous solution and 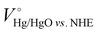 is 0.098 V at 25 °C. NO3− conversion (%), NH3 selectivity (%), faradaic efficiency (%) and NH3 productivity (mg cm−2 h−1) were obtained using the following equations:
is 0.098 V at 25 °C. NO3− conversion (%), NH3 selectivity (%), faradaic efficiency (%) and NH3 productivity (mg cm−2 h−1) were obtained using the following equations:
| Nitrate conversion (%) = ΔCnitrate/C0 × 100% |
| Ammonia selectivity (%) = Cammonia/ΔCnitrate × 100% |
| Faradaic efficiency (%) = (n × F × Cammonia × V)/(M × Q) |
| Ammonia productivity (mg cm−2 h−1) = (Cammonia × V)/(t × S) |
where ΔCnitrate is NO3− concentration difference between before and after the reaction; C0 is the initial NO3− concentration; Cammonia is the concentration of NH3 after reaction; n is the number of electrons to be used in respective reaction; F is the faradaic constant; V is the volume of the cathode compartment (0.015 L); M is the relative molecular mass; Q is the total charge used for electroreduction reaction; t is the reaction time (h); S is the geometric area of the electrode (1 cm2).
Isotope-labelling experiment.
The isotopically-labelled K15NO3 experiments were carried out to trace the source of ammonia during electrochemical nitrate reduction. The coiled Pt wire counter electrode and Hg/HgO reference electrode (filled with 1 M KOH) were used to measure electrochemical nitrate reduction performance with an H-type electrolytic cell separated by a Nafion-117 membrane using Gamry (Reference 3000) workstation. An aqueous electrolyte of 1 M KOH (pH 14) was used as an electrolyte with 5 mM of K15NO3 in the cathode compartment (15 mL) and Ar gas (99.999%) was continuously purged during a hour of reaction time at −0.2 V. After the reaction, 3.25 mL of solution in cathodic counterparts were extracted to mix with 1.5 mL of D2O and 0.25 mL of dimethylsulfoxide (DMSO) before adjusting the pH to about 4 to 5 with 3 M HCl to detect the nitrogen source as ammonium ions. The as-prepared solution was analyzed by 1H NMR for revealing the source of nitrogen in ammonia. Similarly, K14NO3 was used as the nitrogen source of 14NO3− to compare with the 15N isotope experiment.
Product detection.
The amount of NH3 produced was determined by colourimetry using the indophenol blue method.49 2 mL of the electrolyte was taken from the cathodic counterpart, and then, mixed with 2 mL of a 1 M NaOH solution containing 5 wt% of salicylic acid and 5 wt% of sodium citrate. Next, 1 mL of 0.05 M NaClO and 0.2 mL of 1 wt% C5FeN6Na2O (sodium nitroferricyanide) were added to the above solution before storing at room temperature for 2 h. The concentration of NH3 can be detected at a wavelength of about 660 nm using the ultraviolet-visible absorption spectroscopy. The NO2− concentration was detected by dissolving 20 g of p-aminobenzenesulfonamide and 1 g of N-(1-naphthyl) ethylenediamine dihydrochloride into the mixture of 250 mL of DI water and 50 mL of phosphoric acid. Then, this solution was diluted to 500 mL of volume. This colouring agent solution was mixed with electrolyte from the cathodic counterpart. Finally, the concentration of NO2− can be detected at a wavelength of about 540 nm using the ultraviolet-visible absorption spectroscopy.50 The NO3− concentration was determined by adding 1 M HCl (0.2 mL) and 0.8 wt% sulfamic acid solution (0.02 mL) to the NO3− solution (10 mL). The absorption intensities at wavelengths of 220 and 275 nm were measured and calculated by the following equation51 of ‘A = A220nm − 2A275nm.’ A GC-MS (Agilent, GC-7890A and MS-5975C) equipped with a capillary column (Supleco, 30 m × 0.32 mm) and MSD (Mass selective detector, inert triple-axis detector) were used for gas phase product detection.
Conflicts of interest
The authors declare no conflict of interest.
Acknowledgements
This research was supported by the National Research Foundation of Korea (2022M3H4A1A04096482) funded by the Ministry of Science and ICT, and the Basic Science Research Program (2021R1A6A3A14044449) through the National Research Foundation of Korea funded by the Ministry of Education. We also thank SCG Chemicals Public Co. Ltd for support for a Fellowship to C. C.
Notes and references
- Y. Ashida, K. Arashiba, K. Nakajima and Y. Nishibayashi, Nature, 2019, 568, 536–540 CrossRef CAS PubMed.
- S. L. Foster, S. I. Perez Bakovic, R. D. Duda, S. Maheshwari, R. D. Milton, S. D. Minteer, M. J. Janik, J. N. Renner and L. F. Greenlee, Nat. Catal., 2018, 1, 490–500 CrossRef.
- V. Rosca, M. Duca, M. T. de Groot and M. T. M. Koper, Chem. Rev., 2009, 109, 2209–2244 CrossRef CAS PubMed.
- R. F. Service, Science, 2014, 345, 610 CrossRef CAS PubMed.
- Z.-Y. Wu, M. Karamad, X. Yong, Q. Huang, D. A. Cullen, P. Zhu, C. Xia, Q. Xiao, M. Shakouri, F.-Y. Chen, J. Y. Kim, Y. Xia, K. Heck, Y. Hu, M. S. Wong, Q. Li, I. Gates, S. Siahrostami and H. Wang, Nat. Commun., 2021, 12, 2870 CrossRef CAS PubMed.
- T. Kandemir, M. E. Schuster, A. Senyshyn, M. Behrens and R. Schlögl, Angew. Chem., Int. Ed., 2013, 52, 12723–12726 CrossRef CAS.
- S. Z. Andersen, V. Čolić, S. Yang, J. A. Schwalbe, A. C. Nielander, J. M. McEnaney, K. Enemark-Rasmussen, J. G. Baker, A. R. Singh, B. A. Rohr, M. J. Statt, S. J. Blair, S. Mezzavilla, J. Kibsgaard, P. C. K. Vesborg, M. Cargnello, S. F. Bent, T. F. Jaramillo, I. E. L. Stephens, J. K. Nørskov and I. Chorkendorff, Nature, 2019, 570, 504–508 CrossRef CAS PubMed.
- C. Tang and S.-Z. Qiao, Chem. Soc. Rev., 2019, 48, 3166–3180 RSC.
- X. Cui, C. Tang and Q. Zhang, Adv. Energy Mater., 2018, 8, 1800369 CrossRef.
- S. Ye, Z. Chen, G. Zhang, W. Chen, C. Peng, X. Yang, L. Zheng, Y. Li, X. Ren, H. Cao, D. Xue, J. Qiu, Q. Zhang and J. Liu, Energy Environ. Sci., 2022, 15, 760–770 RSC.
- Q. Hu, Y. Qin, X. Wang, Z. Wang, X. Huang, H. Zheng, K. Gao, H. Yang, P. Zhang, M. Shao and C. He, Energy Environ. Sci., 2021, 14, 4989–4997 RSC.
- X. Deng, Y. Yang, L. Wang, X.-Z. Fu and J.-L. Luo, Adv. Sci., 2021, 8, 2004523 CrossRef CAS PubMed.
- X. Zhao, G. Hu, F. Tan, S. Zhang, X. Wang, X. Hu, A. V. Kuklin, G. V. Baryshnikov, H. Ågren, X. Zhou and H. Zhang, J. Mater. Chem. A, 2021, 9, 23675–23686 RSC.
- Y. Guo, R. Zhang, S. Zhang, Y. Zhao, Q. Yang, Z. Huang, B. Dong and C. Zhi, Energy Environ. Sci., 2021, 14, 3938–3944 RSC.
- Z. Fang, Z. Jin, S. Tang, P. Li, P. Wu and G. Yu, ACS Nano, 2022, 16, 1072–1081 CrossRef CAS PubMed.
- Q. Liu, L. Xie, J. Liang, Y. Ren, Y. Wang, L. Zhang, L. Yue, T. Li, Y. Luo, N. Li, B. Tang, Y. Liu, S. Gao, A. A. Alshehri, I. Shakir, P. O. Agboola, Q. Kong, Q. Wang, D. Ma and X. Sun, Small, 2022, 18, 2106961 CrossRef CAS PubMed.
- P. H. van Langevelde, I. Katsounaros and M. T. M. Koper, Joule, 2021, 5, 290–294 CrossRef.
- J. Wang, C. Cai, Y. Wang, X. Yang, D. Wu, Y. Zhu, M. Li, M. Gu and M. Shao, ACS Catal., 2021, 11, 15135–15140 CrossRef CAS.
- J. Li, G. Zhan, J. Yang, F. Quan, C. Mao, Y. Liu, B. Wang, F. Lei, L. Li, A. W. M. Chan, L. Xu, Y. Shi, Y. Du, W. Hao, P. K. Wong, J. Wang, S.-X. Dou, L. Zhang and J. C. Yu, J. Am. Chem. Soc., 2020, 142, 7036–7046 CrossRef CAS PubMed.
- J. Lim, C.-Y. Liu, J. Park, Y.-H. Liu, T. P. Senftle, S. W. Lee and M. C. Hatzell, ACS Catal., 2021, 11, 7568–7577 CrossRef CAS.
- C. L. Ford, Y. J. Park, E. M. Matson, Z. Gordon and A. R. Fout, Science, 2016, 354, 741–743 CrossRef CAS PubMed.
- W. He, J. Zhang, S. Dieckhöfer, S. Varhade, A. C. Brix, A. Lielpetere, S. Seisel, J. R. C. Junqueira and W. Schuhmann, Nat. Commun., 2022, 13, 1129 CrossRef CAS PubMed.
- S. Zhang, M. Li, Q. Song and X. Liu, Proc. Natl. Acad. Sci. U. S. A., 2022, 119, e2115504119 CrossRef CAS PubMed.
- Y. Xu, K. Ren, M. Wang, Z. Wang, X. Li, L. Wang and H. Wang, Appl. Catal., B, 2022, 306, 121094 CrossRef CAS.
- Y. Wang, L. Zhang, Y. Niu, D. Fang, J. Wang, Q. Su and C. Wang, Green Chem., 2021, 23, 7594–7608 RSC.
- G. Chen, T. Wang, J. Zhang, P. Liu, H. Sun, X. Zhuang, M. Chen and X. Feng, Adv. Mater., 2018, 30, 1706279 CrossRef PubMed.
- A. Alobaid, C. Wang and R. A. Adomaitis, J. Electrochem. Soc., 2018, 165, J3395–J3404 CrossRef CAS.
- K.-H. Kim, S. Kim, B. C. Moon, J. W. Choi, H. M. Jeong, Y. Kwon, S. Kwon, H. S. Choi and J. K. Kang, J. Mater. Chem. A, 2017, 5, 8274–8279 RSC.
- K.-H. Kim, J. W. Choi, H. Lee, B. C. Moon, D. G. Park, W. H. Choi and J. K. Kang, J. Mater. Chem. A, 2018, 6, 23283–23288 RSC.
- A. Pastor, C. Chen, G. de Miguel, F. Martin, M. Cruz-Yusta, J.-C. Buffet, D. O’Hare, I. Pavlovic and L. Sánchez, Chem. Eng. J., 2022, 429, 132361 CrossRef CAS.
- W. E. O’Grady, K. I. Pandya, K. E. Swider and D. A. Corrigan, J. Electrochem. Soc., 1996, 143, 1613–1617 CrossRef.
- M. Xu, S. Yao, D. Rao, Y. Niu, N. Liu, M. Peng, P. Zhai, Y. Man, L. Zheng, B. Wang, B. Zhang, D. Ma and M. Wei, J. Am. Chem. Soc., 2018, 140, 11241–11251 CrossRef CAS PubMed.
- X. Fu, X. Zhao, X. Hu, K. He, Y. Yu, T. Li, Q. Tu, X. Qian, Q. Yue, M. R. Wasielewski and Y. Kang, Appl. Mater. Today, 2020, 19, 100620 CrossRef.
- N. S. Gultom, H. Abdullah, C.-N. Hsu and D.-H. Kuo, Chem. Eng. J., 2021, 419, 129608 CrossRef CAS.
- S. Gupta, M. K. Patel, A. Miotello and N. Patel, Adv. Funct. Mater., 2020, 30, 1906481 CrossRef CAS.
- R. Subbaraman, D. Tripkovic, D. Strmcnik, K.-C. Chang, M. Uchimura, A. P. Paulikas, V. Stamenkovic and N. M. Markovic, Science, 2011, 334, 1256–1260 CrossRef CAS PubMed.
- R. Subbaraman, D. Tripkovic, K.-C. Chang, D. Strmcnik, A. P. Paulikas, P. Hirunsit, M. Chan, J. Greeley, V. Stamenkovic and N. M. Markovic, Nat. Mater., 2012, 11, 550–557 CrossRef CAS PubMed.
- M. G. Siebecker and D. L. Sparks, J. Phys. Chem. A, 2017, 121, 6992–6999 CrossRef CAS PubMed.
- Y. Wang, W. Zhou, R. Jia, Y. Yu and B. Zhang, Angew. Chem., Int. Ed., 2020, 59, 5350–5354 CrossRef CAS PubMed.
- P. A. Sermon and G. C. Bond, Catal. Rev., 1974, 8, 211–239 CrossRef.
- Y. Gorbanev, D. O’Connel and V. Chechik, Chem. – Eur. J., 2016, 22, 3496–3505 CrossRef CAS PubMed.
- R. Y. Hodgetts, A. S. Kiryutin, P. Nichols, H.-L. Du, J. M. Bakker, D. R. Macfarlane and A. N. Simonov, ACS Energy Lett., 2020, 5, 736–741 CrossRef CAS.
- F. Du, J. Li, C. Wang, J. Yao, Z. Tan, Z. Yao, C. Li and C. Guo, Chem. Eng. J., 2022, 434, 134641 CrossRef CAS.
- G.-F. Chen, Y. Yuan, H. Jiang, S.-Y. Ren, L.-X. Ding, L. Ma, T. Wu, J. Lu and H. Wang, Nat. Energy, 2020, 5, 605–613 CrossRef CAS.
- R. Jia, Y. Wang, C. Wang, Y. Ling, Y. Yu and B. Zhang, ACS Catal., 2020, 10, 3533–3540 CrossRef CAS.
- C. Wang, Z. Liu, T. Hu, J. Li, L. Dong, F. Du, C. Li and C. Guo, ChemSusChem, 2021, 14, 1825–1829 CrossRef CAS PubMed.
- W. Fu, Z. Hu, Y. Zheng, P. Su, Q. Zhang, Y. Jiao and M. Zhou, Chem. Eng. J., 2022, 433, 133680 CrossRef CAS.
- J. Qin, K. Wu, L. Chen, X. Wang, Q. Zhao, B. Liu and Z. Ye, J. Mater. Chem. A, 2022, 10, 3963–3969 RSC.
- A. Aminot, D. S. Kirkwood and R. Kérouel, Mar. Chem., 1997, 56, 59–75 CrossRef CAS.
- L. C. Green, D. A. Wagner, J. Glogowski, P. L. Skipper, J. S. Wishnok and S. Tannenbaum, Anal. Biochem., 1982, 126, 131–138 CrossRef CAS PubMed.
- Y. Xu, K. Ren, T. Ren, M. Wang, M. Liu, Z. Wang, X. Li, L. Wang and H. Wang, Chem. Commun., 2021, 57, 7525–7528 RSC.
- L. Li, J. Yang, Y. Yun, S. Hu and Y. Huang, Nanomaterials, 2020, 10, 1926 CrossRef CAS PubMed.
- Z. Deng, J. Liang, Q. Liu, C. Ma, L. Xie, L. Yue, Y. Ren, T. Li, Y. Luo, N. Li, B. Tang, A. A. Alshehri, I. Shakir, P. O. Agboola, S. Yan, B. Zheng, J. Du, Q. Kong and X. Sun, Chem. Eng. J., 2022, 435, 135104 CrossRef CAS.
- S. Chatterjee, R. K. Parsapur and K.-W. Huang, ACS Energy Lett., 2021, 6, 4390–4394 CrossRef CAS.
-
I. Chorkendorff and J. W. Niemantsverdriet, Concepts of Modern Catalysis and Kinetics, John Wiley & Sons, 2017 Search PubMed.
- H. Prats and K. Chan, Phys. Chem. Chem. Phys., 2021, 23, 27150–27158 RSC.
- F. Bao, E. Kemppainen, I. Dorbandt, R. Bors, F. Xi, R. Schlatmann, R. van de Krol and S. Calnan, ChemElectroChem, 2021, 8, 195–208 CrossRef CAS.
- B. K. Simpson and D. C. Johnson, Electroanalysis, 2004, 16, 532–538 CrossRef CAS.
- O. Q. Carvalho, R. Marks, H. K. K. Nguyen, M. E. Vitale-Sullivan, S. C. Martinez, L. Árnadóttir and K. A. Stoerzinger, J. Am. Chem. Soc., 2022, 144, 14809–14818 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2023 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *b and
Dermot
O’Hare
*b and
Dermot
O’Hare
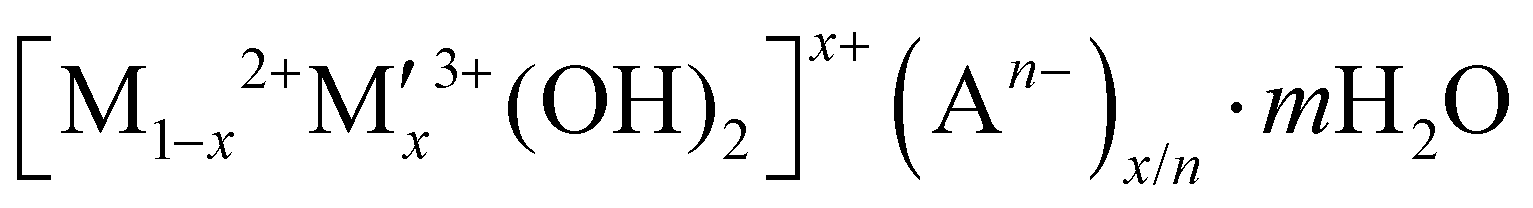 ) could be a unique catalyst platform because of its high overpotential for HER if the Heyrovsky step (Hads* + H2O + e− → H2 + OH) would be suppressed and if the Volmer step (H2O + e− → Hads* + OH−) would be activated. Very few articles have been reported on electrochemical NitRR using an LDH because high voltage should require to drive the cathodic reaction in order to overcome the low inherent electrical conductivity. To date, few LDHs have been used as active materials in electrochemical NitRR, in each case both a high voltage and high nitrate concentration were needed to achieve a high FE which decreases the overall energy efficiency (i.e. [NO3−] = 14 mM [NO3−] for Co1Fe–CO3 LDH;43 [NO3−] = 25 mM for Cu2CoAl–CO3 LDH;52 [NO3−] = 100 mM for Co3AlCO3 LDH53). In this work, we have explored a range of designer LDH decorated Cu foam electrodes (LDH/Cu foam) in order to investigate the role of mixed transition metal combinations to generate hydrogen radicals. We have prepared 12 LDH/Cu foam samples (LDHs = Ni3Al–CO3, Ni3V–CO3, Ni3Fe–CO3, Ni3Co–CO3, Co3Al–CO3, Co3V–CO3, Co3Fe–CO3, Zn3Al–CO3, Zn3V–CO3, Zn3Fe–CO3, Mg3Al–CO3 and Mg3Fe–CO3) which are representative active elements in electrocatalysts and evaluated them for electrochemical NitRR. The ultimate aim of the work is to increase the energy efficiency for selective nitrate reduction at low NO3− concentration. We found that the Ni3Fe–CO3 LDH/Cu foam electrode achieved the best electrochemical NitRR producing a 95.8% NH3 selectivity with a 98.5% NO3− conversion in 5 mM NO3−. Over 1 h the system exhibited a NH3 productivity of 1.261 mg cm−2 h−1 and a 96.8% FE at −0.2 V. This equates to a 36.6% half-cell energy efficiency. As far as we are aware, this is the highest energy efficient electrochemical NitRR to date.
) could be a unique catalyst platform because of its high overpotential for HER if the Heyrovsky step (Hads* + H2O + e− → H2 + OH) would be suppressed and if the Volmer step (H2O + e− → Hads* + OH−) would be activated. Very few articles have been reported on electrochemical NitRR using an LDH because high voltage should require to drive the cathodic reaction in order to overcome the low inherent electrical conductivity. To date, few LDHs have been used as active materials in electrochemical NitRR, in each case both a high voltage and high nitrate concentration were needed to achieve a high FE which decreases the overall energy efficiency (i.e. [NO3−] = 14 mM [NO3−] for Co1Fe–CO3 LDH;43 [NO3−] = 25 mM for Cu2CoAl–CO3 LDH;52 [NO3−] = 100 mM for Co3AlCO3 LDH53). In this work, we have explored a range of designer LDH decorated Cu foam electrodes (LDH/Cu foam) in order to investigate the role of mixed transition metal combinations to generate hydrogen radicals. We have prepared 12 LDH/Cu foam samples (LDHs = Ni3Al–CO3, Ni3V–CO3, Ni3Fe–CO3, Ni3Co–CO3, Co3Al–CO3, Co3V–CO3, Co3Fe–CO3, Zn3Al–CO3, Zn3V–CO3, Zn3Fe–CO3, Mg3Al–CO3 and Mg3Fe–CO3) which are representative active elements in electrocatalysts and evaluated them for electrochemical NitRR. The ultimate aim of the work is to increase the energy efficiency for selective nitrate reduction at low NO3− concentration. We found that the Ni3Fe–CO3 LDH/Cu foam electrode achieved the best electrochemical NitRR producing a 95.8% NH3 selectivity with a 98.5% NO3− conversion in 5 mM NO3−. Over 1 h the system exhibited a NH3 productivity of 1.261 mg cm−2 h−1 and a 96.8% FE at −0.2 V. This equates to a 36.6% half-cell energy efficiency. As far as we are aware, this is the highest energy efficient electrochemical NitRR to date.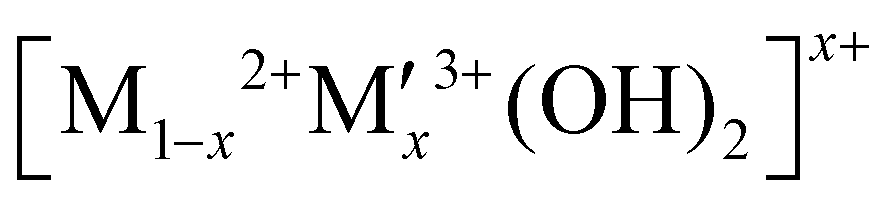 ) are balanced by solvated anionic layers (An−) as shown in Fig. S1 (ESI†).28 Powder X-ray Diffraction (PXRD) patterns for 12 LDHs with different combinations of metal cations (M2+: Ni, Co, Zn, Mg and M′(3+x)+: Al, Fe, Co, V) are shown in Fig. S2 (ESI†), the materials show the Bragg reflections at 2θ = 11° and 22° that can be indexed as (003) and (006) reflections for a prototypical LDH unit cell. In Table S1 (ESI†), calculated interlayer distance (d003) and lattice parameters correspond to a hydrotalcite-3R type LDH with the R3m symmetry.
) are balanced by solvated anionic layers (An−) as shown in Fig. S1 (ESI†).28 Powder X-ray Diffraction (PXRD) patterns for 12 LDHs with different combinations of metal cations (M2+: Ni, Co, Zn, Mg and M′(3+x)+: Al, Fe, Co, V) are shown in Fig. S2 (ESI†), the materials show the Bragg reflections at 2θ = 11° and 22° that can be indexed as (003) and (006) reflections for a prototypical LDH unit cell. In Table S1 (ESI†), calculated interlayer distance (d003) and lattice parameters correspond to a hydrotalcite-3R type LDH with the R3m symmetry.

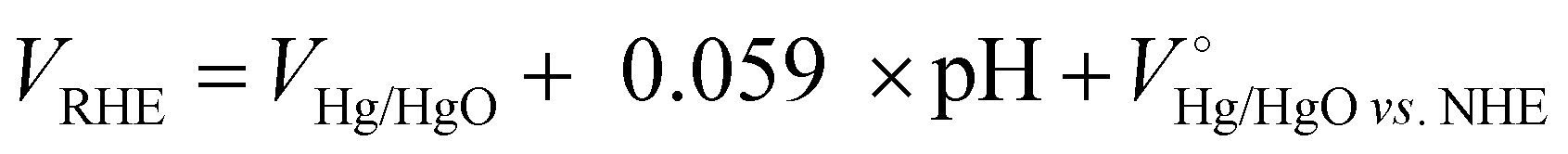
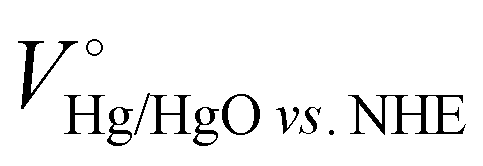 is 0.098 V at 25 °C. NO3− conversion (%), NH3 selectivity (%), faradaic efficiency (%) and NH3 productivity (mg cm−2 h−1) were obtained using the following equations:
is 0.098 V at 25 °C. NO3− conversion (%), NH3 selectivity (%), faradaic efficiency (%) and NH3 productivity (mg cm−2 h−1) were obtained using the following equations: