
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Effect of polar side chains on neutral and p-doped polythiophene†
Peter A.
Finn
a,
Ian E.
Jacobs
 b,
John
Armitage
b,
Ruiheng
Wu
c,
Bryan D.
Paulsen
d,
Mark
Freeley
a,
Matteo
Palma
a,
Jonathan
Rivnay
de,
Henning
Sirringhaus
b and
Christian B.
Nielsen
*a
b,
John
Armitage
b,
Ruiheng
Wu
c,
Bryan D.
Paulsen
d,
Mark
Freeley
a,
Matteo
Palma
a,
Jonathan
Rivnay
de,
Henning
Sirringhaus
b and
Christian B.
Nielsen
*a
aSchool of Biological and Chemical Sciences, Queen Mary University of London, Mile End Road, London, E1 4NS, UK. E-mail: c.b.nielsen@qmul.ac.uk
bOptoelectronics Group, University of Cambridge, Cavendish Laboratory, J J Thomson Avenue, Cambridge, CB3 0HE, UK
cDepartment of Chemistry, Northwestern University, Evanston, IL 60208, USA
dDepartment of Biomedical Engineering, Northwestern University, Evanston, IL 60208-3109, USA
eSimpson Querrey Institute, Northwestern University, Chicago, Illinois 60611, USA
First published on 19th October 2020
Abstract
Incorporation of polar side chains on organic semiconducting materials have been used recently in thermoelectric materials to increase dopant:semiconductor miscibility and stability to further increase the performance and durability of devices. However, investigations into how polar side chains can affect the structure and energetics of polythiophenes compared to non-polar alkyl side chains are usually carried out using materials with no common morphological structure. Within this work we systematically investigate the increase in polar side chain content on poly(3-hexylthiophene) (P3HT) and how the optical, electrochemical, and structural properties are affected. We find a decreasing degree of aggregation with increasing polar side chain content leading to lower charge carrier mobilities. Upon doping with 2,3,5,6-tetrafluoro-7,7,8,8-tetracyanoquinodimethane (F4TCNQ), we find that the electrical conductivity is reduced when incorporating the polar side chain and no stabilising effect is demonstrated when annealing the doped thin films at raised temperatures. This study emphasises that polar functionalities do not always increase dopant:semiconductor interactions and can harm desirable structural and electrical characteristics, and therefore should be incorporated into organic semiconductors with caution.
1. Introduction
Poly(3-hexylthiophene) (P3HT), shown in Fig. 1, is a widely studied organic semiconductor in the field of organic electronics due to its ease of synthesis, commercial availability, solution processability, structural organisation and good charge transport properties. P3HT, a hole transporting semiconducting polymer with a highly regioregular structure, has been used in research for many applications including: organic bioelectronics, organic photovoltaics, organic field effect transistors (OFETs) and organic thermoelectric generators.1–6 The performance of organic semiconductors in electronic devices is often improved by molecular doping which introduces additional positive (p-type doping) or negative (n-type doping) charge carriers.7–11 A well-researched system for molecular doping is P3HT in combination with the oxidant 2,3,5,6-tetrafluoro-7,7,8,8-tetracyanoquinodimethane (F4TCNQ) depicted in Fig. 1. F4TCNQ has a deep lowest unoccupied molecular orbital (LUMO) which makes it energetically favourable to remove an electron from the shallower highest occupied molecular orbital (HOMO) of P3HT thereby introducing additional positive charge carriers in the system. High conductivities have been achieved in this system by different processing techniques, including highly oriented polymer films, vapour doping and sequential doping from solution.12–15 Nevertheless, there is still an issue with dopant stability, since F4TCNQ can diffuse in the solid state, in particular at elevated temperatures, and drift due to applied electric fields, potentially reducing device lifetimes.16,17 Recently, molecular engineering of semiconductors and dopants has been employed as a tool to control dopant diffusion and polymer:dopant interactions. One well-explored strategy is to introduce ethylene glycol motifs into the polymer side chain.18–24 These glycol groups increase the local dielectric constant, stabilizing charged species formed upon doping, and therefore enhancing dopant stability at elevated temperatures. In n-type materials this strategy has been particularly effective, yielding dramatic improvements in conductivity and thermoelectric performance, presumably due to the favourable interactions between glycol groups and cations.20,21 | ||
| Fig. 1 Structure of P3HT (left), P5–P30 (middle) and F4TCNQ (right). | ||
In the work presented herein, to investigate the role of polar side chains in the context of molecular doping of p-type organic semiconductors, we have synthesised and characterised a series of P3HT derivatives with increasing content of ethylene glycol based polar side chains as shown in Fig. 1. P3HT is chosen as the reference system due to its well-studied properties both in its neutral and doped states. The polar side chain contains one ethylene glycol unit separated from the backbone by one carbon atom in order to maintain a six-atom long side chain, and simultaneously diminish resonance effects from the oxygen atoms that will otherwise change frontier molecular orbital energies and doping behaviour drastically.25,26 The random co-polymers were synthesised with 5, 10, 20 and 30% polar side chain content, thus affording P5, P10, P20 and P30 respectively, to systematically cover polar side chain content matching typical doping concentrations used for organic semiconductors. The polymers were subsequently doped with F4TCNQ via sequential doping methods to investigate the electrical properties and thermal stability.13
2. Results and discussion
2.1. Polymer synthesis
P3HT and the random co-polymers were synthesised using the Grignard metathesis method, a well-known polymerisation reaction to produce regioregular P3HT.27,28 The full synthetic method is shown in the ESI.† The general synthesis of the random co-polymers includes trans-metalation of the two 2,5-dibrominated thiophene monomers using iPrMgCl·LiCl in separate flasks, then after combining, using Ni(dppp)Cl2 as the catalyst to afford the polymers in 56–77% yield. Nuclear magnetic resonance spectroscopy confirmed that the monomer feed ratio matches the monomer ratio in the co-polymers as shown in the ESI† (Table S1). As shown in Table 1 and ESI† (Fig. S11), the molecular weights of the polymers determined by gel permeation chromatography in chloroform are similar across the series. Thermogravimetric analysis reveals two thermal degradation events around 300 and 450 °C. The lower temperature event increases in weight percentage loss with increasing polar side chain content indicating that the alkyl side chain is more thermally stable than the ethylene glycol based side chain, as shown in the ESI† (Fig. S12 and Table S2). Differential scanning calorimetry of P3HT reveals melting and crystallisation transitions at 226 and 192 °C respectively, however no strong transitions are observed upon incorporating polar side chains (Fig. S13, S14 and Table S2, ESI†).| Polymer | M w/Mna [kg mol−1] (PDI) | Solution λmaxb [nm] | Thin film λmaxc [nm] |
E
optg
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) [eV]
[eV] |
A 0–0 λ max [eV] |
E
(ox)1/2
[V] vs. Fc/Fc+ |
|---|---|---|---|---|---|---|
| a Measured using gel permeation chromatography in chloroform at 40 °C vs. polystyrene standards. b Measured from 10 μg ml−1 ODCB solutions. c Thin films were spin-cast from 10 mg ml−1 ODCB solutions onto quartz slides at 2000 rpm for 90 seconds. d Calculated from thin film λonset using: E [eV] = 1240/λ [nm]. e Calculated from fittings of normalised thin films UV-vis spectra. f CV recorded vs. Ag/Ag+ (Ag/Ag+ − 0.115 V = Fc/Fc+) of polymer drop-cast onto carbon electrode (0.1 mg ml−1 CHCl3 solution) using 0.1 M TBAPF6 in acetonitrile as supporting electrolyte at 50 mV s−1. E(ox)1/2 = (Epc + Epa)/2. | ||||||
| P3HT | 48.8/38.0 (1.28) | 462 | 557 | 1.90 | 2.05 | 0.50 |
| P5 | 46.2/33.2 (1.39) | 460 | 555 | 1.91 | 2.05 | 0.51 |
| P10 | 53.2/39.1 (1.36) | 456 | 521 | 1.92 | 2.06 | 0.53 |
| P20 | 58.2/40.5 (1.44) | 453 | 515 | 1.92 | 2.07 | 0.64 |
| P30 | 47.3/33.6 (1.41) | 453 | 516 | 1.93 | 2.08 | 0.67 |
2.2. Optical, electrochemical and structural properties
The UV-vis spectra of the polymers dissolved in o-dichlorobenzene (ODCB) show a gradual blue-shift in the absorption maximum (λmax) of ∼9 nm with increasing polar content, as summarised in Table 1 and the ESI† (Fig. S15). UV-vis spectra of the polymer thin films spin-cast from ODCB reveal the degree of structural order in the solid state, as shown in Fig. 2a. P3HT shows the distinct π–π* transition with a λmax at 2.23 eV (557 nm) and a shoulder at 2.06 eV (602 nm) which are assigned the 0–1 and 0–0 vibronic transitions respectively.29 Addition of polar sidechains causes a gradual blue-shift and change in absorption of the 0–1 and 0–0 transitions. Spano calculated that these transitions can be related to J- and H-like aggregates, where the 0–0 absorption feature is ascribed to planarity of the polymer backbone within aggregates.29–31 Therefore a decrease in the ratio between the absorption height of the first two vibronic peaks (A0–0/A0–1) correlates to increased disorder within the aggregates. When incorporating the polar side chain, a gradual blue-shift is observed, along with a significant decrease in A0–0/A0–1, suggesting an increase in disorder within the polymer aggregates as shown in Fig. 2b. | ||
| Fig. 2 (a) Normalised thin film UV-vis spectra of polymer series spin-cast from 10 mg ml−1 ODCB on quartz slides at 2000 rpm for 90 seconds. Greyed areas indicate the 0–0 and 0–1 vibronic transitions. (b) Plot showing degree of crystallinity and A0–0/A0–1 calculated from the fittings of the normalised thin film UV-vis spectra versus polar content of co-polymers. (c) Thin film cyclic voltammograms of drop-cast polymer series vs. Fc/Fc+ (0.1 M TBAPF6 as supporting electrolyte in acetonitrile) at a scan rate of 50 mV s−1. | ||
The solid-state UV-vis spectra were fitted using four Gaussian curves representing the aggregate regions and the experimental solution spectrum representing the amorphous regions as detailed in the ESI† (Fig. S16). From these fittings, a relative degree of aggregation of 64% was estimated for P3HT, with slightly lower values of 63 and 55% for P5 and P10 respectively, dropping further to around 40% for P20 and P30 as depicted in Fig. 2b. Analysis of the Gaussian fitting curves representing the 0–0 and 0–1 transitions reveals that A0–0/A0–1 decreases with increasing polar side chain content, suggesting that incorporation of polar side chains decreases the planarity of the polymer backbone, as discussed earlier. Also shown in Table 1, the onset of absorption (Eoptg) and A0–0 transition energy both blue shift 0.03 eV upon incorporating 30% polar content indicating a slight increase in band gap energy.
The thin film electrochemical properties examined with cyclic voltammetry show a clear and reversible oxidative process for all polymers in the series, as depicted in Fig. 2c and Table 1. The half-wave potential increases by 0.17 V going from P3HT to P30, which we ascribe to the decrease in aggregation with increasing polar content. The electron withdrawing inductive effect from the oxygen atom removed one carbon from the polymer backbone is also expected to contribute to the shift in half-wave potential observed with increasing polar content.25 The major oxidative peak is for all polymers preceded by a minor oxidative event around 0.1–0.2 V which we ascribe to the oxidation of highly ordered crystalline regions; the gradual disappearance of this minor peak with increasing scan rate as depicted in Fig. 2c and Fig. S18 (ESI†) is in agreement with relatively slow ion diffusion into crystalline regions, as would be expected. Interestingly the ascribed crystalline oxidation peak remains at similar potentials across the series, however the major oxidative peak shifts to higher potential, indicating that the polar side chain does not affect the microstructure in a uniform manner.
Density functional theory calculations of gas phase decamers reveal a larger dihedral angle between adjacent thiophene units upon incorporation of polar side chains compared to the corresponding decamer with only alkyl chains, as illustrated in Fig. 3 and the ESI† (Fig. S19–S21). This suggests that the increased disorder with increasing polar content is not only a solid-state effect due to poor packing, but an intrinsic attribute of the mixed alkyl/glycol polymer system, in agreement with the blue-shifted solution UV-vis data.
 | ||
| Fig. 3 Along the chain image of energy-minimised decamers of P3HT and P10 (polar side chain in the middle) in the gas phase using B3LYP/6-31g(d) basis sets. The SCCS dihedral angle between a polar and alkyl side chain unit is indicated by the arrow. | ||
The morphological structure of drop-cast films onto silicon substrates were investigated using X-ray diffraction, revealing a d(100) peak before and after incorporation of polar side chains, as shown in the ESI† (Fig. S22 and Table S4). Interestingly, heating and cooling cycles from differential scanning calorimetry of the polar copolymers show no crystalline melt transitions, however X-ray diffraction reveals the existence of crystalline domains. The structure and orientation of the crystalline polymer domains in spin-cast thin films were probed with grazing incidence wide-angle X-ray scattering (GIWAXS) depicted in Fig. 4 and the ESI† (Fig. S23 and S24). Pristine P3HT thin film structures determined by GIWAXS have previously been reported.32–34 The scattering pattern, depicted in Fig. 4a, shows that P3HT crystallites orient edge-on relative to the substrate with a π–π stack scattering peak (020) in-plane and multiple orders of strong lamellar scattering (h00) out-of-plane, consistent with previous results.33 The structure for newly developed material, poly(3-((2-(2-methoxyethoxy)ethoxy)methyl)thiophene) (P3MEEMT), has been studied as well and has shown to manifest a mix of face-on and edge-on crystallites, with π–π stack scattering peak (020) and lamellar scattering (100) occurring both in- and out-of-plane.35,36
 | ||
| Fig. 4 (a–c) GIWAXS patterns of P3HT, P5 and P30 spin-cast onto silicon substrates at 2000 rpm for 90 s from a 10 mg ml−1 ODCB solution, respectively. (d–f) GIWAXS patterns of P3HT, P5 and P30 thin films doped with F4TCNQ using high doping conditions respectively (1 mg ml−1 F4TCNQ in MeCN then 1 mg ml−1 F4TCNQ in ODCB solutions with penetration times of 60 seconds each). | ||
Incorporation of glycol chains in the polar copolymers presented herein narrows the azimuthal angle distribution of the out-of-plane lamellar scattering (h00) and the near in-plane (020) scattering appears increasingly less arc-like and more rod-like with increasing polar side chain content depicted in Fig. 4 and the ESI† (Fig. S23). These both indicate an improved orientation of the edge-on crystallites. At the same time, the increasing glycol compositions lead to the strengthening of an isotropic lamellar ring shown in Fig. 4b and c, implying the growth of a sub-population of randomly oriented crystallites. In- and out-of-plane line cuts from the 2D plots were fit to extract d-spacings from peak centres and all polymers in the series show similar lamellar d-spacings (16.1–16.2 Å) and π–π stack d-spacings (3.80–3.84 Å) presented in the ESI† (Fig. S23, S24 and Tables S5, S6).
From peak widths, the coherence lengths in the direction of lamellar stacking were extracted from the fitted peaks (Fig. S25, ESI†). For the oriented crystallites, the coherence length increased from 160 Å (∼10 repeat units) to 240 Å (∼15 repeat units) with increasing polar content up to 20%, however at 30% a decrease is observed. For isotropic crystallites, the coherence length continually increased with increasing polar side chain content from a minimum of 48 Å (∼3 repeat units) for P5, to 170 Å (∼10 repeat units) for P30, mirroring the increase in isotropic scattering qualitatively apparent in 2-D scattering plots presented in the ESI† (Fig. S23). Lacking a film thickness normalized absolute measure of incident and scattered photon flux, the GIWAXS data cannot be quantitatively correlated with the 0–1 absorption feature and spectroscopically determined degree of aggregation. However, qualitatively the in-plane π-stack scattering intensity appears to decrease somewhat with increased glycol content in intensity versus the scattering background.
Atomic force microscopy of spin-cast thin films, shown in the ESI† (Fig. S29), reveal surface morphologies of the polymer series in good agreement with reduced aggregation with increasing polar content. Although AFM images cannot unambiguously reveal the crystallinity of films, we observe a steady decrease in RMS roughness from 3.6 nm for P3HT to ∼1 nm for P20 and P30, shown in the ESI† (Fig. S30), consistent with a reduction in aggregation with increasing polar content, as observed by UV-vis spectroscopy.37
2.3. Electrical properties
The charge transport properties were investigated by fabrication of top gate bottom contact organic field-effect transistors (OFETs) with a CYTOP dielectric layer. Output and transfer characteristics are shown in the ESI† (Fig. S33 and Table S7). The saturated hole mobility decreases gradually by approximately one order of magnitude across the series from 3.8 × 10−3 cm2 V−1 s−1 for P3HT to 0.5 × 10−3 cm2 V−1 s−1 for P30. The charge carrier mobility can be related to the free exciton bandwidth (W) which is also related to A0–0/A0–1 of the 0–0 and 0–1 optical transitions as depicted in eqn (1) according to work by Spano et al.29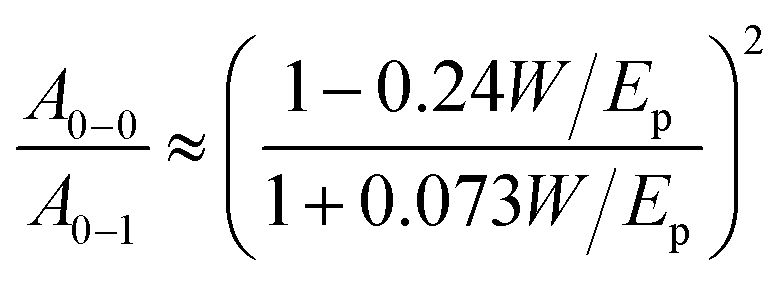 | (1) |
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C vibration (Ep = 0.18 eV). As illustrated in Fig. 5a, the increase in exciton bandwidth with increasing polar content, originating from the decreasing backbone planarity and degree of aggregation, correlates reasonably with a gradual decrease in hole mobility across the series.
C vibration (Ep = 0.18 eV). As illustrated in Fig. 5a, the increase in exciton bandwidth with increasing polar content, originating from the decreasing backbone planarity and degree of aggregation, correlates reasonably with a gradual decrease in hole mobility across the series.
 | ||
| Fig. 5 (a) OFET hole mobility plotted vs. free exciton bandwidth from eqn (1). (b) Electrical conductivity measured on polymer thin films spin-coated (from 10 mg ml−1 ODCB polymer solutions) onto glass slides with 50 nm Gold electrodes in Van de Pauw geometry. Thin film thickness were measured to be between 27–30 nm for P3HT and co-polymers. Co-polymers were doped at high (1 mg ml−1 F4TCNQ in MeCN then 1 mg ml−1 F4TCNQ in ODCB solutions with penetration times of 60 seconds each) and low (0.1 mg ml−1 F4TCNQ MeCN with a penetration time of 10 seconds) doping conditions. (c) π-Stacking distance (d(020)) and lamellar stacking distance (d(h00)) of oriented crystallites from GIWAXS measurements of as-cast (from 10 mg ml−1 ODCB), low doped and high doped polymer thin films. The solid line and the dash line for π-stacking spacing are used for tracking the contracted π-stacking polymorph and the expanded π-stack polymorph, respectively. | ||
The electrical conductivity of the polymer series, using 2,3,5,6-tetrafluorotetracyanoquinodimethane (F4TCNQ) as a molecular p-type dopant, was investigated using a series of different sequential doping conditions. Sequential doping was chosen to afford high doping efficiencies and prevent solution aggregation during thin film fabrication.13,38 The polymers were spin-cast from ODCB and subsequently doped with F4TCNQ in acetonitrile or ODCB at low and high dopant concentrations to emulate the weak and strong doping regimes, respectively, observed for P3HT.13,39,40 A gradual drop in conductivity of an order of magnitude is seen when going from P3HT to P30 for both doping levels as illustrated in Fig. 5b. This is in agreement with the decreasing trend in charge carrier mobilities of the neutral polymer films as discussed below.
2.4. Polymer:dopant interactions
To further examine the doped polymers, we investigated the structural and optical properties using GIWAXS, UV-vis and Fourier-transform infrared (FTIR) spectroscopy. F4TCNQ doped P3HT thin film structures determined by GIWAXS have been widely reported.33,39,40 The 2-D GIWAXS scattering patterns and line cuts are shown in Fig. 4d–f and in the ESI† (Fig. S24, S26 and S27). After doping with F4TCNQ, the backbone peak disappears due to loss of registry.33 For the oriented crystallites, upon low doping the lamellar spacings increase from ∼16.1 to ∼18.0 Å, while at high doping level, the lamellar spacing for P3HT, P5, and P10 increase a further ∼0.3 Å, as shown in Fig. 5c and Table S5 (ESI†). On the other hand, for the isotropic crystallites, upon low doping the lamellar spacing for all copolymers increase by ∼1.7 Å compared to the undoped films, and then increase a further ∼1.0 Å upon high doping (Table S5 and Fig. S28, ESI†). These increases are consistent with penetration of F4TCNQ into the crystalline lamellar stacks.33 For both oriented and isotropic crystallites, the lamellar coherence lengths of all polymers first increase with low F4TCNQ doping, then decrease for high doped samples (Fig. S25, ESI†). Upon low doping, the π-stacking scattering shows the appearance of a shoulder and clearly split into two peaks upon high doping, which is similar to previous reports.33,39,40 On the whole, the GIWAXS data indicate that the structural evolution with doping is similar across the polymer series. However, the in-plane π-stack scattering intensity versus the scattering background saw a bigger decrease with increasing polar content indicating loss in structural order. This result can be correlated with reduction of conductivity.The surface morphology of doped polymer films was probed with AFM as shown in the ESI† (Fig. S31 and S32). After doping, the same trend as seen for undoped films is observed where increasing polar side chain content leads to decreased roughness.
Thin film UV-vis spectroscopy of the high and low doped polymers confirm integer charge transfer between polymer and dopant, in all cases evidenced by bleaching of the neutral polymer π–π* transition and appearance of the distinctive four peaks from the F4TCNQ radical anion at 2.95, 1.78, 1.61, and 1.45 eV.13 The relative intensity of the F4TCNQ radical anion peaks at high doping (Fig. 6a) appear to show a slight reduction with increasing polar group content. However, fits of the high and low doped thin film UV-vis spectra with KF4TCNQ solution spectrum as the F4TCNQ radical anion peaks shown in the ESI† (Fig. S37 and S38) reveal that the F4TCNQ anion content in each sample is similar. In the high doping samples, the fits reveal a slight (∼10%) increase in dopant density for P5 and P10vs. P3HT, while for P20 and P30 the values are within error of P3HT. In the low doping samples, there is a slight (10–20%) increase in dopant density for all polymers compared to P3HT. These carrier density values, along with the measured charge carrier mobilities were then used to estimate an effective doping efficiency (Fig. S39 and Table S9, ESI†). From these calculations we conclude that the doping efficiency is only slightly reduced when introducing polar side chains, and that the drop in conductivity seen in Fig. 5b almost exclusively results from a decrease in mobility (Fig. 5a).
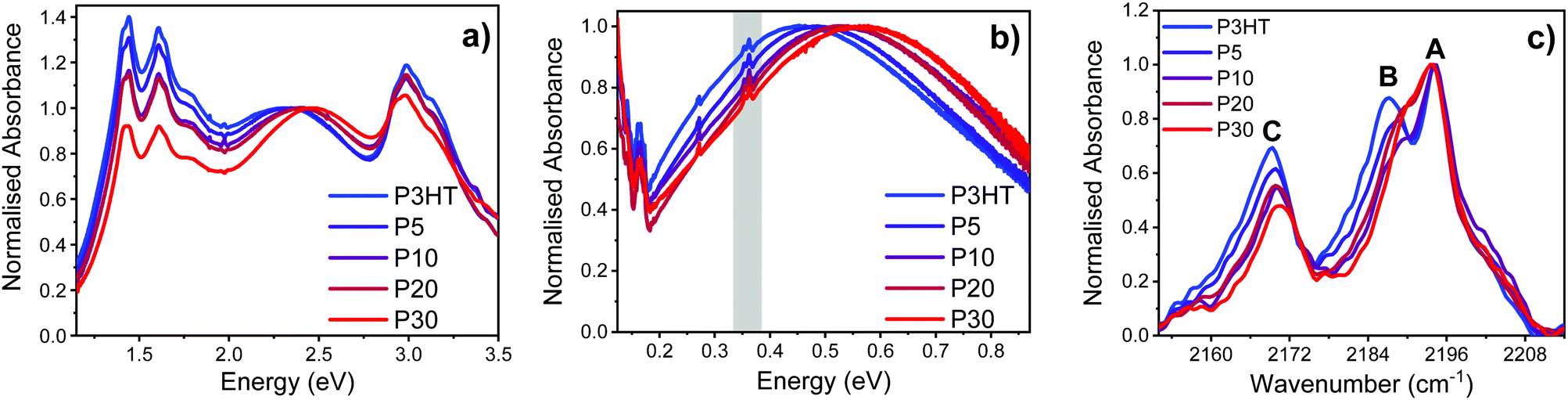 | ||
Fig. 6 (a) UV-vis spectra of high doped polymer thin films normalised to the π–π* transition. (b) Normalised FTIR spectra of high doped polymer thin films (grey area highlighting the region of the F4TCNQ C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N stretches). The spectra are normalised to λmax of the P1 polaron. (c) IR region showing the CN stretches of F4TCNQ where the A, B and C labelled peaks are at 2194, 2187 and 2169 cm−1 respectively. The spectra are normalised to peak A. N stretches). The spectra are normalised to λmax of the P1 polaron. (c) IR region showing the CN stretches of F4TCNQ where the A, B and C labelled peaks are at 2194, 2187 and 2169 cm−1 respectively. The spectra are normalised to peak A. | ||
Since the P2 sub-gap optical transition of the P3HT radical cation at ∼1.5 eV overlaps with the F4TCNQ radical anion peaks in the UV-vis spectrum, FTIR was used to analyse the lower-energy P1 sub-gap transition at ∼0.5 eV.33 The position of this broad peak correlates with the degree of localisation of the polaron, with a shift to lower energies indicative of increased delocalisation.41–43 For both the high and low doped polymer series, a blue-shift of ∼0.13 eV was observed for the P1 transition when going from P3HT (0.45 eV) to P30 (0.55 eV), indicative of increasing localisation of the polaron with increasing polar content as shown in Fig. 6b and ESI† (Fig. S34). This localization signature is reflected in the reduced carrier mobility observed in FET measurements.
In the CN stretching region of the FTIR spectra of high and low doped polymer thin films we see three peaks at 2194, 2187 and 2169 cm−1 which are labelled A, B and C, respectively as illustrated in Fig. 6c (high doping) and Fig. S36 (low doping) (ESI†). The A and B bands are related to the b1u and ag CN stretching modes in the F4TCNQ radical anion, respectively, while the C band is attributed to the b2u and b3g CN stretching modes.44–46 It is hypothesised that the ag (B) and b3g (C) modes, which are not normally IR active, are observed due to a loss of planarity of F4TCNQ when in the polymer film.47 There is a linear relationship between the degree of charge transfer and the change in the IR frequency of the b1u stretching mode (A) from neutral to reduced F4TCNQ.48,49 Since all the polymers herein display a 33 cm−1 shift, we conclude that complete transfer between polymer and dopant takes place for both P3HT and the copolymers with polar side chains.
For high doped polymer films, the relative intensities of the B and C bands decrease with increasing polar content, concurrent with a slight shift to higher wavenumbers. We ascribe this behaviour to the charge transfer complex between the oxidised P3HT and reduced F4TCNQ to be more localised in the case of the polythiophenes with increased polar content. This is in agreement with the blue-shifted P1 transition discussed above and previous work by Pingel et al., where they altered the conjugation length of P3HT through random co-polymerisation with different amounts of the conjugation breaker tetraflurorobenzene (TFB).46 They found that with increasing amounts of TFB, the peak at 2188 cm−1, labelled B herein, decreases in intensity, and they ascribe that to the F4TCNQ charge transfer becoming more localised. A decrease in the intensity of the peaks is also indicative of decreased concentration in integer charge transfer formation.
The FTIR data strongly suggests that the gradual loss of backbone planarity with increasing polar content observed in the neutral polymer films also exists in the doped polymer films.
We subsequently investigated the thermal stability of the doped polymers by annealing the thin films under a nitrogen atmosphere for 20 min at 80 °C. We chose this temperature to observe the effect of diffusion within the film, as it is below the temperature at which F4TCNQ starts to sublimate out of P3HT films.18,50 Conductivity measurements before and after annealing reveal a drop in conductivity across the series for high and low doping conditions as illustrated in Fig. 7. At high doping levels, the conductivity of P3HT increases slightly upon thermal annealing, whereas the relative drop in conductivity upon annealing increases with increasing polar content for the copolymers. Under low doping conditions, a similar trend is observed, however a larger decrease in conductivity around 60% is seen for P30 upon annealing. Under these conditions, there is thus no indication of a stabilising interaction between the dopant and the polar side chain. The reduced thermal stability with increased polar group content may be explained by recent work by Watts et al. who observe thermally induced formation of HF4TCNQ anions in F4TCNQ doped P3HT via proton abstraction of the alpha proton on the side chain.51 Within our system, the carbon spacer with an oxygen adjacent could promote this proton abstraction, leading to a larger decrease in conductivity with increasing polar content.
 | ||
| Fig. 7 Relative change in electrical conductivity of F4TCNQ doped polymer thin films after annealing at 80 °C for 20 min. | ||
3. Conclusions
Polar side chain functionalisation of semiconducting polymers, where an ethylene glycol based motif is directly attached via a C–O bond to the polymer backbone, will have a pronounced effect on frontier energy levels due to resonance effects, and on polymer backbone planarity if attractive intramolecular interactions such as non-covalent S–O interactions can be established. This makes direct comparison with the fully alkylated analogue inaccurate. In this study, we sought to over-come these obstacles by introducing a methylene spacer group between the polymer backbone and the ethylene glycol motif. We find that in this situation, ethylene glycol based side chains have a significant negative impact on polymer microstructure and energetics. This modification is in turn detrimental to properties important in many organic electronic devices, such as field-effect transistors and thermoelectric generators.We have studied in detail the effect of ethylene glycol based polar side chains incorporated in a random fashion into the well-known semiconducting polymer P3HT. Compared to P3HT, introduction of 5 and 10% polar side chain content has a relatively small impact on optical, electrochemical and structural properties, whereas polar content above 10% drastically impairs the polymer crystallinity and shifts the main oxidative event to significantly higher electrochemical potentials. OFET hole mobilities decrease gradually with increasing polar content, initially ascribed to loss of backbone planarity for low polar content, and subsequently further impacted by weakened interchain interactions for higher polar content polymers. Conductivity measurements after doping with F4TCNQ reveal a similar gradual decrease in electrical conductivity with increasing polar content. Changes in dopant concentration and doping efficiency are small across all polymers, indicating that the reduction in carrier mobility dominates the drop in conductivity.
Our study highlights that the incorporation of polar side chains is not a universal solution to e.g. increasing a material's dielectric constant, polymer:dopant miscibility or favourable interactions with ionic species while simultaneously maintaining other desirable properties such as ordered solid-state structural organisation and effective charge carrier transport. We moreover find no evidence in our system that the incorporation of polar side chains enhances the doping stability at elevated temperatures. Upon thermal annealing of F4TCNQ-doped polymer films, conductivity values in fact decrease more with increasing polar content.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge financial support from the Engineering and Physical Sciences Research Council (EP/P007767/1 and EP/R032025/1) and the Materials Research Institute at Queen Mary University of London. P. F. would like to thank Richard Whiteley for measuring XRD of polymer thin films. B. P., R. W., and J. R. gratefully acknowledge support from the National Science Foundation Grant No. NSF DMR-1751308. I. E. J. was supported through a Royal Society Newton International Fellowship. The authors would like to thank Joseph Strzalka and Qingteng Zhang for beam line assistance. This research used resources of the Advanced Photon Source, a U.S. Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Argonne National Laboratory under Contract No. DE-AC02-06CH11357.Notes and references
- B. Lüssem, C. M. Keum, D. Kasemann, B. Naab, Z. Bao and K. Leo, Chem. Rev., 2016, 116, 13714–13751 CrossRef.
- A. Marrocchi, D. Lanari, A. Facchetti and L. Vaccaro, Energy Environ. Sci., 2012, 5, 8457–8474 RSC.
- H. Sirringhaus, Adv. Mater., 2014, 26, 1319–1335 CrossRef CAS.
- O. Bubnova and X. Crispin, Energy Environ. Sci., 2012, 5, 9345–9362 RSC.
- S. Holliday, R. S. Ashraf, A. Wadsworth, D. Baran, S. A. Yousaf, C. B. Nielsen, C. H. Tan, S. D. Dimitrov, Z. Shang, N. Gasparini, M. Alamoudi, F. Laquai, C. J. Brabec, A. Salleo, J. R. Durrant and I. McCulloch, Nat. Commun., 2016, 7, 11585 CrossRef CAS.
- J. Borges-González, C. J. Kousseff and C. B. Nielsen, J. Mater. Chem. C, 2019, 7, 1111–1130 RSC.
- Y. Lin, Y. Firdaus, M. I. Nugraha, F. Liu, S. Karuthedath, A. H. Emwas, W. Zhang, A. Seitkhan, M. Neophytou, H. Faber, E. Yengel, I. McCulloch, L. Tsetseris, F. Laquai and T. D. Anthopoulos, Adv. Sci., 2020, 7, 1–9 Search PubMed.
- K. Kang, S. Watanabe, K. Broch, A. Sepe, A. Brown, I. Nasrallah, M. Nikolka, Z. Fei, M. Heeney, D. Matsumoto, K. Marumoto, H. Tanaka, S. Kuroda and H. Sirringhaus, Nat. Mater., 2016, 1–8 Search PubMed.
- V. Vijayakumar, Y. Zhong, V. Untilova, M. Bahri, L. Herrmann, L. Biniek, N. Leclerc and M. Brinkmann, Adv. Energy Mater., 2019, 1900266, 1900266 CrossRef.
- I. E. Jacobs and A. J. Moulé, Adv. Mater., 2017, 29, 1–39 Search PubMed.
- A. F. Paterson, A. Savva, S. Wustoni, L. Tsetseris, B. D. Paulsen, H. Faber, A. H. Emwas, X. Chen, G. Nikiforidis, T. C. Hidalgo, M. Moser, I. P. Maria, J. Rivnay, I. Mcculloch, T. D. Anthopoulos and S. Inal, Nat. Commun., 2020, 1–11 Search PubMed.
- E. Lim, K. A. Peterson, G. M. Su and M. L. Chabinyc, Chem. Mater., 2018, 30, 998–1010 CrossRef CAS.
- I. E. Jacobs, E. W. Aasen, J. L. Oliveira, T. N. Fonseca, J. D. Roehling, J. Li, G. Zhang, M. P. Augustine, M. Mascal and A. J. Moulé, J. Mater. Chem. C, 2016, 4, 3454–3466 RSC.
- J. Hynynen, D. Kiefer, L. Yu, R. Kroon, R. Munir, A. Amassian, M. Kemerink and C. Müller, Macromolecules, 2017, 50, 8140–8148 CrossRef CAS.
- A. Hamidi-Sakr, L. Biniek, J. L. Bantignies, D. Maurin, L. Herrmann, N. Leclerc, P. Lévêque, V. Vijayakumar, N. Zimmermann and M. Brinkmann, Adv. Funct. Mater., 2017, 27, 1–13 CrossRef.
- J. Li, C. Koshnick, S. O. Diallo, S. Ackling, D. M. Huang, I. E. Jacobs, T. F. Harrelson, K. Hong, G. Zhang, J. Beckett, M. Mascal and A. J. Moulé, Macromolecules, 2017, 50, 5476–5489 CrossRef CAS.
- L. Müller, S. Y. Rhim, V. Sivanesan, D. Wang, S. Hietzschold, P. Reiser, E. Mankel, S. Beck, S. Barlow, S. R. Marder, A. Pucci, W. Kowalsky and R. Lovrincic, Adv. Mater., 2017, 29, 1–7 Search PubMed.
- R. Kroon, D. Kiefer, D. Stegerer, L. Yu, M. Sommer and C. Müller, Adv. Mater., 2017, 1700930, 1700930 CrossRef.
- J. Li, C. W. Rochester, I. E. Jacobs, E. W. Aasen, S. Friedrich, P. Stroeve and A. J. Moulé, Org. Electron., 2016, 33, 23–31 CrossRef CAS.
- D. Kiefer, A. Giovannitti, H. Sun, T. Biskup, A. Hofmann, M. Koopmans, C. Cendra, S. Weber, L. J. Anton Koster, E. Olsson, J. Rivnay, S. Fabiano, I. McCulloch and C. Müller, ACS Energy Lett., 2018, 3, 278–285 CrossRef CAS.
- J. Liu, L. Qiu, G. Portale, M. Koopmans, G. ten Brink, J. C. Hummelen and L. J. A. Koster, Adv. Mater., 2017, 29, 1701641 CrossRef.
- J. Liu, M. P. Garman, J. Dong, B. Van Der Zee, L. Qiu, G. Portale, J. C. Hummelen and L. J. A. Koster, ACS Appl. Energy Mater., 2019, 2, 6664–6671 CrossRef CAS.
- A. I. Hofmann, R. Kroon, L. Yu and C. Müller, J. Mater. Chem. C, 2018, 6, 6905–6910 RSC.
- J. Li, G. Zhang, D. M. Holm, I. E. Jacobs, B. Yin, P. Stroeve, M. Mascal and A. J. Moulé, Chem. Mater., 2015, 27, 5765–5774 CrossRef CAS.
- D. Kiefer, R. Kroon, A. I. Hofmann, H. Sun, X. Liu, A. Giovannitti, D. Stegerer, A. Cano, J. Hynynen, L. Yu, Y. Zhang, D. Nai, T. F. Harrelson, M. Sommer, A. J. Moulé, M. Kemerink, S. R. Marder, I. McCulloch, M. Fahlman, S. Fabiano and C. Müller, Nat. Mater., 2019, 18, 149–155 CrossRef CAS.
- C. B. Nielsen, A. Giovannitti, D.-T. T. Sbircea, E. Bandiello, M. R. Niazi, D. A. Hanifi, M. Sessolo, A. Amassian, G. G. Malliaras, J. Rivnay and I. Mcculloch, J. Am. Chem. Soc., 2016, 138, 10252–10259 CrossRef CAS.
- R. S. Loewe, P. C. Ewbank, J. Liu, L. Zhai and R. D. McCullough, Macromolecules, 2001, 34, 4324–4333 CrossRef CAS.
- R. D. McCullough and R. D. Lowe, J. Chem. Soc., Chem. Commun., 1992, 70–72 RSC.
- F. C. Spano, Acc. Chem. Res., 2009, 43, 429–439 CrossRef.
- J. Clark, J. F. Chang, F. C. Spano, R. H. Friend and C. Silva, Appl. Phys. Lett., 2009, 94, 2007–2010 Search PubMed.
- F. C. Spano and C. Silva, Annu. Rev. Phys. Chem., 2014, 65, 477–500 CrossRef CAS.
- R. J. Kline, M. D. McGehee and M. F. Toney, Nat. Mater., 2006, 5, 222–228 CrossRef.
- D. T. Scholes, P. Y. Yee, J. R. Lindemuth, H. Kang, J. Onorato, R. Ghosh, C. K. Luscombe, F. C. Spano, S. H. Tolbert and B. J. Schwartz, Adv. Funct. Mater., 2017, 27, 1–13 CrossRef.
- N. Kayunkid, S. Uttiya and M. Brinkmann, Macromolecules, 2010, 43, 4961–4967 CrossRef CAS.
- C. G. Bischak, L. Q. Flagg, K. Yan, T. Rehman, D. W. Davies, R. J. Quezada, J. W. Onorato, C. K. Luscombe, Y. Diao, C. Z. Li and D. S. Ginger, J. Am. Chem. Soc., 2020, 142, 7434–7442 CrossRef CAS.
- B. X. Dong, C. Nowak, J. W. Onorato, J. Strzalka, F. A. Escobedo, C. K. Luscombe, P. F. Nealey and S. N. Patel, Chem. Mater., 2019, 31, 1418–1429 CrossRef CAS.
- B. Meng, J. Liu and L. Wang, Polym. Chem., 2020, 11, 1261–1270 RSC.
- D. T. Scholes, S. A. Hawks, P. Y. Yee, H. Wu, J. R. Lindemuth, S. H. Tolbert and B. J. Schwartz, J. Phys. Chem. Lett., 2015, 6, 4786–4793 CrossRef CAS.
- B. Neelamraju, K. E. Watts, J. E. Pemberton and E. L. Ratcliff, J. Phys. Chem. Lett., 2018, 9, 6871–6877 CrossRef CAS.
- D. T. Duong, C. Wang, E. Antono, M. F. Toney and A. Salleo, Org. Electron., 2013, 14, 1330–1336 CrossRef CAS.
- R. Ghosh, C. K. Luscombe, M. Hambsch, S. C. B. Mannsfeld, A. Salleo and F. C. Spano, Chem. Mater., 2019, 31, 7033–7045 CrossRef CAS.
- R. Ghosh, A. R. Chew, J. Onorato, V. Pakhnyuk, C. K. Luscombe, A. Salleo and F. C. Spano, J. Phys. Chem. C, 2018, 122, 18048–18060 CrossRef CAS.
- R. Ghosh, C. M. Pochas and F. C. Spano, J. Phys. Chem. C, 2016, 120, 11394–11406 CrossRef CAS.
- N. L. Haworth, J. Lu, N. Vo, T. H. Le, C. D. Thompson, A. M. Bond and L. L. Martin, ChemPlusChem, 2014, 79, 962–972 CrossRef CAS.
- H. Hase, K. O’Neill, J. Frisch, A. Opitz, N. Koch and I. Salzmann, J. Phys. Chem. C, 2018, 122, 25893–25899 CrossRef CAS.
- P. Pingel, L. Zhu, K. S. Park, J. O. Vogel, S. Janietz, E. G. Kim, J. P. Rabe, J. L. Brédas and N. Koch, J. Phys. Chem. Lett., 2010, 1, 2037–2041 CrossRef CAS.
- E. F. Aziz, A. Vollmer, S. Eisebitt, W. Eberhardt, P. Pingel, D. Neher and N. Koch, Adv. Mater., 2007, 19, 3257–3260 CrossRef CAS.
- H. Méndez, G. Heimel, S. Winkler, J. Frisch, A. Opitz, K. Sauer, B. Wegner, M. Oehzelt, C. Röthel, S. Duhm, D. Többens, N. Koch and I. Salzmann, Nat. Commun., 2015, 6, 8560 CrossRef.
- J. S. Chappell, W. A. Bryden, M. Maxfield, D. O. Cowan, A. N. Bloch and T. O. Poehler, J. Am. Chem. Soc., 1981, 103, 2442–2443 CrossRef CAS.
- K. Kang, S. Schott, D. Venkateshvaran, K. Broch, G. Schweicher, D. Harkin, C. Jellett, C. B. Nielsen, I. McCulloch and H. Sirringhaus, Mater. Today Phys., 2019, 8, 112–122 CrossRef.
- K. E. Watts, B. Neelamraju, M. Moser, I. McCulloch, E. L. Ratcliff and J. E. Pemberton, J. Phys. Chem. Lett., 2020, 11, 6586–6592 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Synthesis, 1H and 13C NMR spectroscopy, UV-vis spectroscopy, UV-vis fittings, FTIR spectroscopy, OFET and conductivity measurements, TGA, DSC, GIXRD, GIWAXS, DFT and AFM data. See DOI: 10.1039/d0tc04290k |
| This journal is © The Royal Society of Chemistry 2020 |