
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceStructure-induced optoelectronic properties of phenothiazine-based materials
Srikanth
Revoju
a,
Anastasia
Matuhina
a,
Laura
Canil
 b,
Henri
Salonen
a,
Arto
Hiltunen
a,
Antonio
Abate
bc and
Paola
Vivo
*a
b,
Henri
Salonen
a,
Arto
Hiltunen
a,
Antonio
Abate
bc and
Paola
Vivo
*a
aFaculty of Engineering and Natural Sciences, Tampere University, P.O. Box 541, FI-33014 Tampere, Finland. E-mail: paola.vivo@tuni.fi
bYoung Investigator Group Active Materials and Interfaces for Stable Perovskite Solar Cells, Helmholtz-Zentrum Berlin für Materialen und Energie, Hahn-Meitner-Platz 1, 14109 Berlin, Germany
cDepartment of Chemical, Materials and Production Engineering, University of Naples, Federico II, Piazzale Tecchio 80, 80125 Fuorigrotta, Naples, Italy
First published on 15th October 2020
Abstract
Phenothiazine (PTZ)-based materials have recently received considerable interest owing to their intriguing optoelectronic properties, low-cost, versatility of functionalization, and commercial availability. The advent of molecular engineering concepts in π-conjugated organic materials, such as the “donor–acceptor” approach, propelled the synthesis of a large number of PTZ-derivatives with tailored properties like low bandgap, tunable energy levels, and reversible redox properties. This resulted in the promising application of PTZs as electron donors or acceptors in organic solar cells or as hole-transporting materials in organic light-emitting diodes and perovskite solar cells. In this review, we discuss the recent and most appealing design strategies of PTZ-based materials for optoelectronics, with emphasis on the impact of the structural modifications on the fundamental physicochemical properties (absorption, emission, Frontier energy levels, charge carrier mobility). We also highlight the key achievements in the development of solar cells, light-emitting diodes, and batteries employing PTZ core semiconductors. Our final goal is to underpin the reasons that still limit the performance of PTZ-based optoelectronics and to outline future research directions for the next-generation PTZ materials with ever enhanced properties.
1. Introduction
During the past few decades, π-conjugated organic materials have attracted great attention from both academic and industrial communities owing to their application in optoelectronic devices, such as light-emitting diodes, field-effect transistors, solar cells, photodetectors, sensors, logical circuits, memory devices, and batteries.1–3 Organic semiconductors display key advantages over their inorganic counterparts, such as the versatility of their chemistry, transparency, low-cost, lightweight, and high-throughput solution-based processing in a wide range of rigid and flexible devices. Importantly, organic electronic devices have witnessed considerable progress during these years in terms of performance, lifetime, and stability.4–9 These developments result from an interdisciplinary research effort from synthetic chemists, physicists, and device engineers leading to innovations in active layers, interfacial materials, and device processing technologies. Typically, organic semiconductor materials lie in the core of the device architecture and their judicious molecular design is the key to achieve optimal optoelectronic properties, which are intimately connected to the structures.10,11 The thorough understanding of the materials’ structure–property relationships is, thus, crucial for realizing high-performance devices.From a molecular engineering point of view, a conjugated molecular semiconductor typically comprises three structural components: the conjugated backbone, the side chains, and the substituents. Although all three parts contribute to the optoelectronic properties in a way or another, the conjugated backbone is the dominant factor in dictating the electronic properties, specifically the energy levels and bandgap.10–12 π-Conjugated molecules inherently possess a bandgap because of bond length alternation between single and double bonds in the backbone. Among the several design strategies developed to decrease the bandgap of conjugated molecules, “donor–acceptor” (D–A) or “push–pull” approach is the most successful and popular one.10–12 The D–A approach involves the alternating incorporation of electron-rich donors and electron-deficient acceptors in the conjugated backbone. The resulting structure possesses a compressed bandgap via molecular hybridization and intramolecular charge transfer (ICT). The most commonly used donor units are thiophene, carbazole, benzodithiophene, phenothiazine, triphenylamine, and porphyrin while the most widely adopted electron acceptors components are fluorene, dibenzosilole, perylene diimide, naphthalimide diimide, diketopyrrolopyrrole, indacenodithiophene, and indacenodithienothiophene. A notable advantage of the D–A approach is that the Frontier energy levels of the conjugated molecule, namely the highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO), can, to a certain degree, be tuned individually as they are located on different parts of the molecule. The HOMO is delocalized on the donor unit whereas the LUMO is predominantly on the acceptor unit.
Moreover, along with the conjugated backbone, side chains and their nature (type, position, length, and shape) exert sizable influence on the material properties, specifically, on the morphological features such as aggregation, packing, stacking, etc., besides their direct influence on the solubility characteristics of the conjugate materials.10–12 For example, the side chain's position on the conjugated backbone has a profound impact on the bandgap of the materials, mainly due to the steric hindrance-induced twist in the adjacent conjugated units. Similarly, several studies have revealed that type, shape, and size of the side chains have a pronounced impact in modulating the intra- and inter-molecular interactions in the solid-state, which in turn influence the device characteristics, for example, the short-circuit current (Jsc), open-circuit voltage (Voc), and fill factor (FF) of solar cells. In comparison to the conjugated backbone or the side-chain modification, the substitution of atoms or the insertion of small functional groups is typically considered as a fine-tuning method to tweak the molecular properties such as energy levels, bandgap, mobility, dipole moment, etc. For example, when an electron donor moiety (e.g. with lone pair on N, O) is introduced in a molecular structure, this raises the HOMO energy while the substitution with an electron acceptor unit (e.g. –C(O)R, –F, –C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N or other withdrawing groups) can lead to a decrease of both HOMO/LUMO.10–12 The application of the above design principles leads to the development of a large variety of novel materials with favorable absorption bandwidths and optical band gaps, and well-matched electronic energy levels for high and balanced charge transport properties.10–12
N or other withdrawing groups) can lead to a decrease of both HOMO/LUMO.10–12 The application of the above design principles leads to the development of a large variety of novel materials with favorable absorption bandwidths and optical band gaps, and well-matched electronic energy levels for high and balanced charge transport properties.10–12
Among the family of heterocyclic donors, PTZ is a well-known building block with distinct features of (i) strong electron-donating character, (ii) a nonplanar “butterfly” type conformation, (iii) tuneable redox properties, (iv) availability of multiple modifiable sites and ease of functionalization, and (iv) cheap and commercial availability.
The presence of electron-rich sulphur and nitrogen heteroatoms and the ionization potential of 6.73 eV is well suited for constructing the D–A materials with enhanced ICT characteristics.13–16 The nonplanar structure of PTZs, with a dihedral angle of 158.5° between the planes of two benzene rings, is responsible for the ability to suppress molecular aggregation and intramolecular excimer formation.15,17,18 For example, PTZ-derivatives substituted at C 3, 7-positions are involved in two-electron reactions whereas N-10 substituted can only donate one electron. The tunable redox properties of PTZ, with reversible and fast electron transfer reactions and stable neutral, mono, and dication radicals, offer electrochemical energy storage application prospects.19–21
Multiple sites of PTZ dye (e.g. N-10, C-2, C-3, C-7, C-8, and sulphur of the thiazine ring) can be functionalized. For example, N-10 active site of the PTZ core can be decorated with suitable solubilizing groups to achieve the desired solubility in organic solvents and solution-processing properties. The easy availability of PTZs and their low-cost (as low as 100 $ per kg, Sigma Aldrich) are quintessential for realizing low-cost molecular devices.22
Exploiting these features, several small molecules and polymers have been designed and synthesized and successfully applied as photoactive materials in organic electronic devices, such as organic light-emitting diodes (OLEDs), organic field-effect transistors (OFETs), dye-sensitized solar cells (DSSCs), organic solar cells (OSCs), perovskite solar cells (PSCs), batteries, sensors, and others.23–26 While the key optoelectronic applications of PTZs, especially related to solar cells, are already reviewed in several good papers,23–26 a comprehensive overview of the use of PTZ chromophores in optoelectronics, with special emphasis on the chemistry of the materials and their structure–property–device performance relationships is not yet thoroughly reported.
In this review, we aim at bridging this knowledge gap by summarizing the most recent development of PTZ-based materials in the active fields of OSCs, PSCs, OLEDs, and batteries. In Section 2, we discuss the synthetic methods and the structural features of the PTZ core along with the intricacies of structural modifications and their effects on the molecular properties of the PTZ-derivatives. In Section 3, we highlight key examples of PTZ-based materials successfully used in solar cells (OSCs and PSCs), OLEDs, and batteries. Finally, we provide an outlook for future research directions in this field, and we suggest how the most promising PTZ molecular designs could be further modified to enhance the performance of optoelectronic devices even further while keeping the synthesis costs low.
Due to a large number of literature reports, we could not assess all the related papers in each application field, but selected representatives are discussed. We believe that this review will inspire and guide numerous researchers in exploring novel PTZ-based molecular designs with intriguing optoelectronic properties beyond the state-of-the-art.
2. Modifications of the phenothiazine core
PTZ is a versatile building block with formula C12H9NS that belongs to the heterocyclic thiazine compounds. The typical PTZ synthesis involves either the reaction of diphenylamine with sulphur in the presence of aluminum chloride as a Lewis acid catalyst or the reaction of halo nitrobenzene precursors with halo thiophenol derivatives via Smiles rearrangement or Ullmann type reaction.17 The PTZ core and its atom numbering are shown in Fig. 1. The functional positions of PTZ core, N-10, C-2, C-3, C-7, and C-8 typically participate in chemical reactions like N-substitution, electrophilic substitutions (halogenation), oxidation, and formylation. Particularly, the halogenation at 3, 7-positions generates the precursor materials for the cross-coupling reactions and opens the synthetic pathways for the construction of diverse π-conjugated materials.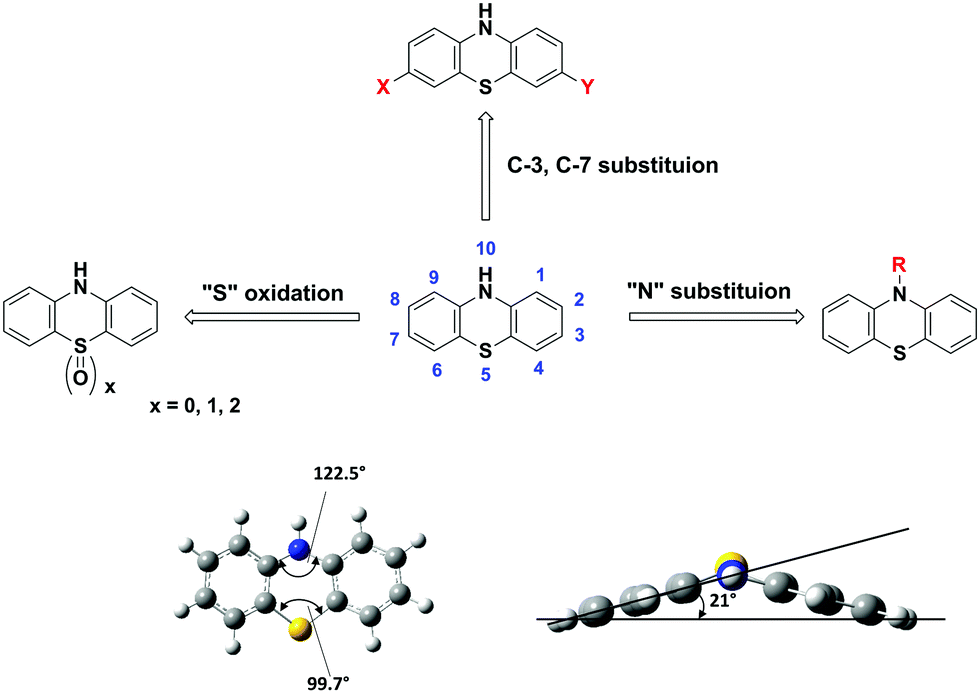 | ||
| Fig. 1 Molecular structure and the key functionalization routes of phenothiazine (PTZ). | ||
The molecular structure of PTZ with its phenylene rings folded along the S⋯N vector and with the aspect angle of 21° is shown in Fig. 1. The PTZ core's unique structural features include a “butterfly” type nonplanar conformation, which impedes the formation of ground state aggregates and excimers. Furthermore, the presence of electron-rich nitrogen and sulphur heteroatoms significantly enhances the electron-donating character of the PTZ unit. This dye has a superior donor character than many of the amines and N-heterocycles such as triphenylamine, phenoxazine, carbazole, etc.26,27 The versatility of its core, derived from the multiple modifiable active sites and the facile tunability into various states (such as neutral, cation radical, and oxygenated sulfoxide forms), allows functionalizing it with a variety of substituents and generate a plethora of derivatives with tuneable optoelectronic properties. For instance, D–A chromophores can be built by covalently attaching electron-withdrawing (EW) or donating groups at C-3 and C-7 positions.16,28 The N-10 active site can be decorated with alkyl, oligo, aryl, or aralkyl groups to induce solution-processable properties and/or to modify the degree of crystallinity and/or to influence the packing motif.22 Similarly, aromatic S-heterocycles can be easily oxidized/reduced to achieve sulphur in different oxidation states (sulphide, sulfoxide, or sulfone) and to affect the redox and spectroscopic properties.29,30 A large variety of such π-conjugated materials with tailor-made properties of low bandgap, high hole mobility, or stable redox system are synthesized and extensively studied for various device applications. Herein, we present a few select examples and elucidate the intricacies of the structural modifications and their influence on the fundamental physicochemical properties and the molecular device performances.
Shinde et al. developed three small molecules, intending to achieve high charge carrier mobilities, by performing subtle structural modifications on the PTZ core.31 The oligomers 1–3 (Fig. 2) contained similar substitutions at C-3 (malononitrile) and N-10 (methoxyphenyl) but they differed in the C-7 position. Although aryl substitutions at C-7 active site showed a weak influence on the spectroscopic characteristics (similar absorption profiles for the 2 and 3), their insertion led to increased steric hindrance in the system. The disorder in the system stimulated molecules 2 and 3 to adopt a more twisted conformation leading to weaker π–π stacking and amorphous films with moderate hole mobilities in the order of 10−6 cm2 V−1 s−1. On the other hand, unsubstituted small molecule 1 adopted a compact and less twisted molecular conformation leading to better π–π stacking and crystalline films, as confirmed by the X-ray powder diffraction (XRD) studies. The small molecule 1 displayed outstanding hole mobility in the order of 10−3 cm2 V−1 s−1 (Fig. 3), which is not only superior to that of 2 and 3 but also, according to the authors, the highest among the reported PTZ-based materials, whose typical values are in the order 10−6 cm2 V−1 s−1.
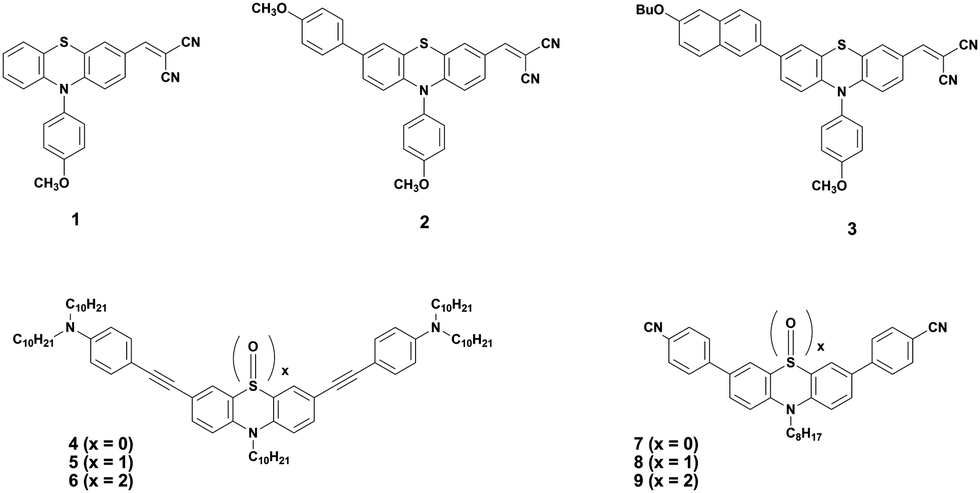 | ||
| Fig. 2 Phenothiazine (PTZ)-based small molecular donor materials. | ||
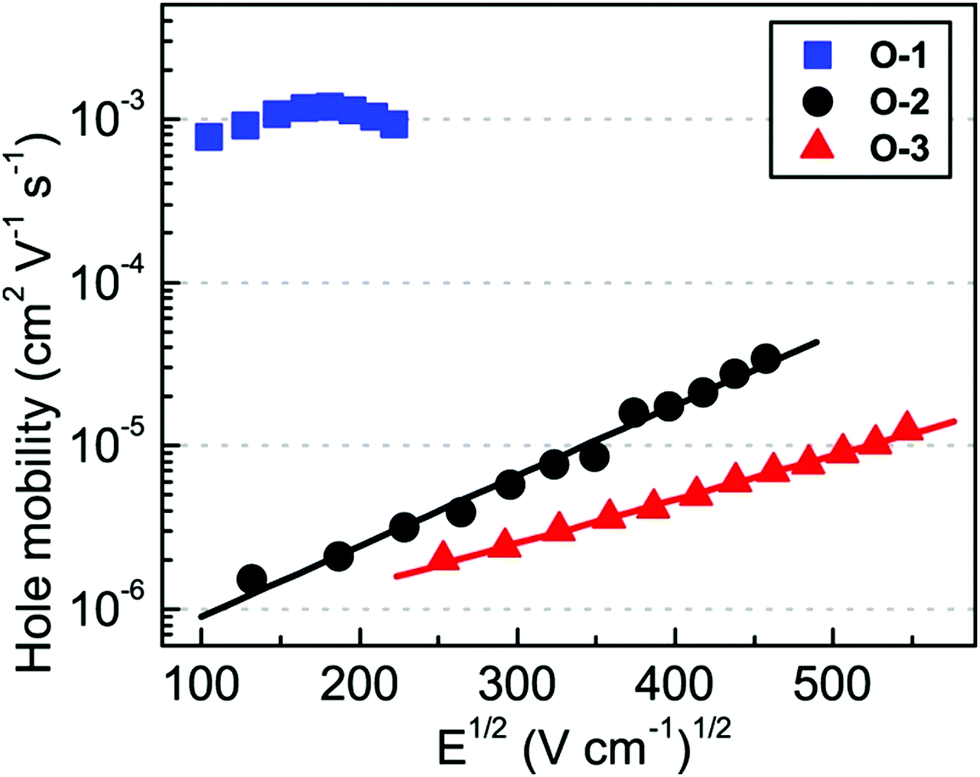 | ||
| Fig. 3 Bulk hole mobility of 1 (O1), 2 (O-2), and 3 (O-3) as a function of the square root of the electric field (figure adapted from ref. 31). | ||
The oxidized counterparts of PTZ moiety offer vastly different molecular properties from those of the parent material, as lone pairs of sulphur are no longer free but are involved in bonding. Sutherland's and Lu's groups systematically investigated the consequences of the insertion of different sulphur oxidation states (sulfide, sulfoxide, and sulfone) into the backbone conjugation using spectroscopic, electrochemical, and density functional theory (DFT) methods. Theriault et al. synthesized three ethynylaniline-PTZ derivatives (4–6) of p-type nature.30 The electrochemical studies of small molecules 4–6 show that the gradual increase in oxidation states from 2+ to 6+ has a profound impact on the HOMO levels, with the oxidation potential changing from 190 mV for 4 to 310 mV for 6, whereas the LUMO levels remained largely unaffected. Absorption and emission profiles of both 5 and 6 exhibited similar features and are quite different from the oligomer 4, as shown in Fig. 4.
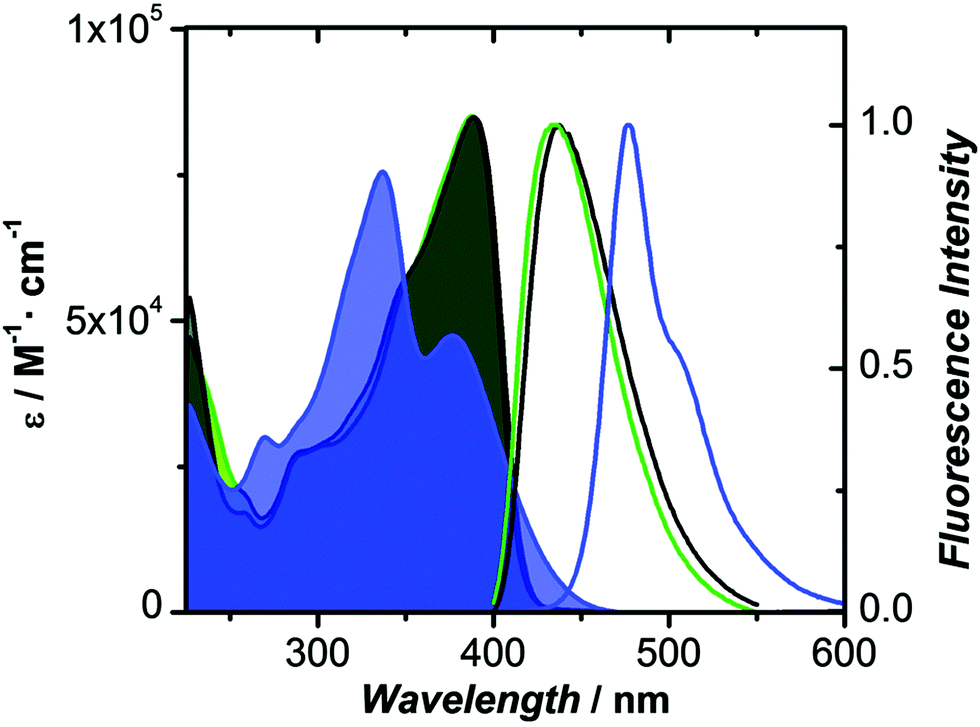 | ||
| Fig. 4 Absorption and fluorescence spectra of 4 (blue), 5 (green) and 6 (black) in CH2Cl2. Reproduced from ref. 30 with permission from The Royal Society of Chemistry. | ||
Notably, a large Stokes shift of 5600 cm−1 was observed for 4 as compared to 5 and 6 with Stokes shifts of 2800 cm−1 and 2900 cm−1, respectively. Such a large shift was attributed to the flattening of the structure in the excited state from a bent ground-state conformation. In another study, Liu et al. designed three molecules (7–9, Fig. 2) having an identical cyano-substituted conjugation system but an aromatic S-heterocycle with varying degrees of sulphur oxidation states. The authors studied this system for application in multilevel memory data storage devices.29 They observed a weaker intramolecular interaction between donor and acceptor units and increased microstructural ordering with the increase in the sulphur oxidation state. Further, a device based on 8 with sulfoxide showed excellent ternary memory behavior, while 7 and 9 with sulphide and sulfone, respectively, showed only binary memory characteristics. To understand the observed memory behavior of the molecules, the energies of the trap states were computed, revealing that the ternary memory characteristics of 8 arise from the presence of two distinct trap states, as shown in Fig. 5.
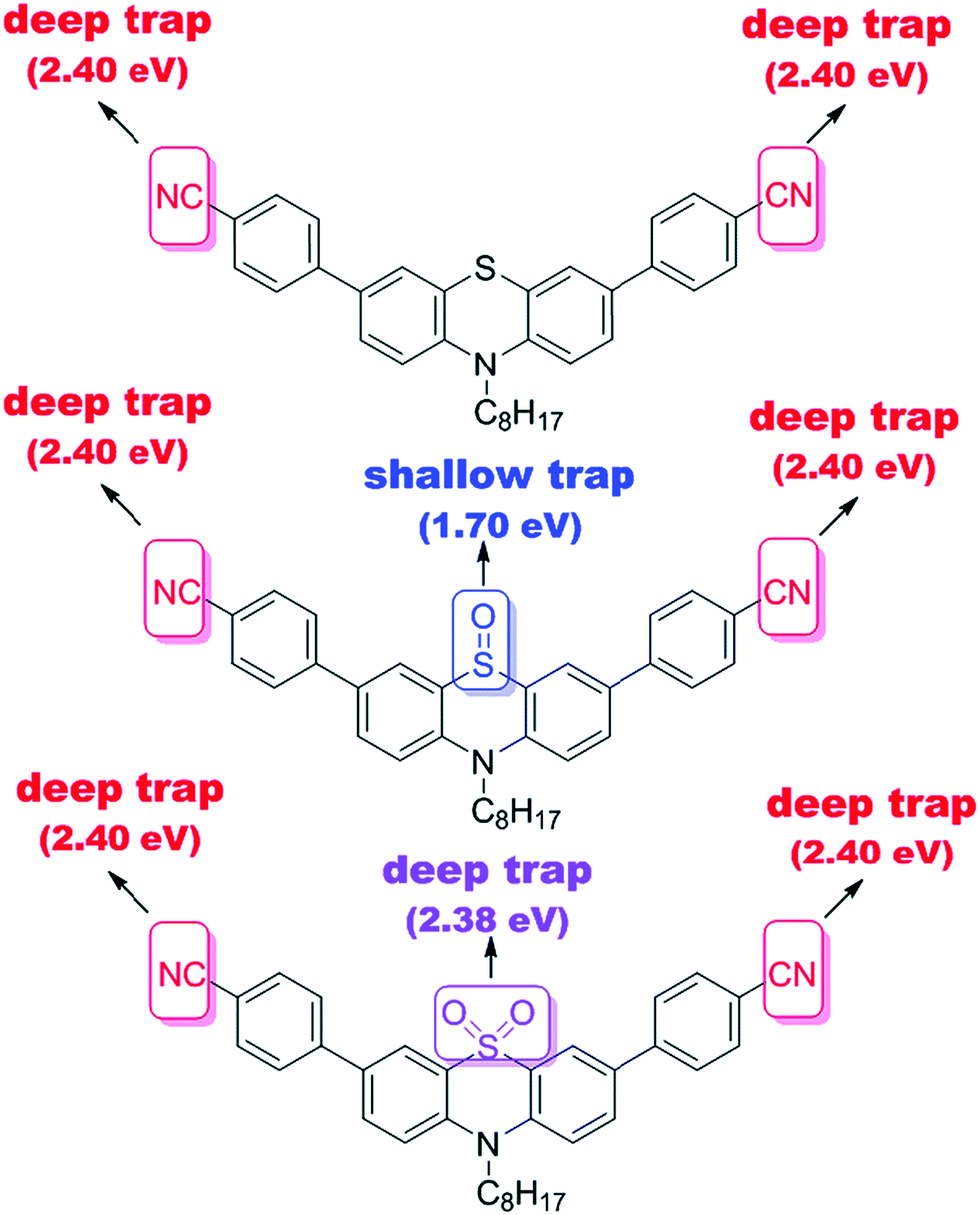 | ||
| Fig. 5 The theoretically calculated trap depths of the functional groups of small molecules 7–9. Reproduced from ref. 29 with permission from The Royal Society of Chemistry. | ||
Although several reports are deciphering the effects of PTZ active sites’ substituents on the molecular properties of the corresponding compounds, a systematic comparative study comprising a considerable number of representative samples is missing. Song and co-workers carried out an extensive work employing DFT and time-dependent functional theory (TDDFT) methods to understand the impact of popular substituents (alkyl chains, aromatic rings, small organic molecules, etc.) on the common C-3/N-7 active sites.32 The authors selected nearly twenty PTZ derivatives from previously published works and computed their absorption, emission, Stokes shift, and oscillator strengths. They observed that the planarity of the ring, the intramolecular charge transfer, and the electron density delocalization are the critical factors affecting the spectral properties of the studied materials. Among them, ICT and electronic delocalization seemed to primarily influence the absorption and emission properties of the derivatives. However, in the systems with limited ICT or delocalization, the absorption characteristics appeared to be more sensitive to planarity than the emission ones. Furthermore, the polarity of the solvent has a greater impact on both Stokes shift and emission than on the absorption profile. For the enhancement of the spectral oscillator strength, substitutions at the C-3 position seem more advantageous than those at the N-10 position. Overall, the simulations pointed out that C-3/N-10 modifications have synergistic effects on the spectral properties, and particularly on the emission.
Very recently, Chen et al. demonstrated how the strategical chemical design of a set of PTZ analogues can significantly boost a typically modest photophysical feature of the parent PTZ compound, i.e. its low photoluminescence quantum yield (PLQY) in nonpolar solvents (<1% in cyclohexane).33 In particular, a highly fluorescent PTZ-based derivative, namely 3-nitrophenothiazine, was achieved with the modification of PTZ with a nitro group (NO2) at the para position with respect to the N atom of the PTZ structure. NO2-modified PTZ chromophore displayed a 100% PLQY in cyclohexane. Furthermore, a significant solvatochromism of the photoluminescence (covering the visible region in the 500–700 nm wavelength range) was observed in polar solvents. These results, supported both experimentally and computationally, are at first sight surprising since NO2 is a well-known emission quencher. However, the introduction of this electron-withdrawing group in the PTZ core leads to the lowering of the LUMO energy level of the parent compound and to the reduction of the mixing of sulphur nonbonding orbitals to ease the allowed π–π* transitions. Furthermore, the introduction of NO2 induces an excited-state charge transfer that pushes towards a planar molecular configuration, thus suppressing the twisting motion, which promotes the nonradiative deactivation and is detrimental to the emission. Overall, this study rationalizes the tuning of the photophysical properties (and particularly the PLQY) with ad hoc structural modifications of the pristine PTZ.
Another interesting structural feature associated with PTZ units, and especially with N-aryl phenothiazines, is the existence of two distinct conformers, “quasi-axial” and “quasi-equatorial”. This is due to the different length of C–N and C–S bonds. Several studies have demonstrated that the type and character of the conformer significantly influence the optoelectronic properties of PTZ derivatives.34–38 Conformationally restricted molecules with charge transfer character are promising candidates as thermally activated delayed fluorescence (TADF) dyes for OLEDs (see Section 3.3), whereas “two-conformation-switchable” molecules with multi-color-changing mechanochromic luminescence are potential candidates for sensing applications.34,35 For example, Adachi et al. reported a D–A molecule containing a PTZ group as donor and 2,4,6-triphenyl-1,3,5-triazine as acceptor that exhibited a dual emission with TADF characteristics.34 DFT and X-ray structure analysis revealed the existence of two ground-state conformers with different energy gap between the lowest singlet excited state and lowest triplet excited state. The quasi-equatorial conformer displayed TADF characteristics owing to small energy difference (0.18 eV) between the singlet and triplet energy states, whereas the quasi-axial conformer showed classical fluorescence emission due to its higher energy difference (1.14 eV). Subsequently, several research groups reported a library of TADF materials with dual stable conformations and steric and electronic properties, as discussed further in OLED Section 3.3.
3. Optoelectronic applications
3.1 Organic solar cells (OSCs)
The organic solar cells (OSCs) are an emerging thin-film photovoltaic (PV) technology holding enormous potential for realizing flexible, lightweight, and low-cost devices via roll-to-roll production process. OSCs typically consists of p-type (polymer or small molecule) and n-type (traditionally fullerene derivatives) semiconductors, blended in a bulk heterojunction (BHJ) configuration. Very recently, an increased interest in OSCs was observed, due to the development of the so-called non-fullerene n-type molecules (or non-fullerene acceptors, NFA) that led to a significant boost in the power conversion efficiency (PCE) values up to 15–18%.39,40 Due to the aforementioned features (Section 2), PTZ is a suitable dye for the design of both polymeric- and small molecule-based active materials, which act as either electron donors (p-type) or acceptors (n-type) in OSCs. The PV properties of key examples of OSCs employing PTZ-based conjugated materials are summarized in Table 1. The corresponding molecular structures are depicted in Fig. 6, 7 and 9.| Molecule | Type | J sc (mA cm−2) | V oc (V) | FF | PCE (%) | Notable features | Ref. |
|---|---|---|---|---|---|---|---|
| 10 | Polymer/donor | 2.38 | 0.78 | 0.29 | 0.53 | – Fluorene and PTZ-based copolymer; band gap of 2.14 eV | 41 |
| – Pure red emission with the chromaticity value of (0.63, 0.36) | |||||||
| 11 | Polymer/donor | 11.9 | 0.51 | 0.34 | 0.20 | – PTZ and bithiophene based regioregular copolymer | 42 |
| – Strong absorption (320–420 nm); emission in blue-green region | |||||||
| 12 | Polymer/donor | 5.37 | 0.80 | 0.43 | 1.85 | – Fluorene and PTZ-based D–A copolymer; bandgap of 1.59 eV | 43 |
| – Broad absorption (400–800 nm) | |||||||
| 13 | Polymer/donor | 4.6 | 0.75 | 0.35 | 1.20 | – LBG polymers with PTZ and benzothiadiazole (BT) units | 44 |
| – Broad absorption (300–700 nm); bandgap of 1.95 eV | |||||||
| 14 | Polymer/donor | 3.14 | 0.85 | 0.29 | 0.80 | – Polythiophene polymer with phenothiazine–vinylene as side chain | 45 |
| – Broad absorption (300–600 nm); high hole mobility (4.7 × 10−3 cm2 V−1 s−1) | |||||||
| 15 | Polymer/donor | 5.75 | 0.77 | 0.38 | 1.69 | – D–A copolymers with PTZ or oxidized PTZ and BT units | 15 |
| 16 | Polymer/donor | 4.11 | 0.92 | 0.32 | 1.22 | – Profound impact of ‘S’ oxidation on optoelectronic properties | |
| – μ16 = 6.9 × 10−4 cm2 V−1 s−1 > μ15 = 9.8 × 10−5 cm2 V−1 s−1 | |||||||
| 17 | Small molecule/donor | 10.3 | 0.97 | 0.32 | 3.25 | – Small molecules with PTZ as the core and dicyanovinyl as end-group | 46 |
| 18 | Small molecule/donor | 5.32 | 0.99 | 0.30 | 1.59 | – A–π–D–π–A type 17 displayed broader absorption, narrower band gap, smoother films, and better PCE than D–π–A type 18 | |
| 19 | Small molecule/donor | 2.9 | 0.84 | 0.29 | 0.71 | – D–A–D type motif with PTZ and DPP units; synthesis by direct heteroarylation; broad absorption (300–700 nm); narrow band gaps (1.50–1.68 eV) | 47 |
| 20 | Small molecule/donor | 3.1 | 0.73 | 0.31 | 0.70 | ||
| 22 | Small molecule/donor | 9.76 | 0.63 | 0.54 | 3.33 | – A–D–A type configuration with PTZ, BODIPY, and alkynyl units | 48 |
| – Broad absorption (320–700 nm); strong PL quenching; high ε (4.7 × 104 cm−1 M−1) | |||||||
| 23 | Small molecule/donor | 6.3 | 0.77 | 0.54 | 2.64 | – A–D–D–A type motif with PTZ core; broad absorption (320–600 nm) | 49 |
| – PV performance improvement with additive DIO | |||||||
| 24 | Small molecule/donor | 10.46 | 0.80 | 0.62 | 5.2 | – D–π–A–π–D motif with DPP and core and PTZ as terminal unit | 50 |
| 25 | Small molecule/donor | 9.42 | 0.79 | 0.53 | 4.0 | – Synthesis by C–H arylation; extended absorption (near IR region) in film | |
| – Strong PL quenching | |||||||
| 26 | Small molecule/donor | 11.18 | 0.99 | 0.56 | 6.20 | – Symmetrical A–π–D–π–A with PTZ as a π-linker and varying EW groups | 16 |
| 27 | Small molecule/donor | 12.06 | 1.04 | 0.60 | 7.45 | – Optoelectronic properties found to vary with acceptor strength | |
| 28 | Small molecule/donor | 9.88 | 0.77 | 0.63 | 4.79 | – Asymmetrical D–D–A motif with PTZ and different EW groups | 28 |
| 29 | Small molecule/donor | 10.68 | 0.89 | 0.65 | 6.18 | – Stronger EW groups found have more pronounced effect on electronic, morphological and PV properties than the weaker ones | |
| 30 | Small molecule/donor | 12.36 | 0.85 | 0.69 | 7.25 | ||
| – Dipole moment seemed to affect the packing pattern and π–π stacking | |||||||
| 31 | Small molecule/donor | 8.73 | 0.95 | 0.58 | 4.81 | – Asymmetrical D–A–D–π–D motif with PTZ as core and varying acceptors | 51 |
| 32 | Small molecule/donor | 11.98 | 0.99 | 0.62 | 7.35 | – Better performance of 32 attributed to the broader absorption and lower bandgap | |
| 33 | Small molecule/donor | 9.38 | 0.81 | 0.52 | 3.95 | – A–A–D–A–A type small molecules with PTZ as core | 22 |
| 34 | Small molecule/donor | 11.94 | 0.93 | 0.59 | 6.55 | – Side chain modification; polar side chains found to play a beneficial role on morphology and device performance | |
| 35 | Small molecule/donor | 12.42 | 0.93 | 0.62 | 7.16 | ||
| 36 | Small molecule/donor | 5.09 | 0.68 | 0.32 | 1.33 | – BODIPY core decorated with PTZ unit at α- or meso positions | 52 |
| 37 | Small molecule/donor | 5.70 | 0.67 | 0.40 | 1.62 | – Broad absorption (300–850 nm); high ε (4.4–6.3 × 104 cm−1 M−1) | |
| 38 | Small molecule/donor | 5.57 | 0.76 | 0.36 | 1.71 | – EW group at meso and PTZ at α-position found to have favorable impact on electronic and PV properties | |
| 39 | Dyad/acceptor | 8.3 | 0.63 | 0.39 | 2.1 | – PTZ–fullerene dyads with fullerene either at C-3 or N-10 position | 53 |
| 40 | Dyad/acceptor | 9.7 | 0.64 | 0.57 | 3.5 | – Efficient photoinduced charge separation between PTZ and fullerene | |
| 41 | Dyad/acceptor | 3.1 | 0.49 | 0.37 | 0.6 | – Position of substitution found to influence charge generation and separation characteristics | |
| 42 | Dyad/acceptor | 4.4 | 0.6 | 0.48 | 1.29 | – PTZ–fullerene dyad; high solubility in organic solvents | 54 |
| 43 | Small molecule/acceptor | 7.28 | 0.98 | 0.56 | 4.16 | – D–A small molecule with N-substituted PTZ unit | 55 |
| – Improved device PCE with the insertion of TiO2 thin layer in between the active layer and the Al electrode |
 | ||
| Fig. 6 Phenothiazine (PTZ)-based polymeric donor materials. The key properties of OSCs employing these dyes in the BHJ blend are listed in Table 1. | ||
 | ||
| Fig. 7 Phenothiazine (PTZ)-based small molecule donor materials. The key properties of OSCs employing these dyes in the BHJ blend are listed in Table 1. | ||
Though PTZ-based organic semiconductors started to be employed as early as 2001, first successful BHJ PVs were reported by Shim and co-workers only in 2006.41 They were based on a series of copolymers (10, Fig. 6) comprising a cyanovinylene-functionalized phenothiazine and fluorene moieties as electron donors with PCEs up to 0.53%. Soon after this first work, Tang et al. reported two series of regioregular and regiorandom alternating copolymers consisting of 10-alkylphenothiazine or 3-pentylthieno[3,2-b]thiophene moieties as electron donor constituents of polymer solar cells.42 Among them, polymer 11-based solar cells recorded a maximum PCE of 0.2% (Table 1). The authors attributed the poor device performance to the weak absorption of the polymers in the near infrared-to-red region of the solar spectrum. In 2008, Lin et al. synthesized a series of D–A low-band-gap (LBG) copolymers derived from arylcyanovinyl-modified-phenothiazine and fluorene units.14 With the tuning of D–A subunit ratios and conjugation lengths, they could induce a stronger intramolecular charge-transfer interaction and broaden the absorption bandwidth of the copolymers to the visible range of 400–800 nm with optical bandgaps of 1.55–2.10 eV. The BHJ device of copolymer 12 produced a modest PCE of 0.51%. Later Chu et al. reported the same polymer with an improved device performance up to 1.84% PCE.43 Further, Lin's group also reported a family of LBG polymers comprising PTZ and various benzodiazole units and investigated the relationship between the chemical structure and the PV properties.44 These polymeric donors showed a broad absorption in the 300–700 nm wavelength region with narrow optical bandgaps of 1.80–1.93 eV. The maximum PCE of 1.20% (Table 1) was obtained for OSCs with phenyl-C71-butyric acid methyl ester (PC71BM) electron acceptor and polymer 13 as a donor. In 2009, Li and co-workers fabricated all-polymeric PV cells based on a polymer blend of donor 14 and acceptor poly(1,4-dioctyloxyl-p-2,5-dicyanophenylenevinylene).45 Authors noticed a strong photoluminescence quenching in the polymer blend indicating an efficient photoinduced charge transfer between the acceptor and the donor components of the device. Further, they studied the influence of thermal annealing on film morphology and PV device performance. Treatment of the devices at 120 °C for 15 min resulted in the formation of an interpenetrating network of D:A domains, which is a prerequisite for efficient solar cells. This led to a performance improvement from 0.41% to 0.8%. Kim et al. carried out an interesting comparative study of two polymers based on PTZ and its oxidized analogue and investigated the effect of insertion of the oxidized form on geometry, charge transporting, electrochemical, photovoltaic, and other characteristics.15 Polymer 16 with oxidized PTZ displayed highly coplanar backbone, blue-shifted absorption, deeper HOMO levels, and higher hole mobility in comparison with its analogue 15. These polymeric donors were tested in both conventional and inverted PV architectures using the fullerene derivates as acceptors. Polymer 15-based devices showed PCEs as high as 1.69% in conventional architectures, whereas polymer 16 showed better performance in inverted solar cells, with a PCE of 1.22%.
PTZ-Based small molecules are comparatively more studied than the polymeric counterparts due to obvious advantages of well-defined molecular architecture, definite molecular weight, facile tunability, easy purifications, and small batch-to-batch variations.56 In 2014, Sun et al. reported two small molecules 17 and 18 as electron donors with PTZ as the core building block and dicyanovinyl as electron-withdrawing end-group.46 The small molecule 17 with A–π–D–π–A motif showed a broader response to solar light and had the narrower bandgap than 18 with D–π–A configuration. Optimized devices based on 17 showed a relatively higher PCE (3.25%) compared to that of 18 (1.59%). Authors attributed the superior performance of 17 to higher Jsc originating from the broader absorption and a better charge carrier mobility. Further, Maglione and co-workers presented three low bandgap D–A–D type small molecules (19–21) with diketopyrrolopyrrole as a core, connected to terminal PTZ via three different electron-rich heteroarenes (thiophene, thiazole, and thienothiophene).47 It is noteworthy to mention here that the molecules were prepared by direct heteroarylation. The BHJ device fabricated with these small molecules as donors and PC71BM as acceptor exhibited moderate PCEs of up to 0.7% with rather low hole-mobilities. In another example, an A–D–A type small molecule with PTZ as the donor, BODIPY as the acceptor, and ethynyl as the bridge were synthesized by Liao et al.48 The donor 22 displayed molecular characteristics of broad optical absorption (320–700 nm) with high attenuation coefficient (ε = 4.7 × 104 cm−1 M−1) and strong fluorescence quenching. The solution-processed OSCs exhibited a reasonable PCE of 3.33% in the optimized conditions. Furthermore, Weng et al. also prepared a series of small molecules based on A–D–A and A–D–D–A type configuration with the PTZ as a core and studied the correlation between different end groups (dicyanovinyl, ethyl 2-cyanoacrylate, and 2-cyano-N-hexalacrylamide) and their photovoltaic performance in inverted OSCs.49 In this study, they observed that the addition of additive 1,8-diiodooctane into the blend film results in microscale segregation of the D:A domains and one-order-of-magnitude increase in hole mobility. BHJ-OSCs with small molecule 23 as donor recorded a maximum PCE of 2.64%. Singh and co-workers constructed two small molecules 24 and 25 with D–π–A–π–D motif bearing thienyl diketopyrrolopyrrole and furanyl diketopyrrolopyrrole as core acceptors and cyano on the π-bridge and PTZ as the terminal donor units, respectively.50 The OSCs fabricated from 24 and 25 with PC71BM exhibited the highest PCEs of 5.2% and 4.0%, respectively. The higher performance of 24 compared to 25 was attributed to the beneficial role of the sulphur heteroatom in the conjugate backbone.
The effect of molecular configuration, optoelectronic properties, carrier mobilities, morphology, and PV properties on the molecular design of oligomers based on PTZ moiety was explored in detail by Revoju et al.16,28 In particular, the authors studied the tuning of the end groups in the molecular structures and its effect on the electron-accepting ability and polarity. In one study, they developed two symmetrical A–π–D–π–A configured oligomers 26 and 27 with a BDT as a central donor core, PTZ as a π-conjugated bridge, and malononitrile or 1,3-indandione as terminal acceptor units.16 Devices based on 27 showed higher PCE than those employing 26 owing to the high hole mobility, better nanoscale morphology, and more controlled interpenetrating network. In another study, the same authors synthesized three asymmetrical small molecules (28–30) with an identical triphenylamine–phenothiazine conjugate backbone but end-capped with different acceptors, namely 3-ethylrhodanine (Rh), malononitrile (MN) and 1,3-indandione (IND).28 In both the studies, oligomers with a stronger electron-withdrawing group (IND in both cases) exhibited broader absorption bandwidth, smaller bandgap, and deeper LUMO levels due to the increased effective conjugation length and a stronger ICT. The optimized solar devices based on 28, 29, and 30 showed overall PCEs of 4.79%, 6.18%, and 7.25%, respectively (Fig. 8). Superior device performance of 30-based devices over others was assigned to the extended absorption, shorted π–π stacking distance, and balanced charge carrier mobilities. The authors also examined the influence of dipole moment on molecular packing and PV performance and found that there is no obvious correlation with efficiency, but it seemed to influence the formation of self-assembled dimer or similar aggregates. In a similar study, Sharma and coworkers prepared two unsymmetrical small molecules 31 and 32 with a D–A–D–π–D architecture, where the central PTZ unit is connected to terminal triphenylamine via electron-withdrawing 1,1,4,4-tetracyanobutadiene and cyclohexa-2,5-diene-1,4-diylidene moieties.51 The optimized OSCs with 32 as the active layer exhibited a high PCE of 7.35%, while the devices with 31 showed a low PCE of 4.81%. Revoju et al. further examined the effect of varying the side chain and π-conjugation length on three A–A–D–A–A type PTZ-based small molecules.22 Small molecules 34 and 35 with polar 2-(2-methoxyethoxy)ethyl side chains exhibited red-shifted absorption spectra, smaller π–π stacking distances, higher dielectric constants, and higher hole mobilities in comparison to N-alkylated 33. The photovoltaic performance followed a similar trend: the devices including 34 (6.55%) or 35 (7.16%) with polar side chain outperformed those based on the nonpolar side-chained molecule 33 (3.95%). These results demonstrate that the nature of the side chains has a profound impact on the molecular characteristics and the PV properties. Recently Cooke's group developed three BODIPY-based donors featuring PTZ moieties as electron-donating units at the α-positions or as meso-linker (36–38).52 All three dyes possess excellent panchromatic absorption covering the spectra from 300 nm to 850 nm with a high attenuation coefficient (4.4–6.3 × 10−4 cm−1 M−1). Among them, small molecule 38 with PTZ unit in the α-position and nitrobenzene in the meso-position led to the highest PCE of 1.71%.
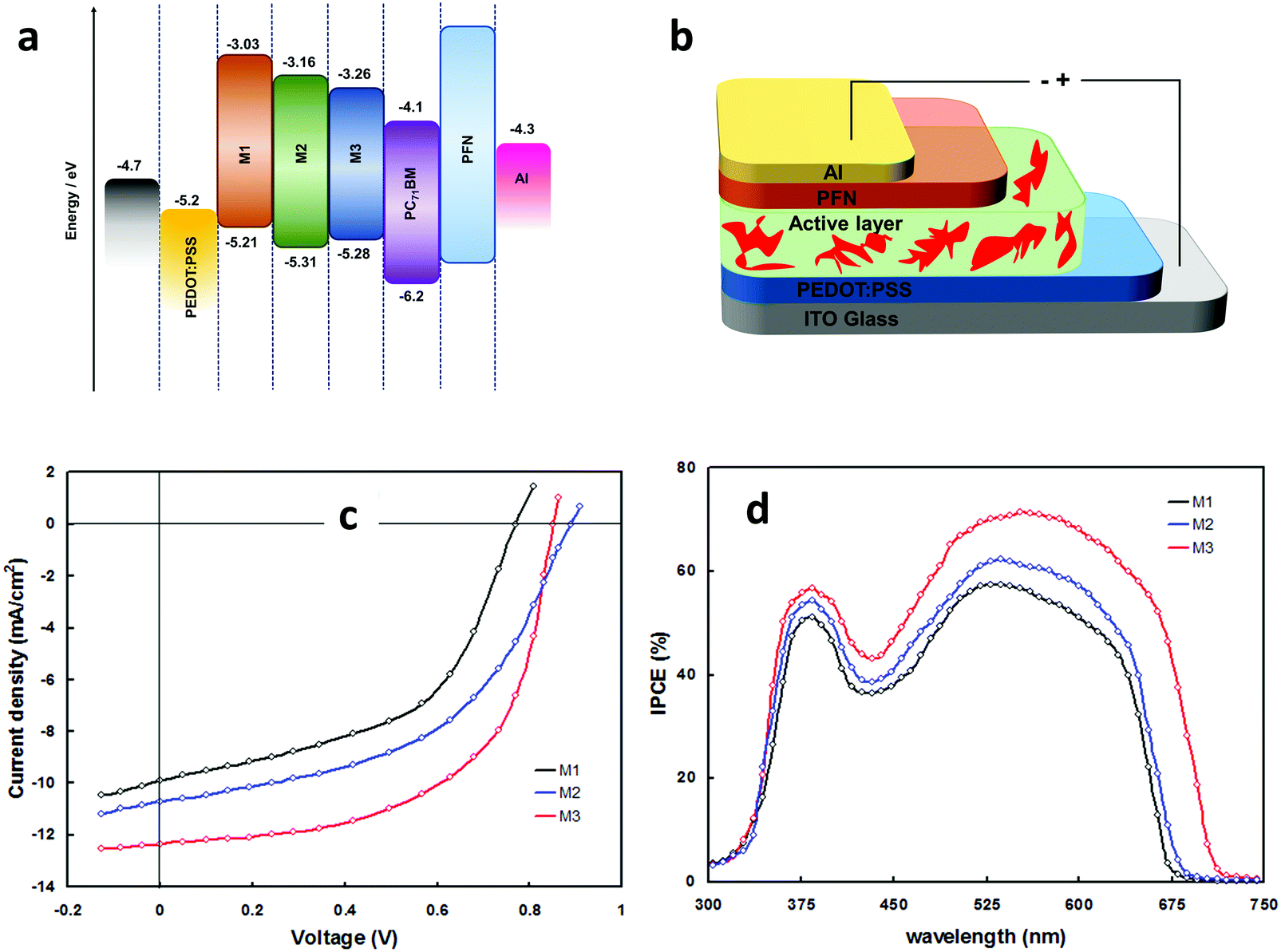 | ||
| Fig. 8 (a) Energy level diagram of the donors 28 (M1), 29 (M2), 30 (M3) (b) device structure of the BHJ-OSCs based on 28–30 blended with PC71BM (c and d) J–V curves and IPCE spectra of the 28–30 based devices. Reproduced from ref. 28 with permission of the PCCP Owner Societies. | ||
In addition to its use in the design of polymeric and small molecule electron donors, the PTZ unit has been seldomly utilized for the functionalization of fullerene and NFA materials (Fig. 9). Only a very few examples of PTZ–fullerene dyads (39–42, Fig. 9) are reported in the literature, possibly owing to the synthetic difficulties.53,54 Fabricated OSCs with PTZ–fullerene dyads as acceptors with P3HT as donor achieved moderate PCEs in the range of 1.3–3.5%. Sharma et al. in 2014 designed and synthesized a carbazole–PTZ dyad 43 and applied it as NFA in OSCs.55 The device PCE was enhanced from 2.80% to 4.16% when a thin layer of TiO2 was inserted between the photoactive layer and the Al electrode. The improved performance was credited to a more balanced charge transport, which increases the charge collection efficiency.
 | ||
| Fig. 9 Phenothiazine (PTZ)-based donor (32–38) and acceptor (39–43) materials. The key properties of OSCs employing these dyes in the BHJ blend are listed in Table 1. | ||
3.2 Perovskite solar cells (PSCs)
Halide perovskite solar cells (PSCs) have rapidly become one of the hottest research topics in the optoelectronic field during the past years.57 Their capability of developing high-performance devices at low cost, together with the high absorption coefficient, tunable bandgap, low-temperature processing, and abundant elemental constituents, make them an attractive alternative to standard solar cells materials.58 Although the stability concerns are still generating extensive discourse, considerable progress was achieved in this area in recent years.59,60 The typical PSC structure includes a light-absorbing layer sandwiched between an electron-transport layer (ETL) and a hole-transport layer (HTL), which have the task of extracting the charges generated in the perovskite and guide them to the contacts.61 A good transport layer should possess photochemical and thermal stability, high hole mobility, appropriate energy levels, and possibly an easy and cheap synthesis procedure.8 Many materials have been tested to fulfill these requirements at best, especially among the HTLs. For instance, we can find examples of hole transport materials (HTMs) based on spirobifluorene, carbazole, fluorene, triphenylamine, tetraphenyl, 3,4-ethylenedioxythiophene, and tetrathiafulvalene.62PTZ-Based HTMs are of particular interest, because, besides the aforementioned optoelectronic properties in Section 2, they can be synthesized through simple and cheap procedures, offering a viable alternative to the commonly used but extremely expensive 2,2′,7,7′-tetrakis[N,N-di(4-methoxyphenyl)amino]-9,9′-spirobifluorene (spiro-OMeTAD).23 Further, multiple functional sites of the PTZ moiety offer the feasibility of using it either as core or as peripheral unit and scope for further functionalization, improving the charge transporting materials. This provides a great advantage compared to other compounds, it is indeed known that small differences in structure can lead to big changes in behaviour.63 Herein, we present a summary of the PTZ-based HTMs that have been developed for applications in PSCs. The hole (h+) mobilities, the position of Frontier energy levels, the decomposition temperature (Td) and the glass transition temperature (Tg), the device performance, and the price of the HTMs are summarized in Table 2.
| HTM | h+ mobility (cm2 V−1 s−1) | HOMOa (eV) | LUMO | TGA (°C) | DSC (°C) | Dopants | PCE (%) | Priceb ($ per g) | Ref. |
|---|---|---|---|---|---|---|---|---|---|
| a Experimental values (cyclic voltammetry). b The price has to be evaluated considering the year of publication. | |||||||||
| Spiro-OMeTAD | 6 × 10−5 | −5.14 | −2.16 | 424 | 122 | Yes | 15–20 | 92–108 | 66 and 69 |
| PTZ as core | |||||||||
| 44 | −4.77 | −1.74 | 359 | 74 | Yes | 2.1 | 111.90 | 63 | |
| 45 | −5.15 | −2.39 | 416 | 135 | Yes | 17.6 | 156.76 | 63 | |
| 46 | 6.75 × 10−4 | −5.21 | −2.51 | 464 | No | 14.3 | 64 | ||
| 47 | 6.18 × 10−5 | −5.39 | −2.83 | 413.1 | 113.8 | Yes | 17.77 | 51.50 | 65 |
| 48 | 6.82 × 10−6 | −5.44 | −2.82 | 406.9 | 111.4 | Yes | 14.65 | 48.54 | 65 |
| 49 | 6.70 × 10−5 | −5.27 | −2.73 | 421.1 | 125.7 | Yes | 19.17 | 45.44 | 65 |
| 50 | 2 × 10−6 | −4.97 | −1.43 | 392 | 85 | Yes | 14.3 | 9 | 66 |
| 51 | 2 × 10−5 | −4.94 | −1.64 | 405 | 120 | Yes | 15.6 | 12 | 66 |
| 52 | 1.74 × 10−4 | −5.25 | −2.67 | 420 | Yes | 16.7 | 62.1791 | 68 | |
| 53 | 5.93 × 10−4 | −5.24 | −2.35 | 420 | Yes | 20.2 | 61.2761 | 68 | |
| PTZ as substituent | |||||||||
| 54 | −5.39 | −2.14 | — | 17 | 69 | ||||
| 54 | 2.08 × 10−3 | −5.08 | −1.93 | 435 | 153 | — | — | — | 70 |
| 55 | 2.76 × 10−4 | −5.20 | −2.04 | 421 | 148 | — | — | — | 70 |
| 56 | 1.29 × 10−4 | −5.33 | −2.18 | 324 | 110 | — | — | — | 70 |
| 57 | −5.29 | −1.75 | 436 | 89 | Yes | 12.14 | 12.98 | 71 | |
| 58 | −5.34 | −2.02 | 448 | 110 | Yes | 4.32 | 3.09 | 71 | |
| 59 | 3.49 × 10−4 | −5.29 | −3.00 | Yes | 7 | 72 | |||
| 60 | 4.80 × 10−4 | −5.30 | −3.04 | Yes | 14.2 | 72 | |||
| 61 | 4.06 × 10−5 | −5.22 | −2.94 | Yes | 10.2 | 72 | |||
| 62 | 9.8 × 10−5 | −5.42 | −2.81 | No | 18.26 | 44.57 | 73 | ||
| 63 | 2.1 × 10−4 | −5.35 | −2.78 | No | 19.16 | 59.22 | 73 | ||
| 64 | 1.09 × 10−4 | −5.30 | −3.17 | 375 | Yes | 3.62 | 74 | ||
| 65 | 1.09 × 10−4 | −5.41 | −3.33 | 381 | 84 | Yes | 17.27 | 74 | |
| 66 | 1.09 × 10−4 | −4.31 | −3.25 | 333 | 135 | Yes | 10.01 | 74 | |
 | ||
| Fig. 10 Molecular structures of PTZ-based HTMs. | ||
The impact of peripheral groups on a PTZ core was also investigated by Zhang et al.,65 who fabricated three HTMs (47–49) with 4,4-dimethyltriphenylamine, N-ethylcarbazole and 4,4-dimethoxyltriphenylamine as side groups connected by double bonds with a synthesis cost of about 50 $ per g (Fig. 10). PSCs with HTM 49 demonstrated to be superior to those based on other PTZ derivates. This can be attributed to its more extended π-conjugation, which assists the hole transport, and the larger molecular weight and stiffness, which help to improve the thermal stability. Moreover, 49-based devices showed higher hydrophobicity resulting in devices with stronger resistance to degradation under ambient conditions. From these results, we deduce that 4,4-dimethoxytriphenylamine can be considered a good option as a peripheral group in PTZ-based HTMs for the development of efficient and stable PSCs.
Alternative PTZ-based HTMs were developed by Salunke et al.66 (50 and 51) with a cost of only 9–12 $ per g (Table 2). The HTMs were prepared via Schiff base chemistry by functionalizing a PTZ core with triarylamine(s) through azomethine bridges. The obtained structures are very similar to the materials mentioned so far, but in this case, it was possible to avoid the use of Pd catalyst in the synthesis, obtaining not only cheap but also eco-friendly HTMs. Also, in this case, the results showed (Fig. 11) n–i–p devices with relatively high efficiency, and better shelf stability (especially 51) and a more efficient hole injection process than both 50 and spiro-OMeTAD. Recently, the same authors reported two more eco-friendly HTMs, AZO III and AZO IV, with a modified molecular design that introduces an aryl spacer between the PTZ and the azomethine bridge and includes a thioethyl group in the 2-position of PTZ unit.67 Although the reported PCE values of the solar cells employing AZO III (9.77%) and AZO IV (11.62%) are inferior to those based on 50 and 51 HTMs, it is worth mentioning that these molecules are processable in a nonchlorinated (toluene) solvent and hence allow a more eco-friendly processing than their predecessors.
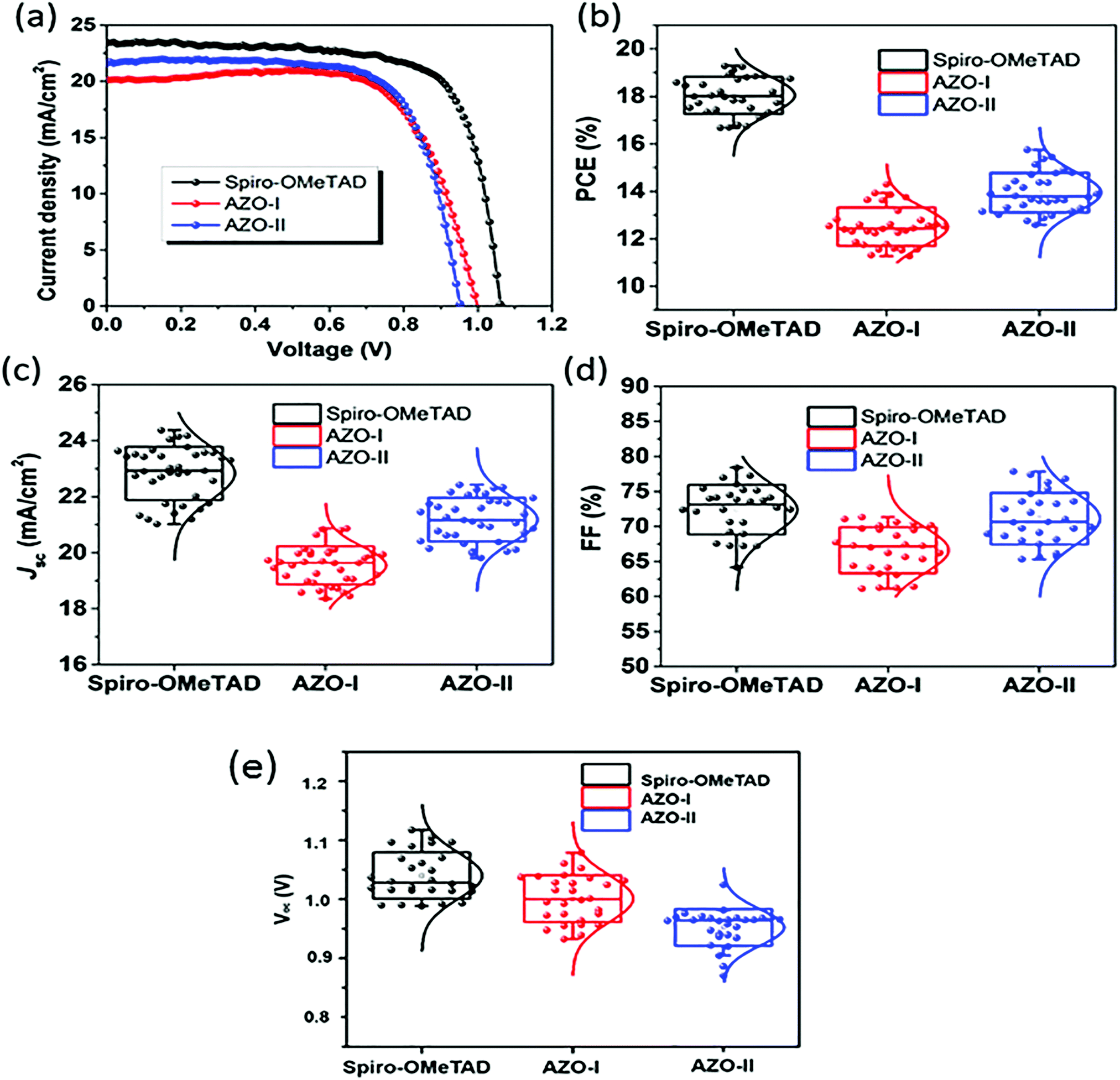 | ||
| Fig. 11 (a) Current density–voltage (J–V) curves for perovskite solar cells (reverse scans) measured with a scan rate of 10 mV s−1 under AM 1.5G simulated solar light illumination by using spiro-OMeTAD, 50 (AZO-I), and 51 (AZO-II) as HTMs. PCE (b), Jsc (c), FF (d), and Voc (e) distributions of PSC devices based on different HTMs (figure adapted from ref. 66). | ||
Recently, Ding et al.68 further exploited the advantage of PTZ by oxidizing it and creating two HTMs (52 and 53) with a novel phenothiazine 5,5-oxide core and anisole or 4,4′-dimethoxyltriphenylamine as the peripheral N-substituent groups (Fig. 10). The oxidation of the electron-donating sulphur atom creates an electron-withdrawing sulfone group. In this way, the charge affinity of the core unit is increased and the HTMs possess a D–A–D structure, which can decrease the HOMO level. It is also noted that the different N-substituent groups, instead, do not influence the charge distribution and the HOMO level position. n–i–p devices prepared with 53 showed an efficiency higher than 20%, overcoming that of reference devices with spiro-OMeTAD as HTM. These results clearly show that not only the adjustment of the peripheral groups can be a useful tool, but also the oxidation of the sulphur atom in PTZ to sulfone can be effective molecular engineering means to enhance the charge carrier mobility and the photovoltaic performance.
 | ||
| Fig. 12 Molecular structures of HTMs with PTZ as a substituent group. | ||
Based on the great advantage of the above-mentioned designs for cost-effective production, Maciejczyk and co-workers continued exploring PTZ as a peripheral group and finally managed to obtain, for the first time in studies about PTZ as a substituent, a soluble HTM.71 They used triphenylbenzene (TPB) as core and PTZ and the commonly used, yet expensive, 4,4′-dimethoxydiphenylamine as a substituent, obtaining two HTMs (57 and 58). n–i–p devices prepared with 57 showed better performance than 58, but the material was four times more expensive 58. This study shows that, by using a TPB core, the solubility of an HTM with PTZ substituents can be largely improved and this opens the way to the employment of its highly attractive properties including ultra-low-cost, a desirable HOMO energy level, good mobility, and thermal stability.
Zhao et al.,72 instead, explored D–A–D type small molecule HTMs. They used dithienopyrrolobenzothiadiazole as acceptor unit and created three new HTMs by crosslinking it with triarylamine (59), PTZ (60), and alkoxytriarylamine (61). The study showed that the strong donor properties of PTZ allowed 60 to outperform 59 and 61. Moreover, the HTM displays also good hydrophobicity, leading to a better shelf-stability of 60-based devices compared to 59, 61, and spiro-OMeTAD.
A similar approach was used by Chen et al.,73 who developed two D–π–D type HTMs 62 and 63, which consist of 4,8-di(hexylthio)benzo[1,2-b:4,5-b′]dithiophene as a π-conjugated linker and, respectively, N-(6-bromohexyl)phenothiazine and N-(6-bromohexyl)phenoxazine as donor units, additionally, the structure contains bromohexyl groups as Lewis bases for perovskite passivation purposes. Both HTMs proved to be thermally stable, to have a high hole extraction efficiency and a hydrophobic nature, which once more provides excellent stability upon humidity exposure for 400 h. The HTMs were tested in dopant-free p–i–n devices leading to performances comparable to the commonly used poly[bis(4-phenyl)(2,4,6-trimethylphenyl)amine] (PTAA) and spiro-OMeTAD, but benefiting from a much lower production cost and higher stability.
Finally, Lu et al. developed three multifunctional PTZ-based HTMs (64, 65, and 66 in Fig. 12) to create a material that embodies all the most important features of an HTM, such as good hole extraction, stability, hydrophobicity, and trap-passivation ability.74 The chemical configuration consists of A–π–D–π–D–π–A architecture backbone end-capped with three different acceptors (Lewis base blocks) on both sides for hole extraction and the passivation of undercoordinated Pb ions. The donor units are PTZ and they can lower the HOMO level, besides having good hole transport and thermal stability and less propensity for aggregation because of their non-planar and butterfly-shaped structure. Moreover, N-alkylation of two dodecyl chains on two PTZ units was carried out to improve solubility and hydrophobicity. In between all the blocks, there are thiophene units to enhance planarity and strengthen the hole transport. The study showed that the end-capped acceptors greatly affected both HOMO/LUMO level positions and solubility, indeed 64 and 66 could not dissolve well in chlorobenzene, resulting in a poor film formation and, therefore, not-optimal hole extraction ability and device performance. 65 gave, instead, successful results both in terms of efficiency and stability, proving that PTZ can be used to create multifunctional materials that satisfy all the requirements for a good HTM.
3.3 Organic light-emitting devices
The emission of PTZ-based derivatives can be easily tuned in a broad wavelength region due to the flexibility of the synthesis process. Owing to the relatively high value of quantum yield (QY) and external quantum efficiency (EQE) up to 81% and 24.3% respectively, nowadays, OLEDs based on PTZ derivatives already cover almost all the CIE color space (Fig. 13).75,76 Generally, they are utilized as the host matrix of the emissive organic layer, playing the role of the energy donor but they could also be applied as the dopant emitter.75,77 Possessing strong electron-donating features, and many favorable properties as pointed out above, PTZs are considered as strong candidates for LED applications. In this section, we summarize the most recently published results on the use of PTZ-derivatives in OLEDs (see Fig. 14 for the molecular structures). The mechanism of emission, time of the delayed decay, QY, EQE, position of the HOMO and LUMO level, energy gap (ΔEST), maximum brightness (Bmax), and color are presented in Table 3.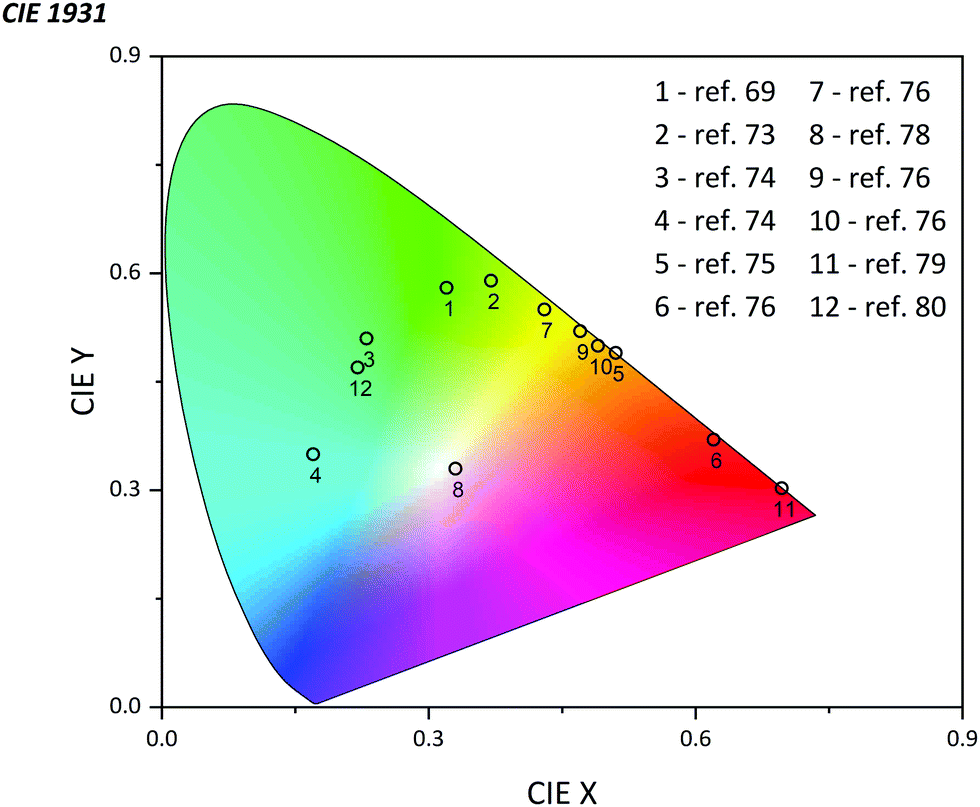 | ||
| Fig. 13 CIE diagram summarizing phenothiazine-based OLEDs. | ||
 | ||
| Fig. 14 PTZ-Based molecules in OLED devices. | ||
| Molecule | Mechanism of emission | Delayed decay, μs | QY, % | EQE, % | HOMO/LUMO | ΔEST, eV | B max, cd m−2 | Color | Ref. |
|---|---|---|---|---|---|---|---|---|---|
| Notes: QY is shown for the films of the emissive material, “—” refers to the value not represented in the work, NA – refers that value is non-applicable for the current work.a Two values represent ΔEST in the different conformations.b Means that QY was measured for the solution. | |||||||||
| 67 | PF | — | 81 | 0.4 | −4.93/−2.27 | N/A | 1071 | Green (0.424, 0.554) | 75 |
| CBP: 10 wt% 67 | PF | — | — | 0.6 | — | N/A | 1365 | Green (0.314, 0.593) | 75 |
| 68 | PF | 1.15 × 10−3 | 3 | — | −5.03/−2.11 | N/A | 499 | Green (0.32, 0.58) | 78 |
| 69 | PF | — | — | — | −4.79/−1.7 | N/A | 2116 | Green (0.37, 0.59) | 79 |
| 70 | PF | — | 10b | — | −5.02/−2.32 | N/A | 2791 | Green (0.23, 0.51) | 80 |
| 71 | PF | — | 12b | — | −4.95/−2.11 | N/A | 754 | Cyan (0.17, 0.35) | 80 |
| FI2rpic: 10 wt% 72 | Ph | — | — | 16.9 | −6.17/−2.68 | N/A | ∼8000 | Blue | 76 |
| FI2rpic: 10 wt% 73 | Ph | — | — | 24.3 | −6.09/−2.80 | N/A | ∼10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 000 |
Blue | 76 |
| FI2rpic: 10 wt% 74 | Ph | — | — | 9.4 | −6.11/−2.82 | N/A | ∼5000 | Blue | 76 |
| PtAu2: 8 wt% 75 | Ph | 3.45 | 73.7 | 13.1 | −5.47/−2.5 | N/A | 8594 | Orange (0.51, 0.49) | 82 |
| Ir(piq)2(acac): 5 wt% 76 | Ph | — | — | 10.3 | — | — | 17000 |
Red (0.62, 0.37) | 83 |
| 76 | AIE/TADF | 1.1 | 37 | 5.4 | −5.55/−2.71 | 0.07 | 49000 |
Yellow-orange (0.43, 0.55) | 83 |
| 77 | TADF | 0.57 | 43 | 11.5 | — | 0.46/0.01a | 10370 |
Green-yellowish | 84 |
| 78 | TADF | — | 11.04 | 2.68 | −5.94/−1.92 | 0.581/0.019 | 100–300 | White (0.33, 0.33) | 85 |
| 79 | TADF | — | 50.83 | 16.34 | −5.52/−2.08 | 0.457/0.005a | 10000 |
Warm white (0.41, 0.47) | 85 |
| 80 | AIE/TADF | 0.8 | 24 | 4.85 | −5.53/−2.68 | 0.02 | 33000 |
Yellow-orange (0.47, 0.52) | 83 |
| 81 | AIE/TADF | 1 | 15 | 5.1 | −5.48/−2.61 | 0.07 | 30000 |
Yellow-orange (0.49, 0.5) | 83 |
| 82 | AIE | 4.39 × 10−2 | 23.4 | — | — | N/A | — | Red (0.6967, 0.3030) | 86 |
| 86 | HLCT | — | — | 4.14 | −5.04/−2.40 | 0.73 | 17166 |
Blue-green (0.22, 0.47). | 87 |
| 87 | HLCT | 0.1 | 60 | 6.62 | −5.14/−2.43 | N/A | 17007 |
Blue | 88 |
OLEDs can be designed to operate based on the molecule's fluorescence (emission from the singlet state) or phosphorescence (emission from the triplet state) (Fig. 15). We first review the fluorescence-based devices. Bodedla et al. synthesized a small molecule 67 (Fig. 14) via one-step Heck cross-coupling and used it in two different devices as a hole-transporting emitting layer and as a 10 wt% dopant emitter in 4,4′-bis(N-carbazolyl)-1,1′-biphenyl (CBP) host, respectively.75 While both OLEDs emitted light in the green region, the first one showed EQE of 0.4% and a slight increase up to 0.6% was observed in the second device, which was credited to efficient Förster energy transfer from the CBP matrix to 67. Shanmugasundaram and co-workers presented 68-based OLED that also worked in the green region with a Bmax of 499 cd m−2.78 Notably, no additional layers (ETL, HTL, or hole-blocking layer) assisting the OLEDs operation were needed. A significantly higher brightness of 2116 cd m−2 was shown by Salunke and co-workers79 using small molecule 69 in the form of tetra-functionalized pyrene derivative as an emissive layer. The improvement was attributed to the low HOMO value of 69 and smooth film morphology assisting in the transport of charge carriers in the recombination zone. In another study, small molecules 70 and 71 were used as dopants in the CBP host matrix emitting green and cyan light respectively.80 The authors associated the considerably higher Bmax of 70 compared with 71 to its better energy levels alignment with the host matrix.
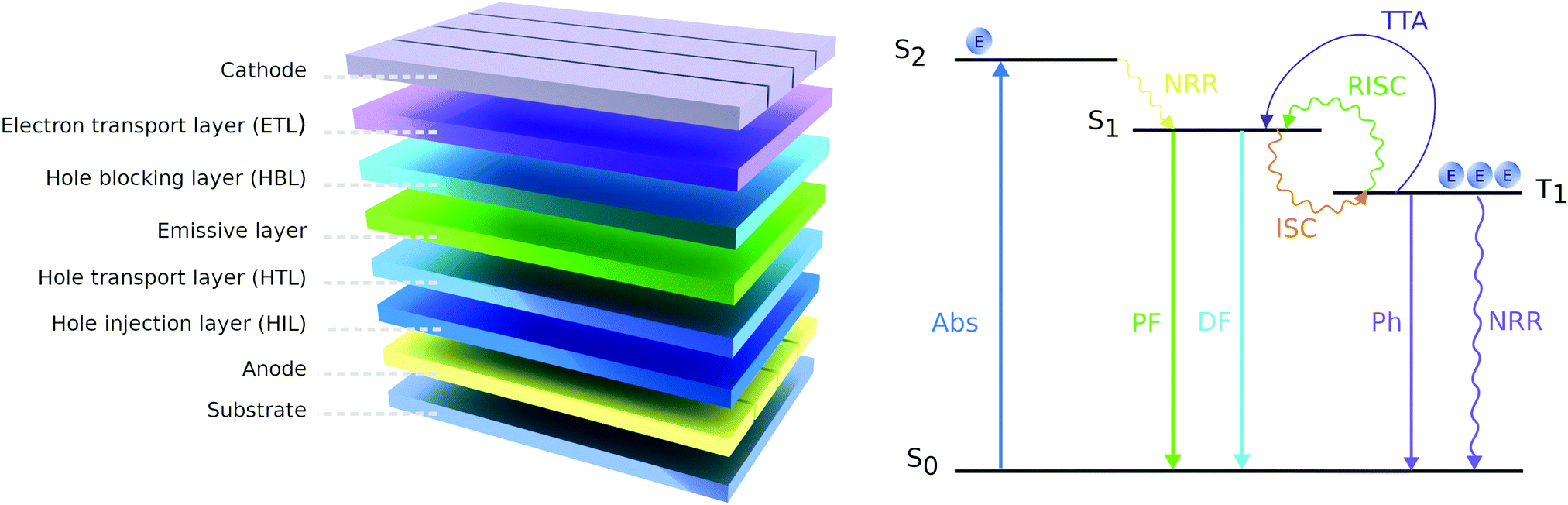 | ||
| Fig. 15 The structure of OLED (left); simplified Jablonski diagram representing emission process in the OLED (right), where E – exciton, Abs – absorption, PF – prompt fluorescence, DF – delayed fluorescence, Ph – phosphorescence, NRR – non-radiative relaxation, ISC – intersystem crossing, RISC – reverse intersystem crossing, TTA – triplet–triplet annihilation. | ||
Theoretically, up to 100% of internal quantum efficiency in the OLED could be obtained via phosphorescence.81 As a downside, this approach is considered more expensive due to the high cost of the dopants. Lee et al. employed small molecules 72, 73, and 74 as electron transport units in the host material.76 Because high triplet energy is essential for blue-colored OLED, the authors emphasized that phenothiazine dioxide had no negative impact on the triplet energy of the backbone structure. The maximum EQEs of the devices using F2Irpic as the dopant were 16.9%, 24.3%, and 9.4% for 72, 73, and 74, respectively. The higher EQE of 73 was explained by the high triplet energy and the balanced carrier density comparing to the other two.
A comparatively high QY of 73.7% and EQE of 13.1% was achieved by Zend and co-workers by dispersing 75 doped by PtAu2 metal complex in poly(methyl methacrylate) matrix. Tan's group used Ir(piq)2(acac) emitters together with bipolar charge transporter 76. As a result, they obtained red OLED with high Bmax (17000 cd m−2) and EQE (10.3%).83
Several studies reported about the TADF observed in phenothiazine derivatives in the last few years.83–85,89–91 As the TADF mechanism allows overcoming the theoretical limit of 25% in the OLEDs due to transformation exciton from triplet state to a singlet state induced by absorption of nearby thermal energy, it has attracted the attention of researchers in the recent years.92 In such devices, PTZ is typically used as an electron donor displaying at the same time low value of the ΔEST between the triplet (T1) and singlet (S1).84 By synthesizing twisted molecular donor–acceptor architectures with minimum HOMO–LUMO overlapping, highly efficient OLEDs were achieved. TADF is usually coupled with dual emission, which was also observed in several works employing PTZ-based materials.84,85,90 It was shown that dual emission arises from two ground-state conformations with different energy gaps between the lowest singlet excited state and lowest triplet excited state, where one of the ΔEST is typically much lower than the other and induces the TADF process. Marghad et al. observed dual emission by incorporating two phenothiazine units to 2-thiophene-1,3,5-triazine 77.84 Authors stated that the proposed method of synthesis, which is based on cobalt catalyzed cross-coupling, is cheaper and simpler compared to other approaches to synthesize materials with TADF features. Notably, the calculated ΔEST values of one of the conformers is much smaller (0.01 eV) than the other (0.46 eV) indicating that to achieve the best TADF performance, careful optimization of the OLED architecture should be done. Wang et al. obtained two molecules 78 and 79via Buchwald–Hartwig cross-coupling reaction and Suzuki reaction, respectively, which were used further to fabricate white OLEDs.85 Two conformations of the molecules were responsible for the observed dual emission. One of the conformers had lower energy levels and TADF characteristics, whereas the other possessed higher energy levels and non TADF characteristics, which led to white emission. A high EQE of 16.34% was realized due to the transferring possibility between orthogonal and planar conformations when originally untapped triplet energy was fully utilized. By changing the D moieties with different flexibility and the interaction between D and A segments, pure white color (0.33, 0.33 CIE) was obtained because of the adjustment of the band gaps and the relative distributions of the two conformations.
Wang and coworkers developed a series of PTZ derivatives and investigated their TADF applications.37,38,93 For example, in one study, they used oxidized form of the PTZ unit as the electron acceptor for the construction of aggregation-induced delayed fluorescence materials.37 The emitter, PXZ2PTO, exhibited both the AIE characteristics and TADF properties. Due to its highly stereoscopic structure and small ΔEST, a green non-doped OLED realized high maximum EQE, CE, and PE of 16.4%, 44.9 cd A−1, and 32.0 lm W−1, respectively. These results demonstrated that phenothiazine oxides can be used as acceptors for the design of novel TADF and AIDF materials. Similarly, in another study, they reported on the design and synthesis of two PTZ-based TADF emitters named PTZ-2PTO and PTZ-AD.93 Both the emitters possessed dual conformations, where quasi-axial and -equatorial conformers were predominant for PTZ-2PTO and PTZ-AD, respectively. The quasi-axial conformers of both emitters showed classical fluorescence, whereas the quasi-equatorial conformers exhibited TADF characteristic. The yellow non-doped TADF-OLED devices based on PTZ-AD (as quasi-equatorial conformers are the predominant) showed high maximum EQE, CE, and PE of 17.08%, 50.48 cd A−1, and 60.04 lm W−1, respectively, which are comparable to the figures of merit of doped devices.
Wang et al. presented two PTZ containing TADF emitters with small ΔEST and relatively long fluorescence lifetimes. OLED devices with the emitters displayed high EQEs values reaching 17.4% with maximum power efficiencies of up to 27.8 lm W−1. Notably, these TADF OLEDs exhibited slight efficiency roll-off with EQEs of 12.9% at a luminescence of 1000 cd m−2.36 In another study, Bryce and coworkers studied the effect of steric hinderance on the structural and photophysical properties of TADF emitters.94 They synthesized a series of D–A–D and D–A molecules with bulky alkyl groups on the donor PTZ unit, which restricts the rotation around the C–N bond linking of D–A units. Compounds with conformational flexibility exhibited efficient TADF whereas sterically hindered analogues showed strong contribution from phosphorescence even at room temperature.
Tan and co-workers also demonstrated TADF-based OLED, emitting in the yellow-orange region, which also possessed aggregation-induced emission (AIE) ability.83 1,3,5-Triazine acceptor was used as the core and 76, 80 or 81 as the donor. The authors reported an improvement of EQE and luminous intensity for all the devices, obtained via the AIE effect. Moreover, an additional increase in the EQE was obtained due to 5% doping by the luminophore. The Improvement was explained by decreasing their ΔEST values. However, compared with the non-doped devices, the doped devices exhibited a shift to the green region and the lower maximum brightness, which was attributed to the misbalance of hole-electron pairs in the light-emitting layers of the doped devices at high electric fields. AIE behavior promoted by the bent geometry was demonstrated in a series of PTZ-based fluorophores obtained via one step Knoevenagal condensation reaction, emitting in red and near-infrared regions. The best performance was shown for molecule 82 with a QY as high as 23.4% (for red emitter in the solid-state).86 Similar examples of AIE type delayed fluorescent emitters were developed by Lee et al. and Gan et al. According to the DFT calculations, the restriction of intramolecular motions are accounted for the AIE characteristics.95,96 Furthermore, Zhang et al. demonstrated three PTZ-triphenylacrylonitrile modified derivatives 83, 84, and 84 that possessed AIE ability and mechanochromism.97 While the as-synthesized materials under UV radiation emitted intense yellow, yellowish-orange, and yellowish-green light, it changed into red, orange-red, and orange, respectively, after the grind. Notably, that observed mechanochromism was reversible – after the fuming with solvents powdered material come into the original state. Different solvents (namely DCM, acetone, toluene, or n-hexane), as well as the temperature and heating time, were tested to optimize the recovery mechanism. Moreover, based on the XRD results authors concluded that mechanochromism could be attributed to the transition between the crystalline and amorphous states.
Another widely utilized mechanism for achieving highly efficient OLEDs is to design hybridized local and charge-transfer excited state (HLCT), which, however, requires a small singlet–triplet energy gap (ΔEST).98,99 Shi and co-workers showed novel blue OLED based on 86 by a Suzuki coupling reaction.87
As a result, HCLT was observed in the OLED with relatively high (for the blue emission) EQE of 4.14%. Further doping of the obtained emitters in the CBP, host material allowed to achieve deep blue emission with EQE of 3.69%. In the following work, they demonstrated blue OLED using 87 as the donor and 1,3,4-oxadiazole as the acceptor (TPEPO) with a higher EQE of 6.62%.88 High ΔEST (0.73 eV) allowed them to estimate that approximately 55% of the radiative excitons attributed to the HLCT process.
3.4 Batteries
The ever-increasing amount of renewable energy, such as wind and solar power requires advancements in energy storage technology to fully realize its potential. Portable electronics are more present in our everyday lives than ever before, and with the additions, such as wearable electronics and advanced smartphones, technology is at present dependent on Li-ion battery technology to provide energy storage for most applications.100,101 Combining organic molecules with the current battery technology could offer a solution for many of the problems, enabling high energy densities and high rates, while improving safety.100,102,103 Organic materials are flexible and tunable for specialized uses but have mostly been able to manage only a limited number of redox cycles when tested in conventional or flow battery cells.101,104 PTZ-Based molecules have shown promise in providing a solution capable of handling redox cycles equivalent to commercial Li-ion batteries.104The first application of polymeric PTZ as cathode active materials in Li-ion batteries was presented by Golriz et al.105 The π–π stacking of the closely packed phenothiazine groups bound by the polymer backbone was suggested to result in improved conductivity. They studied two phenothiazine polymers that had a different distance of the phenothiazine group to the polymer backbone: poly(10-(4-vinylbenzyl)-10H-phenothiazine) (88) and poly(10-(3-(4-vinylphenyl)propyl)-10H-phenothiazine) (89) (Fig. 16). The longer spacer leads to increased long-term stability under cycling. Godet-Bar et al. performed thorough ab initio calculations on PTZ molecules and based on which they synthesized poly-N-propylphenothiazine (90).106 Once the benefits of polymeric π–π stacked phenothiazines had been demonstrated, Kolek et al.107 improved cycling stability even further using poly(3-vinyl-N-methyl-phenothiazine) (91). Developing the ideas presented in the previous works on polymeric PTZ.100,105,106 Peterson et al.108 synthesized cross-linked PTZ-based copolymers that could undergo multiple electron oxidations. This was achieved by adding electron-rich aryl amines – N,N,N′,N′-tetramethyl-p-phenylenediamine (92) and N,N,N′,N′-tetramethylbenzidine (93) – into the main chain. Further development of the copolymerization concept has now reached 30000 charge–discharge cycles while maintaining 97% of the initial capacity.104 This remarkable result was obtained synergically combining the aliphatic redox polymer with the conducting polymers; hence combining the PTZ with bithiophene into a copolymer (94). Various side chains were tested in the same work, best results obtained with aryl ether group at the N-position of the phenothiazine and alkyl chains at the bithiophene. Most recently, the poly(norbornene)s were tested as the polymer backbone with N-methyl-PTZ functionalized side groups (95).101 The performance of the molecules discussed in this paragraph is presented in Table 4 and the molecular structures are described in Fig. 16.
 | ||
| Fig. 16 PTZ-Based molecules used as battery cathodes. | ||
PTZs have also proven to be advantageous in overcharge protection of Li-ion batteries. In brief, the principle is that once the desired battery charge is reached, the protecting molecule begins to oxidize instead, preventing the electrode potential going overcharge. In 2006 and 2007 Buhrmester et al.109 and Moshurchak et al.110 reported a study of several N-alkyl PTZs for overcharge protection of Li-ion batteries showing good initial performance. Almost ten years later, Ergun et al.111 studied N-ethylphenothiazines having electron-withdrawing groups at the 3,7-positions i.e. para to the N-atom. The F3C-substituted, 3,7-bis(trifluoromethyl)-N-ethyl-phenothiazine (BCF3EPT), was found to be the most promising for high voltage applications limiting the potential right below 4.0 V when tested in a graphite/LiFePO4 coin cell battery. In a follow-up work, the same group reported a fully fluorinated alkyl PTZ.102 The N-ethyl-1,2,3,4,6,7,8,9-octafluorophenothiazine (OFEPT) could provide a stable protection onset at 4.3 V when tested together with LiNi0.8Co0.15Al0.05O2 making it the first PTZ molecule to be able to successfully protect a high voltage lithium-ion battery.
On a final note, PTZs have shown promising initial performance in redox flow batteries. Kowalski et al.103 showed that the dication state of a PTZ could be stabilized with methoxy groups. They suggested that the stable two-electron-donating PTZs could advance the design of new redox-based energy storage systems. Zhang et al.112 showed that the methylene blue (3,7-bis(dimethylamino)phenazathionium chloride) had excellent stability up to 900 cycles and high capacity in aqueous redox flow battery giving 71 A h L−1.
4. Conclusions and future outlook
In this review, we provided a broad survey of the most interesting PTZ derivatives for organic and perovskite solar cells, OLEDs, and batteries. In particular, we have summarized the key achievements in PTZ-based optoelectronics from a chemist's perspective, with a closer look at the PTZ-based molecular designs in connection to the performance of the corresponding devices. We believe that with this approach we may inspire many materials chemists in the field, to design the next generation of PTZ-based materials for optoelectronic devices with enhanced performance and stability.The judicious molecular engineering of the PTZ core is an efficient strategy to develop conjugated materials with tunable optoelectronic properties. It is evident from several studies that even a subtle structural modification of the PTZ core or the conjugate backbone can bring sizable alterations to the molecular properties and affects the device performances. For example, the oxidation of sulphur of thiazine ring affects the redox and spectroscopic properties of PTZ chromophores, while the substitutions of N-10 active site of PTZ core with alkyl or polar oligo ether groups influence the solubility characteristics of the material, the solid-state packing patterns, and the charge mobilities. By modulating the electron withdrawing/donating moieties on the conjugate backbone of PTZ-derivatives, a significant effect on the absorption bandwidths, emission profiles, energy levels, and the device performance of the corresponding PTZ-based OSCs is achieved. The thorough molecular engineering approach of the donor/acceptor constituents of the bulk heterojunction blends leads to OSCs with power conversion efficiencies as high as 7.45% (Jsc = 12 mA cm−2, Voc = 1.04 V, FF = 0.65). Good performance in PSCs employing PTZ-core HTMs is also demonstrated, with a performance PCE ∼ 20%, comparable to that of the start-of-the-art devices including the well-known spiro-OMeTAD HTM. PTZ dyes proved to be interesting candidates for OLED applications. Their facile chemistry allows straightforward structural modifications to tune the emission in a broad wavelength range. Typically, PTZ core materials display electron-donor characteristics and have been employed as the host matrix of the emissive organic layer in OLEDs covering almost all the CIE color space. In the field of electrochemical energy storage, the use of PTZ-based molecules allows the known advantages of organic materials and the capability of handling redox cycles similarly as for commercial Li-ion batteries. The promise of these materials lies in their versatile use as cathode active materials in Li-ion batteries, as overcharge protection of Li-ion batteries, or in redox flow batteries.
Despite the recent tremendous advancement in designing PTZ-based materials for various optoelectronic applications, certain challenges remain to be overcome to ensure ever-rising device performances. The bulk hole mobility of these materials, generally in the range of 10−4–10−6 cm2 V−1 s−1, is one of the key issues to be enhanced. The reasons for such modest mobility lie in the typically low dielectric constants of organic semiconductors and to the non-planar structure of PTZ, which inhibits molecular aggregation. In literature, a few successful attempts113,114 to improve the charge mobilities (>10−2 cm2 V−1 s−1) of building blocks other than PTZ were reported. For example, a few recent studies have shown that, with the insertion of polar glycol side chains, it is possible to significantly increase the dielectric constant of the fullerene derivatives without compromising their optical properties, electrical mobility, and energy levels.114 We foresee that similar strategies could be applied also to the PTZ core. The flexible polar glycol side chains enhance the crystallinity of thin films with small stacking distance (Fig. 17), thereby improving the charge carrier mobilities.
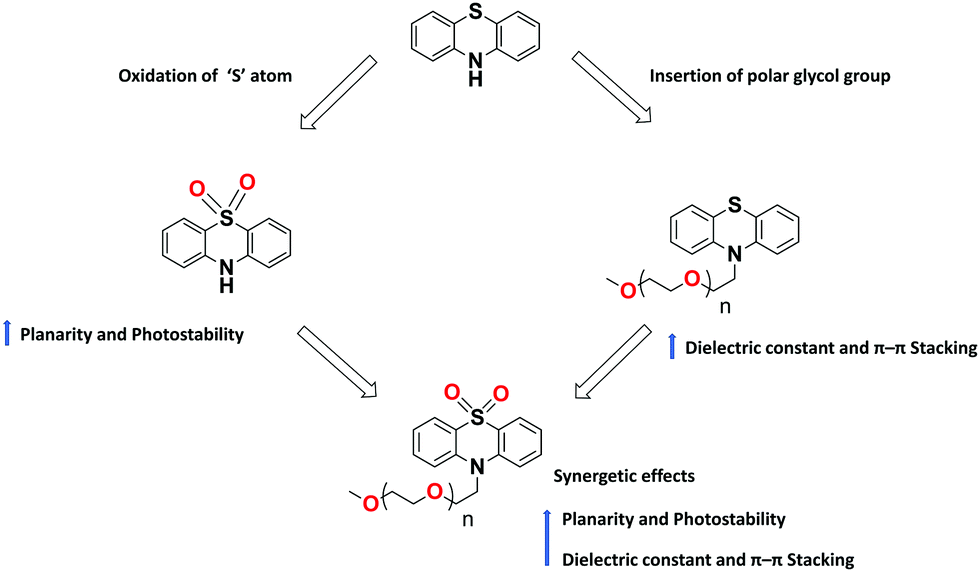 | ||
| Fig. 17 Illustration of structural modifications on PTZ core and their effect on the molecular properties. | ||
PTZ-Based materials demonstrated reasonable thermal stabilities with Tg as high as 153 °C. Further improvements can be achieved by cross-linking the molecules with units, such as vinyl or epoxy groups, that can form insoluble 3D networks under mild processing conditions and resist thermal degradation. Concerning photostability, PTZ dyes are prone to chemical degradation, in particular, to the photo-oxidation of the aromatic sulphur. One way to overcome this is to use the oxidized forms of PTZ (instead of naked PTZ) as substrates for constructing conjugated materials, as shown in Fig. 17.
A wide majority of synthetic schemes of PTZ materials involves reactions of C–C couplings, which use the scarcely available palladium metal catalyst and/or chlorinated solvents, both failing to comply with sustainability requirements, especially important when thinking of an industrial-scale synthesis. Condensation chemistry that leads to eco-friendly by-products or C–H activation that devoid the extensive functionalization of monomers or use of polar chains (such as glycols) to induce water or alcohol soluble characteristics, are among the few potential ways to make the materials synthesis truly environmentally friendly. We believe that there is a great need for new chemistry, innovative design strategies, and a deeper understanding of the connections between engineering at the molecular level and the resulting material properties. This would allow overcoming the current challenges and explore the applications of PTZ dyes in new optoelectronics domains.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank Noora Lamminen for helping with the drawing of the molecular structures. Business Finland and Forschungszentrum Jülich GmbH (project SolarWAVE) are acknowledged for financial support. This work is part of the Academy of Finland Flagship Programme, Photonics Research and Innovation (PREIN, Decision number 320165).References
- A. J. Heeger, Chem. Soc. Rev., 2010, 39, 2354–2371 RSC.
- X. Guo, M. Baumgarten and K. Müllen, Prog. Polym. Sci., 2013, 38, 1832–1908 CrossRef CAS.
- L. Dou, Y. Liu, Z. Hong, G. Li and Y. Yang, Chem. Rev., 2015, 115, 12633–12665 CAS.
- P. C. Y. Chow and T. Someya, Adv. Mater., 2020, 32, 1902045 CAS.
- M. Kim, S. U. Ryu, S. A. Park, K. Choi, T. Kim, D. Chung and T. Park, Adv. Funct. Mater., 2020, 30, 1904545 CrossRef CAS.
- Z. Li, C. C. Chueh and A. K. Y. Jen, Prog. Polym. Sci., 2019, 99, 101175 CrossRef CAS.
- P. Cheng, G. Li, X. Zhan and Y. Yang, Nat. Photonics, 2018, 12, 131–142 CrossRef CAS.
- P. Vivo, J. Salunke and A. Priimagi, Materials, 2017, 10, 1087 CrossRef.
- S. Shahnawaz, S. Sudheendran Swayamprabha, M. R. Nagar, R. A. K. Yadav, S. Gull, D. K. Dubey and J.-H. Jou, J. Mater. Chem. C, 2019, 7, 7144–7158 RSC.
- S. Xiao, Q. Zhang and W. You, Adv. Mater., 2017, 29, 1601391 CrossRef.
- H. Bronstein, C. B. Nielsen, B. C. Schroeder and I. McCulloch, Nat. Rev. Chem., 2020, 4, 66–77 CrossRef CAS.
- J. Mei and Z. Bao, Chem. Mater., 2014, 26, 604–615 CrossRef CAS.
- S. A. Jenekhe, L. Lu and M. M. Alam, Macromolecules, 2001, 34, 7315–7324 CrossRef CAS.
- K. C. Li, Y. C. Hsu, J. T. S. Lin, C. C. Yang, K. H. Wei and H. C. Lin, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 4285–4304 CrossRef CAS.
- G. Kim, H. R. Yeom, S. Cho, J. H. Seo, J. Y. Kim and C. Yang, Macromolecules, 2012, 45, 1847–1857 CrossRef CAS.
- S. Revoju, S. Biswas, B. Eliasson and G. D. Sharma, Dyes Pigm., 2018, 149, 830–842 CrossRef CAS.
- V. Ji Ram, A. Sethi, M. Nath and R. Pratap, Six-Membered Heterocycles, 2019 Search PubMed.
- X. Kong, A. P. Kulkarni and S. A. Jenekhe, Macromolecules, 2003, 36, 8992–8999 CrossRef CAS.
- J. A. Kowalski, M. D. Casselman, A. P. Kaur, J. D. Milshtein, C. F. Elliott, S. Modekrutti, N. H. Attanayake, N. Zhang, S. R. Parkin, C. Risko, F. R. Brushett and S. A. Odom, J. Mater. Chem. A, 2017, 5, 24371–24379 RSC.
- K. A. Narayana, M. D. Casselman, C. F. Elliott, S. Ergun, S. R. Parkin, C. Risko and S. A. Odom, ChemPhysChem, 2015, 16, 1179–1189 CrossRef CAS.
- C. Zhang, Z. Niu, S. Peng, Y. Ding, L. Zhang, X. Guo, Y. Zhao and G. Yu, Adv. Mater., 2019, 31, 1901052 CrossRef.
- S. Revoju, S. Biswas, B. Eliasson and G. D. Sharma, Org. Electron., 2019, 65, 232–242 CrossRef CAS.
- S. Thokala and S. P. Singh, ACS Omega, 2020, 5, 5608–5619 CrossRef CAS.
- J. S. Luo, Z. Q. Wan and C. Y. Jia, Chin. Chem. Lett., 2016, 27, 1304–1318 CrossRef CAS.
- I. J. Al-Busaidi, A. Haque, N. K. Al Rasbi and M. S. Khan, Synth. Met., 2019, 257, 116189 CrossRef CAS.
- Z. S. Huang, H. Meier and D. Cao, J. Mater. Chem. C, 2016, 4, 2404–2426 RSC.
- J. E. Bloor, B. R. Gilson, R. J. Haas and C. L. Zirkle, J. Med. Chem., 1970, 13, 922–925 CrossRef CAS.
- S. Revoju, S. Biswas, B. Eliasson and G. D. Sharma, Phys. Chem. Chem. Phys., 2018, 20, 6390–6400 RSC.
- Z. Liu, E. Shi, Y. Wan, N. Li, D. Chen, Q. Xu, H. Li, J. Lu, K. Zhang and L. Wang, J. Mater. Chem. C, 2015, 3, 2033–2039 RSC.
- K. D. Thériault and T. C. Sutherland, Phys. Chem. Chem. Phys., 2014, 16, 12266–12274 RSC.
- D. B. Shinde, J. K. Salunke, N. R. Candeias, F. Tinti, M. Gazzano, P. P. Wadgaonkar, A. Priimagi, N. Camaioni and P. Vivo, Sci. Rep., 2017, 7, 46268 CrossRef CAS.
- Y. Lu, C. Jiang, X. Li and J. Song, Comput. Theor. Chem., 2019, 1163, 112512 CrossRef.
- M. Chen, Y. Lee, Z. Huang, D. Chen and P. Chou, Chem. – Eur. J., 2020, 26, 7124–7130 CrossRef CAS.
- H. Tanaka, K. Shizu, H. Nakanotani and C. Adachi, J. Phys. Chem. C, 2014, 118, 15985–15994 CrossRef CAS.
- M. Okazaki, Y. Takeda, P. Data, P. Pander, H. Higginbotham, A. P. Monkman and S. Minakata, Chem. Sci., 2017, 8, 2677–2686 RSC.
- B. Wang, X. Qiao, Z. Yang, Y. Wang, S. Liu, D. Ma and Q. Wang, Org. Electron., 2018, 59, 32–38 CrossRef CAS.
- S. Xiang, Z. Huang, S. Sun, X. Lv, L. Fan, S. Ye, H. Chen, R. Guo and L. Wang, J. Mater. Chem. C, 2018, 6, 11436–11443 RSC.
- R. Guo, Y. Wang, Z. Huang, Q. Zhang, S. Xiang, S. Ye, W. Liu and L. Wang, J. Mater. Chem. C, 2020, 8, 3705–3714 RSC.
- L. Meng, Y. Zhang, X. Wan, C. Li, X. Zhang, Y. Wang, X. Ke, Z. Xiao, L. Ding, R. Xia, H. L. Yip, Y. Cao and Y. Chen, Organic and solution-processed tandem solar cells with 17.3% efficiency, 2018, vol. 361 Search PubMed.
- Y. Cui, H. Yao, J. Zhang, K. Xian, T. Zhang, L. Hong, Y. Wang, Y. Xu, K. Ma, C. An, C. He, Z. Wei, F. Gao and J. Hou, Adv. Mater., 2020, 32, 1908205 CrossRef CAS.
- N. S. Cho, J. H. Park, S. K. Lee, J. Lee, H. K. Shim, M. J. Park, D. H. Hwang and B. J. Jung, Macromolecules, 2006, 39, 177–183 CrossRef CAS.
- S. Matsumura, A. R. Hlil, C. Lepiller, J. Gaudet, D. Guay, Z. Shi, S. Holdcroft and A. S. Hay, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 7207–7224 CrossRef.
- J.-H. Huang, K.-C. Li, H.-Y. Wei, P.-Y. Chen, L.-Y. Lin, D. Kekuda, H.-C. Lin, K.-C. Ho and C.-W. Chu, Org. Electron., 2009, 10, 1109–1115 CrossRef CAS.
- H. Padhy, J.-H. Huang, D. Sahu, D. Patra, D. Kekuda, C.-W. Chu and H.-C. Lin, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 4823–4834 CrossRef CAS.
- G. Sang, Y. Zou, Y. Huang, G. Zhao, Y. Yang and Y. Li, Appl. Phys. Lett., 2009, 94, 193302 CrossRef.
- Q. Tan, X. Yang, M. Cheng, H. Wang, X. Wang and L. Sun, J. Phys. Chem. C, 2014, 118, 16851–16855 CrossRef CAS.
- C. Maglione, A. Carella, R. Centore, P. Chávez, P. Lévêque, S. Fall and N. Leclerc, Dyes Pigm., 2017, 141, 169–178 CrossRef CAS.
- J. Liao, Y. Xu, H. Zhao, Q. Zong and Y. Fang, Org. Electron., 2017, 49, 321–333 CrossRef CAS.
- Y.-L. Weng, Y.-C. Li, C.-P. Chen and Y. J. Chang, Dyes Pigm., 2017, 146, 374–385 CrossRef CAS.
- B. Yadagiri, K. Narayanaswamy, R. Srinivasa Rao, A. Bagui, R. Datt, V. Gupta and S. P. Singh, ACS Omega, 2018, 3, 13365–13373 CrossRef CAS.
- Y. Rout, R. Misra, R. Singhal, S. Biswas and G. D. Sharma, Phys. Chem. Chem. Phys., 2018, 20, 6321–6329 RSC.
- J. Marques Dos Santos, L. K. Jagadamma, N. M. Latif, A. Ruseckas, I. D. W. Samuel and G. Cooke, RSC Adv., 2019, 9, 15410–15423 RSC.
- G. D. Blanco, A. J. Hiltunen, G. N. Lim, C. B. KC, K. M. Kaunisto, T. K. Vuorinen, V. N. Nesterov, H. J. Lemmetyinen and F. D’Souza, ACS Appl. Mater. Interfaces, 2016, 8, 8481–8490 CrossRef CAS.
- D. Mi, J. H. Kim, F. Xu, S. H. Lee, S. Cheol Yoon, W. Suk Shin, S. J. Moon, C. Lee and D. H. Hwang, Sol. Energy Mater. Sol. Cells, 2011, 95, 1182–1187 CrossRef CAS.
- G. D. Sharma, M. Anil Reddy, D. V. Ramana and M. Chandrasekharam, RSC Adv., 2014, 4, 33279–33285 RSC.
- S. Revoju, Molecular Design, Synthesis and Performance Evaluation of Phenothiazine-based Small Molecules for Organic Solar Cells, Umea University, Umea, 2018th edn, 2018 Search PubMed.
- M. A. Green, A. Ho-Baillie and H. J. Snaith, Nat. Photonics, 2014, 8, 506–514 CrossRef CAS.
- A. K. Jena, A. Kulkarni and T. Miyasaka, Chem. Rev., 2019, 119, 3036–3103 CrossRef CAS.
- M. Liu, A. Matuhina, H. Zhang and P. Vivo, Materials, 2019, 12, 3733 CrossRef CAS.
- E. V. Ushakova, A. I. Matuhina, A. V. Sokolova, S. A. Cherevkov, A. Dubavik, O. S. Medvedev, A. P. Litvin, D. A. Kurdyukov, V. G. Golubev and A. V. Baranov, Nanotechnology, 2019, 30, 405206 CrossRef CAS.
- I. Hussain, H. P. Tran, J. Jaksik, J. Moore, N. Islam and M. J. Uddin, Emergent Mater., 2018, 1, 133–154 CrossRef CAS.
- L. Calió, S. Kazim, M. Grätzel and S. Ahmad, Angew. Chem., Int. Ed., 2016, 55, 14522–14545 CrossRef.
- R. Grisorio, B. Roose, S. Colella, A. Listorti, G. P. Suranna and A. Abate, ACS Energy Lett., 2017, 2, 1029–1034 CrossRef CAS.
- X. Liu, X. Tan, Q. Chen, H. Shan, C. Liu, J. Xu, Z. K. Chen, W. Huang and Z. X. Xu, RSC Adv., 2017, 7, 53604–53610 RSC.
- F. Zhang, S. Wang, H. Zhu, X. Liu, H. Liu, X. Li, Y. Xiao, S. M. Zakeeruddin and M. Grätzel, ACS Energy Lett., 2018, 3, 1145–1152 CrossRef CAS.
- J. Salunke, X. Guo, Z. Lin, J. R. Vale, N. R. Candeias, M. Nyman, S. Dahlström, R. Österbacka, A. Priimagi, J. Chang and P. Vivo, ACS Appl. Energy Mater., 2019, 2, 3021–3027 CrossRef CAS.
- J. Salunke, X. Guo, M. Liu, Z. Lin, N. R. Candeias, A. Priimagi, J. Chang and P. Vivo, ACS Omega, 2020, 5, 23334–23342 CrossRef.
- X. Ding, C. Chen, L. Sun, H. Li, H. Chen, J. Su, H. Li, H. Li, L. Xu and M. Cheng, J. Mater. Chem. A, 2019, 7, 9510–9516 RSC.
- M. Maciejczyk, A. Ivaturi and N. Robertson, J. Mater. Chem. A, 2016, 4, 4855–4863 RSC.
- X. Liang, C. Wang, M. Wu, Y. Wu, F. Zhang, Z. Han, X. Lu, K. Guo and Y. M. Zhao, Tetrahedron, 2017, 73, 7115–7121 CrossRef CAS.
- M. R. Maciejczyk, R. Chen, A. Brown, N. Zheng and N. Robertson, J. Mater. Chem. C, 2019, 7, 8593–8598 RSC.
- X. Zhao, Y. Quan, H. Pan, Q. Li, Y. Shen, Z. S. Huang and M. Wang, J. Energy Chem., 2019, 32, 85–92 CrossRef.
- Y. Chen, X. Xu, N. Cai, S. Qian, R. Luo, Y. Huo and S. W. Tsang, Adv. Energy Mater., 2019, 9, 1–8 Search PubMed.
- C. Lu, M. Paramasivam, K. Park, C. H. Kim and H. K. Kim, ACS Appl. Mater. Interfaces, 2019, 11, 14011–14022 CrossRef CAS.
- G. B. Bodedla, K. R. Justin Thomas, S. Kumar, J. H. Jou and C. J. Li, RSC Adv., 2015, 5, 87416–87428 RSC.
- I. H. Lee and J. Y. Lee, RSC Adv., 2015, 5, 97903–97909 RSC.
- B. Minaev, G. Baryshnikov and H. Agren, Phys. Chem. Chem. Phys., 2014, 16, 1719–1758 RSC.
- K. Shanmugasundaram, M. S. Subeesh, C. D. Sunesh, R. K. Chitumalla, J. Jang and Y. Choe, J. Phys. Chem. C, 2016, 120, 20247–20253 CrossRef CAS.
- J. K. Salunke, F. L. Wong, K. Feron, S. Manzhos, M. F. Lo, D. Shinde, A. Patil, C. S. Lee, V. A. L. Roy, P. Sonar and P. P. Wadgaonkar, J. Mater. Chem. C, 2016, 4, 1009–1018 RSC.
- R. K. Konidena, K. R. J. Thomas, S. Kumar, Y. C. Wang, C. J. Li and J. H. Jou, J. Org. Chem., 2015, 80, 5812–5823 CrossRef CAS.
- Q. Wei, N. Fei, A. Islam, T. Lei, L. Hong, R. Peng, X. Fan, L. Chen, P. Gao and Z. Ge, Adv. Opt. Mater., 2018, 6, 1800512 CrossRef.
- X. C. Zeng, J. Y. Wang, L. J. Xu, H. M. Wen and Z. N. Chen, J. Mater. Chem. C, 2016, 4, 6096–6103 RSC.
- X.-F. Tan, P.-P. Wang, L. Lu, O. Bezvikonnyi, D. Volyniuk, J. V. Grazulevicius and Q.-H. Zhao, Dyes Pigm., 2020, 173, 107793 CrossRef CAS.
- I. Marghad, F. Bencheikh, C. Wang, S. Manolikakes, A. Rérat, C. Gosmini, D. H. Kim, J. C. Ribierre and C. Adachi, RSC Adv., 2019, 9, 4336–4343 RSC.
- K. Wang, Y. Z. Shi, C. J. Zheng, W. Liu, K. Liang, X. Li, M. Zhang, H. Lin, S. L. Tao, C. S. Lee, X. M. Ou and X. H. Zhang, ACS Appl. Mater. Interfaces, 2018, 10, 31515–31525 CrossRef CAS.
- J. Gong, J. Han, Q. Liu, X. Ren, P. Wei, L. Yang, Y. Zhang, J. Liu, Y. Dong, Y. Wang, X. Song and B. Z. Tang, J. Mater. Chem. C, 2019, 7, 4185–4190 RSC.
- J. Shi, L. Xu, C. Chen, X. Lv, Q. Ding, W. Li, S. Xue and W. Yang, Dyes Pigm., 2019, 160, 962–970 CrossRef CAS.
- J. Shi, W. Cui, Y. Yu, X. Lv, L. Xu, W. Lang, Q. Sun, Y. Zhang, S. Xue and W. Yang, Dyes Pigm., 2020, 172, 107860 CrossRef CAS.
- Z. Lu, D. Fang, Y. Zheng, Y. Jin and B. Wang, Tetrahedron, 2017, 73, 21–29 CrossRef CAS.
- H. Tanaka, K. Shizu, H. Nakanotani and C. Adachi, J. Phys. Chem. C, 2014, 118, 15985–15994 CrossRef CAS.
- P. Xue, J. Ding, P. Chen, P. Wang, B. Yao, J. Sun, J. Sun and R. Lu, J. Mater. Chem. C, 2016, 4, 5275–5280 RSC.
- A. Endo, K. Sato, K. Yoshimura, T. Kai, A. Kawada, H. Miyazaki and C. Adachi, Appl. Phys. Lett., 2011, 98, 083302 CrossRef.
- S. Xiang, R. Guo, Z. Huang, X. Lv, S. Sun, H. Chen, Q. Zhang and L. Wang, Dyes Pigm., 2019, 170, 107636 CrossRef CAS.
- J. S. Ward, R. S. Nobuyasu, A. S. Batsanov, P. Data, A. P. Monkman, F. B. Dias and M. R. Bryce, Chem. Commun., 2016, 52, 2612–2615 RSC.
- I. H. Lee, W. Song and J. Y. Lee, Org. Electron., 2016, 29, 22–26 CrossRef CAS.
- S. Gan, W. Luo, B. He, L. Chen, H. Nie, R. Hu, A. Qin, Z. Zhao and B. Z. Tang, J. Mater. Chem. C, 2016, 4, 3705–3708 RSC.
- G. Zhang, J. Sun, P. Xue, Z. Zhang, P. Gong, J. Peng and R. Lu, J. Mater. Chem. C, 2015, 3, 2925–2932 RSC.
- Y. Y. Pan, J. Huang, Z. M. Wang, S. T. Zhang, D. W. Yu, B. Yang and Y. G. Ma, RSC Adv., 2016, 6, 108404–108410 RSC.
- S. Zhang, L. Yao, Q. Peng, W. Li, Y. Pan, R. Xiao, Y. Gao, C. Gu, Z. Wang, P. Lu, F. Li, S. Su, B. Yang and Y. Ma, Adv. Funct. Mater., 2015, 25, 1755–1762 CrossRef CAS.
- M. Kolek, F. Otteny, P. Schmidt, C. Mück-Lichtenfeld, C. Einholz, J. Becking, E. Schleicher, M. Winter, P. Bieker and B. Esser, Energy Environ. Sci., 2017, 10, 2334–2341 RSC.
- F. Otteny, G. Studer, M. Kolek, P. Bieker, M. Winter and B. Esser, ChemSusChem, 2020, 13, 2232–2238 CrossRef CAS.
- A. P. Kaur, M. D. Casselman, C. F. Elliott, S. R. Parkin, C. Risko and S. A. Odom, J. Mater. Chem. A, 2016, 4, 5410–5414 RSC.
- J. A. Kowalski, M. D. Casselman, A. P. Kaur, J. D. Milshtein, C. F. Elliott, S. Modekrutti, N. H. Attanayake, N. J. Zhang, S. R. Parkin, C. Risko, F. R. Brushett and S. A. Odom, J. Mater. Chem. A, 2017, 5, 24371–24379 RSC.
- P. Acker, L. Rzesny, C. F. N. Marchiori, C. M. Araujo and B. Esser, Adv. Funct. Mater., 2019, 29, 1906436 CrossRef CAS.
- A. A. Golriz, T. Suga, H. Nishide, R. Berger and J. S. Gutmann, RSC Adv., 2015, 5, 22947–22950 RSC.
- T. Godet-Bar, J. C. Lepretre, O. Le Bacq, J. Y. Sanchez, A. Deronzier and A. Pasturel, Phys. Chem. Chem. Phys., 2015, 17, 25283–25296 RSC.
- M. Kolek, F. Otteny, P. Schmidt, C. Mück-Lichtenfeld, C. Einholz, J. Becking, E. Schleicher, M. Winter, P. Bieker and B. Esser, Energy Environ. Sci., 2017, 10, 2334–2341 RSC.
- B. M. Peterson, D. Ren, L. X. Shen, Y. C. M. Wu, B. Ulgut, G. W. Coates, H. D. Abruna and B. P. Fors, ACS Appl. Energy Mater., 2018, 1, 3560–3564 CrossRef CAS.
- C. Buhrmester, L. Moshurchak, R. L. Wang and J. R. Dahn, J. Electrochem. Soc., 2006, 153, A288 CrossRef CAS.
- L. M. Moshurchak, C. Buhrmester, R. L. Wang and J. R. Dahn, Electrochim. Acta, 2007, 52, 3779–3784 CrossRef CAS.
- S. Ergun, C. F. Elliott, A. P. Kaur, S. R. Parkin and S. A. Odom, Chem. Commun., 2014, 50, 5339–5341 RSC.
- C. Zhang, Z. Niu, S. Peng, Y. Ding, L. Zhang, X. Guo, Y. Zhao and G. Yu, Adv. Mater., 2019, 31, 1901052 CrossRef.
- A. Armin, D. M. Stoltzfus, J. E. Donaghey, A. J. Clulow, R. C. R. Nagiri, P. L. Burn, I. R. Gentle and P. Meredith, J. Mater. Chem. C, 2017, 5, 3736–3747 RSC.
- S. Rousseva, H. den Besten, F. S. van Kooij, E. L. Doting, N. Y. Doumon, E. Douvogianni, L. J. Anton Koster and J. C. Hummelen, J. Phys. Chem. C, 2020, 124, 8633–8638 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2020 |