
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe effect of aromatic ring size in electron deficient semiconducting polymers for n-type organic thermoelectrics†
Maryam
Alsufyani
*a,
Rawad K.
Hallani
*a,
Suhao
Wang
b,
Mingfei
Xiao
c,
Xudong
Ji
d,
Bryan D.
Paulsen
d,
Kai
Xu
b,
Helen
Bristow
e,
Hu
Chen
 a,
Xingxing
Chen
a,
Henning
Sirringhaus
c,
Jonathan
Rivnay
d,
Simone
Fabiano
b and
Iain
McCulloch
ae
a,
Xingxing
Chen
a,
Henning
Sirringhaus
c,
Jonathan
Rivnay
d,
Simone
Fabiano
b and
Iain
McCulloch
ae
aKing Abdullah University of Science and Technology (KAUST), Physical Science and Engineering Division, Thuwal, 23955-6900, Saudi Arabia. E-mail: maryam.alsufyani@kaust.edu.sa; rawad.hallani@kaust.edu.sa
bLaboratory of Organic Electronics, Department of Science and Technology, Linköping University, Norrköping, SE-60174, Sweden
cDepartment of Physics, University of Cambridge, Cambridge CB2 1TN, UK
dNorthwestern University, Department of Biomedical Engineering, 2145 Sheridan Rd, Evanston, IL 60208, USA
eDepartment of Chemistry, Chemistry Research Laboratory, University of Oxford, Oxford, OX1 3TA, USA
First published on 7th August 2020
Abstract
N-type semiconducting polymers have been recently utilized in thermoelectric devices, however they have typically exhibited low electrical conductivities and poor device stability, in contrast to p-type semiconductors, which have been much higher performing. This is due in particular to the n-type semiconductor's low doping efficiency, and poor charge carrier mobility. Strategies to enhance the thermoelectric performance of n-type materials include optimizing the electron affinity (EA) with respect to the dopant to improve the doping process and increasing the charge carrier mobility through enhanced molecular packing. Here, we report the design, synthesis and characterization of fused electron-deficient n-type copolymers incorporating the electron withdrawing lactone unit along the backbone. The polymers were synthesized using metal-free aldol condensation conditions to explore the effect of enlarging the central phenyl ring to a naphthalene ring, on the electrical conductivity. When n-doped with N-DMBI, electrical conductivities of up to 0.28 S cm−1, Seebeck coefficients of −75 μV K−1 and maximum Power factors of 0.16 μW m−1 K−2 were observed from the polymer with the largest electron affinity of −4.68 eV. Extending the aromatic ring reduced the electron affinity, due to reducing the density of electron withdrawing groups and subsequently the electrical conductivity reduced by almost two orders of magnitude.
Introduction
Conjugated polymers play an essential role in the development of next-generation organic electronics.1–3 Recently, they have been heavily exploited in the development of cost-competitive organic thermoelectric devices (OTE) that could be employed to convert waste thermal energy collected from mechanical, chemical, and electrical processes, into electricity.4 Although developing OTE materials is still a work in progress especially for n-type OTE, the low cost, ease of fabrication, and high degree of flexibility offer π-conjugated polymers a potential advantage over other type of thermoelectric materials such as inorganic alloys (i.e., lead telluride and bismuth chalcogenide).5 The figure of merit, ZT, expresses the efficiency of heat conversion to electricity by thermoelectric materials, which is determined by electrical conductivity (σ), Seebeck coefficient (S), and thermal conductivity (k), (ZT = S2σT/k). However, due to the low thermal conductivity that conjugated polymers possess,6 their thermoelectric performance is usually evaluated by the power factor (PF) where PF = S2σ. Since the power factor is proportional to the electrical conductivity, which is dependent on carrier concentration n (cm−3), (maximized upon doping), and carrier mobility μ (cm2 V−1 s−1), a polymer with high electron mobility and an efficient doping process would lead to a boost in thermoelectric performance.Hole conducting polymers have demonstrated high electrical conductivities of over 1000 S cm−1. As a result, their thermoelectric figures of merit (ZT) exhibited high values in the range of 0.2–0.4,7–11 comparable to the state-of-the-art inorganic (TE) materials. However, the conductivity of n-type electron-conducting polymers have been trailing behind their p-type counterparts, with few examples having achieved conductivities of over 1 S cm−1,12–15 This is predominantly arising from not exhibiting a large enough electron affinity (too shallow LUMO energy level) to facilitate electron transfer from n-dopant. A deep LUMO level is required to achieve an energy offset that allows electron transfer from the HOMO level of the dopant to the LUMO level of the polymer, thus facilitating the doping mechanism.16,17 Additionally, a low LUMO level is beneficial towards preventing the common redox reactions of water and oxygen with the doped polymer radical anion which could quench the doped state and lead to a rapid decay in electrical conductivities and irreversible chemical degradation.18–20 A consequence of the synthetic schemes for transition metal mediated aromatic coupling polymerization reactions, is the requirement of an electron rich monomer, which is incorporated along the backbone. This hinders the design of very deep LUMO polymers, with large electron affinities, as it dilutes the positive effect on EA of electron deficient co-repeat units. Therefore, it is desirable to introduce new synthetic strategies to couple acceptor–acceptor (A–A) repeat units to afford unipolar n-type semiconducting polymers with large electron affinities and stable ambient operation.21,22
The strategy most adopted to improve the conductivity of semiconducting polymers is electronic doping. To achieve an effective electron doping process, the LUMO energy level of the semiconductor must be deeper than the HOMO energy level of the dopant.17,23 This thermodynamic requirement in effect narrows the choice of dopant to extremely electron rich molecules, which often suffer from ambient oxidation, thus presenting an additional problem. To help alleviate this constraint, it is desirable therefore to design polymers with as large electron affinities (deep LUMO energy levels) as possible. The charge carrier mobility of semiconducting polymers has been shown to increase with decreasing energetic disorder,24 which in turn decreases with increasing backbone rigidity. The conformationally locked molecular design of a series of previously reported electron deficient aldol condensation polymers, can therefore be exploited to facilitate both electron doping and high electron mobility.17,20,25,26 By selecting the appropriate alkyl side chain size,27,28 or by extending the aryl repeat unit size,29,30 intermolecular close contacts can be modified to explore the effect on charge carrier mobility and energy levels.
Herein, replacement of the bis-oxindole monomer with a more electron withdrawing bis-lactone, to synthesize an alternating lactam–lactone copolymer was expected to further deepen the polymer LUMO energy level relative to all previously reported lactams, of which the deepest LUMO was (−4.2 eV). Within the lactam–lactone backbone, sterically accessible sites are promoted by the aryl rings, which will promote intermolecular contacts and thus potentially enhance charge hopping between chains, to ultimately improve the thermoelectric performance (Fig. 1).
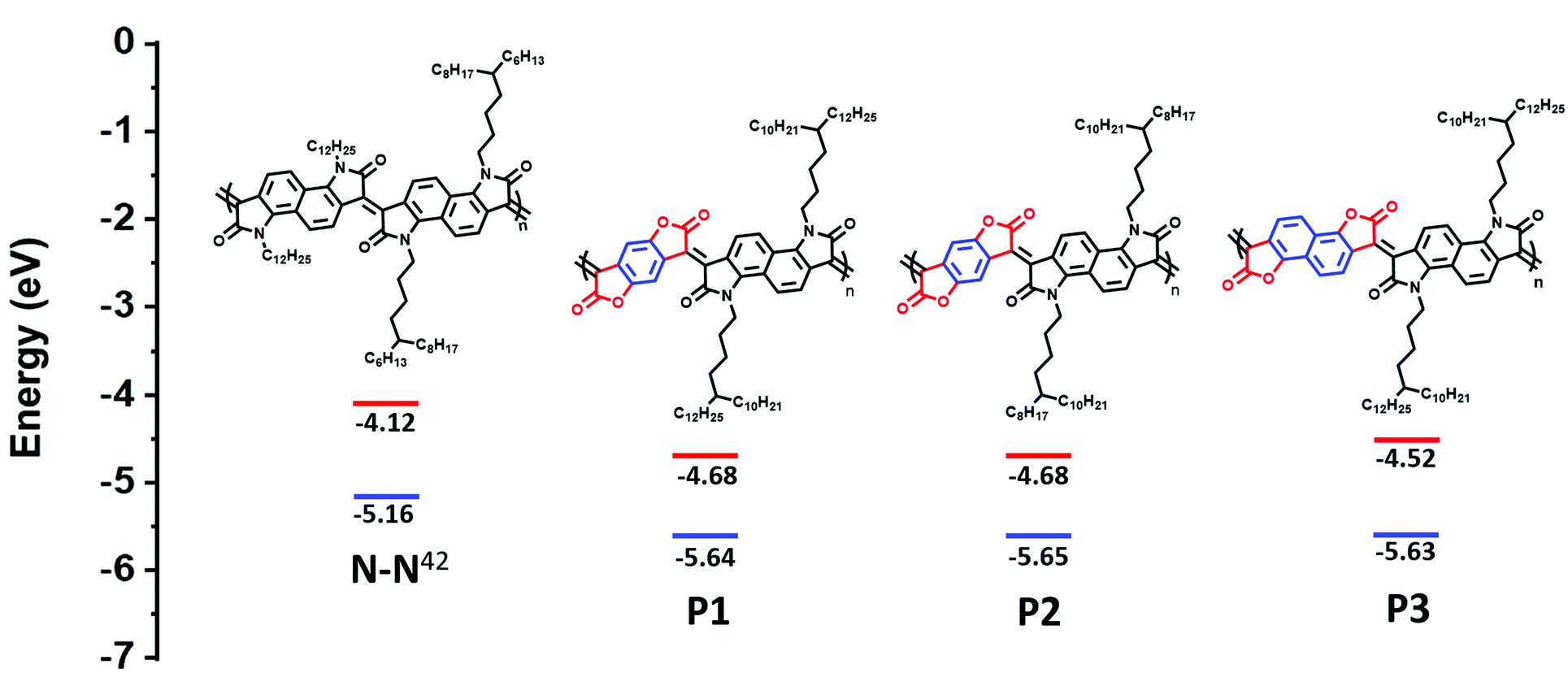 | ||
| Fig. 1 Chemical structures and calculated HOMO/LUMO of previous studied polymer (N–N)42 and new polymers, illustrating the effect of introducing bis-lactone units. HOMO is measured by photoelectron spectroscopy in air (PESA). LUMO is calculated from Eopt.gap and IP. | ||
Results and discussion
Polymers design, synthesis, and characterization
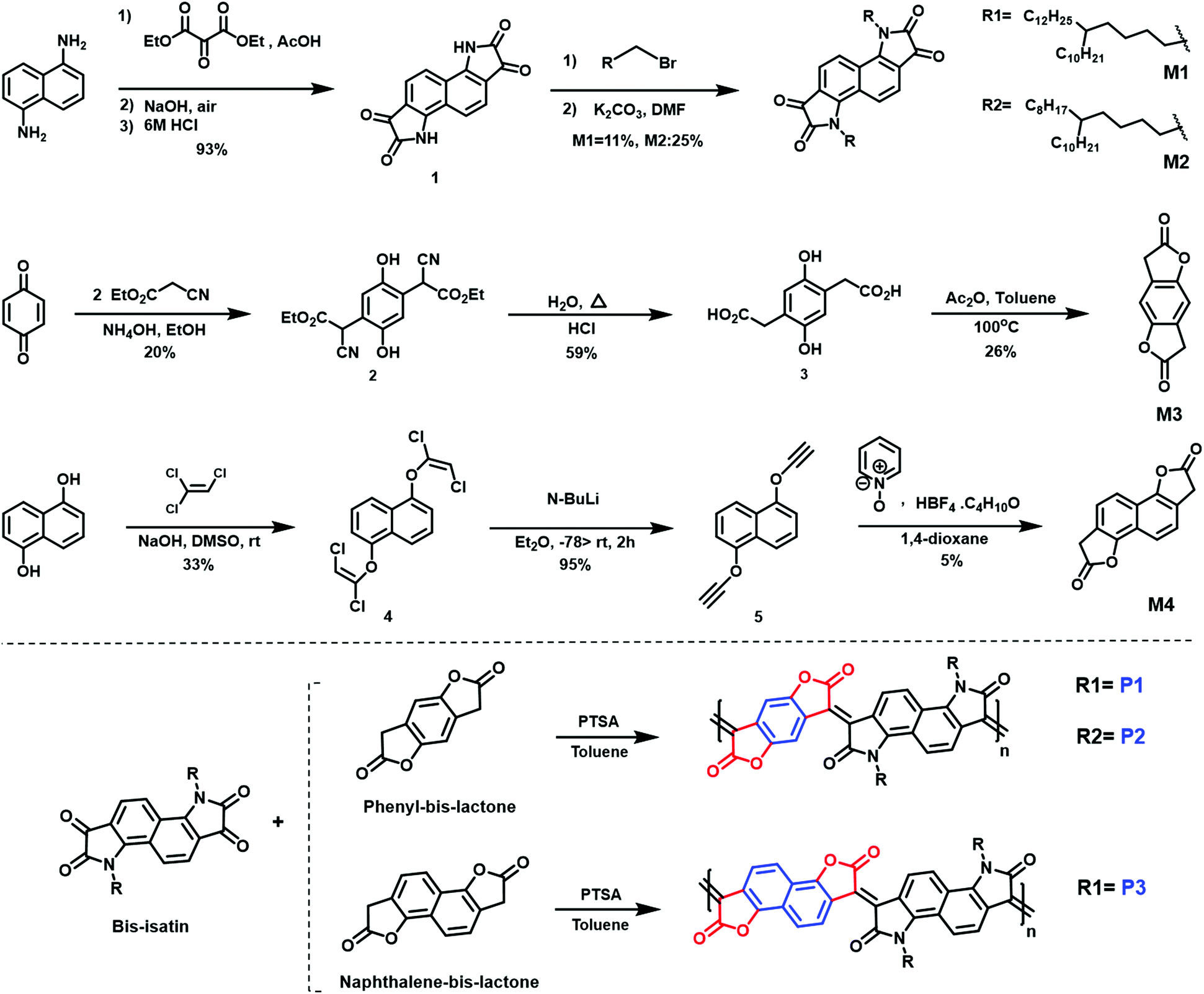 | ||
| Fig. 2 Synthetic approach to monomers (M1, M2, M3, and M4) and polymers (P1, P2, and P3). | ||
Optimizing the doping process depends not only on the electron affinity but also the steric accessibility of the dopant to be effectively accommodated within the polymer microstructure. For polymers P1 and P2, the selection of side chains R1 and R2 involves consideration of polymer solubility, as well the facilitation of intermolecular short contacts by locating the branch points away from the backbone.27 For polymer P3, the phenyl-bis-lactone was replaced with a naphthalene-bis-lactone, anticipating that the enlarged ring size would further promote intermolecular short contacts.30 Through extending the unsubstituted aryl size, it was also expected that the solubility of the resultant polymer would be potentially compromised, hence a longer side chain (R1) was selected.
| Polymer | M n/Mw [kDa] | PDI | IPa [eV] | EAb [eV] | λ (thin film/solution)c [nm] | E opt.gap [eV] | μ e [cm2 V−1 s−1] | σ max [S cm−1] | Seebeck [μV K−1] | PFmax [μW m−1 K−2] |
|---|---|---|---|---|---|---|---|---|---|---|
| a Measured by photoelectron spectroscopy in air (PESA). b Calculated from Eopt.gap and IP. c Thin film and solution absorption onset. d Estimated optical gap calculated using onset of absorption spectra (Eopt.gap = 1240/λonset). e Mobilities measured using a field-effect transistor with a top-gate bottom contact configuration (see ESI, Section S8). | ||||||||||
| P1 | 14/29 | 2.1 | −5.64 | −4.68 | 930/940 | 0.97 | 1 × 10−2 | 0.20 ± 0.05 | −22 ± 4 | 0.12 ± 0.04 |
| P2 | 16/27 | 1.7 | −5.65 | −4.68 | 932/962 | 0.97 | 7 × 10−3 | 0.28 ± 0.01 | −21 ± 5 | 0.16 ± 0.02 |
| P3 | 14/20 | 1.5 | −5.63 | −4.52 | 820/817 | 1.13 | 1 × 10−3 | 0.008 ± 0.001 | −210 ± 14 | 0.034 ± 0.008 |
The thermal properties of the polymers were investigated through thermogravimetric analysis (TGA) and differential scanning calorimetry (DSC). All polymers showed excellent thermal stability with a decomposition temperature of over 350 °C, and no phase transitions in the range from room temperature to 300 °C were observed. ESI† (Section S6 and S7).
Density functional theory (DFT) calculations show that both P1 and P2 polymers exhibited a dihedral angle between the adjacent lactone and lactam cores of (ϕ = 4°), while for P3, the dihedral angle is (ϕ = 9°). (Fig. 3b), as shown in the ESI† (Section S11). As can be seen from the DFT modelling, the replacement of the phenyl ring with naphthalene resulted in an increased backbone twist.
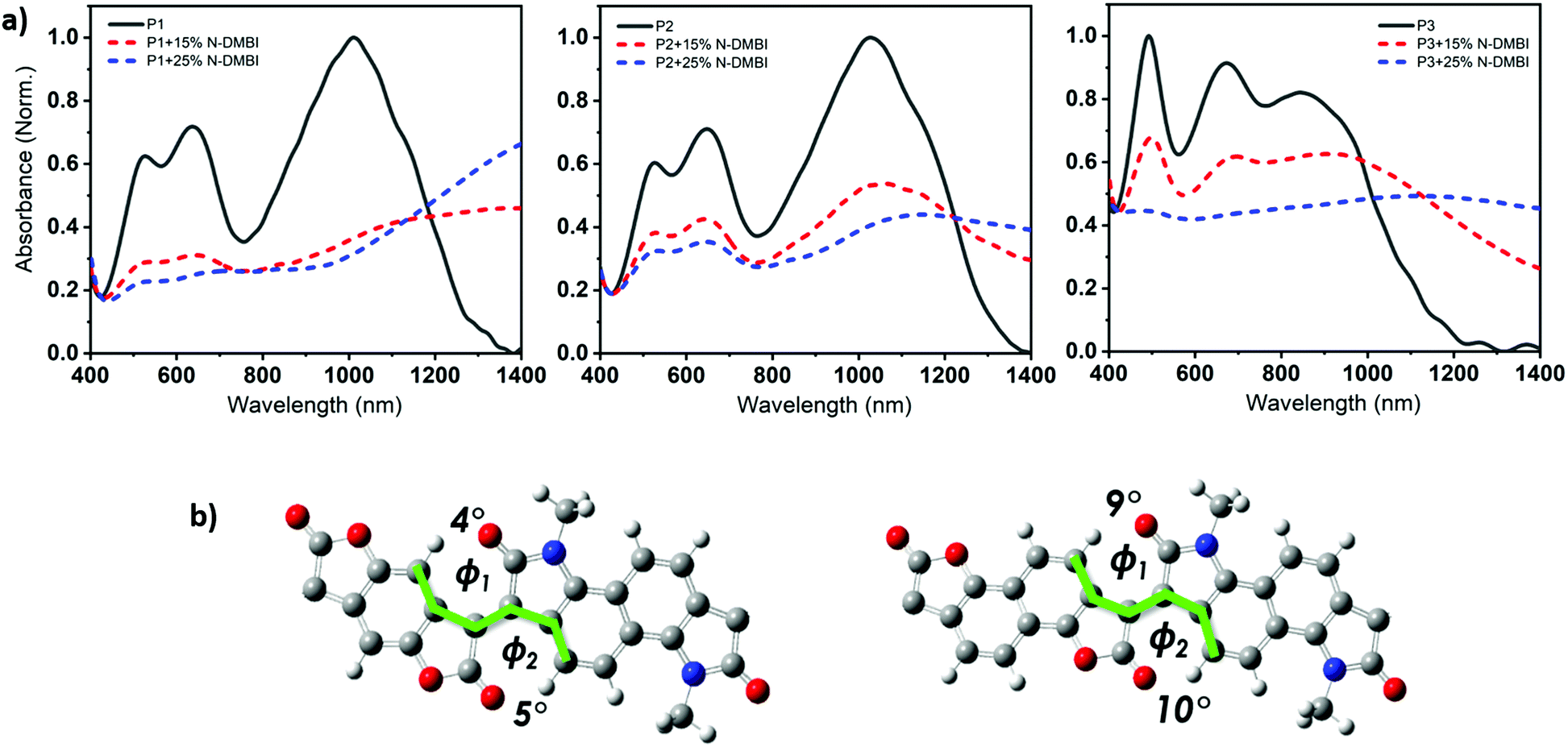 | ||
| Fig. 3 (a) UV-Vis-NIR absorption spectra of the pristine and doped (at 15 and 25 mol% N-DMBI doping ratio) thin films of P1, P2, and P3. (b) DFT-optimized molecular model of the phenyl-bis-lactone and naphthalene-bis-lactone fragments (ωB97XD/6-31G**). | ||
A combination of photo electron spectroscopy in air (PESA) and thin-film absorption spectra (UV-VIS-NIR) were used to determine the energy levels of the polymers. As shown in (Fig. 3a), all pristine polymers exhibit three absorption peaks, two of which are high energy bands in the visible, as well as a broad NIR absorption band. P1 and P2 exhibit similar absorption features, with two absorption peaks in the visible region at 506, 505 nm and 609, 617 nm respectively. Both polymers show a broad NIR with a maximum long-wavelength absorption peaks of 930 and 947 nm respectively. P3, on the other hand featured different absorption characteristics, with two absorption peaks in the visible region of 469 and 647 nm at a shorter wavelength of that of P1 and P2. It also has the shortest wavelength absorption maximum of 820 nm, which could be attributed to backbone twisting that would reduce the pi orbital overlap along the backbone. Upon doping with N-DMBI, the neutral absorption features in the spectrum region at 400–700 nm sharply decrease in intensity, regardless of the polymer. At the same time, the n-doping is accompanied by the rising of new absorption bands at 1100–1200 nm with a tail extending also in the IR region at longer wavelength, which we ascribed to the formation of negative polarons. ESI† (Section S4).
The ionization potential (IP) and the electron affinity (EA) of the polymers are summarized in (Table 1). As indicated, P1 and P2 show similar energy levels with ionization potentials (IP) of −5.65 eV and −5.64 eV, respectively, and an extremely large EA of −4.68 eV, one of the largest reported among all semiconducting polymers. The naphthalene derivative (P3) exhibits an ionization potential (IP) of −5.63 eV, which is slightly lower than that of P1 and P2 (−5.65 eV). However, the electron affinity (EA) of P3 (−4.52 eV) is considerably smaller than that of P1 and P2. This is mainly attributed to the dilution of the density of electron withdrawing lactone units along the conjugated backbone, by increasing the size of the aryl ring. Additionally, having an increased twist in the backbone of P3 will reduce the pi orbital overlap along the backbone and leads to a widening of the bandgap to 1.13 eV, calculated from the absorption spectra, with deepening of the HOMO and a shallowing of the LUMO, and subsequently larger IP and smaller EA. These results are consistent with the absorption spectrum behavior of P3.
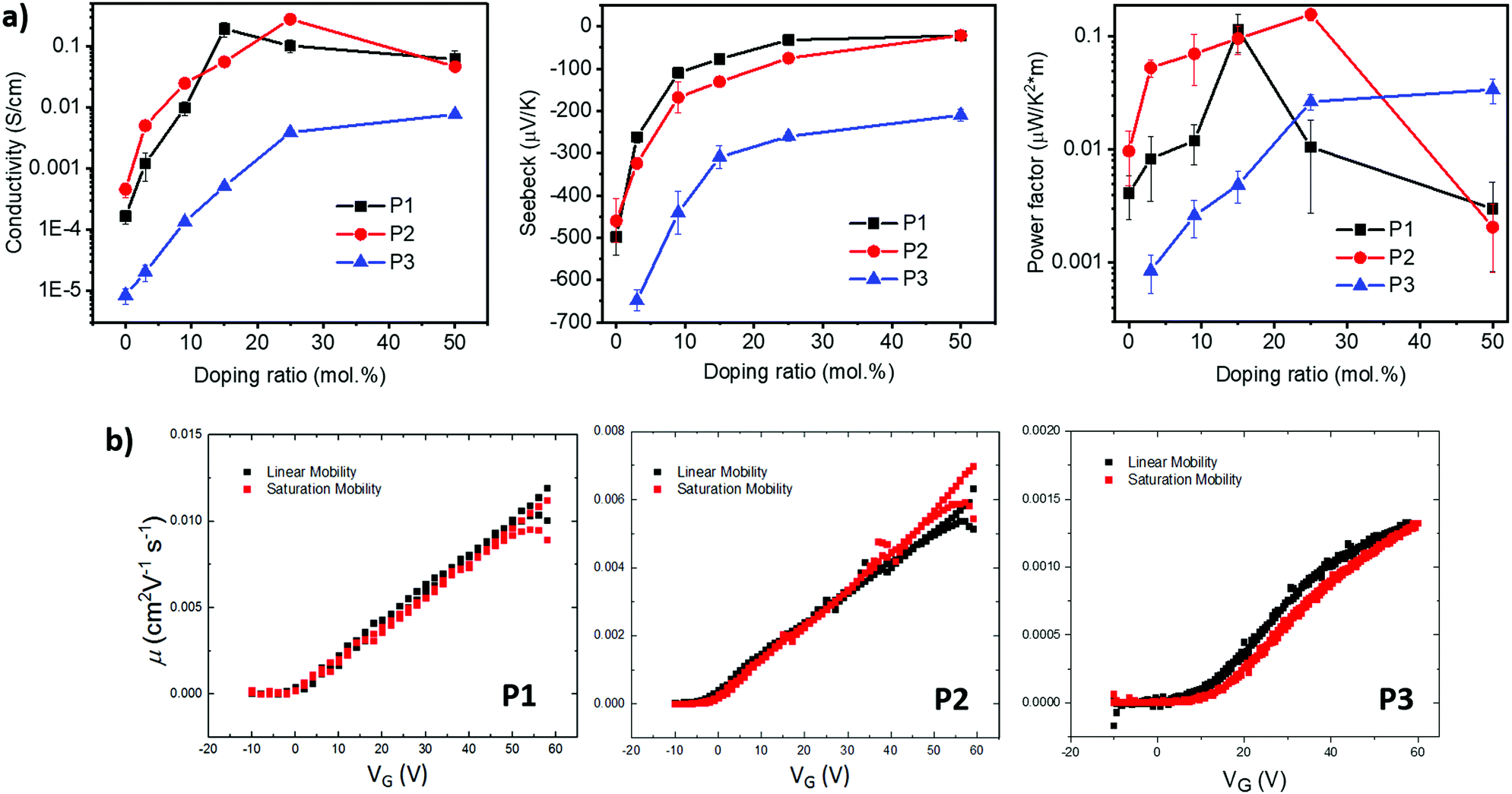 | ||
| Fig. 4 (a) Thermoelectric properties of doped polymers at different doping concentrations including electrical conductivities, Seebeck coefficients and power factors. (b) Transistor properties of pristine P1, P2 and P3. | ||
Grazing-incidence wide-angle X-ray scattering (GIWAXS) were performed to evaluate the molecular packing and the morphologies of the polymer films, (Fig. 5a–c). The lamellar stack (100), π–π stack (010) and amorphous scattering d-spacing were calculated based on the fits of respective peaks from the in-plane (Fig. 5d) and out-of-plane (Fig. 5e) GIWAXS line cuts and listed in Table S1 (ESI†). In general, P1 showed a strong out-of-plane π-scattering with a π–π stacking distances around 3.61 Å while the in-plane π-scattering was relatively weak. The ring-shape scattering pattern observed in the 2D GIWAXS figure represented isotropic amorphous ordering with a characteristic d-spacing of ∼4.7 Å. In addition, multiple orders of lamellar (h00) scattering were present both in and out-of-plane. The lamellar stack d-spacing was calculated to be 31.3 Å from the in-plane (100) peak. The anisotropic nature of the π-scattering of the P1 indicate a predominate crystallite population with a face-on orientation while a minority population with edge-on orientation.
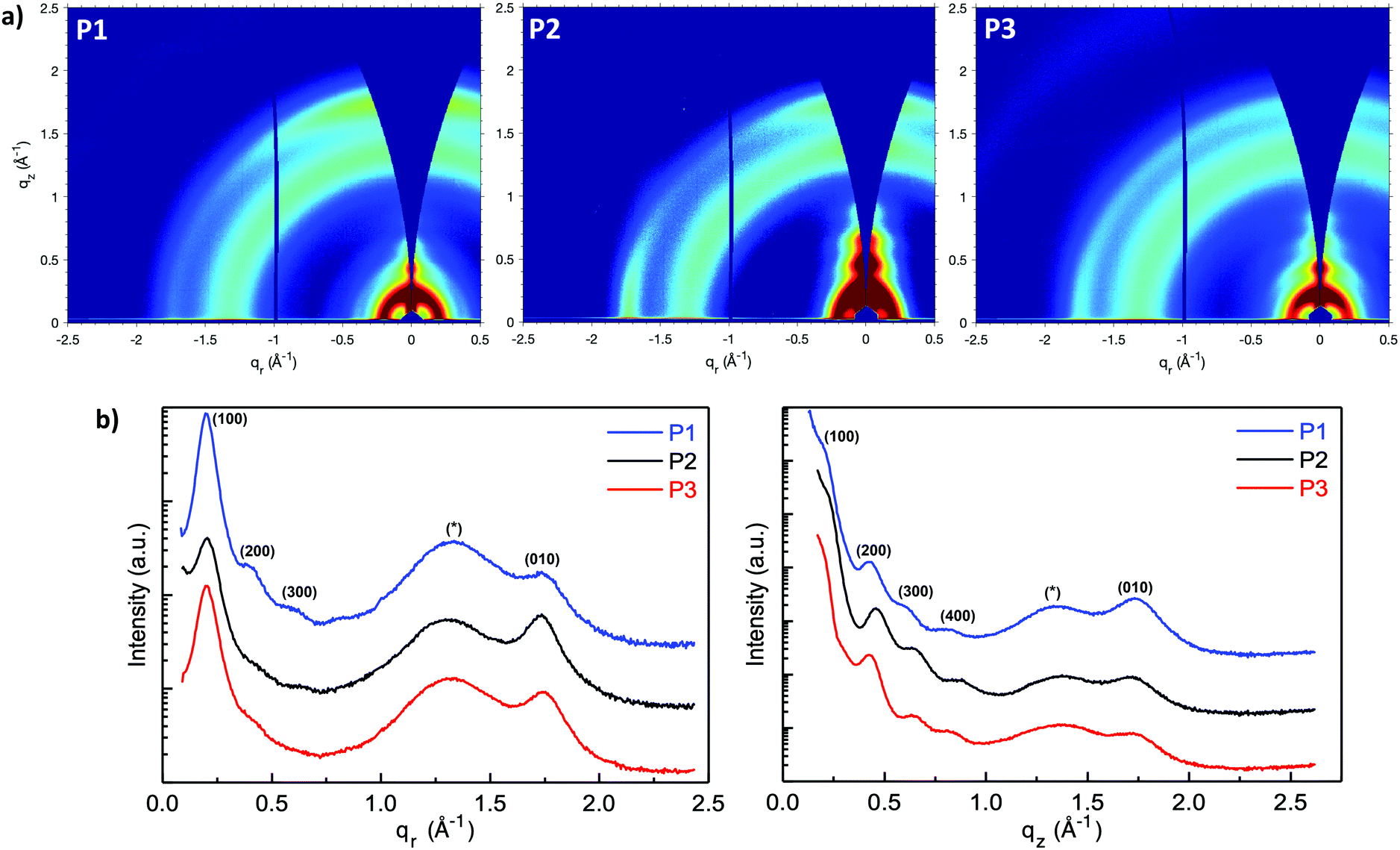 | ||
| Fig. 5 GIWAXS: Two-dimensional grazing incidence X-ray qr–qz scattering map of (a) P1, (b) P2, and (c) P3. Note the π-stack (010) scattering (q ∼ 1.75 Å−1) is strong out of plane for P1, strong both in- and out-of-plane for P2, and completely isotropic for P3. (d) In-plane (qr) and (e) out-of-plane (qz) scattering line cuts from P1, P2, and P3 (offset in intensity for clarity) highlighting the lamellar (h00), π-stack (010), and isotropic amorphous (*) scattering. | ||
Compared with P1, P2 showed stronger in-plane π-scattering, with a π–π stacking distances around 3.63 Å and noticeable out-of-plane π-scattering. Similar to P1, P2 also exhibited as isotropic amorphous scattering ring representing a d-spacing around ∼4.8 Å. In terms of lamellar stacking, four orders of out-of-plane lamellar (h00) scattering were present while only one clear in-plane lamellar (100) scattering was observed that. The decreased lamellar d-spacing (30.7 Å), compared with P1, is consistent with the shorter branched alkyl side chain on the naphthalene-bis-isatin unit of P2. Considering the strong in-plane π-stack scattering, noticeable out-of-plane π-stack scattering and multiple out-of-plane lamellar scattering, P2 is speculated to possess a predominant population of edge-on orientated crystallites and a significant minority population of face-on orientated crystallites. The structure of P1 and P2 was nominally similar to that reported for the all phenyl analogue,41 with pronounced out-of-plane π-stack scattering and an isotropic amorphous scattering ring. However, while increasing the alkyl branch length in the all phenyl analogue diminished ordering and decreased scattering intensity (with an accompanying diminishment in charge transport), here increasing the alkyl branch length did not significantly decrease ordering or scattering intensity, but instead induced increased edge-on texture with minimal change in charge transport.
Comparing P3 with P1, the aryl expansion most dramatically affected film texture. The isotropic (ring-like) π-scattering pattern indicated that crystallite orientation was no longer predominately either edge-on or face-on, as with P1 and P2, but instead randomly oriented. This also contrasted with the all bis-oxindole analogue which lacked a fully isotropic amorphous scattering ring.34 The P3 lamellar scattering was not fully isotropic, indicating more extensive lamellar ordering out-of-plane than in-plane. P3 displayed a similar lamellar d-spacing (31.1 Å) as P1, consistent with their identical side chain architecture. In addition, both the π–π stacking (3.60 Å) and isotropic amorphous scattering (∼4.75 Å) d-spacings of P3 were similar to P1. Beyond the isotropic nature of π-scattering, the intensity of P3 π-scattering intensity was also diminished relative to the amorphous scattering both in- and out-of-plane. This was contrasted with π-scattering that was stronger than amorphous scattering in-plane or out-of-plane, for P1 and P2 respectively.
The weaker and isotropic π–π stacking of the aryl ring extended P3 were coincident with decreased electron mobility and electrical conductivity compared with P1 and P2. The moderate differences in field effect mobility and peak conductivity between P1 and P2 mirrors the similarity in P1 and P2 microstructure, with essentially differ only in ratio edge-on and face-on crystallites. The higher P1 OFET mobility may be in part due to the more uniform orientation of crystallites which could be of benefit in the case of two dimensional charge transport. Likewise, the stronger mix of edge-on and face-one orientated crystallites in P2 may have contributed to the improved peak electrical conductivity as charge transport in doped films follows three dimensional percolated pathways.
Conclusion
New conformationally locked polymers were designed and synthesized, incorporating highly electron deficient bis aryl lactone groups, alternating with alkylated bis-aryl lactam co-repeat units along a conjugated backbone. Derivatives, P1, P2, and P3 that have the deepest LUMO level in all reported lactam rigid-rod building blocks. The partial substitution of the lactam groups with more electron deficient lactone groups along the backbone of conformationally locked aldol condensation polymers, results in extremely electron deficient semiconductors. The large electron affinities facilitated electron transfer from the dopant N-DMBI, achieving conductivities of up to almost 0.3 S cm−1. The polymer conductivity was observed to decrease with decreasing electron affinity, suggesting the doping density was the dominant factor in optimizing conductivity and subsequently the power factor in thermoelectric devices.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The research reported in this publication was supported by funding from King Abdullah University of Science and Technology Office of Sponsored Research (OSR) under awards no. OSR-2018-CARF/CCF-3079, no. OSR-2015-CRG4-2572 and OSR-4106 CPF2019. We acknowledge EC FP7 Project SC2 (610115), EC H2020 (643791), and EPSRC Projects EP/G037515/1, EP/M005143/1, and EP/L016702/1. X. D., B. P., and J. R. gratefully acknowledge support from the National Science Foundation Grant No. NSF DMR-1751308. Special thanks to Joseph Strzalka and Qingteng Zhang for beam line assistance. This research used resources of the Advanced Photon Source, a U.S. Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Argonne National Laboratory under Contract No. DE-AC02-06CH11357. S. F. acknowledges the Swedish Research Council (2016-03979), ÅForsk (18-313, 19-310), Olle Engkvists Stiftelse (204-0256), and the Advanced Functional Materials Center at Linköping University (2009-00971) for financial support.References
- J. Lee, H. F. Chen, T. Batagoda, C. Coburn, P. I. Djurovich, M. E. Thompson and S. R. Forrest, Nat. Mater., 2016, 15, 92–98 CrossRef CAS PubMed
.
- L. Dou, J. You, Z. Hong, Z. Xu, G. Li, R. A. Street and Y. Yang, Adv. Mater., 2013, 25, 6642–6671 CrossRef CAS PubMed
- H. Sirringhaus, Adv. Mater., 2014, 26, 1319–1335 CrossRef CAS PubMed
- B. Russ, A. Glaudell, J. J. Urban, M. L. Chabinyc and R. A. Segalman, Nat. Rev. Mater., 2016, 1, 16050 CrossRef CAS
- J.-L. Brédas, D. Beljonne, V. Coropceanu and J. Cornil, Chem. Rev., 2004, 104, 4971–5004 CrossRef PubMed
- M. Bharti, A. Singh, S. Samanta and D. K. Aswal, Prog. Mater. Sci., 2018, 93, 270–310 CrossRef CAS
- O. Bubnova, Z. U. Khan, A. Malti, S. Braun, M. Fahlman, M. Berggren and X. Crispin, Nat. Mater., 2011, 10, 429–433 CrossRef CAS PubMed
- G. H. Kim, L. Shao, K. Zhang and K. P. Pipe, Nat. Mater., 2013, 12, 719–723 CrossRef CAS PubMed
- Y. Xia, K. Sun and J. Ouyang, Adv. Mater., 2012, 24, 2436–2440 CrossRef CAS PubMed
- N. Kim, S. Kee, S. H. Lee, B. H. Lee, Y. H. Kahng, Y. R. Jo, B. J. Kim and K. Lee, Adv. Mater., 2014, 26(2268–2272), 2109 CrossRef
- S. H. Lee, H. Park, S. Kim, W. Son, I. W. Cheong and J. H. Kim, J. Mater. Chem. A, 2014, 2, 7288–7294 RSC
- C. Y. Yang, W. L. Jin, J. Wang, Y. F. Ding, S. Nong, K. Shi, Y. Lu, Y. Z. Dai, F. D. Zhuang, T. Lei, C. A. Di, D. Zhu, J. Y. Wang and J. Pei, Adv. Mater., 2018, 30, e1802850 CrossRef PubMed
- Y. Wang, M. Nakano, T. Michinobu, Y. Kiyota, T. Mori and K. Takimiya, Macromolecules, 2017, 50, 857–864 CrossRef CAS
- X. Yan, M. Xiong, J. T. Li, S. Zhang, Z. Ahmad, Y. Lu, Z. Y. Wang, Z. F. Yao, J. Y. Wang, X. Gu and T. Lei, J. Am. Chem. Soc., 2019, 141, 20215–20221 CrossRef CAS PubMed
- K. Shi, F. Zhang, C. A. Di, T. W. Yan, Y. Zou, X. Zhou, D. Zhu, J. Y. Wang and J. Pei, J. Am. Chem. Soc., 2015, 137, 6979–6982 CrossRef CAS PubMed
- D. Huang, H. Yao, Y. Cui, Y. Zou, F. Zhang, C. Wang, H. Shen, W. Jin, J. Zhu and Y. Diao, J. Am. Chem. Soc., 2017, 139, 13013–13023 CrossRef CAS PubMed
- Y. Lu, J.-Y. Wang and J. Pei, Chem. Mater., 2019, 31, 6412–6423 CrossRef CAS
- D. De Leeuw, M. Simenon, A. Brown and R. Einerhand, Synth. Met., 1997, 87, 53–59 CrossRef CAS
- M. L. Tietze, B. D. Rose, M. Schwarze, A. Fischer, S. Runge, J. Blochwitz-Nimoth, B. Lüssem, K. Leo and J.-L. Brédas, Adv. Funct. Mater., 2016, 26, 3730–3737 CrossRef CAS
- S. Wang, H. Sun, T. Erdmann, G. Wang, D. Fazzi, U. Lappan, Y. Puttisong, Z. Chen, M. Berggren, X. Crispin, A. Kiriy, B. Voit, T. J. Marks, S. Fabiano and A. Facchetti, Adv. Mater., 2018, 30, e1801898 CrossRef PubMed
- Z. Yuan, B. Fu, S. Thomas, S. Zhang, G. DeLuca, R. Chang, L. Lopez, C. Fares, G. Zhang, J.-L. Bredas and E. Reichmanis, Chem. Mater., 2016, 28, 6045–6049 CrossRef CAS
- D. Di Nuzzo, C. Fontanesi, R. Jones, S. Allard, I. Dumsch, U. Scherf, E. von Hauff, S. Schumacher and E. Da Como, Nat. Commun., 2015, 6, 6460 CrossRef PubMed
- B. D. Naab, S. Guo, S. Olthof, E. G. Evans, P. Wei, G. L. Millhauser, A. Kahn, S. Barlow, S. R. Marder and Z. Bao, J. Am. Chem. Soc., 2013, 135, 15018–15025 CrossRef CAS PubMed
- D. Venkateshvaran, M. Nikolka, A. Sadhanala, V. Lemaur, M. Zelazny, M. Kepa, M. Hurhangee, A. J. Kronemeijer, V. Pecunia and I. Nasrallah, Nature, 2014, 515, 384–388 CrossRef CAS PubMed
- S. Wang, H. Sun, U. Ail, M. Vagin, P. O. Persson, J. W. Andreasen, W. Thiel, M. Berggren, X. Crispin, D. Fazzi and S. Fabiano, Adv. Mater., 2016, 28, 10764–10771 CrossRef CAS PubMed
- P. Prins, F. C. Grozema, J. M. Schins, S. Patil, U. Scherf and L. D. Siebbeles, Phys. Rev. Lett., 2006, 96, 146601 CrossRef CAS PubMed
- I. Meager, R. S. Ashraf, S. Mollinger, B. C. Schroeder, H. Bronstein, D. Beatrup, M. S. Vezie, T. Kirchartz, A. Salleo, J. Nelson and I. McCulloch, J. Am. Chem. Soc., 2013, 135, 11537–11540 CrossRef CAS PubMed
- T. Lei, J. H. Dou and J. Pei, Adv. Mater., 2012, 24, 6457–6461 CrossRef CAS PubMed
- H. Chen, M. Hurhangee, M. Nikolka, W. Zhang, M. Kirkus, M. Neophytou, S. J. Cryer, D. Harkin, P. Hayoz and M. Abdi-Jalebi, Adv. Mater., 2017, 29, 1702523 CrossRef PubMed
- T. H. Thomas, D. J. Harkin, A. J. Gillett, V. Lemaur, M. Nikolka, A. Sadhanala, J. M. Richter, J. Armitage, H. Chen, I. McCulloch, S. M. Menke, Y. Olivier, D. Beljonne and H. Sirringhaus, Nat. Commun., 2019, 10, 2614 CrossRef PubMed
- N. M. Randell, P. C. Boutin and T. L. Kelly, J. Mater. Chem. A, 2016, 4, 6940–6945 RSC
- P. Singla, N. Van Steerteghem, N. Kaur, A. Z. Ashar, P. Kaur, K. Clays, K. S. Narayan and K. Singh, J. Mater. Chem. C, 2017, 5, 697–708 RSC
- Y. Deng, B. Sun, Y. He, J. Quinn, C. Guo and Y. Li, Chem. Commun., 2015, 51, 13515–13518 RSC
- A. Onwubiko, W. Yue, C. Jellett, M. Xiao, H. Y. Chen, M. K. Ravva, D. A. Hanifi, A. C. Knall, B. Purushothaman, M. Nikolka, J. C. Flores, A. Salleo, J. L. Bredas, H. Sirringhaus, P. Hayoz and I. McCulloch, Nat. Commun., 2018, 9, 416 CrossRef PubMed
- P. Wei, J. H. Oh, G. Dong and Z. Bao, J. Am. Chem. Soc., 2010, 132, 8852–8853 CrossRef CAS PubMed
- E. Hasegawa, H. Hirose and K. Sasaki, Heterocycles, 2009, 77, 1147–1161 CrossRef CAS
- X.-Q. Zhu, M.-T. Zhang, A. Yu, C.-H. Wang and J.-P. Cheng, J. Am. Chem. Soc., 2008, 130, 2501–2516 CrossRef CAS PubMed
- R. A. Schlitz, F. G. Brunetti, A. M. Glaudell, P. L. Miller, M. A. Brady, C. J. Takacs, C. J. Hawker and M. L. Chabinyc, Adv. Mater., 2014, 26, 2825–2830 CrossRef CAS PubMed
- D. Kiefer, A. Giovannitti, H. Sun, T. Biskup, A. Hofmann, M. Koopmans, C. Cendra, S. Weber, L. J. Anton Koster, E. Olsson, J. Rivnay, S. Fabiano, I. McCulloch and C. Muller, ACS Energy Lett., 2018, 3, 278–285 CrossRef CAS PubMed
-
G. J. Snyder and E. S. Toberer, Materials for sustainable energy: a collection of peer-reviewed research and review articles from Nature Publishing Group, World Scientific, 2011, pp. 101–110 Search PubMed
- Y. Lu, Z. D. Yu, R. Z. Zhang, Z. F. Yao, H. Y. You, L. Jiang, H. I. Un, B. W. Dong, M. Xiong, J. Y. Wang and J. Pei, Angew. Chem., Int. Ed., 2019, 58, 11390–11394 CrossRef CAS PubMed
- H. Chen, M. Moser, S. Wang, C. Jellet, K. Thorley, G. Harrison, B. Purushothaman, M. Alsufyani, H. Bristow, A. Wadsworth, S. Fabiano and I. McCulloch, under submission.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0tc03347b |
| This journal is © The Royal Society of Chemistry 2020 |