
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceProlonged and efficient near-infrared photoluminescence of a sensitized organic ytterbium-containing molecular composite†
Chen
Lyu
 a,
Hongfei
Li
a,
Peter B.
Wyatt
b,
William P.
Gillin
*ac and
Huanqing
Ye
*ac
a,
Hongfei
Li
a,
Peter B.
Wyatt
b,
William P.
Gillin
*ac and
Huanqing
Ye
*ac
aMaterials Research Institute and School of Physics and Astronomy, Queen Mary University of London, Mile End Road, London E1 4NS, UK. E-mail: h.ye@qmul.ac.uk; w.gillin@qmul.ac.uk
bMaterials Research Institute and School of Biological and Chemical Sciences, Queen Mary University of London, Mile End Road, London E1 4NS, UK
cChromosol Ltd, The Walbrook Building, 25 Walbrook, London, EC4N 8A, UK
First published on 1st July 2020
Abstract
Organic prolonged luminescent materials have attracted attention with various candidates reported providing both UV and visible emission for applications in bio-imaging, light storage, security, etc. However, there is a lack of prolonged near-infrared (NIR) emitting materials, which are highly desirable for many of the proposed applications. NIR-emitting organic lanthanide(III) molecules have suitable wavelengths, but all of them are unsuitable for persistent luminescence due to weak sensitization and the restricted intrinsic ion radiative lifetimes. Herein, this paper demonstrates efficiently sensitized organic Yb(III) compounds with prolonged emission lifetimes up to ∼0.3 s at 1 μm, far exceeding the intrinsic Yb3+ emission lifetime of ∼1 ms. The dynamic equilibrium is studied to demonstrate that this prolonged emission is caused by energy transfer from long-lived organic triplet excitons. Experiment and simulation results suggest a new route to develop bright and ultralong-lived 1 μm emitting materials that could be coupled with other existing organic persistent luminescence materials to shift their emission in to the infrared.
There has been considerable interest in the engineering of inter- or intra-molecular interactions to trap triplet excitons and to use this to demonstrate organic prolonged phosphorescence.1–7 Since triplet excitons in organics mainly result from inter-system crossing (ISC) with a small energy separation from the triplets, the reported phosphoresce usually only red-shifts in the visible, e.g. to ∼500 nm to 600 nm compared to UV-visible fluorescence centred at a wavelength of ∼300 nm to 400 nm.4–6 However, this UV-visible luminescence is not ideal for many of the proposed applications and NIR light, at a wavelength of ∼1 μm (energy of ∼1.1 eV), would be preferred in many biological applications, due to the high transmission in biological systems at this wavelength and in anti-counterfeiting applications as NIR is not visible to the naked eye.8–15
Organic Yb(III) materials with an organic chromophore that strongly absorbs light to sensitize Yb3+ ions can give bright and sharp 1 μm emission under low-power photoexcitation. Various organic Yb(III) complexes or hybrid organic-conjugated Yb3+-doped nanoparticles have been demonstrated to provide sensitized Yb3+ luminescence.16,17 However, a general problem is that Yb3+ emission can be severely quenched by high-energy organic vibrations, e.g. C–H, N–H or O–H bonds, and this causes luminescence lifetimes as short as nanoseconds to microseconds with poor efficiencies of <0.1%.18,19 Even though there are effective protective methods to eliminate quenching, the sensitized Yb3+ emission lifetime is capped by the intrinsic radiative lifetime which is usually shorter than 2 ms and far from the demands of persistent PL.
Our approach utilizes an organic chromophore to sensitize an organic Yb(III) molecule in a molecular composite thin film. The organic Yb(III) molecule we use is a chelate, Yb(F-TPIP)3 that has a Yb3+ ion incorporated with tetrakis-(pentafluorophenyl)imidodiphosphinate, F-TPIP− ligands, and it is inert to visible light.20 The organic chromophore we employ is the zinc salt of 2-(tetrafluoro-2-hydroxyphenyl)tetrafluorobenzothiazole, Zn(F-BTZ)2, which has strong absorption of blue light.21 The chemical properties of the two individual materials have been well studied in previous work.20,21
The 405 nm light photoexcites Zn(F-BTZ)2 to give the Zn(F-BTZ)2 emission broadly spanning from ∼450 nm to 900 nm and sensitizes the sharp Yb3+ emission centred at 975 nm due to the Yb3+:2F5/2 → 2F7/2 transition, shown in Fig. 1. The excitation spectrum for the Yb3+ emission recorded at 1005 nm matches features of the Zn(F-BTZ)2 excitation spectrum at 500 nm, indicating a sensitization effect of Zn(F-BTZ)2 onto Yb3+ excitations (i.e. light is being absorbed by the Zn(F-BTZ)2 and the energy is then transferred to the Yb3+ ions from which it is emitted). Previous studies22 have shown abundant triplet excitons in Zn(F-BTZ)2 and its phosphorescence is centred around 550 nm extending to the NIR beyond 900 nm. This implies the phosphorescence band slightly overlaps the Yb3+ absorption which makes the sensitization from the triplet states possible, probably explaining the observation that higher Yb(F-TPIP)3 doping concentrations selectively quench the phosphorescence to narrow the FWHM of the Zn(F-BTZ)2 emission (Fig. 1).
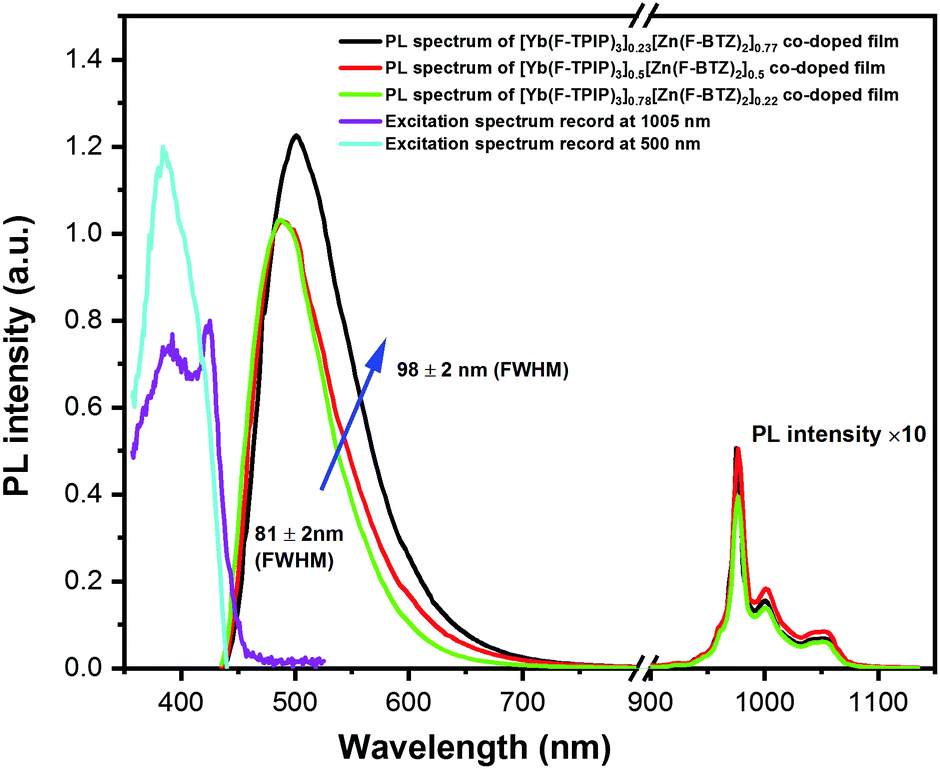 | ||
| Fig. 1 PL spectra of co-doped films of [Yb(F-TPIP)3]x[Zn(F-BTZ)2]1−x (x = 0.23, 0.5, 0.78) and the excitation spectrum of a co-doped film of [Yb(F-TPIP)3]0.5[Zn(F-BTZ)2]0.5. The PL spectra (black, red and green lines) were measured at photoexcitation using a 405 nm laser. The excitation spectra (violet line and light blue line) were recorded at an emission wavelength of 1005 nm and 500 nm, respectively. Both the excitation and PL spectra are optically corrected by the responsivity of photon detectors and spectrometer gratings. | ||
Fig. 2(a) shows the time-resolved PL (TRPL) of excited Yb3+ ions recorded at 975 nm. The directly excited Yb3+ emission lifetime is 0.46 ± 0.12 ms, indicating an internal quantum efficiency of 46%, using a reported intrinsic Yb3+ radiative lifetime of ∼1 ms in Yb(F-TPIP)3.23 The sensitized Yb3+ emission lifetime becomes distinctly longer than those obtained by direct excitation into the Yb3+:2F5/2 level. Four lifetimes are obtained by fitting with their component percentages. The shortest lifetime (τ1) of 0.22 ± 0.11 ms with a component percentage of 6.8% is believed to result from Yb3+ ions that are close to residual quenching centres in the film. The longer lifetime (τ2) of 0.68 ± 0.15 ms (24.83%) corresponds to Yb3+ ions distant from quenching centres. The information about quenching centres and protective techniques is discussed in the ESI.† These demonstrate that the film contains at least two quenching environments, which lead to corresponding internal quantum efficiencies (IQEs) of 21% and 68%, respectively. Interestingly, the lifetime (τ3) of 2.22 ± 0.5 ms (26.59%) and the longest lifetime (τ4) of 10.2 ± 1.5 ms (45.7%) are considerably prolonged compared to that for direct excitation and exceed the intrinsic Yb3+ radiative lifetime (1 ms). Also, these two prolonged lifetimes are comparable to the long phosphorescence lifetimes of 2.2 ± 0.4 ms and 11.9 ± 2.7 ms for the triplet emission at 600 nm (used to reduce the singlet contribution to the decay curve) (Fig. S7 in the ESI†), which implies that energy transfer from the triplets is probably responsible.
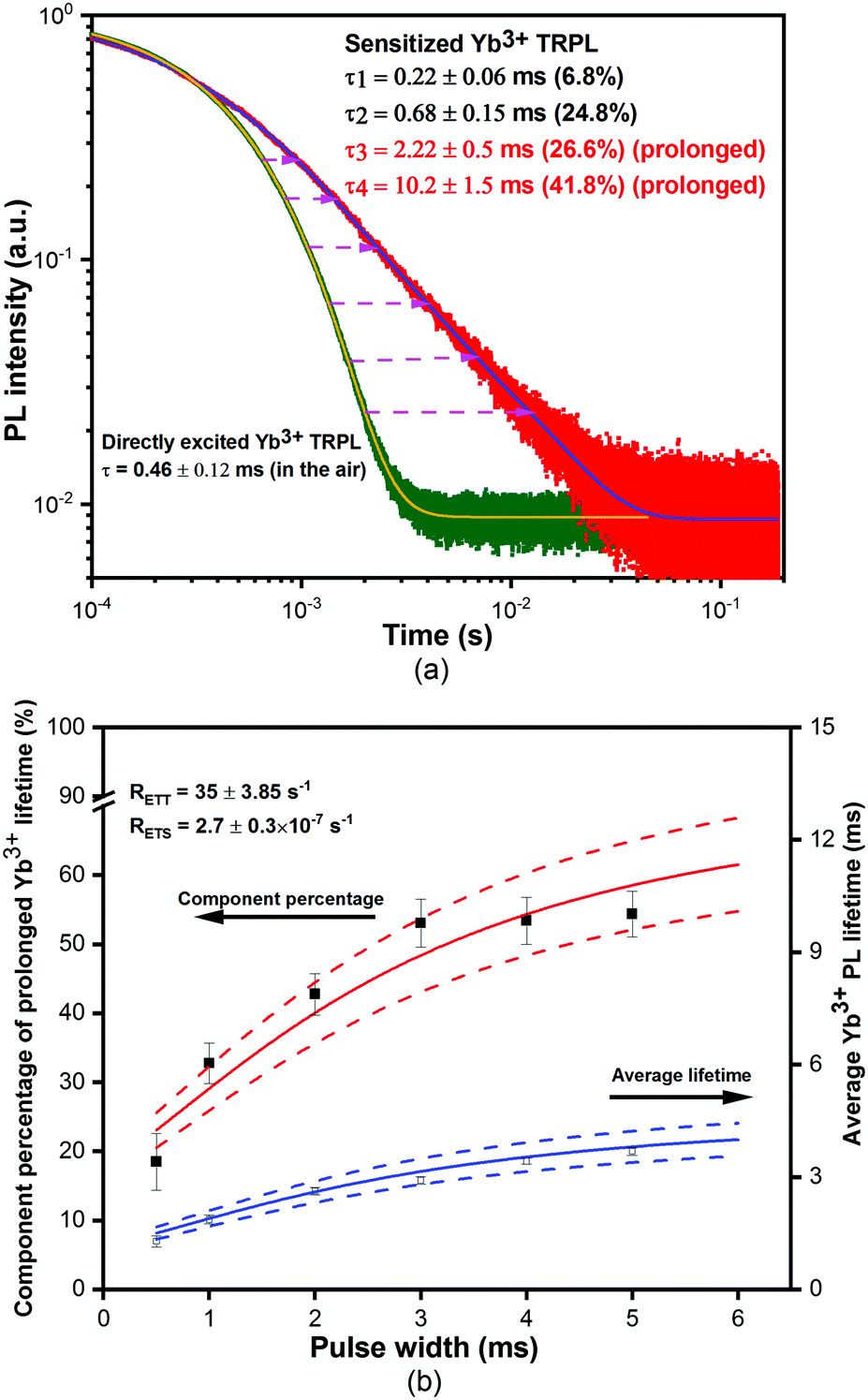 | ||
| Fig. 2 (a) Directly excited and sensitized time-resolved room-temperature PL spectra of the co-doped film of [Yb(F-TPIP)3]0.5[Zn(F-BTZ)2]0.5. The green dots show the directly excited TRPL of the composite film and the red dots show the sensitized TRPL. The orange and blue curves are obtained by fitting the decay traces using exponential decay functions. (b) The component percentages of the prolonged Yb3+ PL lifetimes (solid squares) and the average lifetime of Yb3+ PL (open squares) evolved with photoexcitation pulse widths. The simulated traces for the component percentages and the average Yb3+ PL lifetimes are presented by a red and blue curve, respectively. The error bars of the simulation are given by the dashed lines. | ||
To further investigate those prolonged Yb3+ emission lifetimes, we varied the pulse-width of a 405 nm laser photoexcitation to manipulate the percentages of the four lifetime components, shown in Fig. 2(b). With a longer pulse width, the population of triplet states around each Yb(F-TPIP)3 molecule will accumulate to increase the probability to sensitize the Yb3+ ion. Consequently, the total percentage of the prolonged lifetimes of τ3 and τ4, which appear to be relevant to triplet states, increases with longer pulse widths. The increase in the total percentage makes the average lifetime for the obtained Yb3+ emission become longer with extending pulse width. We resolved a set of rate equations to understand the relationship between the prolonged Yb3+ excitations and triplet states. The construction and deduction of rate equations contain two different time regimes. More details can be found in the ESI.†
The calculation shows that the decay rate for the Yb3+:2F5/2 level is a combination of both the triplet decay rate and the intrinsic decay rate of excited Yb3+ ions. The e−(RETT+RT)×t2 term (see eqn (S17) in the ESI†) is a decay process for the Yb3+ emission that follows the decay rate of the Zn(F-BTZ)2 triplet states. This explains why those prolonged Yb3+ emission lifetimes are similar to the persistent phosphorescence lifetimes of the triplet states. As mentioned above, the presence of either Yb(F-TPIP)3 or Y(F-TPIP)3 makes no difference to the phosphorescence lifetimes within the range of the experimental error bars. Hence, it is reasonable to use the maximum value of the error bars to estimate the ET rate, which is RETT = ∼35 s−1 (see Fig. S7 in the ESI†). Moreover, the calculation can give the average Yb3+ emission lifetime as a function of the photoexcitation pulse width which is shown as the solid lines in Fig. 2(b), with the calculated time-resolved component percentage of the prolonged Yb3+ emission lifetime. The simulation is seen to match the experimental data.
We have used the spectral overlap integral between the triplet phosphorescence spectrum and the Yb3+ absorption spectrum to estimate a Förster resonant energy transfer (FRET) rate, RFRET, which is a conventional method to quantify the Förster energy transfer. The detail of the FRET rate calculation can be found in the ESI.† There are always considerable uncertainties in the parameters used in FRET calculations, not least including the donor quantum yield, orientation factor and the donor–acceptor separation. However, by taking reasonable estimations of the quantum yield as 10%, and the donor–acceptor distance from 7 Å (the radius of single Yb(F-TPIP)3 molecule) to 10 Å (the maximum working distance of FRET),20 the calculated RFRET varies from 64 s−1 to 5 s−1, which are in agreement with our RETT. It is noted that eqn (S21) and (S22) in the ESI† indicate the quantum yield of the energy donor QD would only linearly scale the value of RFRET. Hence, as limited by the maximum and minimum value of QD, the value of RFRET is consistent with RETT to within one order of magnitude.
We present two possible processes to explain the prolonged Yb3+ excitations. Firstly, since the low ET rate keeps triplet excitons long-lived, if an excited Yb3+ ion relaxes to the ground state, it is possible for a neighbouring triplet exciton to re-excite that de-excited Yb3+ ion. Also, long-lived triplet excitons can diffuse via intermolecular interactions among Zn(F-BTZ)2 molecules to reach such an Yb3+ ion. These two routes maintain the population level of excited Yb3+ ions to overcome the intrinsic Yb3+ decay rate.
We have also measured the temperature dependent sensitized-Yb3+ TRPL at 975 nm in a co-doped film of [Yb(F-TPIP)3]0.5[Zn(F-BTZ)]0.5. The contour-colour display of the spectra is shown in Fig. 3(a). It can be clearly seen that prolonged Yb3+ emission becomes dominant with components of 334.5 ± 30.1 ms (87.9%) with 74.4 ± 5.9 ms (11.3%) at 80 K. The values of the fitted lifetimes and their component percentages are shown in Table S3 (ESI†). This prolonged Yb3+ PL implies that a triplet quenching process is suppressed at low temperature. We believe this process is thermally activated reverse-intersystem-crossing (RISC). Using the simple rate model shown in the inset to Fig. 3(b) we can fit the data and obtain an energy gap of 0.17 ± 0.02 eV between the triplet state (T1) and the singlet state (S1) of Zn(F-BTZ)2. The fitted energy gap is of the order of the singlet–triplet energy separation in the chromophore. The result implies the use of a chromophore material with a larger singlet–triplet energy separation would provide a route to exceptionally prolonged room temperature Yb3+ PL lifetimes using this approach.
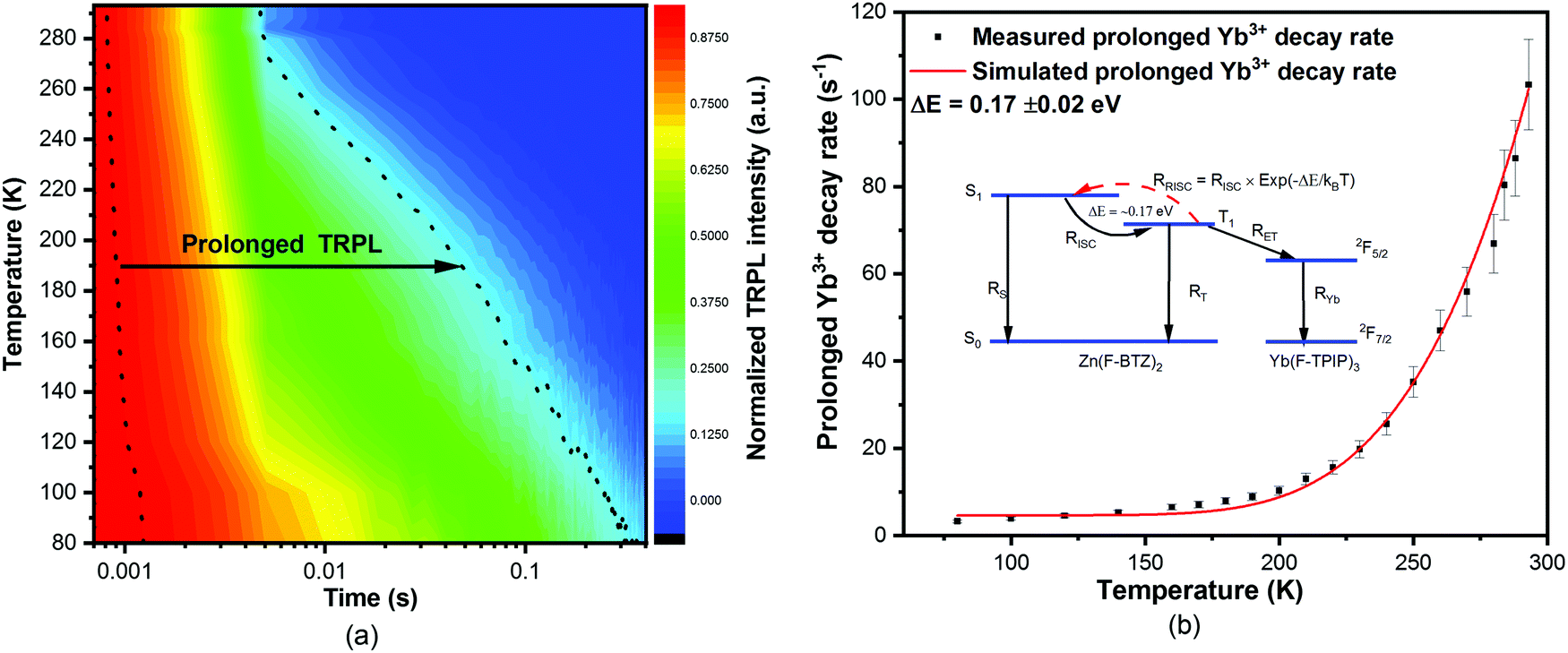 | ||
| Fig. 3 (a) Temperature dependent sensitized Yb3+ TRPL of a co-doped film of [Yb(F-TPIP)3]0.5[Zn(F-BTZ)2]0.5 at 975 nm. (b) The measured and simulated temperature-dependent prolonged Yb3+ decay rates, which resulted from the introduction of a reverse ISC process into the analytical solution of the rate equations shown in the ESI.† | ||
Conclusions
In summary, composite thin films are fabricated to produced sensitized Yb3+ 1 μm photoluminescence. Sensitized time-resolved photoluminescence measurement indicates that the sensitized Yb3+ lifetime is prolonged to exceed its intrinsic lifetime. Simulation of excited state dynamic suggests the prolonged lifetime is due to the energy transfer from long-lived triplet. Additionally, our work demonstrates a potential route to develop bight and ultra-long lived 1 μm emitting material that could be coupled to some known persist luminescence materials to shift their emission wavelength to infrared.Methods
Details of methods, materials and optical measurement techniques can be found in the ESI.† HFTPIP and HFBTZ ligands were supplied by Chromosol Ltd. YbCl3·(H2O)6 (metals basis 99.99% purity) was purchased from Alfa Aesar. Yb3+ salt and the HFTPIP ligand were reacted in methanol:ethanol solution to produce Yb(F-TPIP)3 precipitates. KOH and the HFBTZ ligand were used to form KFBTZ and then reacted with a Zn2+ salt to precipitate Zn(F-BTZ)2. The reaction condition to make Yb(F-TPIP)3 and Zn(F-BTZ)2 are routine and have been reported.20,21 A train vacuum purification system was used to purify the materials before the use. The display of Fig. 2(a) uses logarithmic scales for both the x-axis and y-axes, to fully visualize the details of the decay curves over a wide range time regime, which cannot be seen with a linear x-axis. The composite of two materials in thin films are fabricated using a co-evaporation technique using a Kurt J. Lesker SPECTROS deposition system in a 10 K-class cleanroom. A Hamamatsu R5509-72 nitrogen-cooled detector with a Horiba Triax 550 spectrometer was used to collect and detect photoluminescence signals. Multiple exponential functions are used to fit time-resolved decay curves to obtain TRPL lifetimes. All the film samples were covered by a 100 nm layer of thermally grown aluminium film. The low temperature measurements were taken using an Oxford Instrument continuous flow cryostat.Conflicts of interest
There are no conflicts to declare.Acknowledgements
CL and HL are financially supported by the China Scholarship Council and Queen Mary University of London. WPG acknowledges financial support from EPSRC (EP/L020114/1 and EP/P007767/1).Notes and references
- Y. Su, S. Z. F. Phua, Y. Li, X. Zhou, D. Jana, G. Liu, W. Q. Lim, W. K. Ong, C. Yang and Y. Zhao, Sci. Adv., 2018, 4, eaas9732 CrossRef PubMed
.
- S. Xu, R. Chen, C. Zheng and W. Huang, Adv. Mater., 2016, 28, 9920–9940 CrossRef CAS PubMed
- Q. Miao, C. Xie, X. Zhen, Y. Lyu, H. Duan, X. Liu, J. V. Jokerst and K. Pu, Nat. Biotechnol., 2017, 35, 1102 CrossRef CAS PubMed
- L. Gu, H. Shi, L. Bian, M. Gu, K. Ling, X. Wang, H. Ma, S. Cai, W. Ning and L. Fu, Nat. Photonics, 2019, 13, 406 CrossRef CAS
- Z. An, C. Zheng, Y. Tao, R. Chen, H. Shi, T. Chen, Z. Wang, H. Li, R. Deng and X. Liu, Nat. Mater., 2015, 14, 685 CrossRef CAS PubMed
- Z. Yang, Z. Mao, X. Zhang, D. Ou, Y. Mu, Y. Zhang, C. Zhao, S. Liu, Z. Chi and J. Xu, Angew. Chem., Int. Ed., 2016, 55, 2181–2185 CrossRef CAS PubMed
- W. Zhao, T. S. Cheung, N. Jiang, W. Huang, J. W. Y. Lam, X. Zhang, Z. He and B. Z. Tang, Nat. Commun., 2019, 10, 1595 CrossRef PubMed
- J. Feng and H. Zhang, Chem. Soc. Rev., 2013, 42, 387–410 RSC
- A. D. Aløo, A. Bourdolle, S. Brustlein, T. Fauquier, A. Grichine, A. Duperray, P. L. Baldeck, C. Andraud, S. Brasselet and O. Maury, Angew. Chem., Int. Ed., 2012, 51, 6622–6625 CrossRef PubMed
- M. H. Futscher, A. Rao and B. Ehrler, ACS Energy Lett., 2018, 3, 2587–2592 CrossRef CAS PubMed
- W. Shockley and H. J. Queisser, J. Appl. Phys., 1961, 32, 510–519 CrossRef CAS
- M. B. Smith and J. Michl, Chem. Rev., 2010, 110, 6891–6936 CrossRef CAS PubMed
- A. Rao and R. H. Friend, Nat. Rev. Mater., 2017, 2, 17063 CrossRef CAS
- N. J. L. K. Davis, J. R. Allardice, J. Xiao, A. J. Petty, N. C. Greenham, J. E. Anthony and A. Rao, J. Phys. Chem. Lett., 2018, 9, 1454–1460 CrossRef CAS PubMed
- R. B. Anderson, S. J. Smith, P. S. May and M. T. Berry, J. Phys. Chem. Lett., 2014, 5, 36 CrossRef CAS PubMed
- J.-C. G. Bünzli, Chem. Rev., 2010, 110, 2729–2755 CrossRef PubMed
- I. Hernández, Y. X. Zheng, M. Motevalli, R. H. C. Tan, W. P. Gillin and P. B. Wyatt, Chem. Commun., 2013, 49, 1933–1935 RSC
- C. Bischof, J. Wahsner, J. Scholten, S. Trosien and M. Seitz, J. Am. Chem. Soc., 2010, 132, 14334–14335 CrossRef CAS PubMed
- K. Binnemans, Chem. Rev., 2009, 109, 4283–4374 CrossRef CAS PubMed
- P. B. Glover, A. P. Bassett, P. Nockemann, B. M. Kariuki, R. Van Deun and Z. Pikramenou, Chem. – Eur. J., 2007, 13, 6308–6320 CrossRef CAS PubMed
- Z. Li, A. Dellali, J. Malik, M. Motevalli, R. M. Nix, T. Olukoya, Y. Peng, H. Ye, W. P. Gillin, I. Hernández and P. B. Wyatt, Inorg. Chem., 2013, 52, 1379–1387 CrossRef CAS PubMed
- J. Hu, P. B. Wyatt, W. P. Gillin and H. Ye, J. Phys. Chem. Lett., 2018, 9, 2022–2024 CrossRef CAS PubMed
- H. Ye, V. Bogdanov, S. Liu, S. Vajandar, T. Osipowicz, I. Hernández and Q. Xiong, J. Phys. Chem. Lett., 2017, 8, 5695–5699 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0tc02075c |
| This journal is © The Royal Society of Chemistry 2020 |