
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Investigating the structure–function relationship in triple cation perovskite nanocrystals for light-emitting diode applications†‡
Parth
Vashishtha
 *a,
Sjoerd A.
Veldhuis
b,
Sai S. H.
Dintakurti
acd,
Nicole L.
Kelly
c,
Benjamin E.
Griffith
ac,
Alasdair A. M.
Brown
be,
Mohammed S.
Ansari
a,
Annalisa
Bruno
b,
Nripan
Mathews
ab,
Yanan
Fang
a,
Tim
White
a,
Subodh G.
Mhaisalkar
*ab and
John V.
Hanna
*ac
*a,
Sjoerd A.
Veldhuis
b,
Sai S. H.
Dintakurti
acd,
Nicole L.
Kelly
c,
Benjamin E.
Griffith
ac,
Alasdair A. M.
Brown
be,
Mohammed S.
Ansari
a,
Annalisa
Bruno
b,
Nripan
Mathews
ab,
Yanan
Fang
a,
Tim
White
a,
Subodh G.
Mhaisalkar
*ab and
John V.
Hanna
*ac
aSchool of Materials Science and Engineering, Nanyang Technological University (NTU), 50 Nanyang Avenue, Singapore 639798, Singapore. E-mail: pvashishtha@ntu.edu.sg; subodh@ntu.edu.sg
bEnergy Research Institute@NTU (ERI@N), Research Techno Plaza, X-Frontier Block, Level 5, 50 Nanyang Drive, Singapore 637553, Singapore
cDepartment of Physics, University of Warwick, Gibbet Hill Rd., Coventry CV4 7AL, UK. E-mail: j.v.hanna@warwick.ac.uk
dInterdisciplinary Graduate School, Nanyang Technological University (NTU), 50 Nanyang Avenue, Singapore 639798, Singapore
eSchool of Engineering, Faculty of Engineering and Physical Sciences, University of Southampton, Southampton SO17 1BJ, UK
First published on 3rd August 2020
Abstract
Organic metal halide perovskite nanocrystals are promising candidates for light-emitting diodes due to their narrow emission bandwidth, high photoluminescence quantum yield (PLQY), and color tunability. Nevertheless, these systems suffer from thermal instability, phase impurities, and a sensitivity to processing techniques. This study reports the first synthesis of novel Cs-containing triple cation perovskite nanocrystals with nominal stoichiometry Csx(MA0.17FA0.83)1−xPbBr3 (x = 0–0.15). The effect of Cs+ cation incorporation is thoroughly investigated using diffraction, microscopy and solid state MAS NMR techniques. The solid state 133Cs MAS NMR results reveals the distribution of the Cs+ cations is highly concentration and particle size dependent, with maximized surface/subsurface Cs+ concentrations being achieved with the smaller 5 mol% Cs system. These characteristics directly correlate improved surface passivation and environmental stability of the triple cation system. These triple cation nanocrystals exhibit a maximum photoluminescence quantum yield of ∼93% which upon translation to nanocrystalline LED devices delivers a maximum EQE of 7.4% (30 cd A−1) corresponding to a power efficiency of 34.87 lm W−1. This performance represents a marked improvement compared to CsPbBr3 nanocrystals (PL quantum yield ∼50%; maximum EQE of 2.5% (7.2 cd A−1)) fabricated under similar conditions.
Introduction
Nanocrystals provide several advantages for optoelectronic applications as compared to bulk materials.1–4 They have higher photoluminescence quantum yields (PLQY), narrower emission bandwidth, higher stability, size tuneable optical properties, and the capability for surface functionalisation.3,5–11 Nanocrystal or quantum dot based light emitting diodes (QLEDs) are amongst the best performing LEDs, owing to impressive color rendering, high brightness and higher thermal stability.5,12,13 In the past two decades, several advancements have been made for variety of quantum dots, leading to the fabrication of electrically driven QLEDs with emission throughout the visible range.5,13,14 Currently, state-of-the-art QLEDs use CdSe based core–shell quantum dots as an active layer.15 However, over the last few years, perovskite materials have emerged as promising competitors for optoelectronic applications.Organic–inorganic metal halide perovskite materials have demonstrated attractive properties for applications such as solar cells, LEDs and photodetectors. They have the potential for very cost-effective fabrication, due to both the low precursor material costs and their suitability for scalable, high-throughput, solution-processing techniques. Moreover, the composition tuneability of the perovskite structure offers a powerful approach to optimize the material properties for specific applications.16,17 Since their discovery fifteen years ago, the power conversion efficiency (PCE) of perovskite-based solar cells has improved enormously from 3.6% to 25.2%.18–22 Although methylammonium (MA), formamidinium (FA) and mixed MA/FA based lead halide perovskite solar cell materials demonstrate reasonably high efficiencies, they are very sensitive to processing techniques as a result of thermal and structural instability.23–26 Additionally, MA/FA based perovskite films have revealed difficulties with phase impurity and significant defects and disorder influencing the crystalline framework.23,27 The addition of Cs+ as a third cation into the FA/MA perovskite films has provided a viable solution to these problems as triple cation films exhibit greater stability and higher purity.23,27,28 Furthermore, triple cation perovskites show less sensitivity to processing conditions, thus offering a higher degree of device reproducibility.28–30 As a result, triple cation perovskite devices have shown higher power conversion efficiency in perovskite solar cells.23,27,30
In addition to solar cell devices, perovskites have also attracted significant attention for LED applications. Their tunable emission wavelength has facilitated LEDs of external quantum efficiency (EQE) >20% for green, red and near-IR light.31–34 In order to be viable for LED applications, a material should exhibit high exciton binding energy, narrow PL emission linewidth, and high PLQY.35,36 Hence, prior research has focused on perovskite nanocrystal systems and low dimensional perovskite materials where charge confinement greatly enhances these vital properties.31,35,37–40 Similar to perovskite solar cells, mixed cation systems have proven very useful for LED production. Zhang et al. reported mixed cation FA(1−x)CsxPbBr3 perovskite LEDs and found a significant improvement in EQE from 0.82% to 1.82% upon introducing Cs cations,41 while Xu et al. reported the perovskite LEDs based on mixed cation FAxMA(1−x)PbBr3 quantum dots.42 The influence of cation mixing was elucidated by Borriello et al. who used computational studies to demonstrate that varying distortion of the crystal structure with different A site cations alters the electronic properties of the perovskite.43 This trend encourages research into the formulation of mixed cation based perovskite materials for the next generation of LEDs. Although Kim et al. reported the first LEDs made from triple cation bulk perovskites, achieving EQEs of 1.7%, 7.3% and 9.23% for blue, green, and near infrared emission, respectively,44 LEDs from triple cation perovskite nanocrystal systems have not yet been reported.
In this work, novel triple cation perovskite nanocrystals of nominal stoichiometry Csx(FA0.83MA0.17)1−xPbBr3 (x = 0–0.15) were synthesized using the room temperature ligand-assisted reprecipitation (LARP) method.35,45–49 Octylamine and oleic acid were introduced as surface passivation agents, while dibenzo-21-crown-7-ether effectively solvated the Cs+ cations (from solid CsBr) in DMF solutions. Nanocrystalline systems of varying Cs incorporation have been characterized by HR-TEM, XRD and an extensive multinuclear solid state MAS NMR study to evaluate the long-range order and short-range disorder that characterizes the structural heterogeneity and structure–function aspects of these systems. The photoluminescence quantum yield (PLQY) and external quantum efficiency (EQE) characterizing the optoelectronic performance of these materials, and their potential for LED device fabrication, were also assessed.
Results and discussion
Structural characterization
 | ||
| Fig. 1 High resolution transmission electron microscopy (HR-TEM) images of nanocrystals supporting, (a) 0 mol% Cs, (b) 5 mol% Cs, (c) 10 mol% Cs, and (d) 15 mol% Cs. These nanocrystals were persistent under the beam for ∼45 s. (e) Shows the imaging of the lattice planes in the 5 mol% Cs preparation, while (f)–(h) show the powder XRD data, the expansion of the 30° 2θ region highlighting the peak shift in the (200) plane, and the change in the lattice parameter a with Cs incorporation, respectively. These XRD data pertaining to the d spacing analysis are uncorrected for sample displacement, thus yielding the discrepancy between d spacing and lattice parameter a data in (h). (i) Illustrates the relationship between the relative XRD peak intensities of 〈100〉 vs. 〈200〉 taken from (f) and the average atomic number of the A site cations. In addition, the simulations of the relative intensity I(100)/I(200) vs. the average atomic number of the A site cations from the CsPbBr3, MAPbBr3, and FAPbBr3 systems are plotted for comparison. | ||
These characteristics contrast with the morphological behavior of CsPbBr3 nanoparticles that form highly crystalline nano-cubes (average edge ∼11 nm). As shown in Fig. 2a and b, this system readily exhibits lattice fringes indicating a d spacing of 5.8 Å, in agreement with previous studies.32,50 However, the lower and higher magnification HR-TEM images in Fig. 2c–e show decomposition under shorter electron beam exposure of ∼15 s (i.e. in comparison to similar TEM beam conditions used to study the triple cation systems), consequently inducing a heavily disordered material comprised of various particle shapes and ranges of dimensions.
 | ||
| Fig. 2 HR-TEM micrographs of CsPbBr3 nanoparticles synthesized using LARP method showing (a) the as-synthesized cubic morphology, (b) a higher resolution image emphasizing the cube structure and the lattice fringes, (c) a lower resolution region scan acquired with short beam exposure of ∼5 s, (d) lower resolution scan of the same area after a beam exposure of ∼15 s, (e) a resolution micrograph depicting the disorder caused by beam damage after the same ∼15 s beam exposure, and (f) the XRD pattern of as synthesized CsPbBr3 nanoparticles. | ||
A powder X-ray diffraction study of the triple cation nanocrystal series confirmed the formation of Pm![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m cubic perovskite materials.51,52 While XRD patterns from drop-cast nanocrystal suspensions on zero background substrate holders can exhibit some texture leading to modification of the measured diffraction intensities (see Fig. 1f and g),51,52 those from these nanocrystal precipitates showed no preferred orientation (see Fig. S2a, ESI‡). As evidenced by the HR-TEM image of Fig. 1e for the 5 mol% Cs sample, the nanocrystal lattices were highly ordered as the 〈200〉cubic lattice fringes are visible, with the superimposed FFT match of the 〈200〉 plane obtained from the XRD data (see Fig. 1f–h) corresponding to a d spacing of ∼2.97 Å. As the X-ray scattering power and effective ionic radius of the Cs+, FA+ and MA+ cations both progressively decrease, Fig. 1f–i and Fig. S2b–d (ESI‡) demonstrate that there are systematic changes in unit cell edge and the relative intensity of 〈100〉/〈200〉 with composition in accordance with the nominal compositions.53,54 A continuous solid solution has been previously reported in microcrystalline triple cation perovskite thin films.23 The incorporation of FA and MA was further confirmed by FTIR of Cs0.05(FA0.83MA0.17)0.95PbBr3 nanocrystals (see Fig. S3, ESI‡). TEM-EDXS corroborated that the Cs, Pb and Br molar ratios are in good agreement with the nominal starting precursor proportions (see Tables S1 and S2, ESI‡). This analysis was coupled with further elemental analysis undertaken using solid state 1H and 13C MAS NMR studies (vide infra) to confirm the A-site FA+ and MA+ cation stoichiometry comprising the triple cation systems.
m cubic perovskite materials.51,52 While XRD patterns from drop-cast nanocrystal suspensions on zero background substrate holders can exhibit some texture leading to modification of the measured diffraction intensities (see Fig. 1f and g),51,52 those from these nanocrystal precipitates showed no preferred orientation (see Fig. S2a, ESI‡). As evidenced by the HR-TEM image of Fig. 1e for the 5 mol% Cs sample, the nanocrystal lattices were highly ordered as the 〈200〉cubic lattice fringes are visible, with the superimposed FFT match of the 〈200〉 plane obtained from the XRD data (see Fig. 1f–h) corresponding to a d spacing of ∼2.97 Å. As the X-ray scattering power and effective ionic radius of the Cs+, FA+ and MA+ cations both progressively decrease, Fig. 1f–i and Fig. S2b–d (ESI‡) demonstrate that there are systematic changes in unit cell edge and the relative intensity of 〈100〉/〈200〉 with composition in accordance with the nominal compositions.53,54 A continuous solid solution has been previously reported in microcrystalline triple cation perovskite thin films.23 The incorporation of FA and MA was further confirmed by FTIR of Cs0.05(FA0.83MA0.17)0.95PbBr3 nanocrystals (see Fig. S3, ESI‡). TEM-EDXS corroborated that the Cs, Pb and Br molar ratios are in good agreement with the nominal starting precursor proportions (see Tables S1 and S2, ESI‡). This analysis was coupled with further elemental analysis undertaken using solid state 1H and 13C MAS NMR studies (vide infra) to confirm the A-site FA+ and MA+ cation stoichiometry comprising the triple cation systems.
Multinuclear solid state NMR – nanocrystal core
The 133Cs MAS NMR data for the bulk CsPbBr3, CsPbBr3 nanocrystal (average particle size ∼11 nm) and the triple cation Csx(MA0.17FA0.83)1−xPbBr3 (x = 0.05–0.15) nanocrystal series are shown in Fig. 3a and Fig. S4 (ESI‡), and summarised in Table 1. While the bulk orthorhombic CsPbBr3 phase is characterised by a single resonance at δ 117 ppm and a long T1 value of ∼103 s (B0 = 14.1 T), the data for the CsPbBr3 nanocrystals indicates that more complex Cs speciation describes this system. Broader upfield signals at δ 109 ppm and δ 95 ppm assigned to Cs+ cations residing in the sub-surface and surface nanocrystal layers, respectively, accompany the bulk CsPbBr3 resonance (δ 117 ppm) characterising the nanocrystal core. The layers experience increasing short-range disorder and structural strain associated with the cumulative loss of translational symmetry as these Cs positions approach the particle surface. As evidenced in Table 1, Fig. 3a and Fig. S4 (ESI‡), these assignments are supported by the increasing 133Cs linewidths and markedly reducing T1 values that trend upfield (i.e. towards decreasing 133Cs chemical shift) with increasing proximity of the Cs+ cations to the nanocrystal surface. This effect is most acute for the surface Cs positions experiencing direct contact with the disordered and motional passivation layer stabilising the CsPbBr3 nanocrystal surface. Although 133Cs (I = 7/2) is a quadrupolar nucleus, the very small quadrupole moment Q of −0.343 × 10−30 m2 affords the observation of resonances which are dominated by chemical shift dispersion induced by short-range disorder. These observations mirror the findings from previous 195Pt and 105Pd NMR studies on Pt and Pd metal nanoparticle systems that report similar phenomena where the bulk component is clearly identified as a separate component to the disordered surface and sub-surface speciation of lower translational symmetry.55–57 | ||
| Fig. 3 The (a) 133Cs MAS NMR data (B0 = 14.1 T, νr = 12 kHz), and (b) 207Pb MAS NMR data (B0 = 7.05 T, νr = 20 kHz), characterizing triple cation Csx(MA0.17FA0.83)1−xPbBr3 (x = 0.05–0.15) and CsPbBr3 nanocrystal systems, while (c) depicts the relative motional regimes assumed by these systems as described by their T1 characteristics under the influence of a dipolar relaxation mechanism. | ||
| Sample | 133Cs MAS NMR | 207Pb MAS NMR | 13C MAS NMR | 1H MAS NMR | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| δ iso (ppm) | Δν (Hz) | T 1 (s) (7.05 T) | T 1 (s) (14.1 T) | δ iso (ppm) | 1 J(207Pb,79/81Br) (Hz) | δ iso (ppm) | Integrated intensity (%) | δ iso (ppm) (peak label) | Integrated intensity (%) | Assignments | Octylamine![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :oleic acid ratio :oleic acid ratio |
||
| CsPbBr3 bulk | 117 | 337 | 61.2 ± 3.0 | 103.1 ± 5.0 | 275 ± 10 | 2290 ± 20 | 14.5 | 9.6 | 0.6 (14) | 10.1 | Methyl group | 1.4:1 |
|
| CsPbBr3 nanocrystals | 95 | 1481 | 2.5 ± 0.5 | 5.7 ± 1.0 | 275 ± 10 | 2290 ± 20 | 23.3 | 8.3 | 1.0 (13) | 6.9 | Alkyl chain | ||
| 109 | 847 | 10.2 ± 1.5 | 11.6 ± 1.5 | 25.7 | 4.0 | 1.0 (13) | 46.1 | Alkyl chain | |||||
| 117 | 335 | 28.6 ± 2.9 | 34.8 ± 3.5 | 27.3 | 4.9 | 1.2 (12) | 10.2 | Alkyl chain | |||||
| 27.8 | 7.1 | 1.7 (11) | 7.5 | CH2-vinyl, oleic acid | |||||||||
| 28.2 | 3.9 | 1.9 (10) | 3.1 | α-CH2, oleic acid | |||||||||
| 30.1 | 31.0 | 2.5 (9) | <1.0 | CH2, crown ether | |||||||||
| 30.4 | 10.9 | 3.3 (7) | 4.9 | α-CH2, octylamine | |||||||||
| 32.6 | 8.3 | 5.0 (6) | 3.3 | Vinyl CH, oleic acid | |||||||||
| 35.3 | 2.3 | 6.7 (4) | 7.2 | NH3+, octylamine | |||||||||
| 42.5 | 3.3 | 7.6 (2) | <1.0 | Aromatic CH, crown ether | |||||||||
| 111.4 | 1.1 | ||||||||||||
| 130.1 | 4.1 | ||||||||||||
| 179.1 | 1.4 | ||||||||||||
| Triple cation 15 mol% Cs | 63 | 1047 | 2.9 ± 0.9 | 5.4 ± 1.1 | 445 ± 20 | 14.5 | 2.5 | 0.6 (14) | 10.5 | Methyl group | 6:1 |
||
| 23.3 | 7.6 | 1.0 (13) | 24.1 | Alkyl chain | |||||||||
| 26.2 | 5.1 | 1.2 (12) | 22.9 | Alkyl chain | |||||||||
| 27.4 | 8.9 | 1.8 (10,11) | 8.8 | CH2-vinyl, α-CH2, oleic acid | |||||||||
| 28.5 | 7.7 | 2.0 (9) | <1.0 | CH2, crown ether | |||||||||
| 28.8 | 3.0 | 3.0 (8) | 1.0 | CH3, methylammonium | |||||||||
| 30.1 | 33.8 | 3.3 (7) | 6.8 | α-CH2, octylamine | |||||||||
| 32.7 | 9.1 | 4.9 (6) | 1.5 | Vinyl CH, oleic acid | |||||||||
| 40.6 | 1.1 | 6.1 (5) | 1.2 | NH3+, methylammonium | |||||||||
| 43.0 | 12.5 | 6.6 (4) | 10.2 | NH3+, octylamine | |||||||||
| 130.1 | 1.5 | 7.3 (3) | 8.7 | NH2, formamidinum | |||||||||
| 156.0 | 5.1 | 7.5 (2) | <1.0 | Aromatic CH, crown ether | |||||||||
| 182.2 | 1.5 | 7.9 (1) | 2.2 | CH, formamidinium | |||||||||
| Triple cation 10 mol% Cs | 53 | 810 | 1.2 ± 0.4 | 2.9 ± 0.4 | 445 ± 20 | 14.5 | 2.8 | 0.6 (14) | 10.3 | Methyl group | 6:1 |
||
| 61 | 805 | 4.6 ± 0.8 | 2.8 ± 1.1 | 23.3 | 7.6 | 1.0 (13) | 22.2 | Alkyl chain | |||||
| 26.2 | 3.7 | 1.2 (12) | 25.6 | Alkyl chain | |||||||||
| 27.4 | 7.2 | 1.8 (10,11) | 7.5 | CH2-vinyl, α-CH2, oleic acid | |||||||||
| 27.7 | 7.7 | 2.0 (9) | <1.0 | CH2, crown ether | |||||||||
| 28.5 | 6.4 | 3.0 (8) | 1.2 | CH3, methylammonium | |||||||||
| 28.8 | 4.3 | 3.3 (7) | 7.1 | α-CH2, octylamine | |||||||||
| 30.1 | 31.8 | 4.9 (6) | 1.4 | Vinyl CH, oleic acid | |||||||||
| 32.7 | 9.2 | 6.1 (5) | 10.6 | NH3+, methylammonium | |||||||||
| 40.6 | 1.5 | 6.6 (4) | 0.9 | NH3+, octylamine | |||||||||
| 43.0 | 12.5 | 7.3 (3) | 8.3 | NH2, formamidinum | |||||||||
| 130.1 | 1.6 | 7.5 (2) | <1.0 | Aromatic CH, crown ether | |||||||||
| 156 | 5.4 | 7.9 (1) | 2.3 | CH, formamidinium | |||||||||
| 182 | 1.3 | ||||||||||||
| Triple cation 5 mol% Cs | 51 | 810 | 2.2 ± 0.4 | 9.2 ± 1.3 | 445 ± 20 | 14.5 | 2.8 | 0.6 (14) | 11.1 | Methyl group | 5:1 |
||
| 58 | 663 | 2.6 ± 0.4 | 2.3 ± 0.3 | 23.3 | 7.7 | 1.0 (13) | 24.8 | Alkyl chain | |||||
| 26.2 | 2.6 | 1.2 (12) | 25.2 | Alkyl chain | |||||||||
| 27.4 | 9.2 | 1.8 (10,11) | 8.5 | CH2-vinyl, α-CH2, oleic acid | |||||||||
| 27.7 | 5.7 | 2.0 (9) | <1.0 | CH2, crown ether | |||||||||
| 28.4 | 7.8 | 3.0 (8) | 1.1 | CH3, methylammonium | |||||||||
| 28.8 | 3.5 | 3.3 (7) | 7.4 | α-CH2, octylamine | |||||||||
| 30.1 | 30.6 | 4.9 (6) | 1.6 | Vinyl CH, oleic acid | |||||||||
| 32.6 | 8.9 | 6.1 (5) | 1.0 | NH3+, methylammonium | |||||||||
| 40.6 | 1.3 | 6.6 (4) | 11.2 | NH3+, octylamine | |||||||||
| 43.0 | 12.2 | 7.3 (3) | 8.2 | NH2, formamidinum | |||||||||
| 130.1 | 1.5 | 7.5 (2) | <1.0 | Aromatic CH, crown ether | |||||||||
| 156.0 | 5.5 | 7.9 (1) | 2.0 | CH, formamidinium | |||||||||
| 182.2 | 0.8 | ||||||||||||
Within the nominal stoichiometry of the triple cation Csx(MA0.17FA0.83)1−xPbBr3 (x = 0–0.15) nanocrystal series the Cs+ cation is a dilute species, and a more complex scenario describes the Cs speciation in these systems. In comparison to the CsPbBr3 cases, the 133Cs MAS NMR data in Fig. 3a now exhibits upfield shifted resonances at δ ≤ 65 ppm, however the observed chemical shift and multiplicity of the observed resonances is sensitive to the degree of Cs incorporation. For the highest Cs concentration sample (15 mol%) only a single discrete 133Cs resonance at δ ∼ 65 ppm is observed suggesting that the Cs+ cations in the larger ∼8 nm diameter spheroids generate a Cs-rich core within the nanoparticles, thus generating a ‘pseudo core–shell’ arrangement which is insensitive to the surface and sub-surface layers. In contrast, the smaller 10 mol% and 5 mol% systems supporting reduced Cs incorporation exhibit successive upfield shifts (to lower ppm) in the primary bulk 133Cs resonances to δ ∼61 and δ ∼58 ppm, respectively, and additional upfield resonances at δ ∼51 ppm (10 mol% Cs sample) and δ ∼49 ppm (5 mol% Cs sample) are also observed. These findings suggest that the Cs-rich core of these smaller spheroidal triple cation nanocrystals experiences an increased interaction with the particle surface, and that these systems diverge from a formal pseudo core–shell arrangement. Hence, the relative proportions and influence of the surface/sub-surface layers to the bulk (core) are becoming more apparent as the surface area:volume ratio of these smaller nanocrystals increases. These observations are analogous to the characteristics defining the Cs speciation within the ∼11 nm CsPbBr3 nanocrystal system discussed above (see Fig. 3a), where significant proportions of the Cs positions experience increased structural disorder and altered chemical environments due to proximity to the nanoparticle surface.
The 133Cs T1 data summarised in Table 1, and depicted in Fig. 3c and Fig. S4a–e (ESI‡), yield valuable insights into the motional dynamics of the Cs speciation within the CsPbBr3 bulk and nanocrystal phases, and the triple cation Csx(FA0.83MA0.17)1−xPbBr3 (x = 0–0.15) nanocrystal series. Assuming that the relaxation of these spin systems is dominated by a dipolar relaxation mechanism,58 the long T1 values measured from CsPbBr3 bulk phase (δ 117 ppm; T1 ∼ 61 s at 7.05 T; T1 ∼ 103 s at 14.1 T) demonstrate that Cs+ dynamics reside in the slow motion regime that is characteristic of condensed solids behaviour (see Fig. 3c). In contrast, while the bulk component of the CsPbBr3 nanocrystal phases exhibits similar Cs+ dynamics behaviour although the measured T1 values are not as long (δ 117 ppm; T1 ∼ 29 s at 7.05 T; T1 ∼ 35 s at 14.1 T), the resonances ascribed to sub-surface (δ 109 ppm) and surface (δ 95 ppm) Cs+ cations positions are characterised by significantly shorter T1s of ∼3–12 s which decrease monotonically as the particle surface is approached, and they display reducing magnitude differences between the variable B0 field values. These features indicate that the Cs+ cation migration within these regions is much faster and they reside in a motional regime approaching the extreme narrowing limit, as schematically represented in Fig. 3c. These results corroborate previous computational and electric field driven ion migration studies in mixed metal halide perovskite nanocrystal systems which report increased Cs+ and Br− dynamics in surface species in the presence of increased defect and vacancy structures on the nanocrystal surfaces.32,59
In comparison to the partitioned 133Cs T1 behaviour based on the proximity of the Cs species to the CsPbBr3 nanocrystal surface, the T1 relaxation of the Cs+ cations within the triple cation systems is more complicated as each Cs+ position is now surrounded by MA+ and FA+ positions undergoing their own dynamic processes. Hence, the 133Cs T1 behaviour can no longer be rationalised to slower or faster Cs+ migration governed by a simplistic ‘proximity-to-surface’ phenomenon. As noted from Table 1, Fig. 3c and Fig. S4a–e (ESI‡), the contrast between the magnitudes of the T1 values has largely disappeared. These data show that the triple cation nanocrystals are characterised by consistently shorter T1 values in comparison to the CsPbBr3 nanocrystals. They also exhibit little variation (within experimental error) between the variable B0 field values; i.e. they nearly all reside in motional regimes close to the extreme narrowing limit as depicted in Fig. 3c.58 The only exception to this behaviour is the δ 51 ppm resonance assigned to surface/near-surface Cs+ species in the 5 mol% Cs system which shows clear variable B0 field separation in the measured T1 values (δ 51 ppm; ∼2 s at 7.05 T; ∼9 s at 14.1 T), thus suggesting residence in a slower motion regime associated with decreased Cs+ migration dynamics. It can be assumed that the MA+ and FA+ dynamics modulating the local magnetic fields at the surface, subsurface and bulk Cs+ positions are approximately constant for all the triple cation nanocrystals; hence, the longer, field dependent T1s for the 5 mol% system indicate reduced Cs+ migration linked with the higher Cs+ surface concentration and increased passivation (see Fig. 3a) leading to greater surface environmental stability from that preparation.23
Increased disorder induced by reduced particle size effects is further demonstrated by the 207Pb MAS NMR data of Fig. 3b. The data for bulk CsPbBr3 (δ 275 ppm) exhibits a well-defined 1J(207Pb,79/81Br) coupling of 2290 ± 20 Hz from the cubic PbBr64− framework. Moreover, the corresponding data for the CsPbBr3 nanocrystal is characterised a less resolved/broadened 1J(207Pb,79/81Br) coupling manifold due to the disorder induced by increased structural strain and dislocations as the nanoparticle surface plays a more prominent role. These results represent the first observations of 1J(207Pb,79/81Br) coupling in CsPbBr3 from both bulk and nanocrystalline forms. In contrast, the 207Pb MAS NMR data from the triple cation series represent broad featureless resonances (δ 445 ppm) devoid of any 1J(207Pb,79/81Br) fine structure, thus reflecting the cation disorder that characterises these systems.
The 13C and 1H MAS NMR data from the triple cation Csx(MA0.17FA0.83)1−xPbBr3 (x = 0–0.15) nanocrystal preparations are shown in Fig. 4 and 5. These data describe the H and C speciation emanating from the MA+ and FA+ cations within the nanoparticle, and the more complex speciation associated with the octylamine and oleic acid passivating agents on the nanoparticle surface. An integration of the MA+ and FA+ resonances in both the 13C and 1H MAS NMR data (see Fig. 4a–c) shows that the relative proportions of these cations are close to that of the proposed nominal stoichiometry, and within experimental error this ratio is invariant across the 0–15 mol% Cs compositional range. As summarised in Fig. 4c, these characteristics corroborates the quantitative analysis undertaken using the TEM-EDXS technique. Most importantly, this finding suggests that the trends characterizing the 133Cs MAS NMR data are formally ascribed to the changing nanocrystal size and the proximity of the Cs speciation to the particle surface, and not to other factors linked to variation in the FA+ and MA+ cation concentrations.
 | ||
| Fig. 4 The (a) 13C MAS NMR data (B0 = 14.1 T, νr = 12 kHz), (b) 1H MAS NMR data (B0 = 14.1 T, νr = 60 kHz), (c) quantitative estimates of the MA+ and FA+ A site cation concentrations as determined from the 13C and 1H MAS NMR data, and (d) comparison of the 1H T1ρ data from the triple cation Csx(MA0.17FA0.83)1−xPbBr3 (x = 0.05) and CsPbBr3 nanocrystal systems. The 13C and 1H MAS NMR data both suggest invariant proportions of MA and FA across the compositional range as summarized in (c), which is consistent with the nominal stoichiometric proportions introduced in the synthesis route. | ||
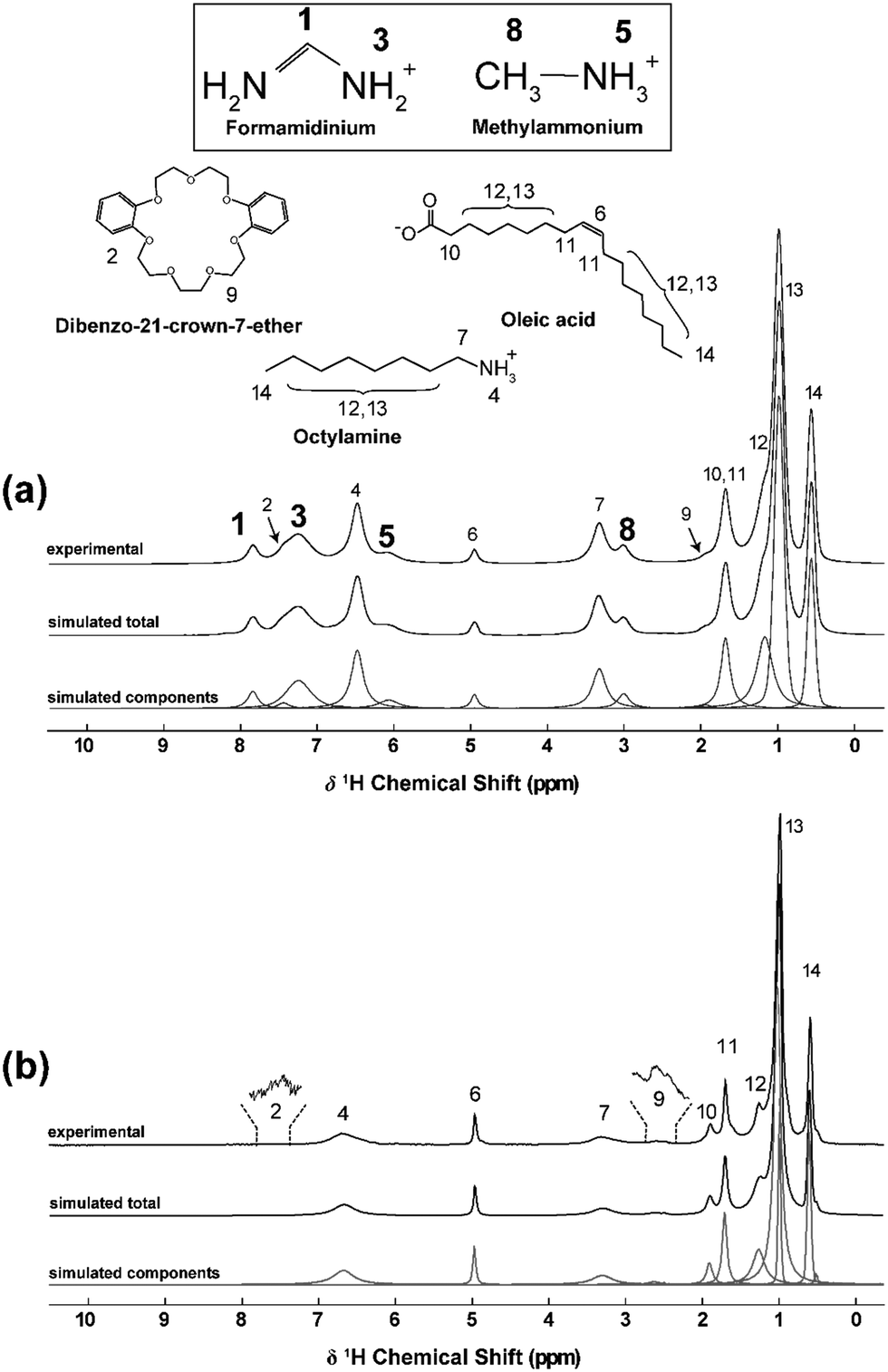 | ||
| Fig. 5 A 1H MAS NMR data (B0 = 14.1 T, νr = 60 kHz) from, (a) the triple cation Csx(MA0.17FA0.83)1−xPbBr3 (x = 0.15) nanocrystal system, and (b) the CsPbBr3 nanocrystal system, highlighting resonances from the FA+ and MA+ A-site cations, the octylamine and oleic acid surface passivating agents, and the trace amounts of the dibenzo-21-crown-7-ether solvent. The data from solid state 2D homonuclear 1H–1H BABA and NOESY experiments assisted these assignments. | ||
Multinuclear solid state NMR – nanocrystal surface
Complete assignments of the 1H MAS NMR spectra characterizing the triple cation Csx(MA0.17FA0.83)1−xPbBr3 (x = 0.15) and CsPbBr3 nanocrystal systems are shown in Fig. 5a and b, respectively. These assignments were assisted by connectivity information drawn from two different solid state 2D homonuclear 1H–1H MAS NMR methodologies. As observed from Fig. 6 and 7, the double quantum/single quantum (DQ/SQ) BAck-to-BAck (BABA) and Nuclear Overhauser Effect SpectroscopY (NOESY) experiments were implemented which provided through-bond and through-space (‘direct’) information, respectively. For the triple cation system (Cs0.15(MA0.17FA0.83)0.85PbBr3) (x = 15 mol% Cs in this case), Fig. 6a shows the 2D DQ/SQ BABA data that clearly defines through-bond connectivity within the FA+ cation, and within the octylamine passivating agent on the nanocrystal surface. These correlations proved to be valuable assignment aids. In contrast, the 2D 1H–1H NOESY data from the same system (see Fig. 6b and c) provides a more complex array of through-space connectivities amongst the FA+, MA+, octylamine and oleic acid species that are both intermolecular and intramolecular in nature. These assignments enabled an unambiguous identification of the octylamine and oleic acid passivating agents populating the nanocrystal surfaces, and they facilitated quantitative estimates to be established.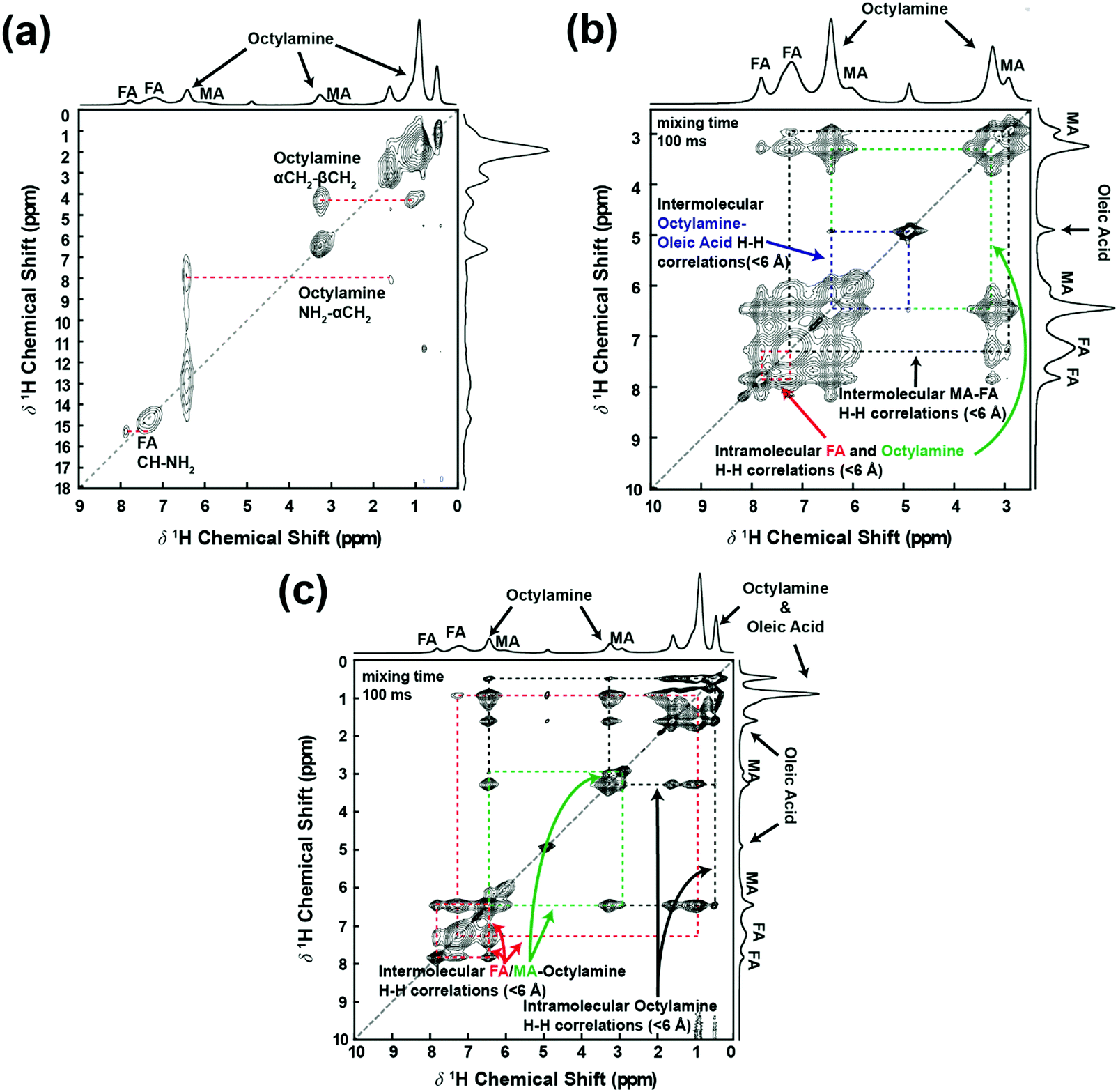 | ||
| Fig. 6 2D homonuclear 1H–1H MAS NMR data (B0 = 14.1 T, νr = 60 kHz) from the triple cation Csx(MA0.17FA0.83)1−xPbBr3 (x = 0.15) system including (a) 2D DQ/SQ BABA data showing through-bond connectivities (indicated with red dashed line) within the FA+ cation and within the octylamine, passivating agent, and (b) 2D NOESY data (mixing time 100 ms) highlighting through-space connectivities associated with intramolecular FA+ (red) and octylamine (green) correlations, and intermolecular MA+/FA+ correlations (black), octylamine/oleic acid correlations (blue). (c) Shows an expanded representation of (b) highlighting additional octylamine/MA+ (green) and octylamine/FA+ (red) correlations, and intramolecular octylamine correlations (black). | ||
 | ||
| Fig. 7 Schematic representation of the surface passivation of the triple cation Csx(MA0.17FA0.83)1−xPbBr3 (x = 0.05–0.15) and CsPbBr3 nanocrystal systems showing, (a) a typical arrangement of the disordered coordination of the octylamine and oleic acid passivating agents on the surface of the 5 mol% Cs (∼6 nm) sample (not to scale, PbBr64− framework excluded), (b) and (c) representations of the 5 mol% Cs (∼6 nm) and CsPbBr3 nanoparticle surfaces supporting different proportions of the octylamine and oleic acid passivating agents, and (d) folded configurations of the octylamine and oleic acid as determined from the solid state 2D 1H–1H NOESY data presented in Fig. 6 and 7. | ||
Although the triple cation precursor solution is comprised of 5% octylamine and 95% oleic acid, the quantitative estimates from deconvolution and simulation of the solid state 1H MAS NMR data of Fig. 5a establish that the bulk of the nanocrystal surface passivation is actually dominated by the lower concentration octylamine component. The quantitative estimate for the 15 mol% Cs sample shows that the surface is populated by ∼86% ± 4 octylamine vs. ∼14% ± 3 oleic acid (i.e. a ratio of ∼6:1). From the similarity exhibited in the triple cation 1D 1H MAS NMR data of Fig. 4b and the corresponding triple cation 2D 1H–1H BABA and 1H–1H NOESY data displayed in Fig. 6a–c and Fig. S6 (ESI‡), the entire triple cation series is characterized by this feature. As summarized in Table 1, the octylamine:oleic acid ratio ranges from ∼1:5–1:6. These observations demonstrates that the sterically smaller octylamine molecule with a positively charged amine group dominates the nanocrystal surface, thus supporting the presence of a residual negative surface charge from the PbBr64− framework.60 Such a scenario can only occur if there exists a predominance of exposed Br− positions undergoing ionic contact with the octylamine NH3+ head group, as depicted in the schematic representations in Fig. 7a and b. The description of the triple cation nanocrystal surface contrasts markedly with that of the CsPbBr3 nanocrystal surface illustrated in Fig. 8c. From the quantitative 1H and 13C MAS NMR analyses describing the CsPbBr3 system it is evident that a greater proportion of oleic acid is present on this surface, however octylamine still represents the larger component (∼60% ± 4 octylamine vs. ∼40% ± 4 oleic acid; i.e. ∼3:2). This implies that more equivalent distributions of Br− and Cs+ species populate this surface (see Fig. 7c). Another important outcome described by the 1H and 13C MAS NMR data from both the CsPbBr3 and triple cation nanocrystal systems is that the dibenzo-21-crown-7-ether is present in trace amounts only (∼1% level), consequently eliminating any likelihood that it exhibits a functional role on these surfaces.
 | ||
| Fig. 8 The photophysical properties from the triple cation Csx(MA0.17FA0.83)1−xPbBr3 (x = 0–0.15) series depicting, (a) a photograph of the 5 mol% Cs sample in hexane under room light and 365 nm wavelength UV light, (b) absorption and PL spectra of 0 mol% Cs, 5 mol% Cs, 10 mol% Cs, 15 mol% Cs samples, and (c) the photostability of a triple cation nanocrystal thin film measured under continuous illumination for 5 h (maximum intensity recorded for PL maximum at 528 nm (λexc = 350 nm)). The inset in (c) shows 2D PL spectra recorded over the first 2 h of continuous illumination emphasizes the absence of intensity changes or spectral drift in this performance. | ||
The different octylamine:oleic acid ratios supported by the triple cation and CsPbBr3 nanocrystal surfaces directly influence the mobility of these surface passivation agents. Fig. 4d summarizes the 1H T1ρ relaxation time measurements associated with each H species populating these surfaces. This relaxation time is sensitive to different modes of molecular motion and reorientation in the millisecond regime.58 These data indicate that (apart from the chain-end CH3 species in both passivating agents that are controlled by methyl rotation), the H species on triple cation surfaces (dominated by octylamine) display consistently longer T1ρ relaxation times emanating from slower segmental motion in the octylamine chain. Conversely, the CsPbBr3 surface which possesses a higher oleic acid population exhibits shorter T1ρ relaxation times associated with the faster segmental motion of the much longer aliphatic chain of this molecule. These observations directly correlate with the higher stability of the triple cation nanocrystals in comparison to their CsPbBr3 counterparts (see the HR-TEM results above), with the former possessing a more homogeneous surface environment characterized by slower modes of motion with lower energetics.
The 2D 1H–1H NOESY experiment is a technique that relies on the transferral of spin polarisation from a specific 1H nucleus to other surrounding 1H nuclei in the structure via the homonuclear 1H–1H dipolar interaction. In cases where H species are sufficiently proximate, an enhancement in the observed NMR signal from the neighboring nuclei can be observed which is proportional to the strength of the 1H–1H dipolar coupling. This phenomenon is limited by the 1/r6 dependence of this interaction, and the dipolar contact between spatially distant H species can only be detected when they are separated by distances of typically less than ∼6 Å.60–62 These characteristics render the sensitivity of this experiment to be limited to H–H distances within one framework cavity (i.e. ranging between ∼2.0–3.0 Å), and intermolecular H–H distances between neighboring framework cavities (i.e. ranging between ∼3.5–6.0 Å). However, MA+ and FA+ cations in next-nearest-neighbour arrangements exhibit H–H distances of >10 Å which are outside the detection limits of this technique. An intermediate mixing time (τ) of 100 ms allows short range and long range correlations to be detected in the 2D MAS NMR data of Fig. 6b and c. A typical analysis of this demarcation of cation H–H distances in a lead halide perovskite is given for the MAPbBr3 system in Fig. S5 (ESI‡).
The sensitivity limits intrinsic to the 2D 1H–1H NOESY experiment have implications for the interpretation of the correlations pertaining to the octylamine and oleic acid passivating agent on the nanocrystal surfaces. From Fig. 6c prominent intermolecular FA+/octylamine correlations are observed, while weaker MA+/octylamine correlations are also evident. The intensity of these correlations demonstrates that the octylamine (with a positively charged NH3+ functional group) exhibits a strong affinity for the PbBr64− surface which supports a large negative charge; i.e. the Br− positions provide the primary point of electrostatic contact. However, the positively charged surface MA+ and FA+ cations (which will be notionally coordinated to the lower concentration oleic acid CO2− functional group) are adjacent to the primary Br− positions and spatially separated by <∼6 Å as per the sensitivity limits of the NOESY experiment. The intensity differences in these correlations are due to the proportionally smaller concentrations of MA+ in the nominal stoichiometry rather than from spatial separation considerations (see Fig. 4c). It is important to recognize that only the surface FA+/octylamine and MA+/octylamine correlations are observed in Fig. 6b and c as the sub-surface FA+ and MA+ cations are >10 Å away (see Fig. 8a). This representation provides an initial depiction of the highly disordered nature of the triple cation nanocrystal surface.
The surface FA+/octylamine and MA+/octylamine correlations of Fig. 6c also provide additional evidence for preferential alignment so that the electrostatic repulsion between the positively charged amine groups and the positively charged octylamine head group is minimized. This phenomenon is evidenced by the observable CH (FA+; δ 7.9 ppm)/NH3+ (octylamine; δ 6.5 ppm) and NH2+ (FA+; δ 7.3 ppm)/CH3 (octylamine; δ 1.0 ppm) correlations marked in red, and the weaker CH3 (MA+; δ 3.0 ppm)/NH3+ (octylamine; δ 6.5 ppm) correlation marked in green (see Fig. 6c). Note that the NH2+ (FA+; δ 7.2 ppm)/NH3+ (octylamine; δ 6.5 ppm) and the NH3+ (MA+; δ 6.1 ppm)/NH3+ (octylamine; δ 6.5 ppm) correlations are absent. As demonstrated in Fig. S6 (ESI‡), these surface structural characteristics are common to all the samples in the 5–15 mol% Cs compositional range of the triple cation nanocrystal system.
In addition, the intramolecular and intermolecular correlations emanating from the 2D 1H–1H NOESY data in Fig. 6b, c and Fig. S7b–e (ESI‡) permit insights into the structural arrangement of the octylamine and oleic acid passivating agents on the triple cation and CsPbBr3 nanocrystal surfaces to be derived. Simple pictorial representations depicting these long-chain aliphatic species as notionally radiating outwards from these nanocrystal surfaces (see Fig. 7d) do not account for the complex array of correlations observed in Fig. 6b, c and Fig. S7b–e (ESI‡) for the triple cation and CsPbBr3 nanocrystal surfaces, respectively. The fully elongated octylamine and oleic acid configurations represented in Fig. 7d are ∼10 and ∼22 Å in length, respectively, suggesting that the CH3 chain ends would not sense the NH3+ and CO2− head group functionalities of the triple cation or CsPbBr3 nanocrystal surfaces with these measurements. However, intramolecular correlations measured from the triple cation system (see Fig. 6c) such as CH3 (octylamine; δ 0.6 ppm)/α-CH2 (octylamine; δ 3.3 ppm) and CH3 (octylamine; δ 0.6 ppm)/NH3+ (octylamine; δ 6.6 ppm) provide direct evidence for dipolar contact between the octylamine CH3 chain end and its polar head group. The only arrangement facilitating these correlations in the 2D 1H–1H NOESY data is when the octylamine folds back on itself towards the nanocrystal surface as depicted in Fig. 7d, promoting a highly disordered and entangled coverage of the triple cation nanocrystal surfaces by the passivation layer. From Fig. 7d a similar yet more pronounced arrangement is observed for the oleic acid passivation of the CsPbBr3 nanocrystal surface. Since octylamine dominates the surface passivation on the triple cation nanocrystal surfaces, a clearer representation of the oleic acid behavior is provided by the 2D 1H–1H NOESY data from the CsPbBr3 nanocrystal surface shown in Fig. S7b–e (ESI‡) where a near-equimixture of the octylamine and oleic acid species exist. These features demonstrate that mid-points of the oleic chain interact with regions near the nanocrystal surface as illustrated in Fig. 7d indicating that a highly folded and entangled arrangement again emerges.
Photophysical properties
The reported photophysical data of Fig. 8a and Table 2 show that the as-synthesized triple cation nanocrystals were highly luminescent under UV excitation and exhibit a high PLQY. As summarized in Table 2, purified samples of the 0 mol% Cs, 5 mol% Cs, 10 mol% Cs and 15 mol% Cs preparations display 80%, 93%, 85% and 82% PLQY, respectively, corresponding to the average carrier lifetime data of 11.7, 14.5, 13.5 and 11.6 ns as detailed in Fig. S8 (ESI‡). The increase in carrier lifetime (and thus increased exciton survival lifetime) measured for the 5 mol% Cs sample indicates the suppression of non-radiative pathways and a concomitant enhancement of the radiative channels in this sample associated with the increase in the observed PLQY. Indeed, the PLQY is proportional to the radiative lifetime (τr), hence perovskite quantum dots with a high PLQY can be realized when the radiative recombination pathways are increased.63,64| Sample name | PLQY (%) | No. of devices | Turn on voltage [Vt] | Peak luminance [cd m−2] | Current efficiency [cd A−1] | Luminous power efficiency [lm W−1] | EQE [%] |
|---|---|---|---|---|---|---|---|
| 0 mol% Cs | 80 | 8 | 2.04 [2.06] | 10218 [9243] |
12.74 [11.48] | 14.64 [12.82] | 3.12 [2.81] |
| 5 mol% Cs | 93 | 8 | 1.98 [2.01] | 10742 [10056] |
30.00 [26.65] | 34.87 [30.38] | 7.36 [6.52] |
| 10 mol% Cs | 85 | 7 | 2.05 [2.11] | 5240 [3400] | 24.62 [22.15] | 27.84 [24.51] | 5.92 [5.31] |
| 15 mol% Cs | 82 | 7 | 2.10 [2.20] | 2159 [1830] | 20.91 [19.17] | 21.58 [19.51] | 5.14 [4.73] |
These properties directly correlate with specific structural elements identified by the solid state NMR study. In particular, as the particle size decreases the 133Cs MAS NMR data reveals the emergence of an upfield 133Cs shift that identifies well-defined surface/subsurface Cs+ cation positions populating nanocrystal surface of the 5 mol% Cs composition. These surface Cs+ cation positions display noticeably longer (and field dependent) T1 relaxation properties in comparison to those predisposed towards the nanocrystal core. As discussed above, the increased Cs+ surface population and concomitant surface passivation induces an overall increased environmental stability and reduced ionic migration that is characterized by longer T1 relaxation values. Only the 5 mol% Cs triple cation composition exhibits the occurrence of these clearly resolved 133Cs shifts in significant concentrations, and this distinctly partitioned T1 behavior. Previous work has postulated that increased populations of surface/subsurface Cs+ cation species are associated with improved surface passivation and environmental stability, which can yield higher PLQYs and longer carrier lifetimes.35,37,40,48
While these characteristics rationalise the trend of increased carrier lifetimes, PLQYs and overall performance of the 5 mol% Cs preparation in relation to the other triple cation samples within the series, they do not explain the absolute increase in performance of the triple cation series in comparison to the CsPbBr3 nanocrystals. Other factors need to be considered to account for the absolute difference in performance between these systems. As observed in Fig. S9 (ESI‡), the CsPbBr3 nanocrystals exhibit a PL peak at 516 nm with PLQY of ∼50%, and the LEDs based on these nanocrystals exhibit an EQE of 2.5% which represents a lower performance in comparison to the triple cation analogues reported Table 2. The lower PLQY and device performance can be attributed to the higher framework Br− and Cs+ cation vacancy concentrations present on the surface of the CsPbBr3 nanocrystals, as reported by recent computational and electric field driven ion migration studies focusing on Br− and Cs+ migration dynamics.32,59 Most importantly, the 1D 13C and 1H MAS NMR, and 2D 1H MAS NMR studies described in Fig. 4–7 and Fig. S7 (ESI‡) clearly suggest that a large negative charge resides on the triple cation nanocrystal surfaces facilitating a predominance of the shorter and more conductive octylamine as the passivation ligand. These data confirm that an octylamine:oleic acid ratio of ∼5:1–6:1 exists throughout the triple cation series in comparison to an octylamine:oleic acid ratio of ∼1.4:1 for CsPbBr3 nanocrystals. In addition to the presence of surface/subsurface Cs+ cation species being associated with improved surface passivation (as discussed above), the exposed (abundant) Br− framework species on the curved triple cation nanocrystal surface are also stabilized through multiple ionic associations to the disordered network of abundant octylamine NH3+ head groups, as depicted in the schematic description of Fig. 7a. Furthermore, the approximate nearest-neighbour Br− site migration distances between vacancy/defect positons on the CsPbBr3 nanocrystal surface involves hops of ∼5.88/4.15 Å, however same process on the triple cation nanocrystal surface is estimated to occur across longer distances of ∼5.98/4.23 Å making this a less probable event, thus enabling longer carrier lifetimes and suppression of non-radiative pathways for improved PLQYs. An alteration of the surface passivation composition could stimulate increased conduction and higher LED efficiencies in the devices based on the triple cation nanocrystals in comparison to the CsPbBr3 nanocrystals-based devices (see Table 1).
Additional consideration also needs to be given to the consequences of the MA+ and FA+ cation dynamics which participate in ultra-fast anisotropic reorientation which are attributes that are absent in the CsPbBr3 system. Quasi elastic neutron scattering, GHz spectroscopy, 14N NMR and electron diffraction studies have measured this phenomenon with the resulting reorientation times (τrot) residing in the picosecond range.62,65–69 Computational and modelling studies focusing on the MA+ cation motion have demonstrated that these fast molecular rotations lead to dynamic modulation of the band structure, with the anisotropic reorientation increasing electronic contact (orbital overlap) with the lead halide cages consequently promoting a larger polaron formation.70 In turn, this enhances the radiative recombination process and thus inducing superior conversion efficiencies and PLQYs.
Fig. 8b describes the absorption and emission characteristics of the as-synthesized triple cation nanocrystals dispersed in hexane; these data show a marginal blue shift in absorption edge with increasing Cs+ incorporation into the cation inventory, similar to that reported for other perovskite systems.10,41,62,71 The PL peak position also shifted to the blue spectral region from 525–518 nm as the Cs content increased from 0 mol% to 15 mol%, although this observation is somewhat mitigated by the subtle and monotonic corresponding increase in nanocrystal diameter which would induce an opposing red shift.10 From Fig. 8c it can be observed that the triple cation nanocrystals revealed promising photostability up to 5 h with constant illumination (λ = 350 nm), with the first 2 h of constant illumination showing no indication of peak shifts and intensity changes. Under ambient conditions, the triple cation samples were also found to be stable in colloidal suspensions for up to ∼30 days, while the non-Cs containing samples were comparatively less stable in similar suspensions and were viable for only ∼10 days. These narrow emission line width, high PLQY and photostability characteristics demonstrate that this material is a suitable candidate for LED applications.
Green-emitting LEDs were fabricated from purified nanocrystals stably dispersed in hexane. The LED device structure comprised: indium tin oxide (ITO)/poly(3,4-ethylenedioxythiophene) polystyrene sulfonate (PEDOT:PSS, 45 nm)/poly(4-butylphenyl-diphenyl-amine) (Poly-TPD, 30 nm)/NCs (45 nm)/2,4,6-tris[3-(diphenylphosphinyl)phenyl]-1,3,5-triazine (POT2T, 45 nm)/LiF (0.8 nm)/Al (80 nm) respectively. PEDOT:PSS and poly-TPD served as a hole injecting layers, while POT2T was an electron injecting layer. A detailed description of the device fabrication process is given in the ESI.‡ The schematic and energy diagram of the LED device structure is presented in Fig. 9a. While energy levels for the organic transport layers were obtained from literature values,5,39 the energy levels of 5 mol% Cs nanocrystals were estimated from photoelectron spectroscopy (PESA) measurements reported in Fig. S10 (ESI‡).
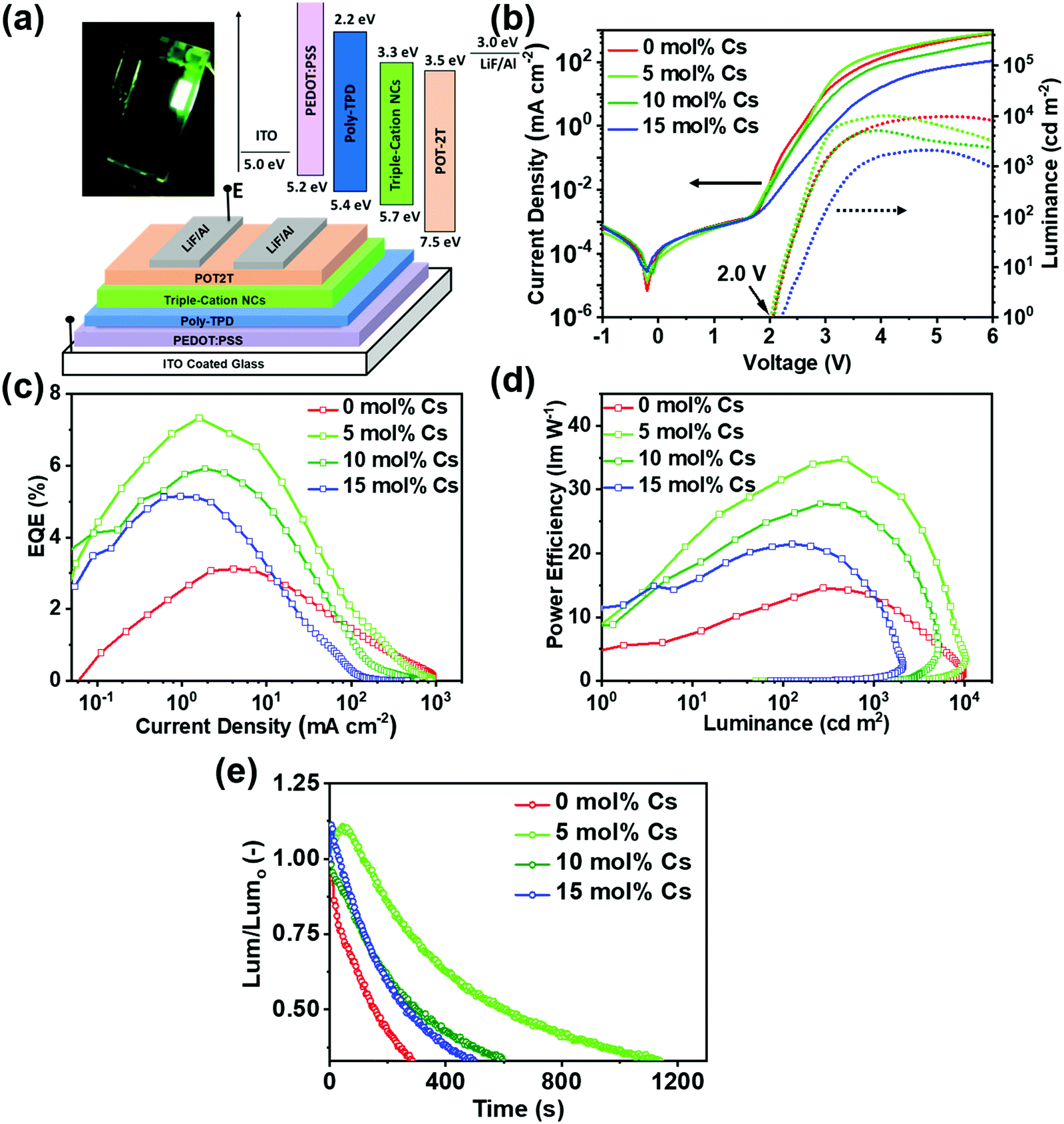 | ||
| Fig. 9 The device characteristics measured from triple cation Csx(MA0.17FA0.83)1−xPbBr3 (x = 0–0.15) series depicting, (a) a schematic diagram and energy comparison of the LED device structure along with a photograph of 5 mol% Cs LED operated at 3.00 V, (b) a plot of current density versus voltage demonstrating that these devices were characterized by low turn-on voltages of ∼2.0 V, (c) a plot of EQE versus current density, (d) a plot of power efficiency vs. luminance, and (e) a study of the device stability at constant current density represented as normalized luminance (Lum/Lumo) as a function of time. For the latter study, the Lumo measurements for the 0 mol%, 5 mol%, 10 mol% and 15 mol% samples were 77.4, 97.7, 107.4 and 110.1 cd m−2, respectively. These Cs-containing devices demonstrate significantly higher device stability with the 5 mol% Cs device exhibiting the highest stability under constant current density. | ||
The LEDs show bright green emission (i.e. in the 520–530 nm spectral region) with a narrow emission linewidth (FWHM = 19 nm). The inset of Fig. 9a shows LEDs fabricated using the 5 mol% Cs sample at 3 V. A marginal red shift of a few nanometres was observed between the electroluminescence (EL) and PL spectra due to the transition from solution to thin-films, wherein Förster resonance energy transfer (FRET) takes place to lower the band gap between neighboring nanocrystals within ∼1 nm (see Fig. S11a, ESI‡).72 As demonstrated in Fig. 9b, all devices exhibit standard diode characteristics with low leakage current and very low turn-on voltages in the range of 2.0–2.1 V, which is minimized in 5 mol% Cs sample. The paired maximum luminance and maximum luminance at peak EQE values were 9662; 276 cd m−2, 10207; 474 cd m−2, 5100; 460 cd m−2 and 2090; 201 cd m−2 measured from the 0 mol% Cs, 5 mol% Cs, 10 mol% Cs and 15 mol% Cs samples, respectively (see Fig. 9b and Fig. S11b, ESI‡). Fig. 9c shows plots of EQE vs. current density for each device, where the EQE increases rapidly and significantly above their turn-on voltage, with roll-off at higher current density. Efficiency roll-off at higher current density is due to Auger recombination.73,74 Overall, the 0 mol% Cs, 5 mol% Cs, 10 mol% Cs and 15 mol% Cs nanocrystal systems deliver maximum EQEs (current efficiencies) of 3.12% (12.53 cd A−1), 7.36% (30.01 cd A−1), 5.92% (24.52 cd A−1) and 5.14% (20.81 cd A−1), respectively (see Fig. 9c and Fig. S11c, ESI‡). From Fig. 9d, a plots of luminous power efficiency vs. luminance displayed indicates that the device employing 5 mol% Cs displays the maximum power efficiency of 34.76 lm W−1 at 478 cd m−2, with a minimum power efficiency of 14.58 lm W−1 at 276 cd m−2 of luminance characterizing the 0 mol% Cs device.
The performance metrics for all LEDs are listed in Table 2. The trend in LED efficiency is in agreement with PLQY and carrier lifetime data. As a small amount of dibenzo-21-crown-7-ether was used in the triple cation nanocrystal synthesis, it was pertinent to rule out any effect of this component on the device efficiency. LED devices were fabricated using the 0 mol% Cs product with and without the incorporation of crown ether; from this comparison no noticeable improvement in device performance was observed (see Fig. S12 and Table S3, ESI‡). This test concludes that the highest EQE achieved from the 5 mol% Cs LED can be attributed to the incorporation of an optimum 5 mol% Cs content into the nanocrystal structure. Furthermore, as demonstrated in Fig. 9e, devices prepared from Cs-containing triple cation nanocrystals displayed improved stability at constant current density, with the 5 mol% Cs device revealing the highest stability at a constant current density of 1.58 mA cm−2. In particular, the 5 mol% Cs device displays a two-fold increase in current stability in comparison the other triple cation devices, corroborating the increased Cs+ passivation and surface environmental stability in this sample.23,73 Nonetheless, a detailed micro-level analysis is warranted to completely investigate the device mechanism for each sample. From Fig. S11d (ESI‡), all these devices show EL peak stability under the applied electric field, which is a crucial factor for future perovskite LED commercialization. It should be noted that no further surface passivation schemes were explored in addition to the octylamine/oleic nanocrystal capping reported here in order to investigate improved conduction properties. Nevertheless, future research will entail the additional optimization of numerous aspects to enhance the device performance. For instance, the implementation of an ‘n–i–p’ device structure and the use of metal nanocrystals in perovskite active layer solutions to enable localized surface plasmon effects were found to be an effective way to improve the EQE and perovskite LED stability.75,76
Conclusion
This study has described the development of a high performance Cs-containing triple cation perovskite nanocrystal system for optoelectronic applications. The room temperature synthesis of triple cation nanocrystals of nominal stoichiometry Csx(MA0.17FA0.83)1−xPbBr3 (x = 0–0.15) was demonstrated, and this new system was able to deliver high PLQY of up to ∼93% while maintaining excellent photostability under continuous UV illumination. An optimized NC purification process performed to obtain high quality nanocrystals ink for LED fabrication. The resultant LEDs displayed green-emitting characteristics, with a maximum EQE of 7.4% (30 cd A−1) corresponding to a power efficiency of 34.87 lm W−1 being achieved from the compositionally optimized Cs0.05(MA0.17FA0.83)0.95PbBr3 (5 mol% Cs) triple cation system. Although the 5 mol% Cs nanocrystal sample was the best performing composition, all samples comprising this suite exhibited improved operational characteristics over the more established CsPbBr3 nanocrystal system. In contrast to the cubic morphology characterizing CsPbBr3 nanocrystals, the triple cation nanocrystal morphology is dominated by spherical particles that exhibit increased thermal stability and durability under the TEM-EDXS beam, and with the specific size range being sensitive to the amount of Cs incorporation.A multinuclear solid state 133Cs, 207Pb 1H and 13C MAS NMR study reveals significant structural disorder within the A-site cation (Cs+, FA+ and MA+) species, and within the octylamine and oleic acid agents passivating the nanocrystal surface, despite the highly ordered PbBr64− perovskite framework underpinning the overall structure (space group Pmm, a = 5.984–5.944 Å). The TEM-EDXS and 133Cs MAS NMR studies suggest that the Cs+ cations are dispersed heterogeneously throughout the spherical nanocrystals; this dispersion appears concentration and particle size dependent. While the Cs speciation is essentially partitioned towards the centre of the larger diameter/higher Cs content (15 mol% Cs) nanocrystals in a ‘pseudo-core–shell’ arrangement, the data from smaller diameter/lower Cs concentration systems (i.e. 10 mol% Cs and 5 mol% Cs) demonstrates an increasing sensitivity to the particle surface with additional upfield (lower ppm) 133Cs resonances being observed. These findings suggest a gradual departure from the ‘pseudo-core–shell’ configuration as the nanocrystal diameter decreases, with the 5 mol% Cs preparation demonstrating the emergence of substantial populations of surface and sub-surface Cs+ cation positions characterized by resolved upfield 133Cs chemical shifts and highly partitioned T1 behavior. This study provides the first solid state NMR evidence linking improved functional properties such as higher PLQYs and surface carrier lifetimes with increased surface passivation and environmental stability, as evidenced by the longer T1 relaxation of the surface Cs+ speciation in the 5 mol% Cs system suggesting slower migration dynamics in comparison to the core Cs+ positions.
The corresponding 1D and 2D 1H and 13C MAS NMR studies demonstrates that the relative FA+:MA+ ratio of ∼4:1 in the initial reaction is maintained throughout this nanocrystal series in close association with the initial nominal stoichiometry. Furthermore, the octylamine surface passivation agent exhibits a far greater binding affinity to the triple cation nanocrystal surface than its oleic acid counterpart (i.e. octylamine:oleic acid ∼6:1 on average throughout the series), despite the ∼20:1 predominance of oleic acid in the precursor mixture. This observation contrasts markedly with similar measurements characterizing the CsPbBr3 nanocrystal surface that shows the support of a near equi-mixture of octylamine and oleic acid (∼1.4:1 ratio). These studies also conclusively prove that other organic solvents such as dibenzo-21-crown-7-ether, DMF and toluene are not transferred from reaction mixtures onto the triple cation and CsPbBr3 nanocrystal surfaces. Although, the crown ether is detected in trace quantities (<1%) it plays no functional role on the surface of either nanocrystalline material. The octylamine dominance suggests that the triple cation nanocrystal surface carries a large negative charge via the exposure of Br− positions on the disordered spherical particle surface. More complex 2D homonuclear 1H–1H NOESY measurements show strong intermolecular FA-octylamine and MA-octylamine correlations to sparse FA+ and MA+ cation surface populations, and much greater FA+ and MA+ sub-surface populations. More importantly, these data also reveal additional correlations that show the local structures of both passivation ligands do not radiate outward from the particle core, but instead fold back towards the nanocrystal surface thus contributing further to the intrinsic disorder of both the triple cation and CsPbBr3 nanocrystal surfaces.
While the superior photophysical properties of the 5 mol% Cs system in comparison to the other triple cation preparations with higher Cs+ cation concentrations can be rationalized in terms of increased surface passivation and reduced Cs+ migration, the superior performance of the triple cation nanocrystal series over its CsPbBr3 counterparts is explained in terms of other contributing factors. As evidenced by the 1D 13C and 1H MAS NMR, and 2D 1H MAS NMR studies indicate that a large negative charge resides on the triple cation nanocrystal surfaces facilitating a predominance of the shorter and more conductive octylamine passivation ligand. These characteristics are coupled with the ultra-fast anisotropic reorientation dynamics of the MA+ and FA+ cations (absent in the CsPbBr3 system) which can induce larger polaron formation, enhanced readiative recombination and superior PLQYs to yield the reported improvements of in this work. Since this study represents the first investigation of triple cation nanocrystal systems, a broadened synthesis strategy is expected to drive improvements in device performance. Further studies will alter the ligand exchange process in order to improve the surface passivation of the nanocrystals, and the conductivity of the nanocrystal thin films. Other approaches could involve the incorporation of different halides (i.e. Cl or I), or novel mixed halide Cl/Br, Br/I triple cation combinations, to stimulate the further tuning of the band gap and emission properties of these nanocrystals for LEDs applications. These materials are also suitable for solar cell applications where hole-transport from the photo absorber perovskite film to the metal electrode can be facilitated. Hence, the nanocrystal series detailed herein could promote a wide range of possibilities towards numerous optoelectronic applications.
Experimental
Materials
Cesium bromide (99.999% trace metal basis), PbBr2 (99.999% trace metal basis, Aldrich), dibenzo-21-crown-7 ether (97%, Aldrich), PbI2 (99.999%, trace metal basis), MABr (Greatcellsolar), FABr (Greatcellsolar), N,N-dimethylformamide (anhydrous, 99.8%, Aldrich), toluene (anhydrous, 99.8%, Aldrich), hexane (anhydrous, 95%, Aldrich), octylamine (99%, Aldrich), and oleic acid (technical grade, 90%, Aldrich).Synthesis
Triple cation nanocrystals incorporating 0, 5, 10 and 15 mol% Cs were prepared using a ligand assisted re-precipitation method (LARP) method.35,45–49 A precursor solution containing CsBr, FABr and MABr (total of 0.1 mmol), PbBr2 (0.1 mmol) and dibenzo-21-crown-7-ether (same mmol as CsBr) was prepared in 1 ml of DMF in argon filled glove box at room temperature. The molar ratio between Cs and (FA0.83MA0.17) was varied from 0:1 to 0.15:0.85 (see Table S1, ESI‡). 150 μl of the precursor solution was added dropwise into a solution containing 5 ml toluene, 21 μl octylamine and 474 μl oleic acid under continuous stirring in ambient conditions. The NCs were instantly precipitated and a bright green-yellow solution was obtained. After the reaction was completed, the NCs were centrifuged at 10000 rpm for 12 min. The resulting supernatant phase was discarded and the precipitate was re-dispersed in 250 μl hexane followed by further centrifugation at 1000 rpm for 12 min. The resultant supernatant was used for further characterization and device fabrication as a NC ink. In order to get a high NC ink concentration, 5 to 6 separate reactions were performed and merged during the washing process.
A similar LARP methodology was adopted to synthesize pure CsPbBr3 nanocrystals, where a 0.1 mmol mixture of CsBr and PbBr2, with 0.1 mmol of dibenzo-21-crown-[7] ether, were dissolved in 1 ml of DMF. 150 μl of this precursor solution was added drop-wise into a 5 ml solution containing toluene, oleic acid and octylamine under stirring. The average size of these nanoparticles was determined to be ∼11 nm from HR-TEM analysis (see Fig. S1e, ESI‡), which is also a typical size of CsPbBr3 nanocrystals synthesized using a conventional (hot-injection) method.10,32 All details describing the nanocrystal characterization, and LED fabrication and measurement, can be found in the ESI.‡
Author contributions
P. V., S. A. V., S. G. M. and J. V. H. designed the initial experiments related to the material synthesis, device fabrication and characterisation. P. V. performed all the synthesis, XRD and TEM characterization, optical measurements, and device fabrication. Valuable assistance for the optical measurements was given by M. S. A., A. B., N. M., and A. A. M. B. and for the crystallographic analyses was given by Y. F. and T. W. J. V. H. designed and guided the all solid state MAS NMR measurements pertaining to the surface and bulk structural characterisation, with experimental and data processing contributions coming from S. H. D., N. L. K. and B. E. G., J. V. H. and P. V. wrote the manuscript and all authors viewed it and contributed to its content. S. G. M., T. W., P. V., and J. V. H. secured the necessary funding for this study.Conflicts of interest
There are no conflicts to declare.Acknowledgements
PV acknowledges a Presidential Postdoctoral Fellowship from Nanyang Technological University (NTU), Singapore via grant M408070000, and SGM, TW and SSHD acknowledge financial support from the Singapore National Research Foundation, Prime Minister's Office, through the Competitive Research Program (CRP Award No. NRF-CRP14-2014-03). NLK thanks EPSRC for a PhD studentship through the EPSRC Centre for Doctoral Training in Molecular Analytical Science (EP/L015307/1). The authors would like to acknowledge the Facility for Analysis, Characterization, Testing and Simulation (FACTS) at NTU, Singapore, for use of their electron microscopy and X-ray diffraction facilities. The authors would also like to thank Dr Bahulayan Damodaran for helpful discussions and assistance. JVH acknowledges financial support for the solid state NMR instrumentation at Warwick used in this research which was funded by EPSRC (grants EP/M028186/1 and EP/K024418/1), the University of Warwick, and the Birmingham Science City AM1 and AM2 projects which were supported by Advantage West Midlands (AWM) and the European Regional Development Fund (ERDF).References
- D. V. Talapin, J.-S. Lee, M. V. Kovalenko and E. V. Shevchenko, Chem. Rev., 2010, 110, 389–458 CrossRef CAS PubMed.
- J. Huang, Y. Yang, S. Xue, B. Yang, S. Liu and J. Shen, Appl. Phys. Lett., 1997, 70, 2335–2337 CrossRef CAS.
- I. Robel, M. Kuno and P. V. Kamat, J. Am. Chem. Soc., 2007, 129, 4136–4137 CrossRef CAS PubMed.
- S. Mehra, S. Bishnoi, A. Jaiswal, M. Jagadeeswararao, A. K. Srivastava, S. N. Sharma and P. Vashishtha, Energy Storage, 2019, e120 Search PubMed.
- P. O. Anikeeva, J. E. Halpert, M. G. Bawendi and V. Bulovic, Nano Lett., 2009, 9, 2532–2536 CrossRef CAS PubMed.
- W. Zou, Z.-j. Du, H.-q. Li and C. Zhang, J. Mater. Chem., 2011, 21, 13276–13282 RSC.
- Z.-S. Guo, L. Zhao, J. Pei, Z.-L. Zhou, G. Gibson, J. Brug, S. Lam and S. S. Mao, Macromolecules, 2010, 43, 1860–1866 CrossRef CAS.
- O. I. Mićić, J. Sprague, Z. Lu and A. J. Nozik, Appl. Phys. Lett., 1996, 68, 3150–3152 CrossRef.
- H. Zhong, S. S. Lo, T. Mirkovic, Y. Li, Y. Ding, Y. Li and G. D. Scholes, ACS Nano, 2010, 4, 5253–5262 CrossRef CAS PubMed.
- J. Butkus, P. Vashishtha, K. Chen, J. K. Gallaher, S. K. Prasad, D. Z. Metin, G. Laufersky, N. Gaston, J. E. Halpert and J. M. Hodgkiss, Chem. Mater., 2017, 29, 3644–3652 CrossRef CAS.
- P. Vashishtha, G. V. Nutan, B. E. Griffith, Y. Fang, D. Giovanni, M. Jagadeeswararao, T. C. Sum, N. Mathews, S. G. Mhaisalkar and J. V. Hanna, Chem. Mater., 2019, 31, 9003–9011 CrossRef CAS.
- L. Qian, Y. Zheng, J. Xue and P. H. Holloway, Nat. Photonics, 2011, 5, 543–549 CrossRef CAS.
- Y. Yang, Y. Zheng, W. Cao, A. Titov, J. Hyvonen, J. R. Manders, J. Xue, P. H. Holloway and L. Qian, Nat. Photonics, 2015, 9, 259–266 CrossRef CAS.
- J. S. Steckel, P. Snee, S. Coe-Sullivan, J. P. Zimmer, J. E. Halpert, P. Anikeeva, L. A. Kim, V. Bulovic and M. G. Bawendi, Angew. Chem., Int. Ed., 2006, 45, 5796–5799 CrossRef CAS PubMed.
- X. Dai, Z. Zhang, Y. Jin, Y. Niu, H. Cao, X. Liang, L. Chen, J. Wang and X. Peng, Nature, 2014, 515, 96–99 CrossRef CAS PubMed.
- P. Vashishtha, D. Z. Metin, M. E. Cryer, K. Chen, J. M. Hodgkiss, N. Gaston and J. E. Halpert, Chem. Mater., 2018, 30, 2973–2982 CrossRef CAS.
- V. A. Hintermayr, A. F. Richter, F. Ehrat, M. Döblinger, W. Vanderlinden, J. A. Sichert, Y. Tong, L. Polavarapu, J. Feldmann and A. S. Urban, Adv. Mater., 2016, 28, 9478–9485 CrossRef CAS PubMed.
- A. Kojima, K. Teshima, Y. Shirai and T. Miyasaka, J. Am. Chem. Soc., 2009, 131, 6050–6051 CrossRef CAS PubMed.
- H.-S. Kim, C.-R. Lee, J.-H. Im, K.-B. Lee, T. Moehl, A. Marchioro, S.-J. Moon, R. Humphry-Baker, J.-H. Yum, J. E. Moser, M. Grätzel and N.-G. Park, Sci. Rep., 2012, 2, 591 CrossRef PubMed.
- Y. Rong, Y. Hu, A. Mei, H. Tan, M. I. Saidaminov, S. I. Seok, M. D. McGehee, E. H. Sargent and H. Han, Science, 2018, 361, eaat8235 CrossRef PubMed.
- E. H. Jung, N. J. Jeon, E. Y. Park, C. S. Moon, T. J. Shin, T.-Y. Yang, J. H. Noh and J. Seo, Nature, 2019, 567, 511–515 CrossRef CAS PubMed.
- Y. Ma, P. Vashishtha, K. Chen, E. L. Peach, D. Ohayon, J. M. Hodgkiss and J. E. Halpert, ChemSusChem, 2017, 10, 2677–2684 CrossRef CAS PubMed.
- M. Saliba, T. Matsui, J.-Y. Seo, K. Domanski, J.-P. Correa-Baena, M. K. Nazeeruddin, S. M. Zakeeruddin, W. Tress, A. Abate and A. Hagfeldt, Energy Environ. Sci., 2016, 9, 1989–1997 RSC.
- J. P. C. Baena, L. Steier, W. Tress, M. Saliba, S. Neutzner, T. Matsui, F. Giordano, T. J. Jacobsson, A. R. S. Kandada and S. M. Zakeeruddin, Energy Environ. Sci., 2015, 8, 2928–2934 RSC.
- M. Saliba, S. Orlandi, T. Matsui, S. Aghazada, M. Cavazzini, J.-P. Correa-Baena, P. Gao, R. Scopelliti, E. Mosconi and K.-H. Dahmen, Nat. Energy, 2016, 1, 15017 CrossRef CAS.
- D. Bi, W. Tress, M. I. Dar, P. Gao, J. Luo, C. Renevier, K. Schenk, A. Abate, F. Giordano and J.-P. C. Baena, Sci. Adv., 2016, 2, e1501170 CrossRef PubMed.
- M. Azam, S. Yue, R. Xu, K. Liu, K. Ren, Y. Sun, J. Liu, Z. Wang, S. Qu and Z. Wang, J. Mater. Chem. A, 2018, 6, 13725–13734 RSC.
- M. Stolterfoht, C. M. Wolff, Y. Amir, A. Paulke, L. Perdigón-Toro, P. Caprioglio and D. Neher, Energy Environ. Sci., 2017, 10, 1530–1539 RSC.
- C. Wang, C. Zhang, S. Wang, G. Liu, H. Xia, S. Tong, J. He, D. Niu, C. Zhou and K. Ding, Sol. RRL, 2018, 2, 1700209 CrossRef.
- Y. Wang, J. Wu, P. Zhang, D. Liu, T. Zhang, L. Ji, X. Gu, Z. D. Chen and S. Li, Nano Energy, 2017, 39, 616–625 CrossRef CAS.
- P. Vashishtha, M. Ng, S. B. Shivarudraiah and J. E. Halpert, Chem. Mater., 2018, 31, 83–89 CrossRef.
- P. Vashishtha and J. E. Halpert, Chem. Mater., 2017, 29, 5965–5973 CrossRef CAS.
- Z. Xiao, R. A. Kerner, L. Zhao, N. L. Tran, K. M. Lee, T.-W. Koh, G. D. Scholes and B. P. Rand, Nat. Photonics, 2017, 11, 108–115 CrossRef CAS.
- K. Lin, J. Xing, L. N. Quan, F. P. G. de Arquer, X. Gong, J. Lu, L. Xie, W. Zhao, D. Zhang and C. Yan, Nature, 2018, 562, 245–248 CrossRef CAS PubMed.
- S. A. Veldhuis, Y. F. Ng, R. Ahmad, A. Bruno, N. F. Jamaludin, B. Damodaran, N. Mathews and S. G. Mhaisalkar, ACS Energy Lett., 2018, 3, 526–531 CrossRef CAS.
- M. Yuan, L. N. Quan, R. Comin, G. Walters, R. Sabatini, O. Voznyy, S. Hoogland, Y. Zhao, E. M. Beauregard and P. Kanjanaboos, Nat. Nanotechnol., 2016, 11, 872–877 CrossRef CAS PubMed.
- G. Li, F. W. R. Rivarola, N. J. Davis, S. Bai, T. C. Jellicoe, F. de la Peña, S. Hou, C. Ducati, F. Gao and R. H. Friend, Adv. Mater., 2016, 28, 3528–3534 CrossRef CAS PubMed.
- F. Yan and H. V. Demir, Nanoscale, 2019, 11, 11402–11412 RSC.
- X. Y. Chin, A. Perumal, A. Bruno, N. Yantara, S. A. Veldhuis, L. Martínez-Sarti, B. Chandran, V. Chirvony, A. S.-Z. Lo and J. So, Energy Environ. Sci., 2018, 11, 1770–1778 RSC.
- A. A. M. Brown, T. J. N. Hooper, S. A. Veldhuis, X. Y. Chin, A. Bruno, P. Vashishtha, J. N. Tey, L. Jiang, B. Damodaran and S. H. Pu, Nanoscale, 2019, 11, 12370–12380 RSC.
- X. Zhang, H. Liu, W. Wang, J. Zhang, B. Xu, K. L. Karen, Y. Zheng, S. Liu, S. Chen and K. Wang, Adv. Mater., 2017, 29, 1606405 CrossRef PubMed.
- B. Xu, W. Cao, X. Zhang, W. Wang, J. Hao, S. Chen, K. Wang and X. Sun, Conference on Lasers and Electro-Optics (CLEO), 2017, 1–2.
- I. Borriello, G. Cantele and D. Ninno, Phys. Rev. B: Condens. Matter Mater. Phys., 2008, 77, 235214 CrossRef.
- H. P. Kim, J. Kim, B. S. Kim, H. M. Kim, J. Kim, A. R. B. M. Yusoff, J. Jang and M. K. Nazeeruddin, Adv. Opt. Mater., 2017, 5, 1600920 CrossRef.
- X. Du, G. Wu, J. Cheng, H. Dang, K. Ma, Y.-W. Zhang, P.-F. Tan and S. Chen, RSC Adv., 2017, 7, 10391–10396 RSC.
- F. Zhang, H. Zhong, C. Chen, X.-G. Wu, X. Hu, H. Huang, J. Han, B. Zou and Y. Dong, ACS Nano, 2015, 9, 4533–4542 CrossRef CAS PubMed.
- I. Levchuk, A. Osvet, X. Tang, M. Brandl, J. D. Perea, F. Hoegl, G. J. Matt, R. Hock, M. Batentschuk and C. J. Brabec, Nano Lett., 2017, 17, 2765–2770 CrossRef CAS PubMed.
- S. Bhaumik, S. A. Veldhuis, Y. F. Ng, M. Li, S. K. Muduli, T. C. Sum, B. Damodaran, S. Mhaisalkar and N. Mathews, Chem. Commun., 2016, 52, 7118–7121 RSC.
- L. Liu, S. Huang, L. Pan, L. J. Shi, B. Zou, L. Deng and H. Zhong, Angew. Chem., Int. Ed., 2017, 56, 1780–1783 CrossRef CAS PubMed.
- L. Protesescu, S. Yakunin, M. I. Bodnarchuk, F. Krieg, R. Caputo, C. H. Hendon, R. X. Yang, A. Walsh and M. V. Kovalenko, Nano Lett., 2015, 15, 3692–3696 CrossRef CAS PubMed.
- S. Govinda, B. P. Kore, D. Swain, A. Hossain, C. De, T. N. Guru Row and D. Sarma, J. Phys. Chem. C, 2018, 122, 13758–13766 CrossRef CAS.
- A. Perumal, S. Shendre, M. Li, Y. K. E. Tay, V. K. Sharma, S. Chen, Z. Wei, Q. Liu, Y. Gao and P. J. S. Buenconsejo, Sci. Rep., 2016, 6, 36733 CrossRef CAS PubMed.
- D. T. Cromer and J. B. Mann, Acta Crystallogr., Sect. A: Cryst. Phys., Diffr., Theor. Gen. Crystallogr., 1968, 24, 321–324 CrossRef CAS.
- D. Creagh and J. Hubbell, Acta Crystallogr., Sect. A: Found. Crystallogr., 1987, 43, 102–112 CrossRef.
- G. J. Rees, S. T. Orr, L. O. Barrett, J. M. Fisher, J. Houghton, G. H. Spikes, B. R. Theobald, D. Thompsett, M. E. Smith and J. V. Hanna, Phys. Chem. Chem. Phys., 2013, 15, 17195–17207 RSC.
- T. J. Hooper, T. A. Partridge, G. J. Rees, D. S. Keeble, N. A. Powell, M. E. Smith, I. P. Mikheenko, L. E. Macaskie, P. T. Bishop and J. V. Hanna, Phys. Chem. Chem. Phys., 2018, 20, 26734–26743 RSC.
- T. Chen, I. Ellis, T. J. Hooper, E. Liberti, L. Ye, B. T. Lo, C. O’Leary, A. A. Sheader, G. T. Martinez, L. Jones, P. L. Ho, P. Zhao, P. J. Cookson, P. T. Bishop, P. Chater, J. V. Hanna, P. Nellist and S. C. E. Tsang, J. Am. Chem. Soc., 2019, 141, 19616–19624 CrossRef CAS PubMed.
- T. C. Farrar and E. D. Becker, Pulse and Fourier Transform NMR: Introduction to Theory and Methods, Elsevier, 2012 Search PubMed.
- B. B. Zhang, F. Wang, H. Zhang, B. Xiao, Q. Sun, J. Guo, A. B. Hafsia, A. Shao, Y. Xu and J. Zhou, Appl. Phys. Lett., 2020, 116, 063505 CrossRef.
- J. De Roo, M. Ibáñez, P. Geiregat, G. Nedelcu, W. Walravens, J. Maes, J. C. Martins, I. Van Driessche, M. V. Kovalenko and Z. Hens, ACS Nano, 2016, 10, 2071–2081 CrossRef CAS PubMed.
- D. Neuhaus, eMagRes, 2007, 1–16 Search PubMed.
- T. Baikie, N. S. Barrow, Y. Fang, P. J. Keenan, P. R. Slater, R. O. Piltz, M. Gutmann, S. G. Mhaisalkar and T. J. White, J. Mater. Chem. A, 2015, 3, 9298–9307 RSC.
- J. Shi, F. Li, J. Yuan, X. Ling, S. Zhou, Y. Qian and W. Ma, J. Mater. Chem. A, 2019, 7, 20936–20944 RSC.
- C. H. Tien, L. C. Chen, K. Y. Lee, Z. L. Tseng, Y. S. Dong and Z. J. Lin, Energies, 2019, 12, 3507 CrossRef CAS.
- A. M. A. Leguy, J. M. Frost, A. P. McMahon, V. G. Sakai, W. Kochelmann, C. H. Law, X. E. Li, F. Foglia, A. Walsh, B. C. O'Regan, J. Nelson, J. T. Cabral and P. R. F. Barnes, Nat. Commun., 2015, 6 Search PubMed.
- D. H. Fabini, T. A. Siaw, C. C. Stoumpos, G. Laurita, D. Olds, K. Page, J. G. Hu, M. G. Kanatzidis, S. Han and R. Seshadri, J. Am. Chem. Soc., 2017, 139, 16875–16884 CrossRef CAS PubMed.
- X. Wu, L. Z. Tan, X. Shen, T. Hu, K. Miyata, M. T. Trinh, R. Li, R. Coffee, S. Liu, D. A. Egger, I. Makasyuk, Q. Zheng, A. Fry, J. S. Robinson, M. D. Smith, B. Guzelturk, H. I. Karunadasa, X. Wang, X. Zhu, L. Kronik, A. M. Rappe and A. M. Lindenberg, Sci. Adv., 2017, 3, e1602388 CrossRef PubMed.
- G. M. Bernard, R. E. Wasylishen, C. I. Ratcliffe, V. Terskikh, Q. Wu, J. M. Buriak and T. Hauger, J. Phys. Chem. A, 2018, 122, 1560–1573 CrossRef CAS PubMed.
- D. J. Kubicki, D. Prochowicz, A. Pinon, G. Stevanato, A. Hofstetter, S. M. Zakeeruddin, M. Gratzel and L. Emsley, J. Mater. Chem. A, 2018, 7, 2326–2333 RSC.
- C. Motta1, F. El-Mellouhi, S. Kais, N. Tabet, F. Alharbi and S. Sanvito, Nat. Commun., 2014, 1 Search PubMed.
- L. Protesescu, S. Yakunin, M. I. Bodnarchuk, F. Bertolotti, N. Masciocchi, A. Guagliardi and M. V. Kovalenko, J. Am. Chem. Soc., 2016, 138, 14202–14205 CrossRef CAS PubMed.
- P. O. Anikeeva, J. E. Halpert, M. G. Bawendi and V. Bulović, Nano Lett., 2007, 7, 2196–2200 CrossRef CAS PubMed.
- M. Yang, N. Wang, S. Zhang, W. Zou, Y. He, Y. Wei, M. Xu, J. Wang and W. Huang, J. Phys. Chem. Lett., 2018, 9, 2038–2042 CrossRef CAS PubMed.
- Y. Shirasaki, G. J. Supran, W. A. Tisdale and V. Bulović, Phys. Rev. Lett., 2013, 110, 217403 CrossRef PubMed.
- Z. Shi, Y. Li, S. Li, X. Li, D. Wu, T. Xu, Y. Tian, Y. Chen, Y. Zhang and B. Zhang, Adv. Funct. Mater., 2018, 28, 1707031 CrossRef.
- Z. Shi, Y. Li, Y. Zhang, Y. Chen, X. Li, D. Wu, T. Xu, C. Shan and G. Du, Nano Lett., 2017, 17, 313–321 CrossRef CAS PubMed.
Footnotes |
| † The experimental data for this study are provided as a supporting dataset from WRAP, the Warwick Research Archive Portal at https://wrap.warwick.ac.uk/134281. |
| ‡ Electronic supplementary information (ESI) available: Experimental details of the device fabrication and materials characterization including XRD, TEM, NMR, UV-vis, and PL methodologies. Also shown are TEM micrographs for size and lattice spacing estimation, powder XRD, FTIR, 133Cs T1 data acquired from MAS NMR, a graph describing the relationship between H–H distance vs. the number of protons in MAPbBr3 system, 2D homonuclear 1H–1H MAS NMR data from triple cation and CsPbBr3 systems, graphs showing the optoelectronic properties of the CsPbBr3 nanocrystals, PESA measurements, and remaining device characterisation for triple cation systems including the effect of crown ether, a table outlining the precursor concentrations, a table for elemental analysis using EDXS, and a table describing the effect of crown LED performance. See DOI: 10.1039/d0tc02038a |
| This journal is © The Royal Society of Chemistry 2020 |