
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBenzothiazolium-functionalized NU-1000: a versatile material for carbon dioxide adsorption and cyanide luminescence sensing†
Lapo
Luconi
a,
Giorgio
Mercuri
a,
Timur
Islamoglu
 b,
Andrea
Fermi
*c,
Giacomo
Bergamini
c,
Giuliano
Giambastiani
*ade and
Andrea
Rossin
*a
b,
Andrea
Fermi
*c,
Giacomo
Bergamini
c,
Giuliano
Giambastiani
*ade and
Andrea
Rossin
*a
aIstituto di Chimica dei Composti Organometallici (ICCOM-CNR), Via Madonna del Piano 10, 50019 Sesto Fiorentino, Italy. E-mail: giuliano.giambastiani@iccom.cnr.it; a.rossin@iccom.cnr.it
bDepartment of Chemistry, Northwestern University, 2145 Sheridan Road, Evanston, IL 60208, USA
cDipartimento di Chimica “G. Ciamician”, Università di Bologna, via Selmi 2, Bologna 40126, Italy. E-mail: andrea.fermi2@unibo.it
dInstitute of Chemistry and Processes for Energy, Environment and Health (ICPEES), UMR 7515 CNRS-University of Strasbourg (UdS), 25, rue Becquerel, 67087 Strasbourg Cedex 02, France
eKazan Federal University, Alexander Butlerov Institute of Chemistry, Kazan 420008, Russian Federation
First published on 16th April 2020
Abstract
A tailor-made benzothiazolium bromide salt functionality (BzTz) is introduced via solvent-assisted ligand incorporation (SALI) into the mesoporous Zr-based metal–organic framework NU-1000. The resulting NU-1000-BzTz composite has been thoroughly characterized in the solid state. The functional group loading has been determined through 1H NMR analysis of the digested sample (5% HF-DMSO-d6): a maximum value of 1.7 BzTz ligand per [Zr6] node is achieved. The material preserves its pristine crystallinity after SALI, as witnessed by powder X-ray diffraction. The functionalized MOF has a slightly lower thermal stability than its parent material (Tdec = 780 vs. 800 K, respectively). The N2 adsorption isotherm collected at 77 K disclosed that its BET specific surface area (1530 m2 g−1) is lower than that of prisitine NU-1000 (2140 m2 g−1), because of the space taken and weight added by the dangling benzothiazolium groups inside the pores. A total CO2 uptake of 2.0 mmol g−1 (8.7 wt% CO2) has been calculated from the CO2 adsoprtion isotherm collected at T = 298 K and pCO2 = 1 bar. Despite the lower BET area, NU-1000-BzTz shows an increased thermodynamic affinity for CO2 (isosteric heat of adsorption Qst = 25 kJ mol−1) if compared with NU-1000 (Qst = 17 kJ mol−1), confirming that the presence of a polar functional group in the MOF pores improves the interaction with carbon dioxide. Finally, NU-1000-BzTz has been exploited as a luminescent sensor for polluting anions (CN−, SCN−, OCN−, and SeCN− as sodium or potassium salts) in aqueous solutions, after bromide exchange. A marked reversible blue shift of its emission band from 490 to 450 nm is observed in all cases, with the associated emission color change from light green to blue under a UV lamp. The detection limit of CN− (1.08 × 10−6 M) is much lower than that measured for the other “stick-like” anions considered in this study. The process occurs efficiently even in the presence of other competing ions (i.e. in ordinary tap water), opening promising application perspectives in cyanide luminescence sensing in drinking water.
Introduction
Metal–organic frameworks (MOFs) undoubtedly represent the most widely studied coordination compounds in contemporary materials chemistry research.1–4 Crystalline and often highly porous, their chemical diversity and typically high surface areas give them strength for a broad range of potential applications including gas storage and separation,5–9 catalysis,10–13 sensing14–16 and light harvesting.17,18 Besides the infinite metal node-linker combinations conceivable for the construction of previously unknown crystalline scaffolds, their versatile nature can be further improved through the post-synthetic modification of already known structures. The introduction of new chemical entities into the MOF framework can be achieved through various approaches like covalent linker modification19,20 or partial replacement of the organic linkers using the so-called solvent-assisted linker exchange (SALE) technique.21–23 Another promising experimental protocol in this context (especially for the zirconium-based MOFs) is metal node functionalization through solvent-assisted ligand incorporation (SALI), i.e. the anchorage of dangling functional groups to the metal clusters at the corners of the MOF scaffold via an acid–base reaction between a monocarboxylic acid and the hydroxo groups present in the pristine MOF metallic secondary building units.24,25 MOFs derived from oxophilic [ZrIV6] nodes from the UiO family (UiO = University of Oslo) have been particularly exploited due to their high thermal (up to 500 °C), chemical (pH 1–pH 11) and mechanical stability.26 One example belonging to this class is NU-1000 (NU = Northwestern University),27 a mesoporous MOF of general formula [Zr6(μ3-OH)4(μ3-O)4(H2O)4(OH)4(TBAPy)2] containing [Zr6(μ3-OH)4(μ3-O)4(H2O)4(OH)4]8+ nodes where eight of the twelve coordination sites are occupied by TBAPy4− linkers [H4TBAPy = 1,3,6,8-tetrakis(p-benzoic-acid)pyrene, Fig. 1]. The four terminal –OH groups are reactive and present opportunities for channel chemical modification. Functionalization via SALI leads to a daughter material where the charge compensating –COO− ligands are strongly bound to the NU-1000 node in a chelating fashion. From the metal node stoichiometric composition, a maximum loading of four functional groups per [Zr6] node is thus possible. The as-synthesised samples retain the same crystallinity of the parent NU-1000 but they generally have a slightly lower surface area, because of the empty volume reduction and added weight upon functionalization. Previous work by some of us has already shown the great potentiality of this technique for the preparation of novel NU-1000-FG (FG = functional group) materials. Some of these compounds have shown excellent performances in carbon dioxide storage,28 with high absolute uptakes at ambient temperature and pressure conditions and enhanced thermodynamic affinity compared to their parent NU-1000.29 The improvement of the CO2 isosteric heat of adsorption (Qst) values has been achieved through the introduction of perfluorinated alkyl chains (because of the known affinity of supercritical CO2 for perfluoroalkanes)24,25,30 or decoration of the MOF pores with di- or tripeptides that present charge distributions complementary to those of quadrupolar CO2.31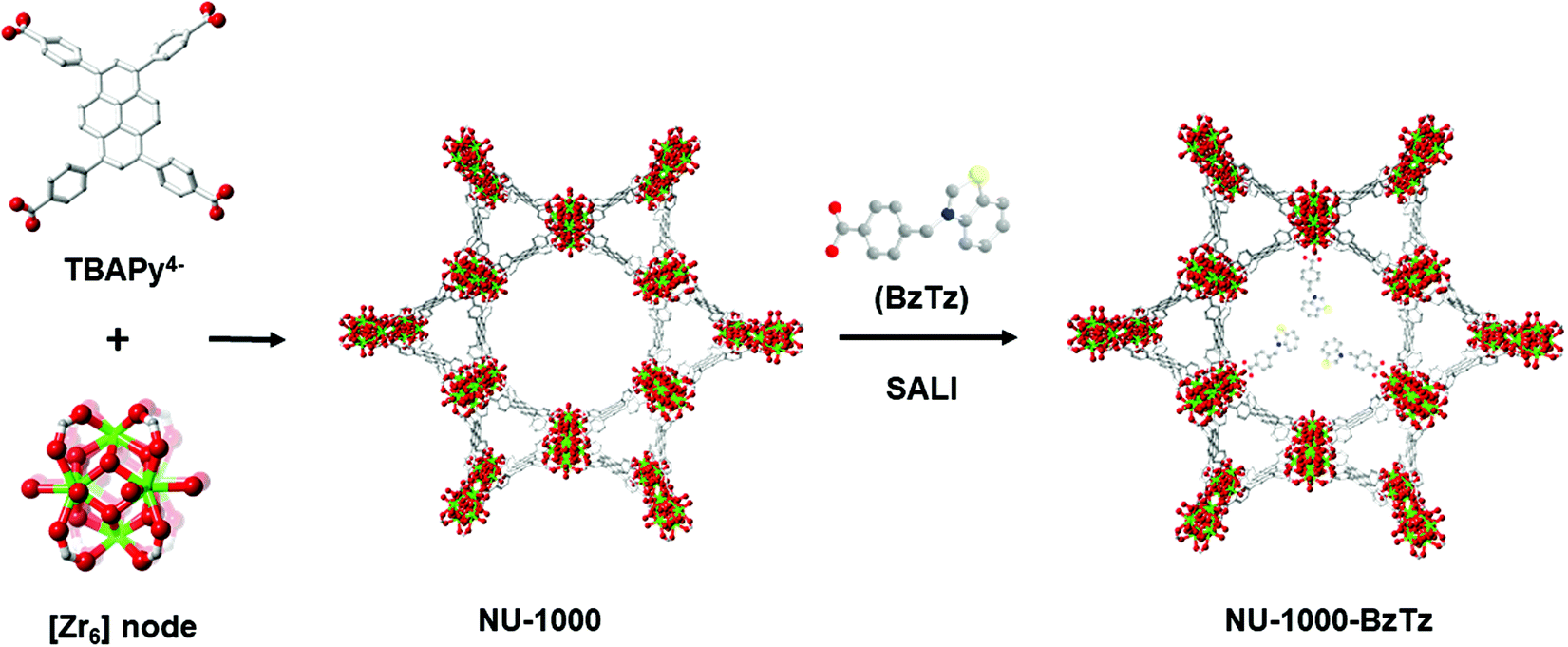 | ||
| Fig. 1 Molecular representations of NU-1000 and schematic representation of its SALI functionalization with a benzothiazolium salt to afford the new material NU-1000-BzTz (idealized structure only, no X-ray data from real sample available). | ||
Searching for additional applicative contexts of NU-1000-FG materials besides CO2 storage, we considered luminescent ion sensing. As chemical sensors, luminescent MOFs possess a number of advantages over other luminescent materials.32 Analyte adsorption within MOF pores allows for its pre-concentration, increasing sensor sensitivity. Selectivity can be achieved by tuning pore dimension and/or by proper functionalization of the linkers or the metallic nodes. In the most interesting cases, the guest target molecule induces a shift in the MOF emission frequency. This signal transduction is highly desirable because analyte uptake can be read out as a marked color change by the naked eye (if the emission band falls in the visible region). NU-1000-FG MOFs have already found applications in photocatalysis or in light harvesting.33–35 Only recently, a few examples focused on luminescence sensing of dibenzo-p-dioxins,36 cholesterol37 and 1-hydroxypyrene38 by NU-1000 or NU-1000-FG materials have started to appear in the literature. Our previous experience in MOF synthesis for CO2 storage39–41 and luminescence sensing42–44 prompted us to design a single NU-1000-FG material suitable for both targets. As (N,S)-containing and intrinsically luminescent heterocycles with basic sites that can interact with acidic CO2, thiazoles and thiazolium salts are the optimal choice in this context.45 In this work, we present the SALI functionalization of NU-1000 with a monocarboxylic acid decorated with a benzothiazolium tail: BzTz = 3-(4-carboxybenzyl)benzo[d]thiazolium bromide (Fig. 1). The resulting NU-1000-BzTz composite has been thoroughly characterized in the solid state and successfully exploited in two different applicative contexts: carbon dioxide adsorption and luminescence sensing of polluting anions in aqueous solutions.
Experimental section
Materials and methods
All the chemicals and reagents employed were purchased from commercial suppliers and used as received without further purification. NU-1000 was prepared according to the published procedures.46 For the organic syntheses, solvents were purified through standard distillation techniques. Deuterated solvents (Sigma Aldrich) were stored over 4 Å molecular sieves and degassed by three freeze–pump–thaw cycles before use. NMR spectra were recorded on a BRUKER AVANCE 400 MHz spectrometer. 1H and 13C{1H} NMR chemical shifts are reported in parts per million (ppm) downfield of tetramethylsilane (TMS) and were calibrated against the residual resonance of the protiated part of the deuterated solvent. FT-IR spectra (KBr pellets) were recorded on a PerkinElmer Spectrum BX Series FTIR spectrometer, in the 4000–400 cm−1 range, with a 2 cm−1 resolution. Thermogravimetric analysis measurements were performed under a N2 atmosphere (100 mL min−1) at a heating rate of 10 K min−1 on an EXSTAR Thermo Gravimetric Analyzer (TG-DTG) Seiko 6200. The C, H, N, and S elemental analyses were carried out at ICCOM-CNR using a Thermo FlashEA 1112 Series CHNS-O elemental analyzer with an accepted tolerance of ±2% for carbon (C), hydrogen (H), nitrogen (N) and sulfur (S). ESI-MS spectra were recorded using direct introduction (10 μL min−1) on a FinniganLTQ mass spectrometer (Thermo, San Jose, CA). The instrument was equipped with a conventional ESI source. The working conditions were the following: positive polarity: spray voltage 5 kV, capillary voltage 35 V, capillary temperature 548 K and tube lens 110 V. Sheath gas was set at 10 a.u. and auxiliary gas was kept at 3 a.u. For acquisition, Xcalibur 2.0 software (Thermo) was used. Aqueous sample solutions of BzTz were diluted to 20 ng μL−1. X-ray powder diffraction (PXRD) qualitative measurements were carried out using a Panalytical X’PERT PRO powder diffractometer equipped with a diffracted beam Ni filter and a PIXcel© solid state detector in the 4–50° 2θ region, operating with Cu Kα radiation (λ = 1.5418 Å). Anti-scatter slits were used both on the incident (0.25° and 0.5° divergence) and the diffracted (7.5 mm height) beams.Synthesis of 3-(4-carboxybenzyl)benzo[d]thiazolium bromide (BzTz)
4-(Bromomethyl)benzoic acid (0.950 g, 4.43 mmol, 1.2 eq.) was added to a solution of benzo[d]thiazole (0.400 mL, 3.7 mmol, 1.0 eq.) in dry and degassed acetonitrile (17 mL). The reaction mixture was left under solvent reflux for 48 h with stirring. During this time, formation of a white precipitate was observed. Afterwards, the mixture was cooled to room temperature and the precipitate was filtrated on a Büchner funnel and washed with fresh acetonitrile (3 × 10 mL) and diethyl ether (3 × 10 mL). The white solid residue was recovered and dried under vacuum (1.100 g, yield: 84.1%). Crystals suitable for X-ray diffraction were obtained from a concentrated methanolic solution at 277 K. The single-crystal X-ray diffraction data acquisition and treatment and the molecular structure are reported in the ESI† (Fig. S1 and Table S1). 1H NMR (400 MHz, DMSO-d6, 298 K, Fig. S2, ESI†): δ 6.26 (s, 2H, CH2, H8), 2.59 (d, 3JHH = 8.2 Hz, 2H, CH Ar, H10,14), 7.83 (m, 2H, CH Ar, H4,5), 7.94 (d, 3JHH = 8.2 Hz, 2H, CH Ar, H11,13), 8.23 (d, 3JHH = 8.1 Hz, 1H, CH Ar, H3), 8.56 (d, 3JHH = 8.1 Hz, 1H, CH Ar, H6), 10.87 (s, 1H, CH Ar, H1), 13.10 (brs, 1H, COOH). 13C{1H} NMR (100 MHz, DMSO-d6, 298 K, Fig. S3, ESI†): δ 54.7 (CH2, C8), 117.3 (CH Ar, C3), 125.5 (CH Ar, C6), 128.3 (CH Ar, C10,14), 128.5 (CH Ar, C4), 129.7 (CH Ar, C5), 129.9 (CH Ar, C11,13), 131.2 (C Ar, C2), 131.8 (C Ar, C12), 137.6 (C Ar, C9), 140.0 (C Ar, C7), 165.8 (CH Ar, C1), 166.7 (COOH, C15). Elem. anal. calc. (%) for C15H12BrNO2S (FW = 350.23 g mol−1): C 51.44, H 3.45, N 4.00, S 9.16; found: C 51.21, H 3.39 N 3.98, S 8.75. IR (KBr pellet, cm−1): ν = 2996 [s, ν(C–H)], 1701 [s, ν(COO)], 1609 [m, ν(C
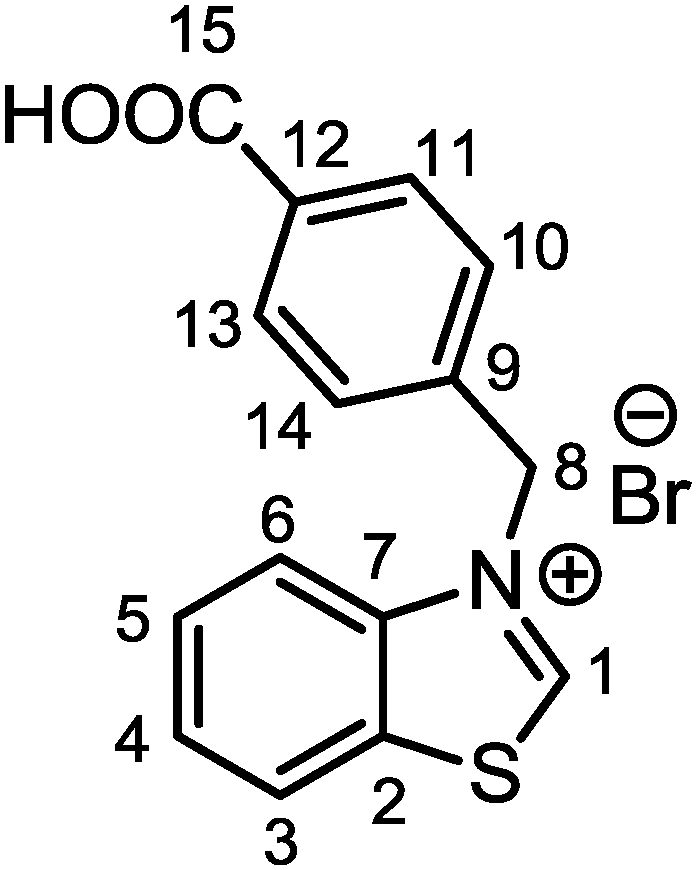
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C)], 1417 (m), 1380 (s), 1233 [s, δ(O–H)], 1177 (m), 1115 (m), 771 [s, γ(C–H)]. ESI-MS: m/z = 270.3 (M − Br)+.
C)], 1417 (m), 1380 (s), 1233 [s, δ(O–H)], 1177 (m), 1115 (m), 771 [s, γ(C–H)]. ESI-MS: m/z = 270.3 (M − Br)+.
Synthesis of NU-1000-BzTz
Following the general SALI procedure reported by Hupp, Farha et al.,24,25 the benzothiazolium salt BzTz (0.163 g, 0.46 mmol, 10 eq.) was added to a suspension of benzoate-free47NU-1000 (0.100 g, 0.046 mmol) in a dry and degassed polar solvent mixture (total volume 45 mL, acetonitrile![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :dimethylsulfoxide = 90:10). The reaction mixture was heated at 353 K for 24 h with occasional gentle swirling. After that time, the mixture was brought back to room temperature and the precipitate was filtered over a 0.2 μm PTFE filter. The bright yellow solid residue was sequentially washed with hot acetonitrile, acetone and dichlorometane (3 × 20 mL each) and finally dried in air. The extent of benzothiazolium salt incorporation was estimated through signal integration of the 1H NMR spectrum of the solution obtained after digesting the sample in a 5% HF-DMSO-d6 mixture and heating to 343 K for 30 min (see ESI† and Fig. S4). The maximum loading achieved was 1.7(BzTz) per [Zr6] node. IR (KBr pellet, cm−1): 3387 (w, br) [ν(N–H)], 3095 (w) [ν(C–Haromatic)], 2928 (w) [ν(C–Haliphatic)], 1655 (vs) [ν(CO)], 1500 (s) [ν(CC + CN)], 1435 (w), 1384 (s), 1275 (m), 1250 (m), 1204 (w), 1163 (w), 1123 (s), 1093 (m), 1056 (s), 1014 (w), 946 (s), 839 (m).
:dimethylsulfoxide = 90:10). The reaction mixture was heated at 353 K for 24 h with occasional gentle swirling. After that time, the mixture was brought back to room temperature and the precipitate was filtered over a 0.2 μm PTFE filter. The bright yellow solid residue was sequentially washed with hot acetonitrile, acetone and dichlorometane (3 × 20 mL each) and finally dried in air. The extent of benzothiazolium salt incorporation was estimated through signal integration of the 1H NMR spectrum of the solution obtained after digesting the sample in a 5% HF-DMSO-d6 mixture and heating to 343 K for 30 min (see ESI† and Fig. S4). The maximum loading achieved was 1.7(BzTz) per [Zr6] node. IR (KBr pellet, cm−1): 3387 (w, br) [ν(N–H)], 3095 (w) [ν(C–Haromatic)], 2928 (w) [ν(C–Haliphatic)], 1655 (vs) [ν(CO)], 1500 (s) [ν(CC + CN)], 1435 (w), 1384 (s), 1275 (m), 1250 (m), 1204 (w), 1163 (w), 1123 (s), 1093 (m), 1056 (s), 1014 (w), 946 (s), 839 (m).
Gas adsorption
NU-1000-BzTz (∼40 mg) was activated at 393 K under high vacuum (10−6 Torr) for 12 h before each measurement. The textural properties were estimated by volumetric adsorption carried out using an ASAP 2020 Micromeritics instrument, using N2 as adsorbate at 77 K. For the Brunauer–Emmett–Teller (BET) specific surface area calculation, the 0.01–0.1 p/p0 pressure range of the isotherm was used to fit the data. Within this range, all the Rouquerol consistency criteria are satisfied.48 The pore size distribution was determined on the basis of the NLDFT method (Tarazona model for cylindrical pores). CO2 adsorption isotherms were recorded at 273 and 298 K at a maximum pressure of 1.2 bar. The isosteric heat of adsorption (Qst) was calculated from the CO2 isotherms measured at 273 and 298 K according to a variant of the Clausius–Clapeyron equation:49,50 | (1) |
Luminescence measurements and anion sensing experiments
The experiments were carried out in air-equilibrated water suspensions at 298 K unless otherwise noted. UV-vis absorption spectra were recorded using a PerkinElmer λ40 spectrophotometer using quartz cells with path length of 1.0 cm. Luminescence spectra were recorded using a PerkinElmer LS-50 or an Edinburgh FLS920 spectrofluorimeter equipped with a Hamamatsu R928 phototube. Lifetimes shorter than 10 μs were measured by the above-mentioned Edinburgh FLS920 spectrofluorimeter equipped with a TCC900 card for data acquisition in time-correlated single-photon counting experiments (0.5 ns time resolution). For solid samples, the emission quantum yield was calculated from the corrected emission spectra registered using an Edinburgh FLS920 spectrofluorimeter equipped with a barium sulfate coated integrating sphere (4 inch), a 450 W Xe lamp (λ excitation tunable by a monochromator supplied with the instrument) as light source, and an R928 photomultiplier tube, following the procedure described by De Mello et al.51 The estimated experimental errors are 2 nm for the band maximum, 5% for the molar absorption coefficient and luminescence lifetime, and 20% for the emission quantum yield of a solid.Results and discussion
Synthesis of NU-1000-BzTz, solid state characterization and CO2 adsorption
After the straightforward preparation of the benzothiazolium salt BzTz through benzothiazole N-alkylation by 4-(bromomethyl)benzoic acid (Scheme S1, ESI†), the new monocarboxylic ligand was incorporated into activated microcrystalline samples of NU-1000via SALI by exposing the MOF to solutions of 10 eq. of BzTz per [Zr6] node in a polar solvent mixture of dimethyl sulfoxide (DMSO) and acetonitrile (MeCN) at 353 K for 48 h (see Experimental section for a detailed description). As already noticed for other FGs,24 SALI was unsuccessful in solvent mixtures containing water. Therefore, it is essential to employ anhydrous solvents to get a good conversion in this step. The extent of FG incorporation was estimated by 1H NMR spectroscopy after dissolving NU-1000-BzTz samples in a 5% HF-DMSO-d6 mixture heated to 343 K to promote the digestion process. The corresponding signals of the incorporated FG (Fig. S4, ESI†) were integrated against that of the TBAPy ligand. In our hands and under the same (optimized) experimental conditions, the amount of BzTz incorporated into NU-1000 is reproducible for all the batches prepared. A maximum loading of 1.7 BzTz ligand per [Zr6] node was obtained. This stoichiometry is slightly lower than that observed for other NU-1000-FG samples in the literature (between 2 and 4),24,25 possibly due to the big steric hindrance of BzTz that hampers further functionalization of the [Zr6] cluster. The as-obtained NU-1000-BzTz has been thoroughly characterized in the solid state. Powder X-ray diffraction (PXRD) analysis (Fig. 2a) showed that the parent framework remains intact after functionalization; only slight differences in relative diffraction peak intensities and positions were observed, due to changes in the electron density introduced by BzTz and to the modification of the unit cell lattice parameters after inclusion of the bulky benzothiazolium groups. The IR spectra (Fig. S5, ESI†) are not informative of the occurred transformation, the main adsorption bands being superimposable for NU-1000 and NU-1000-BzTz in the 2000–400 cm−1 wavenumber range. A more detailed analysis can be made by looking at the difference spectrum [(NU-1000-BzTz)–(NU-1000)] compared with that of pure BzTz (Fig. S6, ESI†). Some of the typical benzothiazolium normal vibrational modes52 can be identified at 1700 cm−1 [ν(COO)], 1609 cm−1 [ν(CC)], 1417 cm−1 [δ(CH2)] and 729 cm−1 [γ(CH)]. Thermogravimetric analysis (TGA, Fig. 2b) showed that the thermal stability of NU-1000-BzTz is slightly lower than that of NU-1000 (Tdec = 780 vs. 800 K, respectively), possibly because of the (destabilizing) electronic effect caused by the dangling group attached to the metal nodes. An initial weight loss of ca. 22.0 wt% (in line with the stoichiometric 1:1.7 [Zr6]:BzTz ratio found through 1H NMR analysis) can be reasonably ascribed to the BzTz dangling group decomposition. In fact, the DTG peak found in this range falls at T = 532 K, a value that is very close to that found for the isolated BzTz decomposition occurring at T = 510 K (Fig. S7, ESI†). In line with the higher mass unit of the functionalized MOF, the solid residue at the end of the decomposition process is higher for NU-1000-BzTz if compared with that obtained with the parent MOF (47.6 vs. 44.727 wt%, respectively). No precise chemical composition can be provided for the solid residue obtained from NU-1000-BzTz. The heavier residue compared to NU-1000 suggests that heavier atoms (not present in the pristine MOF; most likely sulfur) are present. The PXRD pattern collected on the solid obtained after heating at T ≈ 1080 K did not show any crystalline phase in the sample. According to previous information collected on zirconium MOFs with thiazole linkers,39 we can tentatively propose that a complex mixture of heavy zirconium-containing (amorphous) inorganic compounds (like sulfates, sulphides and oxides) is formed at the end of the thermal decomposition.
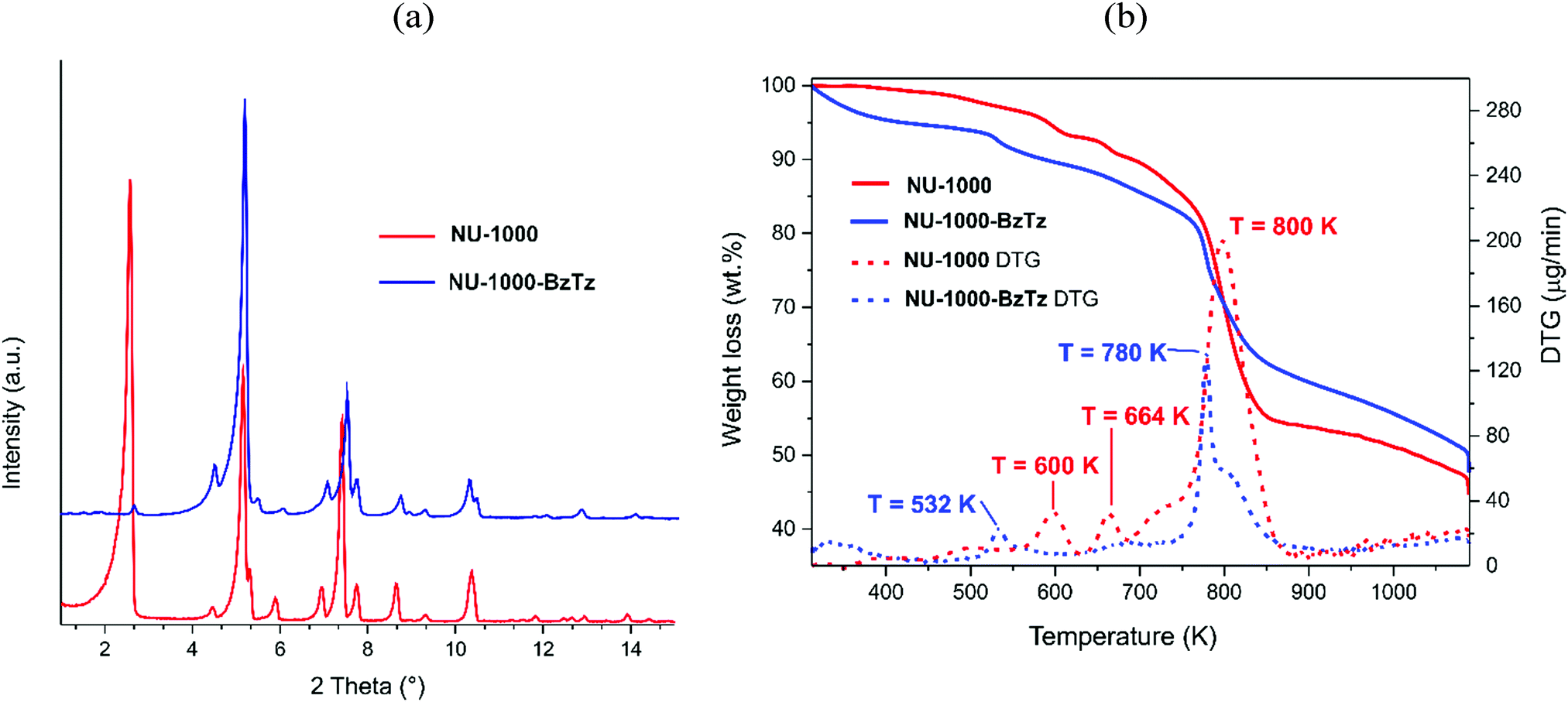 | ||
| Fig. 2 (a) PXRD patterns of NU-1000 and NU-1000-BzTz for comparison; (b) TGA-DTG profiles of NU-1000 and NU-1000-BzTz for comparison. | ||
The porosity of NU-1000-BzTz was evaluated by recording the N2 adsorption isotherm at 77 K on desolvated samples (Fig. 3a). The isotherm shape is the same as that of NU-1000, confirming its mesoporous texture. An obvious reduction in gas uptake and the BET surface area in comparison to that of the parent NU-1000 material was recorded (from 2140 to 1530 m2 g−1), because of the reduced empty pore volume after functionalization (the total pore volume at p/p0 = 0.98 equals 1.53 and 0.93 cm3 g−1 for NU-1000 and NU-1000-BzTz, respectively). In line with this finding, the pore size distribution evaluated through the NLDFT model (Fig. S8, ESI†) shows a slightly reduced mesopore size for the functionalized material (w = 33 vs. 28 Å for NU-1000 and NU-1000-BzTz, respectively), while the micropore size is practically unchanged. This reveals that SALI functionalization occurs in the mesopores only, similar to other NU-1000-FG materials reported in the literature. NU-1000-BzTz showed an increased affinity for carbon dioxide compared with its parent MOF. The total CO2 uptake at pCO2 = 1 bar and T = 298 K and 273 K is 8.7 wt% (2.0 mmol g−1) and 10.8 wt% (2.5 mmol g−1), respectively (Fig. 3b). The absolute gas uptake falls in the same range as that found for other thiazole-containing MOFs like Zr6(O)4(OH)4(TzTz)6 (7.5 wt%, TzTz2− = [2,2′-bithiazole]-5,5′-dicarboxylate)39 or Cu(5-Tz)2 (9.0 wt%, 5-Tz− = thiazole-5-carboxylate),40 but it is lower than that measured for NU-1000 (ca. 3.0 mmol g−1),25 this value being proportional to the respective BET areas. Nevertheless, when the CO2 isosteric heat of adsorption at zero coverage (Qst) is considered, the benzothiazolium-functionalized MOF has a higher Qst value than that found for its parent analogue (25 vs. 1725 kJ mol−1, respectively). The isosteric heat of adsorption reflects the interaction strength between CO2 and the inner MOF pore walls; the introduction of a polar molecule like a benzothiazolium salt into the MOF channels is beneficial for the MOF–CO2 interaction. The Qst value found for NU-1000-BzTz is of the same order of magnitude as that calculated for other perfluoroalkane-functionalized25 or peptide-functionalized31NU-1000 samples of the literature (between 24 and 34 kJ mol−1).
 | ||
| Fig. 3 (a) N2 adsorption isotherms of NU-1000 (red diamonds) and NU-1000-BzTz (blue diamonds) for comparison. The desorption branch is depicted with empty symbols. (b) CO2 adsorption isotherms of NU-1000-BzTz at T = 273 (blue squares) and 298 K (blue circles). | ||
Luminescence anion sensing with NU-1000-BzTz
Before testing NU-1000-BzTz for luminescence ion sensing in aqueous solutions, its water stability was preliminarily assessed. Like its parent MOF NU-1000,53,54 the functionalized MOF maintains its crystallinity degree and structural texture after soaking in water for 24 h, as witnessed by the PXRD control pattern recorded after this time (Fig. S9, ESI†). As observed for NU-1000,27NU-1000-BzTz shows very weak solubility in pure water, even after heating and thorough sonication. As a consequence, the dissolution of small amounts of the two MOFs in water gives opaque dispersions whose normalized UV-vis absorption spectra display a significant offset and tailed trace due to the particulate scattering (Fig. 4a).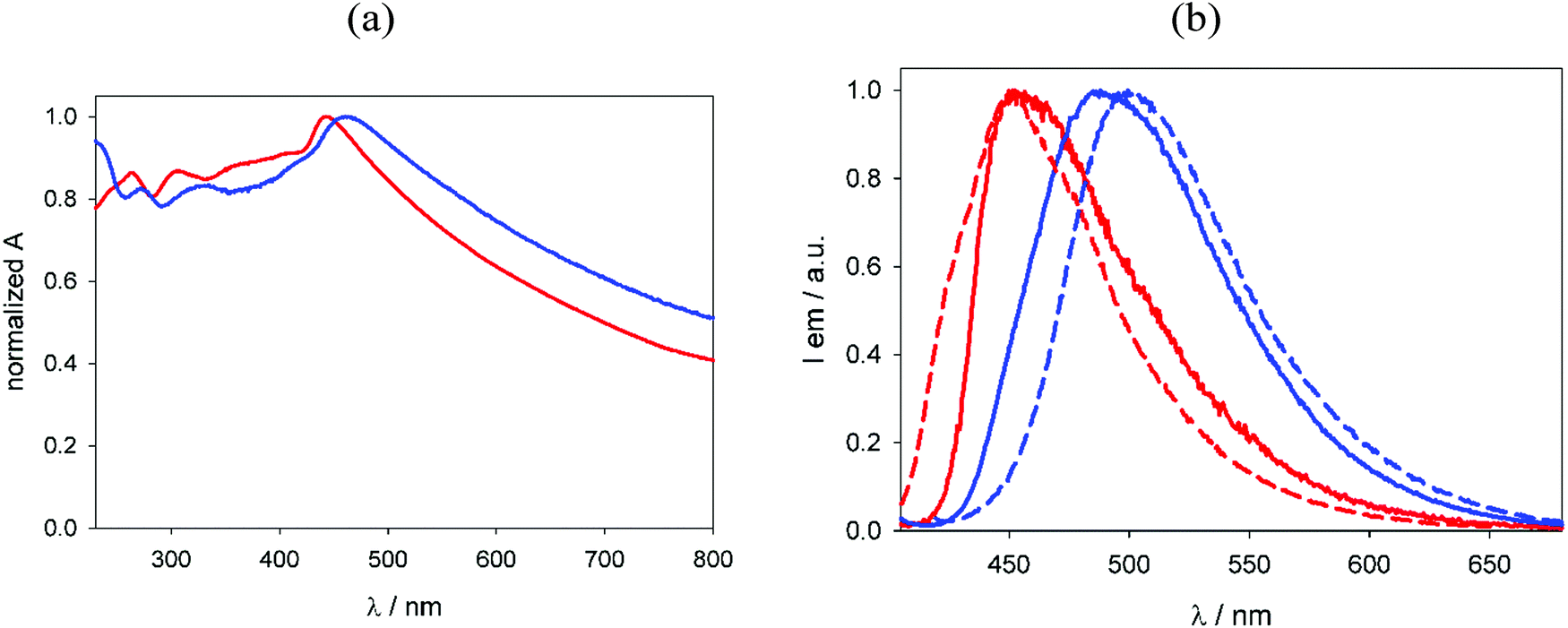 | ||
| Fig. 4 (a) Normalized absorption spectra recorded for suspensions of NU-1000 (red line) and NU-1000-BzTz (blue) in H2O at r.t. (b) Comparison between normalized emission spectra of NU-1000 (red lines) and NU-1000-BzTz (blue lines) recorded on suspensions in H2O at r.t. (solid) and on powders at r.t. (dashed). | ||
Interestingly, in the absorption spectrum of NU-1000-BzTz, the characteristic absorption band at λ = 280 nm registered in H2O for the model compound BzTz (Fig. S10, ESI†) does not appear. However, significant differences between the two MOFs are observed in their luminescence properties (e.g. in suspensions, λmax,em = 450 nm and 490 nm for NU-1000 and NU-1000-BzTz, respectively; Fig. 4b). Moreover, both the MOFs show appreciable emission quantum yields in the visible spectrum as powders in the solid phase (Φem = 0.06 and 0.04 for NU-1000 and NU-1000-BzTz, respectively) and in aqueous suspensions, with small discrepancies in the emission maxima and profiles recorded under the two experimental conditions. The small redshift in the absorption and emission spectra of NU-1000-BzTz compared to NU-1000 could be explained by the presence of the positively charged BzTz subunit, which perturbs the electronic environment of the emitting pyrene ligand and slightly stabilizes its excited state.55,56 Analysis of the luminescence lifetimes on powders and aqueous suspensions of both MOFs gives evidence of nanosecond-scale multiexponential decays, corresponding to singlet excited state paths of deactivation (Fig. S11, ESI†).
Given the confinement properties of several other MOFs, which can be modulated by the preferential interactions between the charged cavities of the MOFs and anions,57 we decided to test the uptake of NU-1000 and NU-1000-BzTz towards highly toxic cyanide in aqueous solutions. Initially, the sensing ability and the emission behaviour of bare BzTz in the presence of aqueous cyanide were checked. Consecutive additions of increasing amounts of aqueous KCN (30.5 mM) to a solution of BzTz in H2O (1.05 × 10−4 M) led to small changes in the absorption spectra and to the progressive quenching of its fluorescence. Moreover, we did not record any changes in the emission profile, i.e. no lambda shift in the fluorescence band occurs upon cyanide addition (Fig. S12, ESI†). Hence, the anchoring of the benzothiazolium emitter to a solid MOF support is of fundamental importance to have a better sensing response. In fact, upon addition of KCN, we observed changes in the emission intensity in both MOFs, with NU-1000 exhibiting a steadily increasing emission quantum yield without showing any saturation in the presence of high CN− concentrations (Fig. 5a). On the other hand, the emission spectrum of NU-1000-BzTz shifts to shorter wavelengths with increasing the concentration of cyanide, restoring the luminescence of the parent MOF NU-1000. This is probably due to the anionic exchange within the framework, promoted by the presence of the positively charged BzTz fragment (Fig. 5b). Analysis of the excitation spectra demonstrates that the strong emission centered at 450 nm likely originates from the tetrasubstituted pyrene subunit (Fig. S13, ESI†).58 A very similar behaviour was observed when increasing concentrations of analogous “stick-like” anions such as cyanate (OCN−), thiocyanate (SCN−) and selenocyanate (SeCN−) are added to suspensions of NU-1000-BzTz (Fig. S14, ESI†). By comparing the increments of emission intensities at the emission maxima as functions of the anion concentrations, cyanide was found to give the strongest change (Fig. 5c) with an estimated limit of detection (LOD) of 1.08 × 10−6 M (Fig. S15, ESI†), making the MOF suitable for practical application in the detection of cyanide in drinking water (LOD ≤ 2 × 10−6 M). The luminescence response is proportional to the relative basicity of the anions, following the order CN− ≫ OCN− > SCN−.59 The direct experimental relationship between LOD and anion basicity leads to the hypothesis of the existence of strong hydrogen bonding interactions between the anion and the H atom in the 2-position of the benzothiazolium unit (the most acidic of the whole dangling group) as the origin of the MOF emission behaviour. This assumption is also supported by a literature example of a carbazole-based UiO-67 MOF, whose strong emission intensity variation in the presence of cyanide stems from the linker deprotonation by CN−.60 Given the lower C–H vs. N–H proton acidity, in the present case, no complete deprotonation could be achieved.
 | ||
| Fig. 5 (a) Evolution of the emission spectra recorded for a suspension of NU-1000 upon addition of KCN (from 0 to ca. 1.4 mM, red to orange line, respectively) in H2O at r.t. λex = 385 nm; A385nm ≈ 0.38. Inset: Dependence between [CN−] and the emission intensity at 450 nm. (b) Evolution of the emission spectra recorded for a suspension of NU-1000-BzTz upon addition of KCN (from 0 to ca. 1.0 mM, blue to green line, respectively) in H2O at r.t. λex = 385 nm; 0.32 < A385nm < 0.35. Inset: Dependence between [CN−] and the emission intensity at 430 nm. (c) Evolution of the emission intensities recorded at 430 nm for suspensions of NU-1000-BzTz upon addition of KCN, KOCN and NaSCN (λex = 385 nm). | ||
To test the reversibility of the NU-1000-BzTz sensing in water, the cyanide-containing aqueous suspensions were treated with progressive additions of silver triflate [Ag(CF3SO3)] in order to form the insoluble AgCN and remove cyanide from the solution. Interestingly, upon increasing Ag+ concentration, the MOF emission decreases in intensity and shifts back to its pristine maximum around 490 nm. This is proof of the cyanide removal and reversible luminescence sensing of NU-1000BzTz (Fig. 6a); upon repeated additions of KCN and Ag(CF3SO3), the same MOF showed good reversibility, while its fluorescence emission was monitored at 430 nm (Fig. S16–S19 and attached Movie in the ESI†). Finally, NU-1000-BzTz was found to be effectively sensitive to cyanide also in experiments run in ordinary tap water (Fig. 6b), demonstrating its capability to act like a selective cyanide luminescent probe even in complex ion matrices.
 | ||
| Fig. 6 (a) Evolution of the emission spectra recorded for a suspension of NU-1000-BzTz upon addition of KCN (from 0 to ca. 0.33 mM, blue to turquoise line, respectively) in H2O at r.t. followed by addition of Ag(CF3SO3) (up to ca. 1.3 mM, dark green line). λex = 385 nm; 0.25 < A385nm < 0.44. Inset: Photos taken of three suspensions of NU-1000-BzTz in pure water (left), 0.5 mM KCN (centre) and 0.5 mM KCN plus excess of Ag(CF3SO3) (right), under ambient light (top) and under UV excitation (bottom). (b) Evolution of the emission spectra recorded for a suspension of NU-1000-BzTz upon addition of KCN (from 0 to ca. 1.6 mM, blue to turquoise line, respectively) in tap water at r.t. λex = 385 nm; 0.18 < A385nm < 0.21. | ||
Conclusions
The SALI technique has been successfully employed to create a new MOF derived from NU-1000 through incorporation of a tailor-made benzothiazolium salt inside its pores. The as-obtained NU-1000-BzTz is a stable and multifunctional material, which can be exploited in either carbon dioxide storage (owing to its enhanced thermodynamic affinity for CO2 if compared with its parent MOF) or luminescence polluting ion sensing in aqueous solutions. In particular, its emission response is very sensitive to cyanide, and it is totally reversible and selective in aqueous solutions containing other competing anions (complex matrices like ordinary tap water). The emission colour change from light green to blue under a UV lamp in the presence of trace amounts of CN− makes the detection process very straightforward. The extremely low cyanide limit of detection of NU-1000-BzTz opens new horizons to its practical utilization in drinking water samples. To the best of our knowledge, this is the first example of the employment of an NU-1000-derived MOF as a cyanide luminescence sensor. New thiazole-based MOF materials are currently being prepared in our laboratories with the aim of testing them in CO2 storage and luminescence sensing.Conflicts of interest
The authors have no conflicts of interest to declare.Acknowledgements
G. G. thanks the Italian MIUR through the PRIN 2017 project MULTI-e (20179337R7) “Multielectron transfer for the conversion of small molecules: an enabling technology for the chemical use of renewable energy” and the TRAINER project “Catalysts for Transition to Renewable Energy Future” (Ref. ANR-17-MPGA-0017) for financial support.References
- L. R. MacGillivray and C. M. Lukehart, Metal-Organic Framework Materials, John Wiley & Sons, New York, 2014 Search PubMed.
- B. Seyyedi, Metal–Organic Frameworks: a New Class of Crystalline Porous Materials, Lambert Academic Publishing, Saarbrücken, 2014 Search PubMed.
- D. Farrusseng, Metal-Organic Frameworks: Applications from Catalysis to Gas Storage, Wiley-VCH Verlag, Weinheim, 2011 Search PubMed.
- M. Schroeder, Functional Metal-organic Frameworks: Gas Storage, Separation and Catalysis, Springer-Verlag, Berlin, Heidelberg, 2010 Search PubMed.
- A. Sturluson, M. T. Huynh, A. R. Kaija, C. Laird, S. Yoon, F. Hou, Z. Feng, C. E. Wilmer, Y. J. Colon, Y. G. Chung, D. W. Siderius and C. M. Simon, Mol. Simul., 2019, 45, 1082–1121 CrossRef PubMed.
- X. Yang and X. Qiang, Cryst. Growth Des., 2017, 17, 1450–1455 CrossRef.
- H.-C. Zhou, J. R. Long and O. M. Yaghi, Chem. Rev., 2012, 112, 673–674 CrossRef PubMed.
- M. P. Suh, H. J. Park, T. K. Prasad and D. W. Lim, Chem. Rev., 2012, 112, 782–835 CrossRef PubMed.
- O. K. Farha, A. O. Yazaydın, I. Eryazici, C. D. Malliakas, B. G. Hauser, M. G. Kanatzidis, S. T. Nguyen, R. Q. Snurr and J. T. Hupp, Nat. Chem., 2010, 2, 944–948 CrossRef CAS PubMed.
- A. Rossin, G. Tuci, L. Luconi and G. Giambastiani, ACS Catal., 2017, 7, 5035–5045 CrossRef CAS.
- A. H. Chughtai, N. Ahmad, H. A. Younus, A. Laypkovc and F. Verpoort, Chem. Soc. Rev., 2015, 44, 6804–6849 RSC.
- J. Liu, L. Chen, H. Cui, J. Zhang, L. Zhang and C.-Y. Su, Chem. Soc. Rev., 2014, 43, 6011–6061 RSC.
- J. Gascon, A. Corma, F. Kapteijn and F. X. Llabrés i Xamena, ACS Catal., 2014, 4, 361–378 CrossRef CAS.
- Y. Zhang, S. Yuan, G. Day, X. Wang, X. Yang and H.-C. Zhou, Coord. Chem. Rev., 2018, 354, 28–45 CrossRef CAS.
- F.-Y. Yi, D. Chen, M.-K. Wu, L. Han and H.-L. Jiang, ChemPlusChem, 2016, 81, 675–690 CrossRef CAS PubMed.
- Z. Hu, B. J. Deibert and L. Jing, Chem. Soc. Rev., 2014, 43, 5815–5840 RSC.
- S. Jin, H.-J. Son, O. K. Farha, G. P. Wiederrecht and J. T. Hupp, J. Am. Chem. Soc., 2013, 135, 955–958 CrossRef CAS.
- H.-J. Son, S. Jin, S. Patwardhan, S. J. Wezenberg, N. C. Jeong, M. So, C. E. Wilmer, A. A. Sarjeant, G. C. Schatz, R. Q. Snurr, O. K. Farha, G. P. Wiederrecht and J. T. Hupp, J. Am. Chem. Soc., 2013, 135, 862–869 CrossRef CAS.
- G. Tuci, A. Rossin, X. Xu, M. Ranocchiari, J. A. van Bokhoven, L. Luconi, I. Manet, M. Melucci and G. Giambastiani, Chem. Mater., 2013, 25, 2297–2308 CrossRef CAS.
- S. M. Cohen, Chem. Rev., 2012, 112, 970–1000 CrossRef CAS PubMed.
- W. Bury, D. Fairen-Jimenez, M. B. Lalonde, R. Q. Snurr, O. K. Farha and J. T. Hupp, Chem. Mater., 2013, 25, 739–744 CrossRef CAS.
- T. Li, M. T. Kozlowski, E. A. Doud, M. N. Blakely and N. L. Rosi, J. Am. Chem. Soc., 2013, 135, 11688–11691 CrossRef CAS.
- M. Kim, J. F. Cahill, Y. Su, K. A. Prather and S. M. Cohen, Chem. Sci., 2012, 3, 126–130 RSC.
- P. Deria, W. Bury, J. T. Hupp and O. K. Farha, Chem. Commun., 2014, 50, 1965–1968 RSC.
- P. Deria, J. E. Mondloch, E. Tylianakis, P. Ghosh, W. Bury, R. Q. Snurr, J. T. Hupp and O. K. Farha, J. Am. Chem. Soc., 2013, 135, 16801–16804 CrossRef CAS PubMed.
- J. H. Cavka, S. Jakobsen, U. Olsbye, N. Guillou, C. Lamberti, S. Bordiga and K. P. Lillerud, J. Am. Chem. Soc., 2008, 130, 13850–13851 CrossRef PubMed.
- J. E. Mondloch, W. Bury, D. Fairen-Jimenez, S. Kwon, E. J. DeMarco, M. H. Weston, A. A. Sarjeant, S. T. Nguyen, P. C. Stair, R. Q. Snurr, O. K. Farha and J. T. Hupp, J. Am. Chem. Soc., 2013, 135, 10294–10297 CrossRef CAS PubMed.
- J.-R. Li, J. Yu, W. Lu, L.-B. Sun, J. Sculley, P. B. Balbuena and H.-C. Zhou, Nat. Commun., 2013, 4, 1538 CrossRef PubMed.
- T. Islamoglu, S. Goswami, Z. Li, A. J. Howarth, O. K. Farha and J. T. Hupp, Acc. Chem. Res., 2017, 50, 805–813 CrossRef CAS.
- A. Atilgan, T. Islamoglu, A. J. Howarth, J. T. Hupp and O. K. Farha, ACS Appl. Mater. Interfaces, 2017, 9, 24555–24560 CrossRef CAS PubMed.
- P. Deria, S. Li, H. Zhang, R. Q. Snurr, J. T. Hupp and O. K. Farha, Chem. Commun., 2015, 51, 12478–12481 RSC.
- B. Wang, X.-L. Lv, D. Feng, L.-H. Xie, J. Zhang, M. Li, Y. Xie, J.-R. Li and H.-C. Zhou, J. Am. Chem. Soc., 2016, 138, 6204–6216 CrossRef CAS.
- J. Yu, X. Li and P. Deria, ACS Sustainable Chem. Eng., 2019, 7, 1841–1854 CrossRef CAS.
- A. Van Wyk, T. Smith, J. Park and P. Deria, J. Am. Chem. Soc., 2018, 140, 2756–2760 CrossRef CAS.
- A. W. Peters, Z. Li, O. K. Farha and J. T. Hupp, ACS Appl. Mater. Interfaces, 2016, 8, 20675–20681 CrossRef CAS.
- B. Wang, P. Wang, L.-H. Xie, R.-B. Lin, J. Lv, J.-R. Li and B. Chen, Nat. Commun., 2019, 10, 3861 CrossRef.
- M. Gong, J. Yang, Y. Li, Q. Zhuang and J. Gu, J. Mater. Chem. C, 2019, 7, 12674–12681 RSC.
- Y. Zhou, Q. Yang, J. Cuan, Y. Wang, N. Gan, Y. Cao and T. Li, Analyst, 2018, 143, 3628–3634 RSC.
- P. Müller, B. Bucior, G. Tuci, L. Luconi, J. Getzschmann, S. Kaskel, R. Q. Snurr, G. Giambastiani and A. Rossin, Mol. Syst. Des. Eng., 2019, 4, 1000–1013 RSC.
- A. Rossin, G. Tuci, G. Giambastiani and M. Peruzzini, ChemPlusChem, 2014, 79, 406–412 CrossRef CAS PubMed.
- A. Rossin, B. Di Credico, G. Giambastiani, A. Peruzzini, G. Pescitelli, G. Reginato, E. Borfecchia, D. Gianolio, C. Lamberti and S. Bordiga, J. Mater. Chem., 2012, 22, 10335–10344 RSC.
- G. Mercuri, G. Giambastiani and A. Rossin, Inorganics, 2019, 7, 144 CrossRef.
- S. Staderini, G. Tuci, L. Luconi, P. Müller, S. Kaskel, A. Eychmüller, F. Eichler, G. Giambastiani and A. Rossin, Eur. J. Inorg. Chem., 2017, 4909–4918 CrossRef CAS.
- S. Staderini, G. Tuci, M. D’Angelantonio, F. Manoli, I. Manet, G. Giambastiani, M. Peruzzini and A. Rossin, ChemistrySelect, 2016, 6, 1123–1131 CrossRef.
- A. Rossin and G. Giambastiani, CrystEngComm, 2015, 17, 218–228 RSC.
- T. Islamoglu, K. Otake, P. Li, C. T. Buru, A. W. Peters, I. Akpinar, S. J. Garibaya and O. K. Farha, CrystEngComm, 2018, 20, 5913–5918 RSC.
- The as-synthesized form of NU-1000 contains residual benzoate ligands at the node sites. Benzoate is present because benzoic acid was used as a modulator in the synthesis of NU-1000. SALI is possible only after removal of coordinated benzoate by extended treatment of the as-synthesized material with aq. HCl in DMF at 353 K.
- J. Rouquerol, P. Llewellyn and F. Rouquerol, in Studies in Surface Science and Catalysis, ed. P. L. Llewellyn, F. Rodriquez-Reinoso, J. Rouquerol and N. Seaton, Elsevier, Amsterdam, 2007, vol. 160, p. 49 Search PubMed.
- X. Zhu, C. Tian, G. M. Veith, C. W. Abney, J. Dehaudt and S. Dai, J. Am. Chem. Soc., 2016, 138, 11497–11500 CrossRef CAS PubMed.
- X. Zhu, S. M. Mahurin, S.-H. An, C.-L. Do-Thanh, C. Tian, Y. Li, L. W. Gill, E. W. Hagaman, Z. Bian, J.-H. Zhou, J. Hu, H. Liu and S. Dai, Chem. Commun., 2014, 50, 7933–7936 RSC.
- J. C. de Mello, H. F. Wittmann and R. H. Friend, Adv. Mater., 1997, 9, 230–232 CrossRef CAS.
- I. Culak, V. Sutoris and V. Sekerka, Chem. Pap., 1990, 44, 389–397 CAS.
- P. Deria, Y. G. Chung, R. Q. Snurr, J. T. Hupp and O. K. Farha, Chem. Sci., 2015, 6, 5172–5176 RSC.
- J. E. Mondloch, M. J. Katz, N. Planas, D. Semrouni, L. Gagliardi, J. T. Hupp and O. K. Farha, Chem. Commun., 2014, 50, 8944–8946 RSC.
- E. A. Dolgopolova, A. M. Rice, C. R. Martin and N. B. Shustova, Chem. Soc. Rev., 2018, 47, 4710–4728 RSC.
- C. H. Hendon, D. Tiana, M. Fontecave, C. Sanchez, L. D’arras, C. Sassoye, L. Rozes, C. Mellot-Draznieks and A. Walsh, J. Am. Chem. Soc., 2013, 135, 10942–10945 CrossRef CAS PubMed.
- A. Karmakar, P. Samanta, A. V. Desai and S. K. Ghosh, Acc. Chem. Res., 2017, 50, 2457–2469 CrossRef CAS PubMed.
- S. Yamaguchi, I. Yoshikawa, T. Mutai and K. Araki, J. Mater. Chem., 2012, 22, 20065–20070 RSC.
- J. H. Boughton and R. N. Keller, J. Inorg. Nucl. Chem., 1966, 28, 2851–2859 CrossRef CAS.
- A. Das and S. Biswas, Sens. Actuators, B, 2017, 250, 121–131 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Additional characterization data for BzTz (crystal structure and structure refinement details, multinuclear 1H and 13C{1H} NMR spectroscopy, IR spectroscopy, and TG-DTG analysis), 1H NMR spectrum of the digested NU-1000-BzTz for the quantification of the functionalization degree, pore size distributions of pristine and functionalized materials for comparison, water stability tests for NU-1000-BzTz, additional absorption and emission spectra of BzTz and NU-1000-BzTz, fluorescence decay in aqueous suspensions, behavior in the presence of KOCN, NaSCN and KSeCN and reversibility tests with repeated cyclic additions of KCN and Ag(CF3SO3). CCDC 1988747. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0tc01436b |
| This journal is © The Royal Society of Chemistry 2020 |