
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNanoporous gold peel-and-stick biosensors created with etching inkjet maskless lithography for electrochemical pesticide monitoring with microfluidics†
John A.
Hondred
,
Zachary T.
Johnson
and
Jonathan C.
Claussen
 *
*
Department of Mechanical Engineering, Iowa State University, Ames, IA 50011, USA. E-mail: jcclauss@iastate.edu; Tel: 515-294-4690
First published on 8th June 2020
Abstract
Nanoporous gold leaf (NPGL) is an attractive material for flexible electrochemical biosensors due its high and tunable surface area, electrical conductivity, biocompatibility/non-reactive nature, and rich surface chemistry. However, NPGL synthesis and patterning protocols are complex, costly and consequently not suited for flexible electronic fabrication. This work develops a new manufacturing technique coined Etching Inkjet Maskless Lithography (E-IML) to synthesize and pattern NPGL and metal leaf materials (thickness ∼100 nm) for flexible electronics. E-IML utilizes the versatility of an inkjet printer to pattern electrodes for rapid prototyping, even on flexible substrates (polyimide). We demonstrate how NPGL electrodes, with feature and pores sizes down to approximately 25 nm and 5 nm respectively, can be synthesized and patterned from gold/silver leaf material with E-IML and dealloyed (silver removed) via electrochemical etching. Additionally, a pseudo-reference electrode was E-IML patterned from silver leaf and chlorinated with a diluted bleach solution to make a Ag/AgCl electrode for use in 3-electrode electrochemical biosensing with NPGL working and counter electrodes. These 3-electrode electrochemical biosensors were patterned on adhesive polyimide films for use as disposable peel-and-stick tape biosensors or wearable sticker biosensors. In order to demonstrate the utility of the peel-and-stick biosensors, a disposable tape pesticide biosensor and reusable 3D printed flow cell were developed for organophosphate detection in soil samples. Multiple NPGL working electrodes were fabricated on single devices so that that each electrode could be functionalized with distinct concentrations of the enzyme acetylcholinesterase. Paraoxon (a model organophosphate) sensing results demonstrated a low detection limit (0.53 pM) and high sensitivity (376 nA nM−1). The unique multi-electrode enzyme functionalization protocol allowed for a wider paraoxon sensing range (four orders of magnitude: 1 nM–10 μM) than one electrode alone. The flow cell platform biosensor was also tested in real-world samples (soil slurry) and demonstrated a signal recovery of approximately 93.5% and 91.5% for soil slurry samples spiked with 10 nM and 1 μM paraoxon concentration respectively. Hence these thin-film E-IML NPGL patterning and synthesis techniques could be useful for a wide variety of applications including electrochemical sensors, supercapacitors, batteries, fuel cells, energy harvesters, triboelectric nanogenerators, and membranes.
Introduction
Flexible biosensors for rapid in-field or point-of-care testing has recently gained tremendous traction with a predicted market value of $5.5 billion by 2025 due to their low-cost, disposability, low weight, portability, and ability to conform to curvilinear features such as those found on the body.1–3 Electrochemical biosensors in particular offer a sensing modality capable of quantifying chemical/biochemical analytes in complex and/or turbid field samples with high specificity and sensitivity for a wide variety of applications4 including those associated with healthcare and physical fitness,5,6 food quality,7–9 security/national defense,10–12 drinking water safety,13,14 and farmland pollution.15,16 Recent advances in high precision micromachining, printing, and lithography have permitted the development of flexible electrochemical biosensors that can be incorporated into previously inconceivable devices including eyeglasses,17 contact lenses,18,19 wristbands,20 mouthguards,21 gloves,22 and tattoos.23,24 Moreover electrochemical biosensors have been incorporated into microfluidics or flow cells to improve their performance by enhancing analyte diffusion and particle filtration as well as simplifying testing procedures (e.g., automated sample bifurcation and mixing). Such microfluidic electrochemical biosensors can help produce highly sensitive biosensors capable of multiplexed analyte monitoring within complex and small volume biological samples such as multiplexed detection of cancer proteins in serum.25–28 The more recent concomitance of flexible electrochemical biosensors and microfluidics has opened the door to autonomous, lab-on-a-chip devices that can, for example, produce devices that are capable of real-time continuous sampling and monitoring of sweat metabolites (glucose and lactate) and pH.27,29In terms of materials and fabrication, flexible electrochemical biosensors generally consist of flexible substrates (e.g., paper, polyimide, polyethylene terephthalate) that are patterned with an electroactive, conductive material that is consequently functionalized with a biorecognition agent (e.g., antibody, enzyme, DNA/aptamer, ionophore).30 A wide variety of nanomaterials of distinct size and morphology including carbon nanomaterials (e.g., nanotubes, graphene/graphene oxide, buckypaper),31–33 metal and metal oxide nanoparticles,34,35 and nanoporous metals36 have been incorporated into electrode materials to improve the electrical conductivity and surface area of the electrode. Both improvements, electrical conductivity and surface area, can lead to higher biosensor sensitivities and detection limits due to increased solid-state electron transport within the electrode, increased heterogenous electron transport from the electrode to reactant in solution, and increased loading of biorecognition agents. Moreover, one of the most thoroughly researched electrode transduction materials is gold due to its high electrical conductivity, wide electrochemical window, facile functionalization through thiol groups, biocompatibility, non-reactive nature, and capability with various electrode synthesis techniques (e.g., metal evaporation and chemical etching).37–41 However, the expensive material and fabrication costs of gold have hindered its use in flexible and disposable sensors. Furthermore, gold electrodes typically exhibit low electrochemical surface area due to an extremely smooth surface (roughness factor ∼1) which can effectually limit their electrochemical reactivity, biorecognition agent loading, and sensitivity. However, nanoporous gold leaf (NPGL) electrodes fabricated via de-alloying gold/silver leaf material could circumvent these drawbacks.
Metallic leaf materials, fabricated through compressing/stamping of metal foil, are extremely thin (∼100 nm) and cost effective (e.g. 100 mm × 100 mm gold leaf costs less than $1).42 Recently, Ding et al. demonstrated the fabrication of NPGL by dealloying a gold/silver alloy.43 The porosity of the resultant NPGL could be tuned to create an extremely high surface area with pore sizes reaching less than 10 nm by selectively etching silver from the alloy. Other researchers have applied similarly constructed NPGL to a variety of applications including those involving nanoporous membranes, solar collectors,44 battery and super capacitor electrodes, fuel cells,45 and catalysts.44,46 Several researchers have also utilized NPGL to modify electrode surfaces for electrochemical sensing. For example, Hu et al. modified glassy carbon with NPGL to increase the electrode surface area by nine fold and to construct a sensitive electrochemical DNA biosensor.47 Meng et al. modified a glassy carbon electrode with NPGL to fabricate a non-enzymatic hydrogen peroxide sensor.48 Santos et al. demonstrated the creation of gold leaf nanoband electrodes (∼100 nm in width) that was constructed by sandwiching gold leaf between two layers of Kapton tape and exposing the cross-section of the laminate layers to form the electrode.49 This study characterized the electrochemical behavior of solid gold leaf nanobands through ferricyanide cyclic voltammetry but did not create nor characterize NPGL nanobands. Hence, to our knowledge there has not been a demonstration of a technique that is capable of selectively patterning gold leaf for flexible biosensors nor a technique shown to create flexible electrodes with NPGL. Moreover, there are no published research articles that have utilized NPGL as a stand-alone electrode for electrochemical sensing.
Herein, we present a new manufacturing method coined Etching Inkjet Maskless Lithography (E-IML) to selectively dealloy and pattern gold/silver leaf into NPGL electrodes with high resolution feature sizes (down to 25 nm). This method uses an inkjet printed polymer layer that acts as a protective layer for chemical etching, which consequently, can be used to pattern a variety of metal films (copper and aluminum) as well as leaf materials (defined as metal films ≤100 nm thick) such as gold, silver, and gold/silver alloys. We demonstrate how complex geometries including interdigitated electrodes (IDEs), inductance loop antennas, and various high diffusion/entropy designs can be created via computer aided design (CAD) software and consequently synthesized and patterned through the E-IML technique. Moreover, this work demonstrates how such E-IML NPGL flexible electrodes can be patterned directly onto adhesive films which could be used for wearable tattoo sensors or for integration into microfluidic flow cells, the latter which is expanded upon in this work.
Moreover, we demonstrate how the E-IML process can be used to create 3-electrode electrochemical pesticide biosensors. A stable Ag/AgCl reference, for the 3-electrode set-up, can be created by patterning and then chlorinating pure silver leaf (SL). Working and counter electrodes comprised of E-IML patterned NPGL and a reference electrode comprised of E-IML patterned SL can be fabricated on adhesive polyimide tape. These electrodes can be functionalized to create disposable tape biosensors which can adhere to a reusable 3D printed flow cell/microfluidic cartridge. We demonstrate how these peel-and-stick disposable tape biosensors can be used for pesticide or nerve agent detection with a very broad detection range across five orders of magnitude for the organophosphate paraoxon. To achieve this broad sensing range, five different working NPGL electrodes were developed with E-IML and four were functionalized with distinct concentrations (1.56, 3.125, 6.25, 12.5 U) of acetylcholinesterase (ACHE) while the fifth was not enzymatically functionalized (used as a sentinel electrode to normalize the data to endogenous electroactive molecular interferences). These microfluidic peel-and-stick disposable tape biosensors exhibited a detection limit of 0.53 pM and a linear sensing range of 1 nM to 10 uM. The biosensors were capable of accurately monitoring organophosphate in soil slurry (±10%).
Results and discussion
Overview of E-IML process
The E-IML manufacturing protocol developed herein uses a four-step process to pattern the thin film metal electrodes (Fig. 1 and Movie S1, ESI†). First, metal foils or leaf materials were adhered to the surface of silicon adhesive polyimide tape to form a metal laminate. A gold/silver alloy leaf (working and counter electrodes) and pure silver leaf (reference electrode) laminate were also created for 3-electrode pesticide biosensing (Fig. 1a-i). Next, a protective polymer sacrificial layer was inkjet printed directly onto the gold laminate in the pattern of the electrode configuration (Fig. 1a-ii). Third, the entire laminate was placed in an etching solution to remove the metal foil/leaf that was not protected by the inkjet polymer layer (Fig. 1a-iii). Finally, the polymer protective layer was removed by an acetone wash (Fig. 1a-iv). This simple process allows for rapid prototyping of high resolution thin film metal circuits. The versatility of the E-IML process to pattern a variety of different materials such as copper, aluminum, silver, and gold was also explored (Fig. 1b). Simply by altering the etching solution, different thin film laminates can by patterned. For example, iron chloride was used to etch copper and aluminum; iron nitride etched silver; and iodine solution (gold etch) etched gold, silver, or gold/silver alloy as noted in more detail in the Materials and methods section. Therefore, E-IML is a useful method for patterning a variety of thin film materials which could be adopted to numerous applications.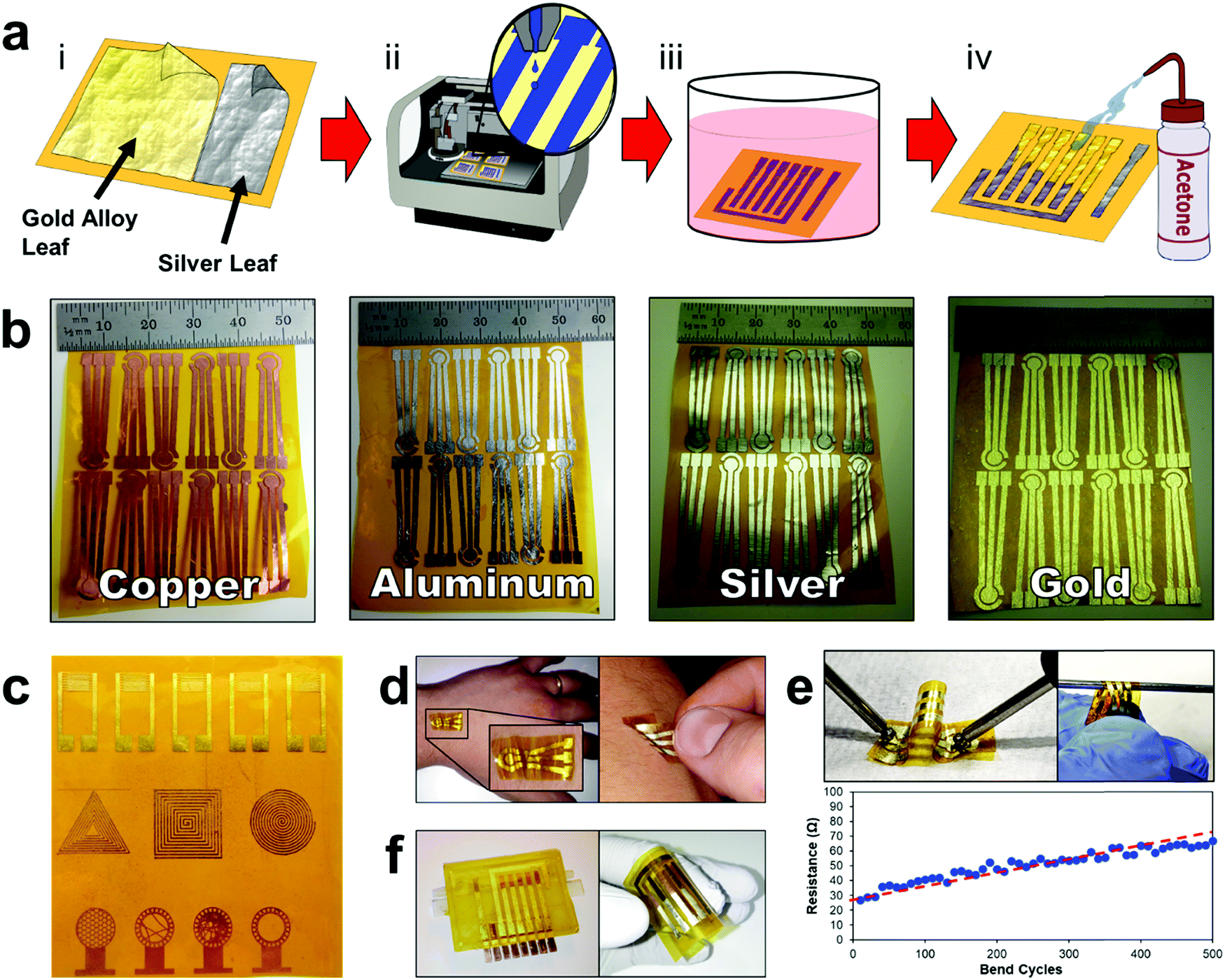 | ||
| Fig. 1 E-IML manufacturing process steps and resultant metal leaf circuit designs. (a) Schematic depicting the four-step process of E-IML electrode fabrication. (i) Metal leaf laminates are fabricated by adhering gold/silver alloy and pure silver leaf material to polyimide tape. (ii) A protective polymer layer is inkjet printed onto the surface of the laminate. (iii) Material not covered by the polymer is selectively etched. (iv) Polymer protective layer is removed using acetone wash. (b) Various materials patterned through E-IML (copper, aluminum, silver, gold). (c) E-IML prototype circuits of distinct geometries including IDEs, antennas, and various high diffusion/entropy designs. (d) Demonstration of ability to adhere E-IML circuits to skin. Inset showing E-IML circuit wrinkles and E-IML circuit conforming to the skin. (e) Bending test demonstrating the flexibility of patterned metal gold leaf electrodes. (f) Disposable and flexible tape biosensor that adheres to and seals the microfluidic channels of a 3D printed flow cell. | ||
To further demonstrate the versatility of the E-IML manufacturing technique, various pure gold electrodes of distinct geometries were designed and patterned including: interdigitated electrodes (IDEs) for impedance sensing,50,51 inductance loop antennas for passive radio frequency (RF) applications,52 and high entropy electrodes for enhanced sensing via increased analyte-to-electrode diffusion (Fig. 1c).50 E-IML can print with microscale resolution and small feature size as the achievable IDE finger width and spacing dimensions were both 150 μm. Additionally, as E-IML can pattern electrodes using laminates with adhesive film backings, the adhesive layer can remain ‘sticky’ during the fabrication process and consequently be used for different tape sensor applications. For example, tattoo sensors that are able to stick and conform to the skin were fabricated (Fig. 1d). These tattoo sensors can adhere to and wrinkle with the skin during the natural flexing and movement of the body. These patterned electrodes have excellent flexibility while retaining their conductivity. As seen in Fig. 1e, even after flexing around a 1 mm diameter rod 500 times, the electrical resistance of the electrodes only slighlty increased (∼40 Ω). Note, that the increase in resistance is probably due to damaging the electrode surface while measuring with the multimeter. Disposable tape biosensors were also fabricated, Fig. 1f, that integrate directly to reusable 3D printed flow cells and form microfluidic channels for contact with test fluids by sealing the open top of the 3D printed channels. These tape sensors are functionalized with enzymes and converted to pesticide biosensors as demonstrated in subsequent sections of this manuscript.
Nanoporous gold leaf (NPGL) electrode fabrication
Gold/silver leaf electrodes that have been patterned through the E-IML process can be subsequently converted into NPGL electrodes with tunable pore size through a dealloying, electrochemical etching technique that effectively removes the silver from the gold/silver leaf composite. It is important to note here that the ability to control the material morphology and nanoparticle feature size to the nanometer scale provides the opportunity to utilize unique surface characteristics not found in the bulk material.53,54 Specifically for electrochemical systems, morphology with nanoscale features increases the interfacial area between the electrode and the electrolyte that consequently enhances electroactivity, improves thermodynamics, and increases scaffold area for biological agent immobilization.55,56 These attributes increase the sensitivity and lower the detection limit of electrochemical biosensors. Nanoporous gold in particular is an exceptional electrochemical material as it has a wide electrochemical window, excellent structural continuity, high conductivity, facile biorecognition agent functionalization through gold–thiol chemistry, and good biocompatibility.57Typically, the dealloying of gold/silver leaf to make NPGL is performed by floating the alloy leaf material on concentrated nitric acid which often takes hours to completely remove the silver from the composite.45,58,59 During these long etching times the gold self-diffuses, meaning gold atoms migrate and locally rearrange into a network. This gold self-diffusing process results in a “coarsening effect” that fills in void space. Hence, nanoporous gold feature sizes can increase from ∼5 nm to over 100 nm through such ‘coarsening effects,’ which consequently, significantly decreases the electrochemical surface area.43 To counteract this coarsening effect, Xu et al. demonstrated that by applying an oxidation potential (0.9 V) in concentrated nitric acid, silver can be dealloyed in minutes instead of hours.60 Erlebacher's group further expanded upon this work to demonstrate that small pore sizes (∼5 nm) can be formed with high purity levels (>99%) via electrochemical etching with concentrated nitric acid.61 In our experience the use of concentrated nitric acid still prevents this process utilization as it is extremely corrosive and damages most flexible substrates including various polymers such as polyimide. However, by applying a higher oxidation potential (1.2 V), silver can be selectively removed from the alloy even in diluted nitric acid solutions (Fig. 2a). This less harsh electrode synthesis environment permits the use of a wider range of substrate materials. Moreover, NPGL fabricated by this technique is conducive to batch chemical processing techniques and hence amenable to high throughput manufacturing. Additionally, using a diluted nitric acid solution (35%) significantly decreases the silver etching rate; therefore, electrodes can be selectively etched through this oxidation potential (see Movie S2, ESI†). During the dealloying process, the electrode changes to a yellow-brown color signifying the conversion to pure gold, Fig. 2b. To confirm this change, Energy Dispersive X-ray Spectroscopy (EDS) was performed (Fig. 2c). Initially the alloy displays strong energy dispersive peaks from the gold and silver, accounting for 45.6 and 45.8% respectively. Additionally, there are minor peaks for carbon, oxygen, and silicon (∼4, 1, and 3%, respectively), which correspond to the polyimide film and the underlying silicon adhesive. After dealloying into NPGL, the silver dispersion peak completely disappears (less than 1%). As the material becomes more porous, the weight percent of carbon, oxygen, and silicon increases due to the stronger interaction with the adhesive and polyimide film.
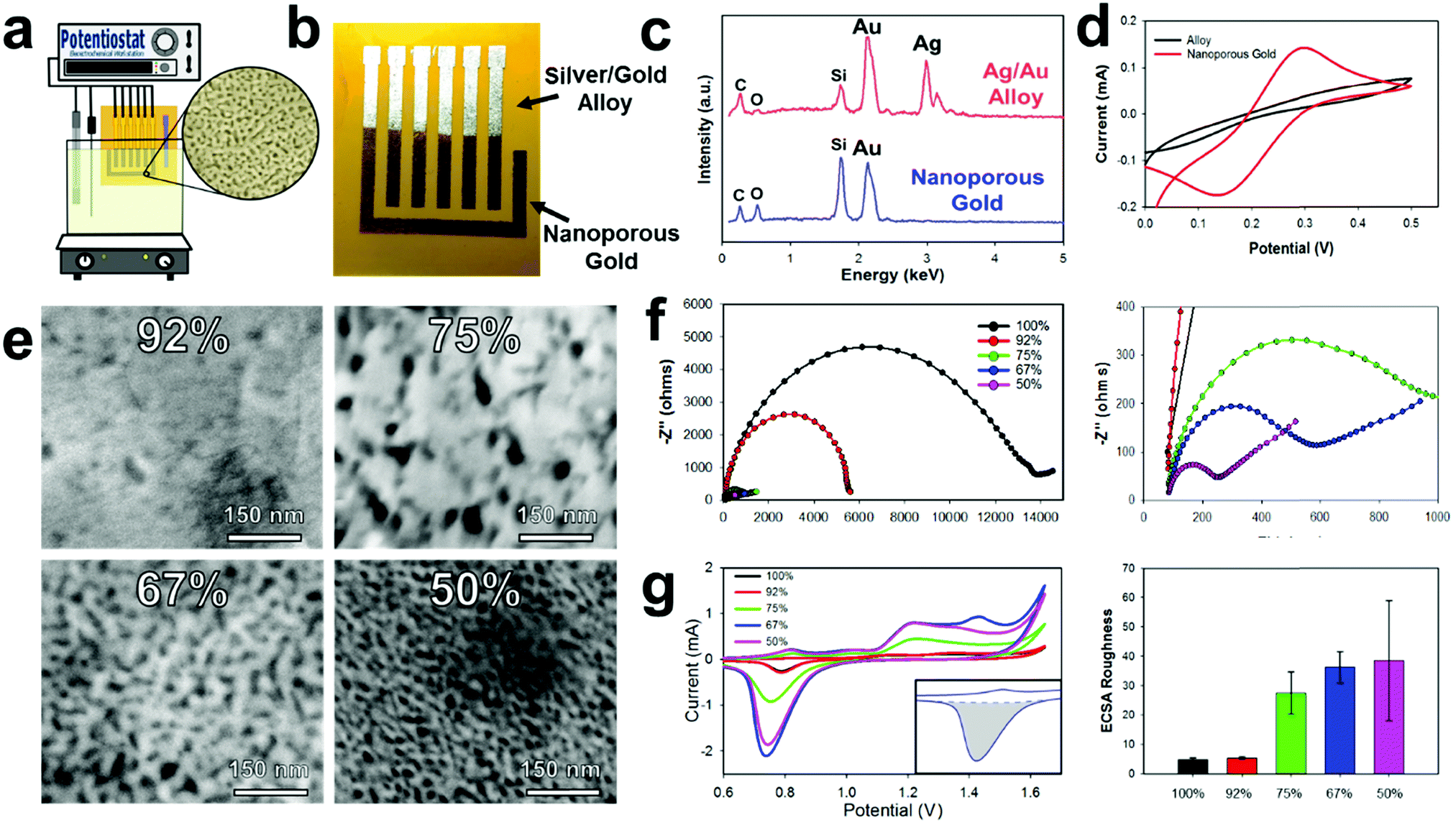 | ||
| Fig. 2 NPGL electrode characterization. (a) Schematic illustrating dealloying silver using diluted nitric acid (35%) and an oxidative potential (1.2 V). (b) Optical imaging depicting NPGL after silver has been removed through de-alloying. (c) EDS analysis of gold/silver alloy and NPGL, demonstrating removal of all silver. (d) CV in Fe(CN)63−/4− of gold alloy before and after silver removal (e) SEM images obtained from dealloying various gold/silver leaf ratios. (f) Nyquist impedance graphs of NPGL using different gold/silver leaf ratios. Right: Magnification of Nyquist plot. (g) CV of NPGL in 10 mM sulfuric acid forming and reducing a gold-oxide monolayer. Right: Bar graph extracted from CV showing the Electrochemical Surface Area (ECSA) roughness or ECSR, scale bars from 5 repeat measurements. | ||
As the silver is etched away from the leaf materials, the electrochemical area and electroactivity increases making the resultant working electrode more sensitive. Initially the E-IML patterned electrodes are inefficient at oxidizing and reducing ferro/ferricyanide (Fe(CN)63−/4−, redox probe) (Fig. 2d). After de-alloying in diluted nitric acid, distinct oxidation and reduction peaks appear due to the electroactive three-dimensional nanoporous structure. Due to the extremely thin film thickness (<100 nm) and high electroactive surface area, the NPGL transduction layer displays highly reversible redox cycling of Fe(CN)63−/4−, with peak-to-peak distances of 130 mV. While larger than the theoretical 59 mV of a perfectly reversible reaction, the NPGL electrodes demonstrate excellent cycling ability or high current response capability with limited electrical resistance.62,63 Various alloy leaf materials with different gold/silver concentrations were dealloyed to characterize and analyze the resulting transduction layer characteristics with distinct nanoporous morphologies (Fig. 2e). Generally, the porosity of the NPGL increases with higher starting concentrations of silver. With only 8 percent silver, limited porosity is developed as nitric acid is unable to etch through the dense gold surface. Increasing the silver concentration to 25% formed large pores (∼25 nm) that were sparse and irregular. Further increasing the concentration of silver formed a denser network of pores; interestingly, the pore size decreased (down to ∼5 nm) as well (Fig. 2e).
Next, electrochemical impedance spectroscopy (EIS) was conducted to evaluate the conductivity of the NPGL electrodes and the interfacial resistance between the electrode and electrolyte. All electrodes exhibited a resistance of roughly 80 Ω, corresponding to a sheet resistance of ∼5 Ω sq−1 (Fig. 2f with magnified scale to the right). When the concentration of silver was increased to 60%, the NPGL no longer formed a continuous network and the electrodes cracked. Initially the gold electrodes exhibited very high interfacial resistance (∼14 kΩ). After dealloying, the interfacial resistance of the NPGL reduces significantly. As the porosity of the gold increases due to smaller feature sizes, the charge transfer resistance between the electrode and electrolyte increases by roughly 2 orders of magnitude (interfacial resistance of ∼200 Ω at 50% silver). Note that the acid etching process most likely cleaned the gold surface (e.g., removed superficial organics) and hence improved electroactivity.64,65
The electrochemical surface of the NPGL was also characterized by cycling the electrodes in diluted sulfuric acid (10 mM). Typical electrochemical surface area calculations using cyclic voltammetry and various scan rates in Fe(CN)63−/4− are unable to estimate the surface area of the NPGL due to the growth of the diffusion layer which is orders of magnitude larger than the nanoscale feature sizes of the NPGL. Therefore, the electrochemical surface area roughness or ECSR was calculated instead by using the following protocols. At potentials above 1 V vs. Ag/AgCl, gold oxidizes forming a monolayer during the anodic scan. During the subsequent cathodic scan, the oxide layer is stripped creating a sharp reduction peak centered around 0.75 V vs. Ag/AgCl (Fig. 2g). The integration of this peak (minus the area of the surface charge) is proportional to the electrochemical surface area. For 100% gold leaf, the charge consumed during the reduction corresponded to 752 μC cm−2 (illustrated area greyed in Fig. 2g inset). Assuming pure planar gold corresponds to 559 μC cm−2,66 the patterned pure gold leaf material has an ECSR factor of 1.35 (Fig. 2g right). The pure gold surface is slightly higher than the projected geometric surface area due to wrinkling of the leaf material and defects in the surface. Also, gold leaf/silver alloy with 92% gold exhibited a slightly higher surface area (ECSR of 2.4) which corroborates the fact that some large pores were visualized in the gold during SEM image analysis (Fig. 2e). As the concentration of silver is increased, the surface roughness significantly increases to 27.5, 36.3, and 38.5 for leaf material containing 75%, 67%, and 50% gold, respectively. While 50% silver/50% gold leaf had the highest electrochemical surface area, it also had the largest standard deviation in ECSR (±20.4). This high deviation occurs because the gold becomes very porous without forming a continuous conductive network (some of the superficial gold is not electroactive). In order to increase the repeatability and robustness of the electrodes, gold leaf/silver alloys containing 67% gold were used which had an ECSR of 36.3 ± 5.3. Therefore, an electrode with a 4.5 mm2 geometric area has an approximate electrochemical surface area of 163 mm2.
Ag/AgCl pseudoreference leaf electrode fabrication
Most electrochemical setups, especially sensor related, need three separate electrodes (e.g. working, counter, and reference). The use of a reference electrode provides a constant oxidation–reduction potential which is used as a “reference” for the working electrode. In the past decade, there has been a significant push to decrease the size of reference electrodes and integrate them directly onto the substrate.67 One of the most common reference electrode materials is Ag/AgCl due to its compatibility with miniaturization, simple construction, safety, and reliability.68,69 E-IML presents a new manufacturing method for constructing Ag/AgCl reference electrodes. A silver leaf reference electrode can be patterned simultaneously with the gold or gold/silver alloy counter and working electrodes (gold etch effectively removes both gold and silver). The silver electrode is then chlorinated into a solid-state reference electrode. Therefore, E-IML presents a low-cost and facile technique for rapid prototyping a stable reference electrode.The solid-state Ag/AgCl pseudoreference electrode was fabricated by chlorinating an E-IML silver leaf electrode with 1% bleach solution for 5 min, using a PDMS mold as a reservoir (Fig. 3a). Bleach is a concentrated sodium hypochlorite solution which effectively chlorinates silver as reported in literature.70–72 After chlorination, the luster/color of the silver changed to a dull grey indicating the formation of a AgCl layer (Fig. 3a). SEMs before and after chlorination displayed the formation of AgCl nanoparticles on the surface of the electrode. After 5 minutes, a homogenous dense array of AgCl nanoparticles (∼250 nm) formed (Fig. 3b). The formation of a AgCl layer was confirmed through EDS (Fig. 3c). For the silver leaf electrode, there were strong and distinct elemental silver peaks at 3 keV and EDS quantitative analysis showed the presence of 100% silver without any contaminations. After chlorination, a sharp peak at 2.7 keV appeared with an atomic percentage of 30% Cl and 70% Ag.
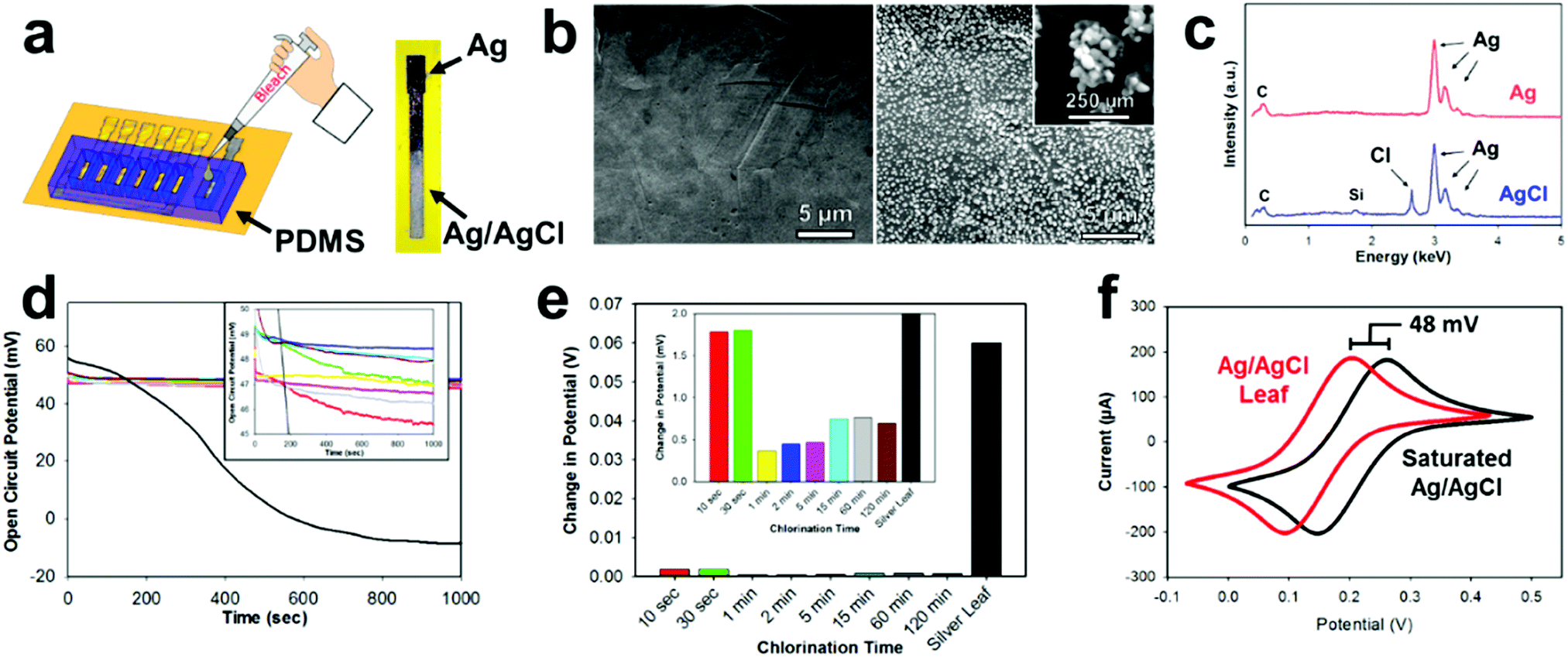 | ||
| Fig. 3 Ag/AgCl pseudoreference electrode characterization. (a) Schematic illustrating PDMS mold and silver chlorination using 1% concentrated bleach solution. (b) SEM images obtained from silver leaf electrode before and after chlorination for 5 min. Inset: Magnification of AgCl nanoparticle. (c) EDS analysis of silver leaf before and after chlorination demonstrating the formation of AgCl nanoparticles. (d) Open-circuit potential graphs of silver and different silver chlorination times. Inset: Magnification demonstrating less than 5 mV difference between chlorination times. (e) Bar graph of open-circuit potential showing change in potential over time. (f) Cyclic voltammograms of saturated single membrane Ag/AgCl reference (black) and chlorinated silver leaf (red) within solution containing the Fe(CN)63−/4− redox probe. | ||
After chlorination, the Ag/AgCl leaf electrodes demonstrated stable oxidation–reduction potential (Fig. 3d). When an open circuit potential was applied to a pure silver leaf electrode, the potential dropped from approximately 50 mV to −10 mV, demonstrating the unstable nature of using pure silver as a reference. Various chlorination times (10 s to 120 min) of E-IML silver electrodes were tested. All electrodes, irrelevant of their chlorination time, exhibited an open circuit of 5 mV from each other. The electrodes had an average open circuit potential of 48 mV vs. saturated single membrane Ag/AgCl reference, demonstrating that the fabricated Ag/AgCl leaf electrodes are not fully saturated with an AgCl layer. Nevertheless, the fabricated pseudoreference electrodes maintained stability with only minor changes (<5 mV) in potential for over 1000 seconds (Fig. 3e). To test the Ag/AgCl pseudoreference leaf electrode, an electrode was placed in 5 mM Fe(CN)63−/4− and cyclical voltammographs were collected for both the single membrane saturated Ag/AgCl and Ag/AgCl leaf electrodes (Fig. 3f). Both voltammographs displayed identical structure with a shift of 48 mV.
Disposable tape pesticide biosensor fabrication
The disposable tape electrodes were functionalized with enzymes for subsequent pesticide biosensors via thiolated polymers that are able to covalently bind the enzyme to the gold surface for consequent pesticide biosensing (Fig. 4a). The same PDMS mold used for chlorinating the silver electrodes was also utilized to isolate the working electrode for biofunctionalization with enzyme. Having separate wells for each working electrode provided a simple way to isolate and modify different working electrodes. For example, using this process, a multiplexed sensor array could be made with different biorecognition agents; or as demonstrated later in this manuscript, different enzyme concentrations can be immobilized to make a larger linear sensing range (high enzyme concentration) or lower detection limits (low enzyme concentration). Hence, the NPGL electrodes provide easy functionalization through thiol groups which self-assemble into monolayers (SAMs). L-Cysteine was used instead of alkanethiols (e.g., 11 mercaptoundecanoic acid (11-mua)), as the former could be dissolved and functionalized onto the gold surface in an aqueous solution instead of the latter which would require an ethanolic solution.73 While the electrodes are stable in ethanol or acetone solution, the adhesive tends to swell/degrade in the ethanol solution as opposed to the aqueous solution. Moreover, L-cysteine conjugation to gold is more rapid than 11-mua (10 min instead of multiple hours)—an attribute which further lessens the likelihood of adhesive degradation viaL-cysteine functionalization. L-Cysteine also contains amine functional groups that increases the rate of functionalization to gold; these amine–gold interactions are short lived and are quickly replaced by thiol–gold bonding.74 In terms of biofunctionalization, L-cysteine monolayer exposes carboxylic acids which provide unique immobilization sites for functionalization through EDC/NHS deprotonation mechanisms (Fig. 4b).75 This “zero order” functionalization strategy is water soluble and provides excellent stability for enzymes.76 First, EDC reacts with carboxyl groups to make an O-acylisourea intermediate ester. Second, NHS is added to form a stable amine-reactive NHS ester, which increases immobilization efficiency of the EDC coupling reaction. In our case, the enzyme ACHE is then bound to the ester by a free amine in the enzyme.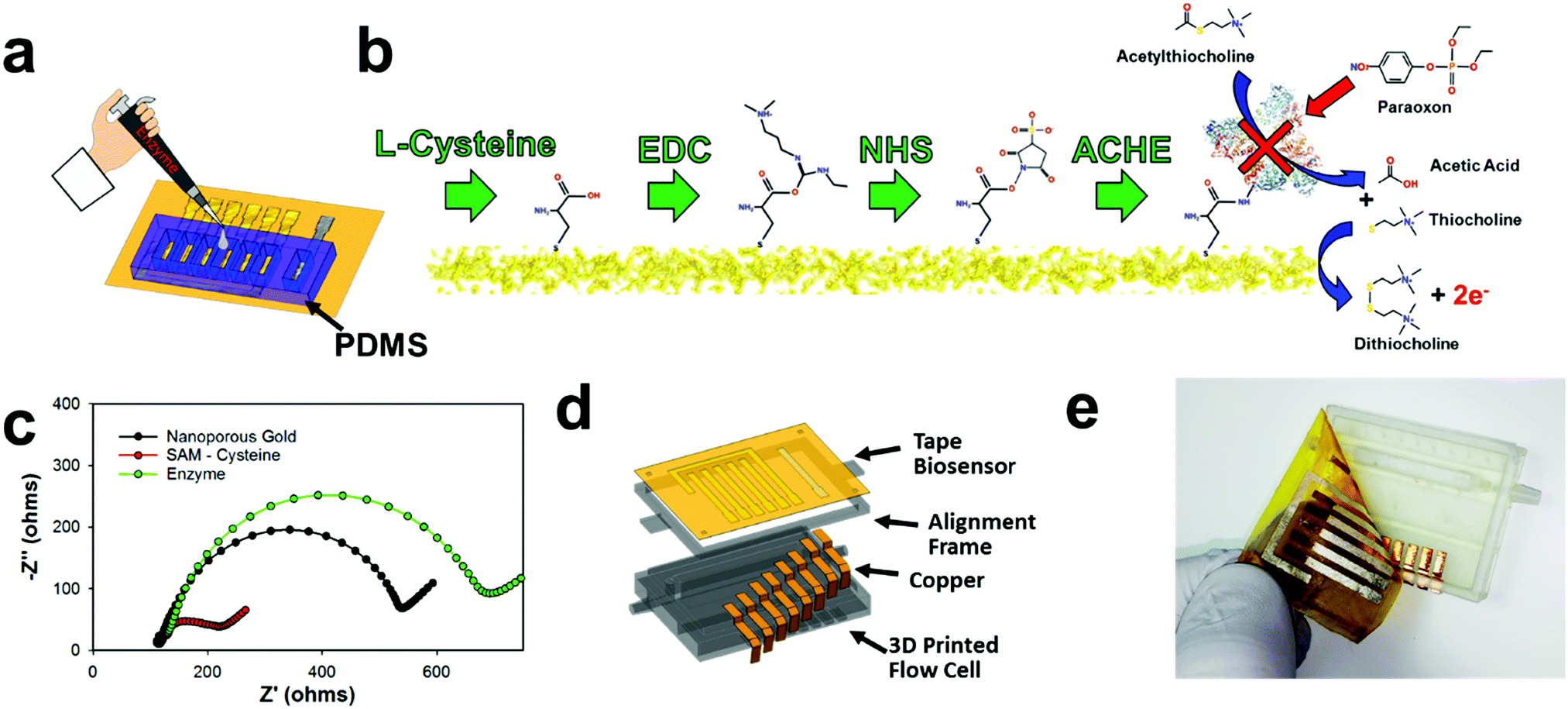 | ||
| Fig. 4 Functionalization of NPGL into a disposable and sensitive pesticide sensor with integrated flow cell. (a) Schematic illustrating PDMS mold to isolate and modify individual working electrodes. (b) Diagram of functionalization strategy for pesticide biosensing using L-cysteine and EDC/NHS. From left to right: L-cysteine forms SAM onto gold with thiol groups. EDC reacts with carboxyl groups to make an O-acylisourea intermediate ester. NHS reacts to form a stable amine-reactive ester. Finally, NHS ester binds ACHE to the gold surface through available amines in the enzyme. Basic operations of the ACHE pesticide biosensor. From top to bottom: ACTH reacts with ACHE and produces acetic acid and thiocholine. Thiocholine (2×) is oxidized by the NPGL electrode and produces dithiocholine and two electrons. Paraoxon (model pesticide/organophosphate) inhibits ACHE and prevents the catalysis of ACTH. Note: size not to scale. (c) Change in impedance with response to functionalization steps. (d) Schematic showing CAD design of fabricated flow cell and disposable tape sensor. (e) Photo of completed 3D printed flow cell with tape sensor being pealed back with fingers. | ||
It should be noted that ACHE biosensors are capable of detecting insecticide pesticides through enzyme inhibition which consequently inhibits the enzyme from continuously hydrolyzing the molecule acetylthiocholine (ACTH) (Fig. 4b right). This hydrolysis forms acetic acid and thiocholine which is an electroactive molecule that can be oxidized at the working electrodes with an applied potential of 450 mV vs. Ag/AgCl. This subsequent reaction produces dithiocholine and two electrons. These electrons consequently transfer to the electrode surface heterogeneously and are detected through an amperometric sensing modality to monitor distinct concentrations of insecticides. More specifically, organophosphates (such as paraoxon) irreversibly bind to ACHE and prevent the hydrolysis of ACTH. This results in the inhibition of ACHE that consequently decreases the amperometric current.
Next, interfacial resistance between the electrode and electrolyte was characterized through electrochemical impedance spectroscopy (EIS) employing Fe(CN)63−/4− which acts as a redox probe for the different functionalization steps (Fig. 4c). The NPGL has low interfacial resistance of roughly 450 Ω (diameter of semicircle portion of Nyquist plot). However, after being functionalized with a SAM of L-cysteine, the resistance significantly reduced to approximately 100 Ω. This decrease in charge transfer resistance due to L-cysteine modification is attributed to the fact that the monolayer (rich in carboxylic acids) promotes faster Fe(CN)63−/4− redox cycling.77 After immobilizing ACHE through EDC/NHS chemistry, the impedance significantly increased with interfacial resistance of roughly 600 Ω. As the large protein was attached to the surface, carboxylic acid groups and the high electroactive NPGL electrode was blocked from oxidizing and reducing Fe(CN)63−/4−, which confirms that ACHE was functionalized to the surface of the nanoporous gold.
An E-IML NPGL tape pesticide biosensor was next designed and integrated with a 3D printed microfluidic flow cell (Fig. 4d). The inlet and outlet channels were attached with 1/8-inch internal diameter tubing. The flow was then directed to an open channel that was sealed with the disposable tape biosensor. The open channel was constructed to be 3 mm wide and 150 μm deep. Copper leads were fabricated using thin cut copper foil tape to electrically connect the five NPGL electrodes (four working electrode and one counter electrode) and one chlorinated SL electrode (reference electrode). This multiplexed tape biosensor adhered well to the 3D printed flow cell as fluid did not leak during flow cell operation and electrical contact was maintained with the potentiostat during electrochemical sensing (Fig. 4e).
Characterization of microfluidic pesticide biosensor
The sealing capability of the NPGL tape biosensors with the 3D printed microfluidic was characterized by pumping water (dyed blue) through the device with a syringe pump (Fig. 5a and Movie S3, ESI†). The 3D printed open channel microfluidic was completely sealed by the tape biosensor without any leaks. Additionally, the liquid properly wet the entire fluidic and did not trap any bubbles along its flow. For electrochemical measurements, small alligator clips were attached to the copper leads. Electrically insulative plastic films were placed between clips to prevent shorting of the electrode.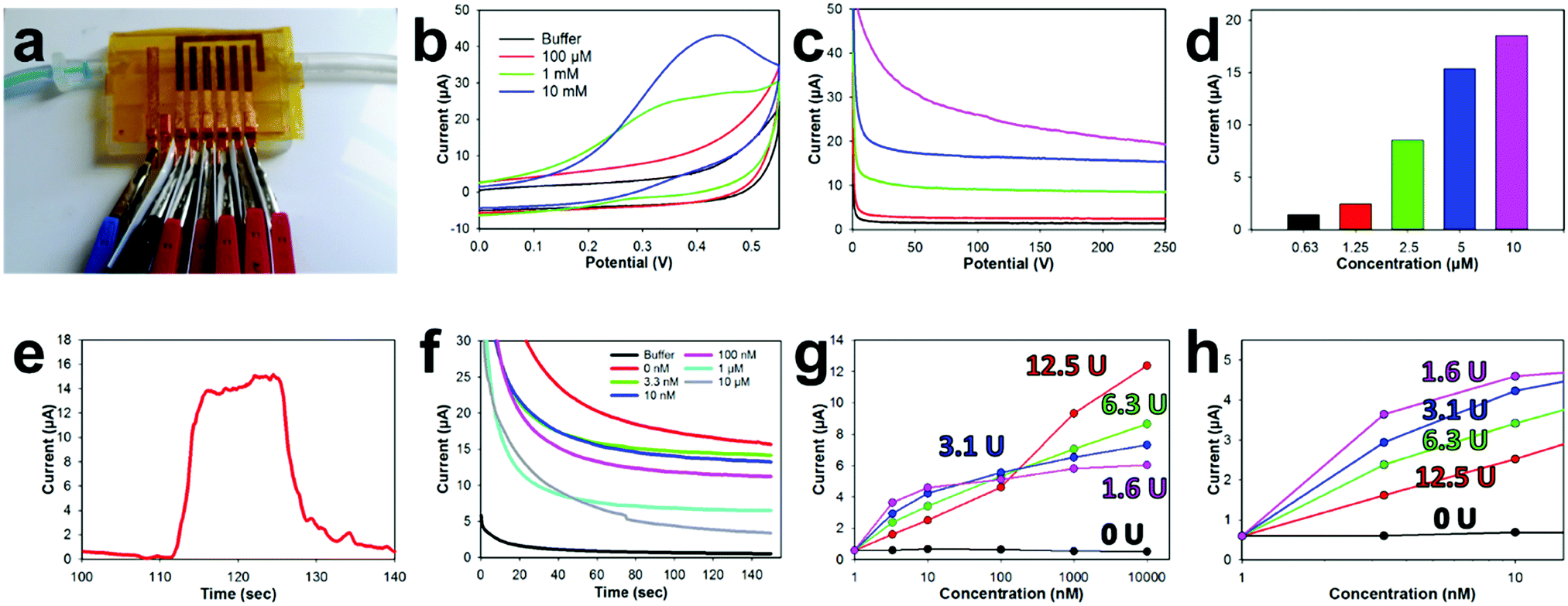 | ||
| Fig. 5 Performance of the designed disposable tape pesticide biosensor and microfluidic flow cell. (a) Photo of the experimental setup. (b) CVs with bare NPGL electrode within different concentrations of ACTH. (c) Sensitivity response of NPGL electrode to ACTH and (d) bar graph showing sensitivity to distinct concentration of ACTH. (e) Amperometric response obtained with NPGL electrode attached to flow cell while flowing buffer solution was spiked for 20 s with ACTH. (f) Amperometric sensitivity to various concentrations of paraoxon. (g) Concentrations vs. current graph showing pesticide sensitivity across five different working electrodes. (h) Magnified view of concentration vs. current graph. Enzyme concentration used to functionalize each electrode is highlighted in (g) and (h). | ||
Various concentrations of ACTH, were then pumped into the flow channel and a static CV was obtained (Fig. 5b) from the attached ACHE functionalized biosensors. A thiolcholine electroactive oxidation peak at 0.4 V was formed in these CVs as the concentration of ACTH increased to 10 mM. This potential is slightly shifted (∼50 mV) lower compared to the 0.45 V oxidation potential of thiocholine previously noted in the literature.78 This potential shift is most likely due to the unique electrochemical characteristics of the E-IML chlorinated silver leaf reference. Therefore, an applied potential of 0.4 V was used, for the corresponding amperometry tests with the ACHE functionalized electrodes. Increasing concentrations of ACTH were then pumped through the microfluidic with a flow rate of 0.25 mL min−1. The developed sensor has a sensitivity to ACTH of 1.8 μA μM−1. As the concentration of ACTH was increased, there was an increase in oxidation current (Fig. 5c and d). A baseline substrate concentration of 5 μM was used for subsequent pesticide measurements as the amperometric signal response was clear at this concentration and did not contain the noise that was experienced at higher substrate concentrations (10 μM). The response time of this sensor towards ACTH was tested by spiking a buffer solution with 5 μM ACTH at the 110 s mark for 20 s (Fig. 5d). Almost instantaneously there was a dramatic increase in current which held relatively steady during the substrate addition. The current quickly decreased when ACTH was removed.
The disposable tape ACHE biosensor was developed by functionalizing different working electrodes with distinct concentrations of ACHE (0, 1.56, 3.125, 6.25, 12.5 U). Incorporating a sensor without any ACHE provided a sentinel working electrode that could be used to detect potential interference oxidation and reduction molecules in the testing solution. Having different concentration of ACHE for various electrodes on the tape biosensor not only provided repetitive measurements for improved validation but provided a unique opportunity to have different LODs and linear sensing ranges. A calibration plot for each working electrode was developed by flowing increasing concentrations of paraoxon across the sensor surfaces through the microfluidic (Fig. 5f). As the concentration of paraoxon was increased, ACHE was inhibited which lowered the oxidation current. Electrodes functionalized with low enzyme concentrations (1.56 U, Fig. 5g and h, fuchsia) needed only a small amount of pesticide to completely inhibit the sensor (almost completely inhibited at 10 nM).78 However, such a small amount of functionalized enzyme provided a more favorable biosensor signal-to-noise ratio which led to a higher biosensor sensitivity and lower detection limit (376 nA nM−1 and 0.53 pM, respectively). Comparatively, having a large enzyme concentration (12.5 U, red) on the electrode of the biosensor required a large amount of pesticide to inhibit the sensor. This electrode functionalized with a large enzyme concentration experienced a large linear sensing range (roughly 4 orders of magnitude (1 nM–10 μM)) but a lower sensitivity and higher detection limit (197 nA nM−1 and 10.1 pM, respectively).
Finally, the developed microfluidic biosensors were tested in an actual complex biological solution. Soil from a local field was mixed with tap water at a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio by mass. This soil slurry was then spiked with 10 nM or 1 μM concentrations of the pesticide paraoxon (a model organophosphate pesticide16), vortex mixed for 1 min, and allowed to rest/settle for 5 min. Next, the slurry was filtered through a 0.45 μm syringe filter, adjusted to a pH of 8 with NaOH, and the ionic concentration of the solution was increased by adding 10 mM NaCl. The solution was then flowed through the 3D printed cartridge that was sealed with the tape biosensor with a syringe pump. This biosensor system was able to detect the pesticide in the filtered soil slurry within 10% accuracy (Table 1). It should be noted that using multiple working electrodes provides additional measurements for improved precision, but also provides different accuracy ranges. The biosensors with lower concentrations of ACHE (1.56 and 3.125 U) were more accurate at low concentrations but underestimated higher concentrations (Table 1). Conversely, high ACHE electrodes were not as accurate at low concentrations but were within about 1% accuracy at higher concentrations (1 μM). Interestingly, almost all sensors under projected the pesticide concentration. We hypothesis this low measurement was due to the pesticide binding to soil and clay particles, which would decrease the concentration in the final filtered slurry mixture and hence lower the signal response.
:1 ratio by mass. This soil slurry was then spiked with 10 nM or 1 μM concentrations of the pesticide paraoxon (a model organophosphate pesticide16), vortex mixed for 1 min, and allowed to rest/settle for 5 min. Next, the slurry was filtered through a 0.45 μm syringe filter, adjusted to a pH of 8 with NaOH, and the ionic concentration of the solution was increased by adding 10 mM NaCl. The solution was then flowed through the 3D printed cartridge that was sealed with the tape biosensor with a syringe pump. This biosensor system was able to detect the pesticide in the filtered soil slurry within 10% accuracy (Table 1). It should be noted that using multiple working electrodes provides additional measurements for improved precision, but also provides different accuracy ranges. The biosensors with lower concentrations of ACHE (1.56 and 3.125 U) were more accurate at low concentrations but underestimated higher concentrations (Table 1). Conversely, high ACHE electrodes were not as accurate at low concentrations but were within about 1% accuracy at higher concentrations (1 μM). Interestingly, almost all sensors under projected the pesticide concentration. We hypothesis this low measurement was due to the pesticide binding to soil and clay particles, which would decrease the concentration in the final filtered slurry mixture and hence lower the signal response.
| Conc. spike | 1.56 U | 3.125 U | 6.25 U | 12.5 U | Average | Accuracy |
|---|---|---|---|---|---|---|
| 10 nM | 12.8 nM | 13.2 nM | 6.9 nM | 4.5 nM | 9.35 nM | 93.5% |
| 1 μM | 0.72 μM | 0.89 μM | 0.93 μM | 1.079 μM | 0.915 μM | 91.5% |
Conclusions
In conclusion, a facile amperometric biosensor platform was developed for the detection and measurement of organophosphates (paraoxon), using a disposable nanoporous gold leaf (NPGL) tape biosensor that interfaces and sealed a 3D printed microfluidic flow cartridge. The tape biosensor was fabricated with a newly developed thin film patterning technique coined Etching Inkjet Maskless Lithography (E-IML). The resulting biosensor was equipped with 4 different working electrodes biofunctionalized with different concentrations of the enzyme acetylcholinesterase (ACHE) to provide increased sensing precision including a larger sensing window for organophosphate concentrations. The E-IML NPGL electrode functionalized with a low concentration of ACHE (1.56 U) exhibited a low paraoxon detection limit of 0.53 pM and a sensitivity of 376 nA nM−1 while the electrode functionalized with a high ACHE enzyme concentration (12.5 U) exhibited a much wider linear sensing range for paraoxon from 1 nM to 10 μM (four orders of magnitude). The biosensor flow cell platform was tested in a soil slurry that was spiked with paraoxon (10 nM and 1 μM) to demonstrate in-field testing. This biosensor flow cell exhibited a signal recovery of approximately of 93.5% and 91.5% for the low and high paraoxon concentrations respectively.E-IML NPGL provides an excellent transduction layer exhibiting an extremely high electroactive surface area (roughness factor of 36), low sheet resistance (∼5 Ohms sq−1), and excellent electroactivity. We show that NPGL has nearly completely reversible oxidation and reduction reactions for Fe(CN)63−/4−, provides a large scaffold platform for enzyme immobilization through facile thiol SAM conjugation, and therefore is an exceptional transduction layer for ACHE biosensors. Additionally, we show for the first time that pure silver leaf (SL) can be fabricated into stable solid-state reference electrodes. Using E-IML, the electrodes can be patterned simultaneously with the NPGL electrodes and chlorinated by simply drop casting a diluted bleach solution onto the SL. The developed biosensor exhibited stable potential with less than 5 mV potential changes over 1000 s. Therefore E-IML, provides a facile method for patterning all electrodes needed (working, counter, and reference) on the same film simultaneously for standard 3-electrode electrochemistry.
This work also demonstrated the versatility of the E-IML technique in that the technique can be used to pattern a variety of different geometries such as interdigitated electrodes (IDEs), antennas, and high entropy designs and be used to pattern a variety of distinct leaf materials (copper, aluminum, gold, silver, and gold/silver alloys). Hence this metallic leaf patterning technique could have wide utility across different application fields that use thin film materials, such as electrochemical sensors, supercapacitors, batteries, fuel cells, energy harvesters, triboelectric nanogenerators, and membranes.
Materials and methods
Chemicals, reagents, and materials
Leaf materials were purchased from Sepp Leaf and polyimide (Kapton tape) with silicon adhesive was purchased from Amazon. Acetylcholinesterase (ACHE) derived from Electrophorus electricus was purchased from Sigma Aldrich as lyophilized powder (200–1000 units per mg) and was used without any further purification. ACHE ink aliquots were made by dissolving the enzyme (2 mg) in 1 mL of 50% glycerol and 50% 1× phosphate-buffer solution (0.1 M PBS, pH 7.0), aliquoted into 25 μL, and stored in a −80 °C freezer until use. All pesticides (including paraoxon) were obtained from Chem Service. All other chemicals and solvents were purchased from Sigma-Aldrich and were used without any other purification unless otherwise stated. Water was filtered through a B-Pure Water Purification system (resistivity, 18.2 MΩ cm−2).Gold, silver, and alloy leaf materials (∼100 nm thick) were ordered from Sepp Leaf and manufactured by Manetti. Polyimide tape (2.5 mill, Kapton Tape) with a silicon adhesive layer was ordered from Grainger. The metal laminate is fabricated by simply adhering a metal foil or leaf material to silicon adhesive polyimide tape. For ease of manufacturing and improved laminate quality the leaf material is transferred with “patent paper” (a special tissue paper loosely adhered to the metal leaf). The metal laminate is then rinsed with acetone, isopropanol and DI water to remove any surface contaminates.
Inkjet printed protective polymer layer
The physical properties of the protective polymer ink and the printer settings are critically important to create a high-resolution pattern. Similar to the sacrificial ink that we previously developed to create high resolution printed graphene electrodes,78,79 the protective layer consists of an acrylic lacquer and an organic solvent. In short, 2 mL of an acrylic formaldehyde lacquer and 8 mL of cyclohexanone were vortexed together. The ink was then placed in a desiccator for 2 min to remove any air bubbles induced during mixing. The polymer ink was printed using a Dimatix Material Printer (DMP 2800 series). The polymer ink was loaded into a 3 mL printer cartridge and printed with 10 pL nozzles. The waveform was adjusted to print at a temperature of 40 °C with a 25 μm drop spacing while the patent table was adjusted to a temperature of 60 °C. These parameters yielded consistent droplets that adhered to the surface without pooling or creating a “coffee ring” effect. Five consecutive layers of polymer ink were patterned as it insured that no pinholes were present in the protective layer.Metal wet etch patterning
Next, the foils are then immersed in an etching solution to remove any metal not protected by the inkjet printed polymer layer. For copper and aluminum foils, an iron chloride etching solution was made by dissolving 300 g of iron (3+) chloride hexahydrate in 1 L of DI water. It should be noted that a copper foil that is 3 mm in thickness requires about 30 minutes of wet etching before the patterns are complete (some undercut etching will be visible). Gold, silver, and gold/silver alloy materials are etched in 30% gold etchant (Sigma 651818). As leaf material is extremely thin (∼100 nm), a diluted gold etchant is able to remove the unprotected gold and silver in under one minute with no visible undercut.Dealloy gold/silver leaf to create nanoporous gold leaf (NPGL)
NPGL electrodes were fabricated by dealloying silver out of 12k (50% gold and 50% silver) alloy leaf. After patterning through E-IML, the electrodes were placed in diluted nitric acid (35% concentrated) and exposed to an oxidative potential of 1.2 V vs. Ag/AgCl for 30 s. Without an oxidation potential being applied, the silver etches much slower. This allows for the working and counter electrodes to be selectively etched as exhibited in Movie S2 (ESI†). This high potential quickly etched the majority of the silver from the electrodes. Next, a lower potential of 0.9 V vs. Ag/AgCl was applied for 300 s to remove any remaining silver. Note, that an acrylic polymer layer is brush coated over the pure silver reference electrode to prevent any etching of the pseudoreference electrode during NPGL dealloying.Ag/AgCl pseudoreference electrode fabrication
Similar to the gold electrode fabrication protocol, a Ag/AgCl reference electrode was fabricated using pure silver leaf material and the E-IML printing method. First, a silver laminate was constructed by adhering pure silver leaf material onto silicon adhesive Kapton tape. Next, a protective polymer layer is inkjet printed (5 layers, 25 μm drop spacing) in the dimensions of the reference electrode directly onto the silver leaf laminate. The metal laminate is then placed in gold etch which etched and removed the leaf material that is not protected by the polymer layer. The remaining electrode is then exposed by rinsing with acetone to remove the protective polymer layer. A 1% concentrated bleach solution is pipetted onto this electrode for 5 min to cholorinate the silver electrode and form a superficial layer of AgCl on the electrode surface. A PDMS mold was used to isolate the reference electrode for chlorination.ACHE ink preparation and deposition
NPGL electrodes were functionalized with a self-assembled monolayer (SAM). A PDMS mold was used to isolate the working electrodes and then a 10 mM solution of L-cysteine in 1× PBS was pipetted into the wells for 10 min. L-Cysteine forms a SAM with the thiol group leaving an exposed carboxyl group. After functionalization with L-cysteine, the electrodes were thoroughly rinsed with 1× PBS to remove any unbound particles.The L-cysteine functionalized NPGL electrodes are then bioconjugated with the enzyme ACHE using 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide and N-hydroxysuccinimide (EDC/NHS) chemistry, which facilitated the functionalization of carboxyl groups to the amines within the ACHE enzyme.80 In summary, a PDMS mold was placed onto the NPGL electrodes to isolate the working electrodes, a solution of EDC/NHS mixture (50 mM EDC, 50 mM NHS) was then pipetted into the wells, and placed on a tilter plate for 30 min. The electrodes were then rinsed with 1× PBS. Excess water was blown off but not completely dried as the surface remained “wetted.” Next, 5 μL of the pre-aliquoted ACHE solution was immediately pipetted into the PDMS wells and incubated overnight. Electrodes were washed 3 times in 1× PBS to remove any unbound ACHE before testing. To create the four distinct working electrodes, a series of diluted concentrations (50, 75, 87.5, and 93.8%) of ACHE was immobilized on the four electrode surfaces.
Electro-analytical measurements
All electrochemical biosensor measurements and procedures were conducted on a CH instrument potentiostat (600E series) with a standard three electrode setup (single membrane Ag/AgCl reference electrode, platinum wire counter electrode, and NPGL working electrodes). For pesticide biosensing, a multiplexed tape biosensor with E-IML printed NPGL working and counter electrodes and a bleach chlorinated silver leaf reference electrode was used.Unless specified, each flow cell/tape biosensor test was performed with 15 mL of 1× PBS buffer pumped through the flow cell with a syringe pump with a flow rate of 0.25 mL min−1. For pesticide sensing, the biosensor was first tested in buffer to achieve a baseline or background noise measurement and ensure that solution did not contain contaminates that oxidize at the working potential of 400 mV. The sensor was then tested in 5 mM ACTH to acquire a base sensitivity to ACTH. The biosensor was then incubated in an aqueous test solution with the pesticide for a minimum of 300 s. During this incubation step, the pesticide permanently binds to ACHE, preventing it from catalyzing ACTH and therefore the product thiocholine from oxidizing on the electrode surface. The sensor is then retested in the same 5 mM ACTH to acquire the decrease in amperometric response (referred to henceforth as inhibition sensitivity).
Electrochemical surface area (ECSA) calculations
ECSA of the NPGL was estimated by cycling the electrodes in 100 mM of sulfuric acid with a scan rate of 100 mV s−1 between −0.2 and 1.6 V. During the forward oxidation cycle oxygen adsorption/chemisorption occurs on the gold surface; during the consecutive anodic cycle, the oxide layer is reduced. The area under the reduction peak (1.3–1.6 V) is proportional to the electrochemical surface area. Pure planar gold typically has an electrochemical surface area value between 0.4–0.6 μC cm−2, with most researchers assuming a value of 559 μC cm−2.81 Therefore, the electrochemical surface area roughness was calculated by dividing the integrated reduction peak by 559 μC cm−2. ECSA was then calculated by multiplying the surface roughness by the geometric area.Flow cell
The microfluidic flow cell was designed in CAD software and was printed with a FormsLab 2 stereolithography printer using UV curing resin (FLGPWH03). Copper foil was adhered to the 3D printed fluidic to provide conductive tabs for electrical connection with the potentiostat. To increase the contact between the copper electrodes and the E-IML gold foil electrodes, tabs 250 μm high were added under electrode leads. An alignment frame was also designed that could help align the disposable tape biosensor but also provide some tension across the very flexible sensor to prevent uneven adhesion to the flow cell.Field emission SEM images
Field emissions scanning electron microscopy (FESEM) images were acquired using a FEI Quanta 250 FESEM. All images were magnified to 500×, 1500×, 5000×, or 15000× using secondary electrons, spot size of 3.0, and with a 10 kV potential.
Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
J. C. C. gratefully acknowledges funding support for this work by the National Institute of Food and Agriculture, U.S. Department of Agriculture, under award number 2018-67011-27989 and 2016-67021-25038.References
- A. Economou, C. Kokkinos and M. Prodromidis, Lab Chip, 2018, 18, 1812–1830 RSC.
- M. Xu, D. Obodo and V. K. Yadavalli, Biosens. Bioelectron., 2019, 124-125, 96–114 CrossRef CAS PubMed.
- https://www.marketsandmarkets.com/Market-Reports/wearable-sensor-market-158101489.html?gclid=CjwKCAiAmb7RBRATEiwA7kS8VBfPfEHB04iFOH0H-Ldx6CN1YfnLNtTWhD3wXYIXtWyNtuB0bO2yjBoCVbMQAvD_BwE .
- N. J. Ronkainen, H. B. Halsall and W. R. Heineman, Chem. Soc. Rev., 2010, 39, 1747–1763 RSC.
- J. Kim, A. S. Campbell, B. E.-F. de Ávila and J. Wang, Nat. Biotechnol., 2019, 37, 389–406 CrossRef CAS PubMed.
- A. C. Sun, C. Yao, A. Venkatesh and D. A. Hall, Sens. Actuators, B, 2016, 235, 126–135 CrossRef CAS PubMed.
- E. B. Setterington and E. C. Alocilja, Biosens. Bioelectron., 2011, 26, 2208–2214 CrossRef CAS PubMed.
- D. Vanegas, L. Patiño, C. Mendez, D. Oliveira, A. Torres, C. Gomes and E. McLamore, Biosensors, 2018, 8, 42 CrossRef PubMed.
- Y. Ye, W. Yan, Y. Liu, S. He, X. Cao, X. Xu, H. Zheng and S. Gunasekaran, Anal. Chim. Acta, 2019, 1074, 80–88 CrossRef CAS PubMed.
- X. Cetó, A. M. O’Mahony, I. A. Samek, J. R. Windmiller, M. del Valle and J. Wang, Anal. Chem., 2012, 84, 10306–10314 CrossRef PubMed.
- J. Erickson, L. Shriver-Lake, D. Zabetakis, D. Stenger and S. Trammell, Sensors, 2017, 17, 1769 CrossRef PubMed.
- S. A. Trammell, L. C. Shriver-Lake and W. J. Dressick, Sens. Actuators, B, 2017, 239, 951–961 CrossRef CAS.
- J. Barton, M. B. G. García, D. H. Santos, P. Fanjul-Bolado, A. Ribotti, M. McCaul, D. Diamond and P. Magni, Microchim. Acta, 2016, 183, 503–517 CrossRef CAS.
- Y. Qin, H.-J. Kwon, M. M. Howlader and M. J. Deen, RSC Adv., 2015, 5, 69086–69109 RSC.
- N. T. Garland, E. S. McLamore, N. D. Cavallaro, D. Mendivelso-Perez, E. A. Smith, D. Jing and J. C. Claussen, ACS Appl. Mater. Interfaces, 2018, 10, 39124–39133 CrossRef CAS PubMed.
- J. A. Hondred, J. C. Breger, N. J. Alves, S. A. Trammell, S. A. Walper, I. L. Medintz and J. C. Claussen, ACS Appl. Mater. Interfaces, 2018, 10, 11125–11134 CrossRef CAS PubMed.
- J. R. Sempionatto, T. Nakagawa, A. Pavinatto, S. T. Mensah, S. Imani, P. Mercier and J. Wang, Lab Chip, 2017, 17, 1834–1842 RSC.
- M. X. Chu, K. Miyajima, D. Takahashi, T. Arakawa, K. Sano, S.-I. Sawada, H. Kudo, Y. Iwasaki, K. Akiyoshi, M. Mochizuki and K. Mitsubayashi, Talanta, 2011, 83, 960–965 CrossRef CAS PubMed.
- J. Kim, M. Kim, M.-S. Lee, K. Kim, S. Ji, Y.-T. Kim, J. Park, K. Na, K.-H. Bae and H. K. Kim, Nat. Commun., 2017, 8, 14997 CrossRef PubMed.
- W. Gao, S. Emaminejad, H. Y. Y. Nyein, S. Challa, K. Chen, A. Peck, H. M. Fahad, H. Ota, H. Shiraki and D. Kiriya, Nature, 2016, 529, 509 CrossRef CAS PubMed.
- J. Kim, S. Imani, W. R. de Araujo, J. Warchall, G. Valdés-Ramírez, T. R. Paixão, P. P. Mercier and J. Wang, Biosens. Bioelectron., 2015, 74, 1061–1068 CrossRef CAS PubMed.
- R. K. Mishra, L. J. Hubble, A. Martín, R. Kumar, A. Barfidokht, J. Kim, M. M. Musameh, I. L. Kyratzis and J. Wang, ACS Sens., 2017, 2, 553–561 CrossRef CAS PubMed.
- A. J. Bandodkar, W. Jia and J. Wang, Electroanalysis, 2015, 27, 562–572 CrossRef CAS.
- W. Jia, A. J. Bandodkar, G. Valdés-Ramírez, J. R. Windmiller, Z. Yang, J. Ramírez, G. Chan and J. Wang, Anal. Chem., 2013, 85, 6553–6560 CrossRef CAS PubMed.
- B. V. Chikkaveeraiah, V. Mani, V. Patel, J. S. Gutkind and J. F. Rusling, Biosens. Bioelectron., 2011, 26, 4477–4483 CrossRef CAS PubMed.
- L. Gervais, N. De Rooij and E. Delamarche, Adv. Mater., 2011, 23, H151–H176 CrossRef CAS PubMed.
- A. Martín, J. Kim, J. F. Kurniawan, J. R. Sempionatto, J. R. Moreto, G. Tang, A. S. Campbell, A. Shin, M. Y. Lee, X. Liu and J. Wang, ACS Sens., 2017, 2, 1860–1868 CrossRef PubMed.
- C. Rivet, H. Lee, A. Hirsch, S. Hamilton and H. Lu, Chem. Eng. Sci., 2011, 66, 1490–1507 CrossRef CAS.
- Y. Lei, W. Zhao, Y. Zhang, Q. Jiang, J.-H. He, A. J. Baeumner, O. S. Wolfbeis, Z. L. Wang, K. N. Salama and H. N. Alshareef, Small, 2019, 15, 1901190 CrossRef PubMed.
- S. Iguchi, H. Kudo, T. Saito, M. Ogawa, H. Saito, K. Otsuka, A. Funakubo and K. Mitsubayashi, Biomed. Microdevices, 2007, 9, 603–609 CrossRef CAS PubMed.
- J. C. Claussen, J. Shi, A. R. Diggs, D. M. Porterfield and T. S. Fisher, in Nanotechnologies for the Life Sciences, ed. C. Kumar, 2012, vol. 9 Search PubMed.
- J. N. Tiwari, V. Vij, K. C. Kemp and K. S. Kim, ACS Nano, 2015, 10, 46–80 CrossRef PubMed.
- J. Wang, Electroanalysis, 2005, 17, 7–14 CrossRef CAS.
- J. M. George, A. Antony and B. Mathew, Microchim. Acta, 2018, 185, 358 CrossRef PubMed.
- W. Siangproh, W. Dungchai, P. Rattanarat and O. Chailapakul, Anal. Chim. Acta, 2011, 690, 10–25 CrossRef CAS PubMed.
- H.-J. Qiu, X. Li, H.-T. Xu, H.-J. Zhang and Y. Wang, J. Mater. Chem. C, 2014, 2, 9788–9799 RSC.
- J. M. Pingarrón, P. Yañez-Sedeño and A. González-Cortés, Electrochim. Acta, 2008, 53, 5848–5866 CrossRef.
- L. Wang, W. Mao, D. Ni, J. Di, Y. Wu and Y. Tu, Electrochem. Commun., 2008, 10, 673–676 CrossRef CAS.
- S. Guo and E. Wang, Anal. Chim. Acta, 2007, 598, 181–192 CrossRef CAS PubMed.
- X. Zhang, D. Li, L. Bourgeois, H. Wang and P. A. Webley, ChemPhysChem, 2009, 10, 436–441 CrossRef CAS PubMed.
- S. M. Woo, C. M. Gabardo and L. Soleymani, Anal. Chem., 2014, 86, 12341–12347 CrossRef CAS PubMed.
- S. Products, SeppLeaf, https://www.seppleaf.com/.
- Y. Ding, Y. J. Kim and J. Erlebacher, Adv. Mater., 2004, 16, 1897–1900 CrossRef CAS.
- P. N. Ciesielski, A. M. Scott, C. J. Faulkner, B. J. Berron, D. E. Cliffel and G. K. Jennings, ACS Nano, 2008, 2, 2465–2472 CrossRef CAS PubMed.
- R. Zeis, A. Mathur, G. Fritz, J. Lee and J. Erlebacher, J. Power Sources, 2007, 165, 65–72 CrossRef CAS.
- X. Ge, R. Wang, P. Liu and Y. Ding, Chem. Mater., 2007, 19, 5827–5829 CrossRef CAS.
- K. Hu, D. Lan, X. Li and S. Zhang, Anal. Chem., 2008, 80, 9124–9130 CrossRef CAS PubMed.
- F. Meng, X. Yan, J. Liu, J. Gu and Z. Zou, Electrochim. Acta, 2011, 56, 4657–4662 CrossRef CAS.
- M. S. F. Santos, W. A. Ameku, I. G. R. Gutz and T. R. L. C. Paixão, Talanta, 2018, 179, 507–511 CrossRef CAS PubMed.
- R. Sidhu, Y. Rong, D. C. Vanegas, J. Claussen, E. S. McLamore and C. Gomes, Smart Biomedical and Physiological Sensor Technology XIII, 2016, vol. 9863 Search PubMed.
- S. Ding, S. R. Das, B. J. Brownlee, K. Parate, T. M. Davis, L. R. Stromberg, E. K. L. Chan, J. Katz, B. D. Iverson and J. C. Claussen, Biosens. Bioelectron., 2018, 117, 68–74 CrossRef CAS PubMed.
- S. Charkhabi, A. M. Beierle, M. D. McDaniel and N. F. Reuel, ACS Sens., 2018, 3, 1489–1498 CrossRef CAS PubMed.
- C. B. Murray, A. C. R. Kagan and M. G. Bawendi, Annu. Rev. Mater. Sci., 2000, 30, 545–610 CrossRef CAS.
- A. J. Marenco, D. B. Pedersen, S. Wang, M. W. P. Petryk and H.-B. Kraatz, Analyst, 2009, 134, 2021–2027 RSC.
- J. C. Claussen, A. Kumar, D. B. Jaroch, M. H. Khawaja, A. B. Hibbard, D. M. Porterfield and T. S. Fisher, Adv. Funct. Mater., 2012, 22, 3399–3405 CrossRef CAS.
- S. Sabury, S. H. Kazemi and F. Sharif, Mater. Sci. Eng., C, 2015, 49, 297–304 CrossRef CAS PubMed.
- F. Jia, C. Yu, Z. Ai and L. Zhang, Chem. Mater., 2007, 19, 3648–3653 CrossRef CAS.
- R. Zeis, T. Lei, K. Sieradzki, J. Snyder and J. Erlebacher, J. Catal., 2008, 253, 132–138 CrossRef CAS.
- D. Lee, X. Wei, X. Chen, M. Zhao, S. C. Jun, J. Hone, E. G. Herbert, W. C. Oliver and J. W. Kysar, Scr. Mater., 2007, 56, 437–440 CrossRef CAS.
- C. Xu, J. Su, X. Xu, P. Liu, H. Zhao, F. Tian and Y. Ding, J. Am. Chem. Soc., 2007, 129, 42–43 CrossRef CAS PubMed.
- J. Snyder, K. Livi and J. Erlebacher, J. Electrochem. Soc., 2008, 155, C464–C473 CrossRef CAS.
- L. Chen, Y. Tang, K. Wang, C. Liu and S. Luo, Electrochem. Commun., 2011, 13, 133–137 CrossRef CAS.
- S. Petrovic, Chem. Educ., 2000, 5, 231–235 CrossRef CAS.
- R. F. Carvalhal, R. Sanches Freire and L. T. Kubota, Electroanalysis, 2005, 17, 1251–1259 CrossRef CAS.
- L. M. Fischer, M. Tenje, A. R. Heiskanen, N. Masuda, J. Castillo, A. Bentien, J. Émneus, M. H. Jakobsen and A. Boisen, Microelectron. Eng., 2009, 86, 1282–1285 CrossRef CAS.
- Y. Xia, W. Huang, J. Zheng, Z. Niu and Z. Li, Biosens. Bioelectron., 2011, 26, 3555–3561 CrossRef CAS PubMed.
- T. Matsumoto, Electrochemical sensor having a reference electrode, US Pat., US6773563B2, 2004 Search PubMed.
- J. Zhou, K. Ren, Y. Zheng, J. Su, Y. Zhao, D. Ryan and H. Wu, Electrophoresis, 2010, 31, 3083–3089 CrossRef CAS PubMed.
- B. J. E. Merten, D. Battocchi, D. Tallman and G. Bierwagen, ECS Trans., 2009, 19, 223–232 CrossRef CAS.
- E. T. S. G. N. da Silva, S. Miserere, L. T. Kubota and A. Merkoçi, Anal. Chem., 2014, 86, 10531–10534 CrossRef CAS PubMed.
- A. Määttänen, U. Vanamo, P. Ihalainen, P. Pulkkinen, H. Tenhu, J. Bobacka and J. Peltonen, Sens. Actuators, B, 2013, 177, 153–162 CrossRef.
- J. Dickinson, M. Bromley, F. Andrieux and C. Boxall, Sensors, 2013, 13, 3635–3651 CrossRef CAS.
- J. C. Claussen, M. M. Wickner, T. S. Fisher and D. M. Porterfield, ACS Appl. Mater. Interfaces, 2011, 3, 1765–1770 CrossRef CAS PubMed.
- N. Tawil, A. Hatef, E. Sacher, M. Maisonneuve, T. Gervais, R. Mandeville and M. Meunier, J. Phys. Chem. C, 2013, 117, 6712–6718 CrossRef CAS.
- C. Fernández-Sánchez, E. Pellicer, J. Orozco, C. Jiménez-Jorquera, L. M. Lechuga and E. Mendoza, Nanotechnol., 2009, 20, 335501 CrossRef PubMed.
- V. A. Pedrosa, S. Paliwal, S. Balasubramanian, D. Nepal, V. Davis, J. Wild, E. Ramanculov and A. Simonian, Colloids Surf., B, 2010, 77, 69–74 CrossRef CAS PubMed.
- I. Feliciano-Ramos, M. Caban-Acevedo, M. A. Scibioh and C. R. Cabrera, J. Electroanal. Chem., 2010, 650, 98–104 CrossRef CAS.
- J. A. Hondred, I. L. Medintz and J. C. Claussen, Nanoscale Horiz., 2019, 4, 735–746 RSC.
- J. A. Hondred, L. R. Stromberg, C. L. Mosher and J. C. Claussen, ACS Nano, 2017, 11, 9836–9845 CrossRef CAS PubMed.
- I. V. Pavlidis, M. Patila, U. T. Bornscheuer, D. Gournis and H. Stamatis, Trends Biotechnol., 2014, 32, 312–320 CrossRef CAS PubMed.
- J. Tkac and J. J. Davis, J. Electroanal. Chem., 2008, 621, 117–120 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0tc01423k |
| This journal is © The Royal Society of Chemistry 2020 |