
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceLanthanide-based inorganic–organic hybrid materials for photon-upconversion
Muhammad
Safdar
 a,
Amr
Ghazy
a,
Mika
Lastusaari
b and
Maarit
Karppinen
*a
a,
Amr
Ghazy
a,
Mika
Lastusaari
b and
Maarit
Karppinen
*a
aDepartment of Chemistry and Materials Science, Aalto University, FI-00076 Espoo, Finland. E-mail: maarit.karppinen@aalto.fi
bDepartment of Chemistry, University of Turku, FI-20014 Turku, Finland
First published on 8th May 2020
Abstract
Photon-upconversion materials are capable of converting low energy infrared light into higher energy visible or ultraviolet light. Such materials are demanded for applications such as deep-tissue imaging, cancer therapy, nano-thermometry, biosensing, display and solar-cell technologies, and beyond. Trivalent lanthanide ions are promising materials for upconversion due to their suitable f-orbital energy levels allowing absorption in the near-infrared and emission in the visible wavelength range. The major obstacle in realizing the full potential of the Ln-based upconverters is their characteristically small absorption cross-sections. As many organic molecules possess much larger absorption cross-sections, their combination with Ln3+ ions could bring about remarkable mutual benefits. Additionally, the organic ligands can also function as spacers to yield metal–organic framework type upconverting materials. Indeed, superior upconverting properties have already been reported for a diverse family of Ln-based inorganic–organic hybrids. Here we present an account of the recent developments in the field of Ln-based inorganic–organic upconverting materials and their emerging applications.
1 Introduction
1.1 Photon upconversion
Photon-upconversion luminescence is a property of certain materials capable of converting low energy light, typically in the infrared (IR) range, into higher energy visible (Vis) or ultraviolet (UV) light. This lower energy-to-higher energy or so-called anti-Stokes luminescence process has great potential for applications such as deep-tissue imaging, cancer therapy, nano-thermometry, biosensing, display technologies, and solar cells requiring or utilizing IR radiation (Fig. 1).1–6 For example, lanthanide-based upconverting materials are excellent as biological labels because the detection wavelength is far from the excitation wavelength, and accordingly there is no autofluorescence from the medium to disturb the signal from the label.7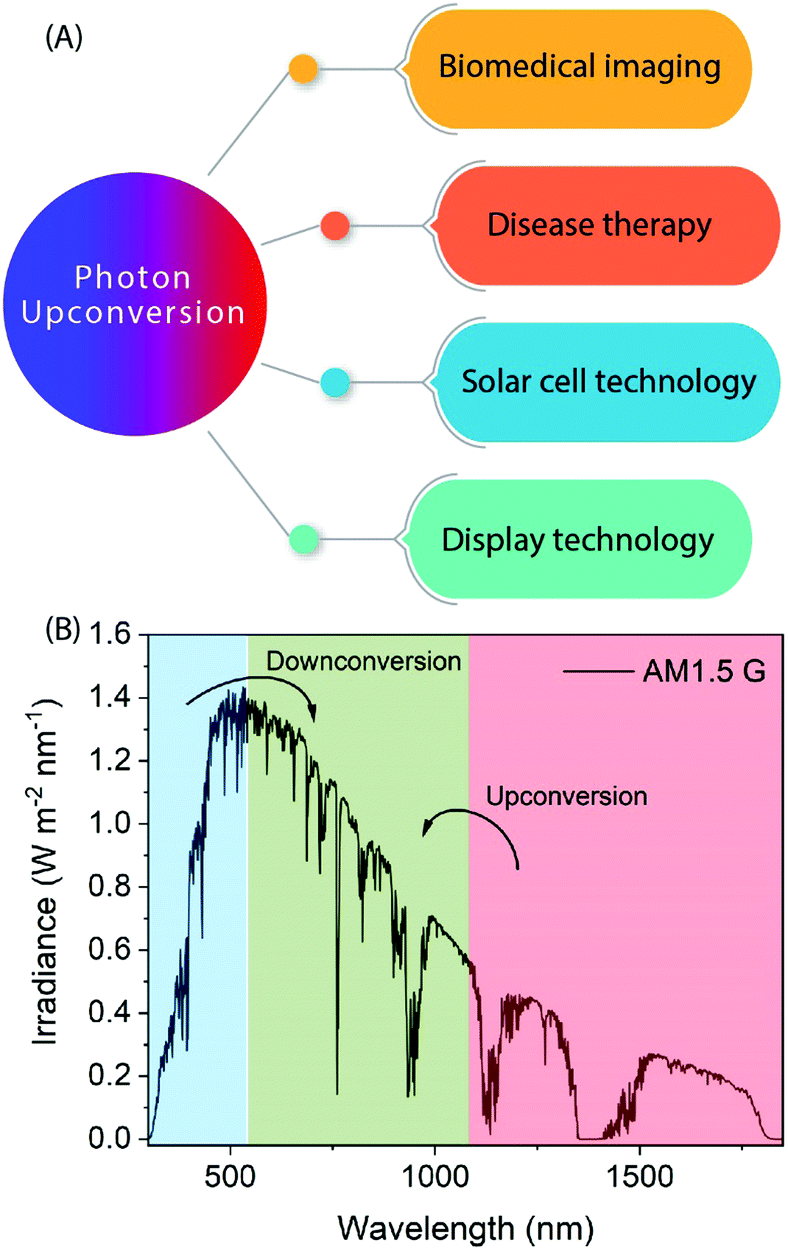 | ||
| Fig. 1 (A) Examples of the prominent application areas of photon upconversion. (B) AM1.5 Global solar spectrum with highlighted parts that are unusable but could be made usable with the help of down- or upconverting materials. | ||
The upconversion (UC) process is based on sequential stacking of two or more successively absorbed low energy photons. The thus excited material then emits a higher energy photon through radiative relaxation. There are two other anti-Stokes processes known, which should not be mixed with the UC process, i.e. so-called two-photon and hot-band absorption processes.8,9 In the former process the stacking of photons is not successive but simultaneous (i.e. it does not involve intermediate energy levels and occurs within a much shorter time domain),10 while in the latter case a single absorbed photon is combined with higher vibrational states.11
Considering the upconverting material systems, the UC processes can be divided into three major groups: lanthanide (Ln) based processes, d-block transition metal based processes, and triplet–triplet annihilation (TTA) processes seen e.g. in anthracene.10,12–14 Here we focus on the Ln-based processes, and in particular on the currently strongly emerging materials where Ln ions are combined with organic components for either better harvesting of photons, or to construct metal–organic hybrid type UC materials. These inorganic–organic materials are gaining increasing interest in IR-to-Vis UC applications, as they combine in synergistic manner the unique spectroscopic properties of Ln ions and the diverse functionalities of organic molecules.
There are several excellent review papers on the topic of photon-upconversion, but mostly focusing either on purely inorganic materials, or organic TTA-based material systems. In 2017, Wang et al.15 reviewed dye-sensitized UC nanoparticles and their underlying energy-transfer mechanisms, while Wen et al.16 very recently highlighted the potential of different types of hybrid UC materials and the challenges these materials currently face. Here in this review, we exclusively focus on lanthanide-based inorganic–organic hybrid materials where the organic ligands/linkers intimately bound to the Ln ions play an intrinsically important role.
1.2 Lanthanide-based upconverters
Trivalent lanthanide (Ln3+) ions possess exciting luminescence properties due to f–f transitions in their partially filled 4f-orbitals. The filled 5s2 and 5p6 orbitals shield the inner 4f orbitals from a drastic influence of ligands and subsequently only a minimal perturbation of electronic configuration is experienced by the Ln3+ ion. Because of the shielding, Ln3+ ions exhibit characteristic narrow-band emission and since the f–f transitions are forbidden, the excited states have long lifetimes.A conventional Ln-based inorganic upconverter consists of a host lattice in which the upconverting Ln3+ dopant ions are embedded. Both components have a profound impact on the UC properties of the material. For instance, specific energy transfer properties of the host lattice may strongly alter the emission properties of the dopant Ln3+ ion.13 The most common host lattice materials used to produce Ln-based UC systems are NaYF4, NaGdF4 and NaLuF4.17 The basic criteria for the selection of a host lattice material are that, (i) its crystal structure should allow the easy incorporation of the Ln dopant, (ii) it should be chemically and thermally stable, (iii) possess low phonon energies, and (iv) be sufficiently transparent to the impinging IR photons. The other task in the design of an efficient UC system is the optimization of the Ln3+ dopant ion composition for the maximal IR photon harvesting and UC luminescence. To make the IR photon harvesting more efficient, additional Ln3+ co-dopants or organic molecules can be used for the purpose of sensitization.
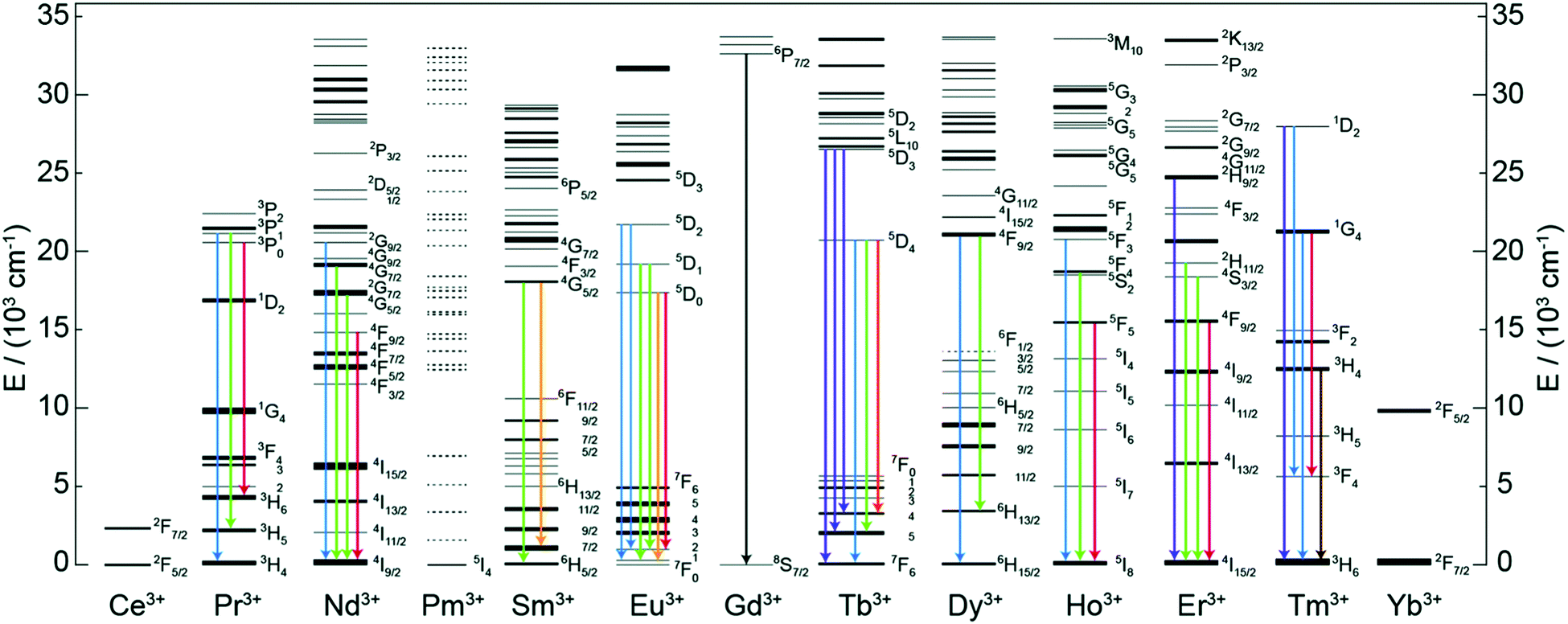
2 Energy transfer and UC mechanisms
The existence of an efficient energy transfer process between the organic antenna molecule, the sensitizer and the activator ions is crucial for the UC luminescence. The antenna–sensitizer–activator system can be imagined as a donor/acceptor pair. There should be an optimal distance between the sensitizer and activator ions to allow for an efficient energy exchange, which typically takes place following electrostatic interaction or non-radiative exchange mechanisms.29,30 For a detailed explanation of these processes, the reader is referred to some excellent recent reviews from theoretical perspective.31,32The Ln3+ UC is a very complex process and is generally not considered to be very efficient, as the typical normalized efficiencies of different two-photon mechanisms range from 10−13 to 10−3 cm2 W−1.10 In practice, the lower limit of excitation power density that results in detectable UC depends not only on the system studied but also on the sensitivity of the detection setup or the practical lower limit of the excitation source. For example, Joseph et al.33 report 4 mW cm−2 as the lower limit for microcrystalline NaYF4, YF3, La2O3 and YCl3, all doped with 18% Yb3+ and 2% Er3+. However, such low excitation powers are very rarely used in published studies, whereas lower limits in the range of ca. 0.1 to 0.5 mW cm−2 are more commonly reported34,35 Moreover, because UC is a nonlinear process, the dependence between excitation power (P) and UC intensity (I) is typically not linear: for the overall UC process involving the stacking of n photons I ∝ Pn at low power densities and drops to I ∝ P1 or below at higher densities.36 There are many possible UC mechanisms depending upon the UC system, such as excited-state absorption (ESA), energy transfer upconversion (ETU) and photon avalanche (PA). Some of the key processes are illustrated in Fig. 3 and briefly discussed below.
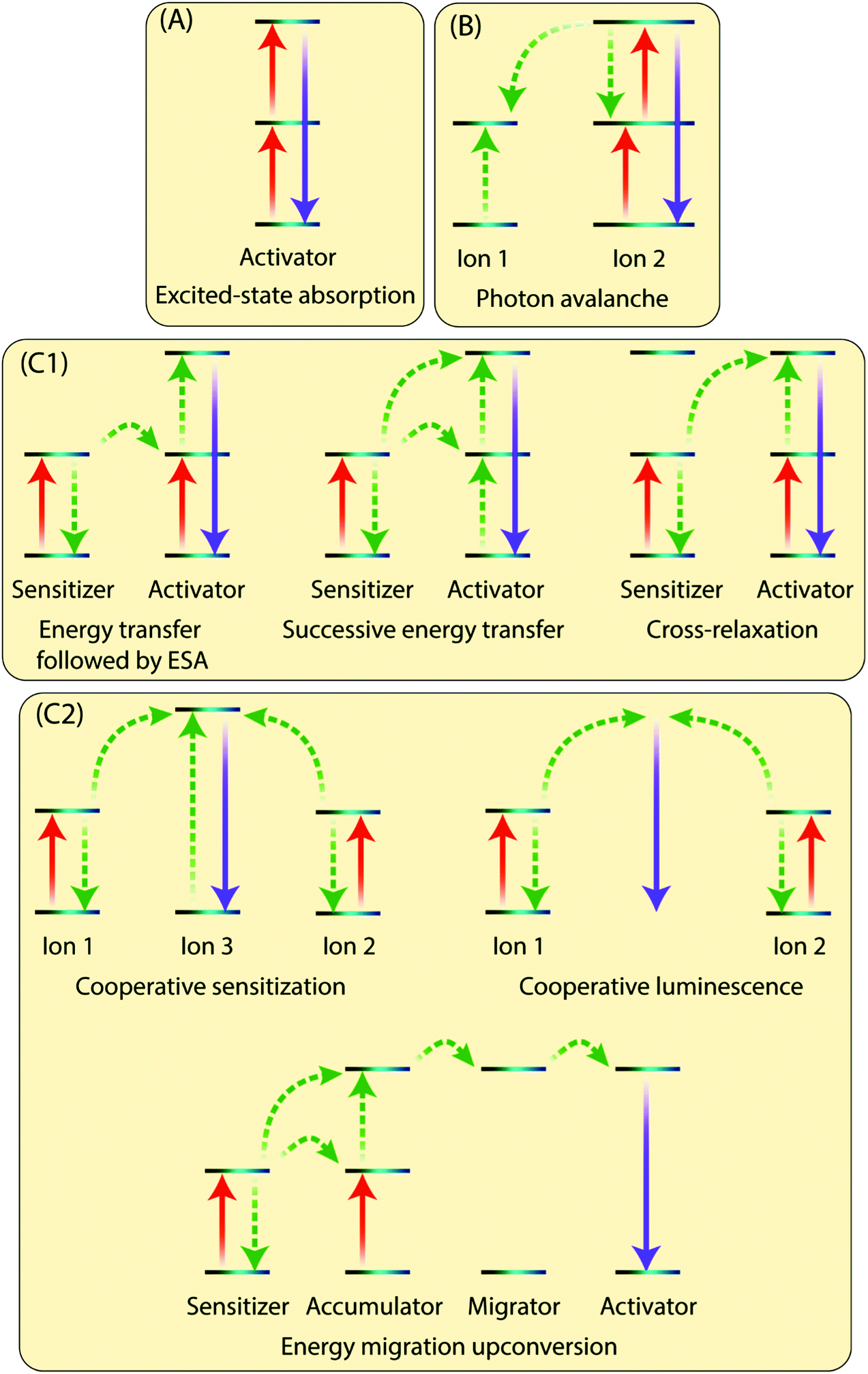 | ||
| Fig. 3 Schematic illustration of different absorption and energy transfer processes. The red, purple and dotted green arrows illustrate direct excitation, radiative emission and energy transfer processes, respectively. | ||
2.1 Excited-state absorption (ESA)
In ESA mechanism (Fig. 3A), a ground state Ln3+ ion sequentially absorbs two photons to emit a single higher energy photon. The absorption of first photon promotes the ion from the ground state to the metastable excited state, which absorbs the second impinging photon to reach the second excited state. A relaxation of the second excited state back to the ground state emits an upconverted, higher energy photon. ESA takes places on singly doped UC materials with low enough concentration of the active ions to prevent interaction between them. This process has a typical relative efficiency of 10−5.102.2 Photon avalanche (PA)
This process has a strong dependence on cross-relaxation between luminescent centres and the excitation power density. The PA upconversion emission is produced only above a certain threshold of the excitation power. Below that threshold, negligible luminescence is observed. This mechanism is not common in nanostructured Ln-based UC materials (Fig. 3B).2.3 Energy transfer upconversion (ETU)
The ETU mechanism resembles ESA, except that it requires at least two Ln3+ ions in a proximity, one being the sensitizer and other acting as the activator. The ETU mechanism may involve (i) non-cooperative sensitization and (ii) cooperative processes. The non-cooperative sensitization involves only one sensitizer ion (Fig. 3C1), and can be further categorized as follows:3 Selection of organic ligand/linker molecules
There are a few common requirements for the organic ligand/linker to fulfil the basic criteria to be considered for its use in a hybrid inorganic–organic UC material:• efficient absorption in the NIR range
• emission in a range that overlaps with the absorption spectrum of the Ln3+ sensitizer/activator ions to enable the energy transfer
• no absorption of the upconversion emission
• functional/reactive groups such as carboxylic or sulphate groups to enable coordination to the sensitizer/activator Ln3+ ions
• photostability
The organic ligands/linkers typically used with Ln3+ ions have their absorption and emission peaks positioned around 780–820 nm and 750–1100 nm, respectively.24,26 Other small organic molecules have also been used, for example as spacer ligands to create MOF-type upconverting materials.37,38 Molecular structures of the most common organic components employed are displayed in Fig. 4 and summarized in Table 1.
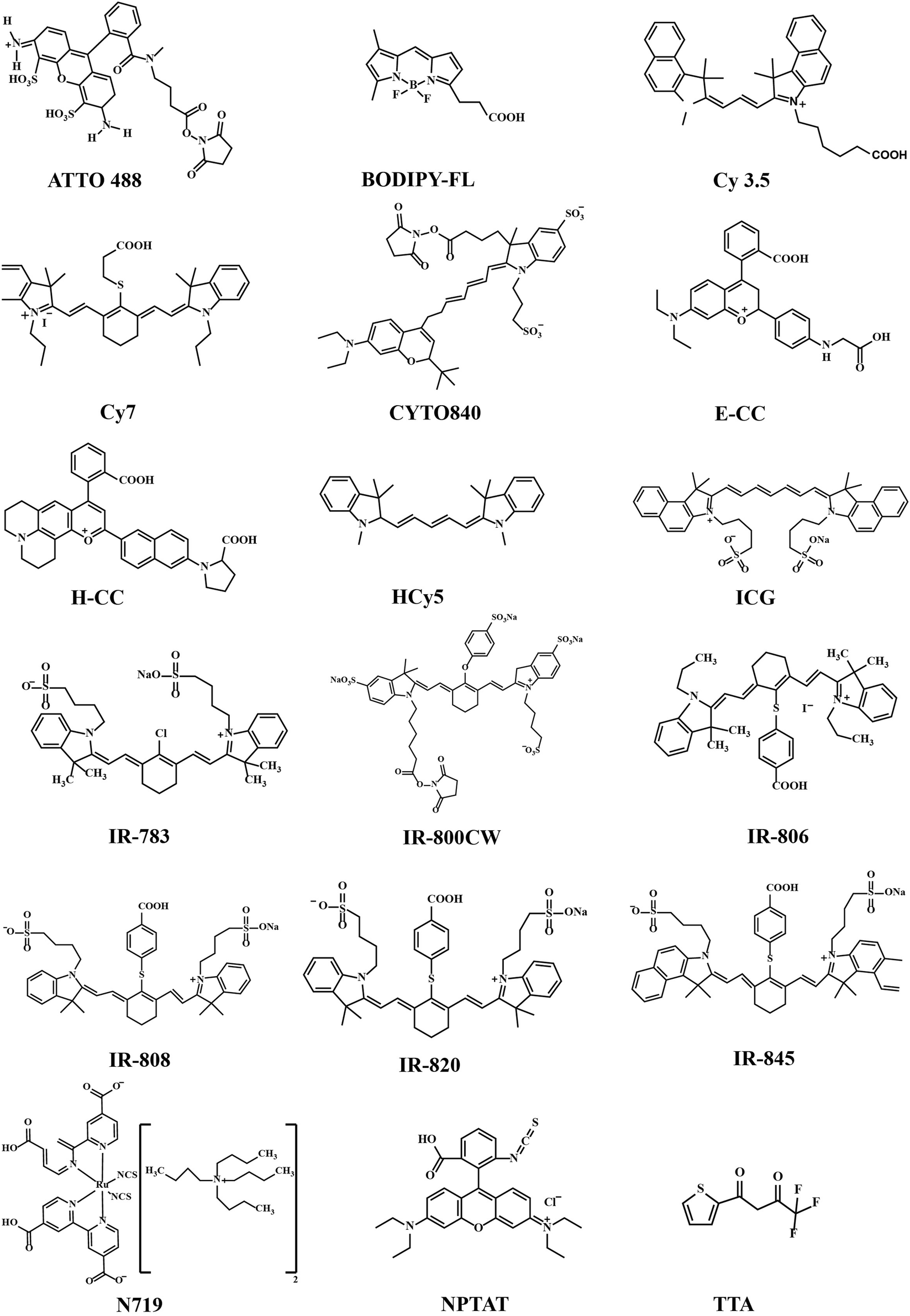 | ||
| Fig. 4 Chemical structures of the most commonly used organic molecules; it should be noted that the naming scheme for many of the molecules is based on their absorption maxima, and it is possible that several similar but not identical molecules have the same absorption maxima and have been thus given the same name in literature. | ||
| Organic ligand | Inorganic composition | Excitation (nm) | Emission range (nm) | Ref. | |
|---|---|---|---|---|---|
| Core-only | IR-806 | β-NaYF4:20%Yb3+/2%Er3+ | 800 | 510–530 | 24 |
| 530–570 | |||||
| 630–680 | |||||
| DyL800 | NaYF4:Yb3+/Tm3+ | 801, 975 | 520–570 | 58 | |
| NaYF4:Yb3+/Ho3+ | |||||
| DY-831 | NaYF4:Yb3+/Er3+ | ||||
| NaYF4:Er3+ | |||||
| IR-783 | β-NaYF4:20%Yb,2%Er | 790–850 | 500–685 | 59 | |
| IR-808 | |||||
| IR-820 | |||||
| IR-845 | |||||
| BODIPY-FL | β-NaYF4:20%Yb3+, 0.5%Tm3+ | 450–975 | 330–480 | 60 | |
| Cy3.5 | |||||
| IR-806 | |||||
| Cyto 840 | NaYF4:Tb3+,Tm3+ | 810 | 430–675 | 61 | |
| ATTO 488 | |||||
| ATTO 542 | Na(Y/Gd/Lu)0.8 F4:Yb0.18,Er0.02 | 980 | 580 | 62 | |
| Indocyanine green (ICG) | NaYF4:Nd3+ | 800 | 400–700 | 63 | |
| Phenanthroline (Phen), thenoyltrifluoroacetone (TTA) | NaYbF4:15%Tb3+,15%Eu3+ | 980 | 450–500 | 64 | |
| 510–575 | |||||
| 580–700 | |||||
| IR-806 | β-NaY0.78−x,Gd0.x,Yb0.2,Er0.02,F4 | 808 | 510–565 | 65 | |
| 640–680 | |||||
| Core/shell | IR-808 | (NaYbF4:Tm3+ 0.5%)/NaYF4:Nd3+ | 800 | 320–680 | 44 |
| IR-820 | |||||
| Cy7 | NaYF4:Yb3+,Nd3+,Er3+/NaYF4:Nd3+ | 800 | 500–675 | 66 | |
| ICG | NaYF4:Yb3+,X3+/NaYbF4/NaYF4:Nd3+ (X = null, Er, Ho, Tm, or Pr) | 800 | 1000–1700 | 67 | |
| IR-808 | NaGdF4:Yb3+,Er3+/NaGdF4:Yb3+/NaNdF4:Yb3+ | 808 | 510–530 | 68 | |
| 530–570 | |||||
| 630–680 | |||||
| IR-820 | NaLuF4:Gd3+,Yb3+,Tm3+/NaLuF4:Gd3+,Yb3+/NaNdF4:Yb3+ | 808 | 450–500 | 69 | |
| 630–670 | |||||
| IR-808 | NaGdF4:Yb3+,Er3+/NaGdF4:Nd3+,Yb3+ | 808 | 500–575 | 70 | |
| 625–680 | |||||
| IR-806 | β-NaYF4:20%Yb3+,2%Er3+/β-NaYF4:10%Yb3+ | 800 | 450–700 | 71 | |
| IR-820 | NaLuF4:Gd3+,Yb3+,Er3+/NaLuF4:Yb3+,Pr3+ | 820 | 475–565 | 72 | |
| 635–675 | |||||
| IR-806 | β-NaYF4:20%Yb3+,2%Er3+/NaYF4:10%Yb3+ | 800 | 500–550 | 73 | |
| 630–680 | |||||
| ICG | NaYF4:Yb3+,X3+/NaYF4:Nd3+,Yb3+ (X = Er, Tm, and Ho), | 800 | 325–700 | 74 | |
| IR-783 | NaYF4:10%Yb3+,2%Er3+/NaYF4:30%Nd3+ | 800 | 500–575 | 43 | |
| 625–675 | |||||
| IR-806 | NaYF4:Yb3+,Er3+/NaYF4:Yb3+,Nd3+ | 808 | 510–570 | 75 | |
| 630–680 | |||||
| Heptamethine cyanine | NaYF4:20%Yb3+,2%Er3+/NaYF4:20% Nd3+ | 808 | 510–570 | 76 | |
| IR-806 | NaYF4:25%Yb,2%Er/NaYF4:Nd | 980 | 500–560 | 77 | |
| 630–675 | |||||
| NPTAT* | β-NaGdF4:20%Yb,2% Er/NaGdF4 | 980 | 450–675 | 46 | |
| Rhodamine B isothiocyanate | β-NaGdF4:20%Yb,0.2% Tm/NaGdF4 | ||||
| α-NaYbF4:10% Er@NaYF4 | |||||
| IR-808 | NaGdF4:Yb,Er/NaGdF4:Yb | 808 | 500–600 | 45 | |
| 980 | |||||
| E-CC | NaYF4:Yb,Er,Tm/NaYF4 | 980 | 500–850 | 78 | |
| H-CC | 575–850 | ||||
| HCy5 | NaGdF4:Yb,Er,Tm (20,2,1%)/NaGdF4 | 980 | 500–800 | 79 | |
| Cy7 | |||||
| 800CW | NaYF4:Yb,Er/NaYF4:Nd,Yb | 808 | 515–560 | 80 | |
| Cy3 | |||||
| N719 | |||||
| Metal–organic network-type structures | 2-Aminotere-phthalic acid | NaYF4:Er3+,Tm3+/NaYF4 | 980 | 520–570 | 81 |
| 1,4-Benzodi-carboxylic acid, biphenyl-4,4′-dicarboxylate | NaGdF4:Yb3+,Tm3+/NaGdF4 | 635–675 | |||
| 2,3-Pyrazine-dicarboxylic acid | Y0.92,Yb0.04,Er0.04(thd)3* | 974 | 400–425 | 37 | |
| 500–600 | |||||
| 640–680 | |||||
| IR-806 | Yb3+,Er3+ | 973 | 510–560 | 82 | |
| 640–680 | |||||
| 1,4-Dicarboxy-benzene, biphenyl-2,5,2′,5′-tetracarboxylate | Yb3+,Er3+ | 980 | 500–550 | 38 | |
| 625–685 | |||||
Most of the organic components employed are strongly delocalized conjugated systems built up with methine groups. In general, for the π-conjugated polymers and their oligomers, the absorption wavelength of the lowest excited state increases with increasing polymethine chain length.39 This has been shown for example for the polymethine cyanine dyes composed of two aromatic or heterocyclic rings linked by a double bond conjugated polymethine chain.40,41 Heptamethinecyanine dyes contain N-alkyl substituents, and absorb in the range of 730–820 nm in solution. Their solubility in organic solvents is strongly affected by the nature of the N-alkyl substituent, the heteroaromatic ring, and the counter anion.42 Examples of this group include the so-called IR-783, IR-806, IR-808 and IR-820 dyes, simply named based on their maximum absorption wavelengths (λmax).24,43,44
The photostability of these organic dyes is an important requirement to achieve efficient UC luminescence. The photobleaching of organic dye molecules upon exposure to light can cause degradation of their optical properties, which may pose deleterious effects to UC emission. This can particularly be the case when dye molecules are decorated onto the outer surface of UCNPs.45 A solution for such issue could be to isolate the dye molecules from the environment by encapsulation inside inert layers. For instance, Zhou et al.46 studied photostability of NPTAT and Rhodamine B isothiocyanate dyes doped into a SiO2 layer of UCNPs, and found out that the dyes were photostable under 980 nm NIR laser or visible light irradiation.
4 Design of UC material geometries
Current UC research is concentrated on materials prepared in the form of colloidal nanoparticles (NPs), which can be categorized as core-only NPs and core–shell NPs (Fig. 5A and B). Besides the different NP configurations, upconverting inorganic–organic thin films have recently started to emerge as a new promising geometry; these thin-film upconverters may require entirely different design considerations to achieve optimal UC luminescence (Fig. 5C).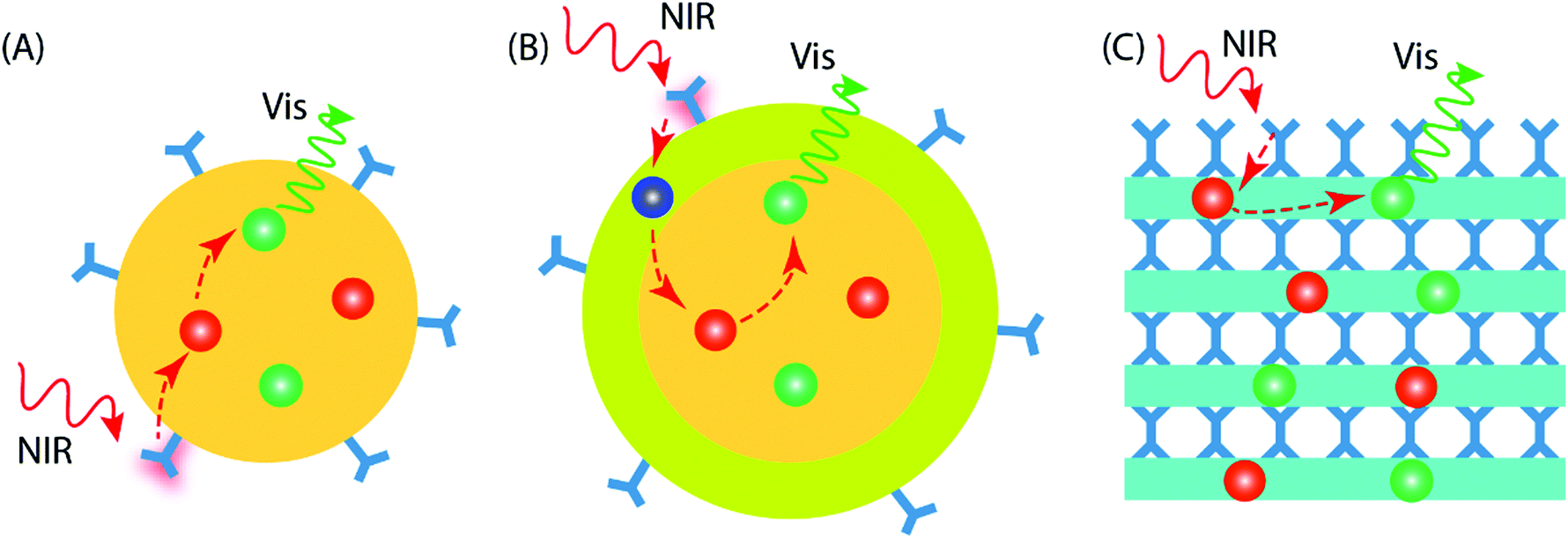 | ||
| Fig. 5 Different geometry designs of inorganic–organic UC materials: (A) core-only NP with organic antenna on the surface, (B) core–shell NP with organic antenna on the shell surface, and (C) thin film with inorganic and organic interlayers. The organic ligands (blue) form metal–organic molecular network-type structures and may also contribute to energy absorption and transfer to sensitizer ions (red). The sensitizer further transfers the energy to the activator ions (green) for a subsequent UC emission. | ||
The design of core-only NPs for photon-upconversion involves the incorporation of sensitizer and activator ions into the inorganic host lattice core. The surface of the core is functionalized with desired organic antenna that absorbs and transfers the energy to the sensitizer ions (typically Yb3+). Then, the energy transfer from sensitizer to activator ions (e.g. Ho3+, Er3+, Tm3+) takes place through the afore-described energy transfer UC mechanism(s). The core-only NPs take benefit of a simple energy transfer process from the organic antenna to the activator via the sensitizer ions. However, the presence of surface defects on the core-only NPs may act as quenchers of the UC luminescence.47 Additional factors that may deleteriously influence the UC luminescence, are solvent effects and the loosely bound ligands, which may cause multiphonon relaxation/deactivation of the excited Ln3+ ions.48,49
In core–shell NPs, the host lattice core with embedded sensitizer and activator ions is coated with a suitable material to minimize the energy losses on the particle surface. To enable this, the core/shell interface must possess significantly less defects/quenching sites compared to the core-only NP case. The chemistry of shell structure can be engineered by adopting suitable materials.50 For example, (i) the shell may be passivating in nature to minimize energy transfer to the particle surface. For this, the bare host material without doping can be a simple and convenient choice;51 the shell can be made of (ii) the host material doped with suitable Ln3+ ions to enhance the energy transfer or emission processes,52 or (iii) a material that facilitates functionalization of the outer surface for various practical applications.53
The thickness of the shell is an important factor to be considered while designing core/shell NPs. A thicker shell can be helpful to overcome surface-induced quenching effect of the core and hence boost the UC emission. However, as the energy transfer process from the dye → sensitizer → activator is strongly dependent on distance, an increase in shell thickness may reduce the energy transfer efficiency of the system.
Compared to the colloidal UCNPs, preparation of the upconverting inorganic–organic materials in thin-film form is a relatively new approach. Thin films are particularly interesting and important due to their compatibility towards solid-state optical devices and applications. The UCNPs can be certainly applied as thin films using methods such as spray drying, dip coating, dispersion casting, etc. Nevertheless, the films may contain traces of solvent impurities that can cause poor film adhesion or more seriously, degrade device performance and its lifetime. A gas-phase deposition method can provide an attractive way to produce thickness-controlled inorganic–organic hybrid UC thin films on a variety of substrates. The organic molecules may serve as bridging ligands between two inorganic layers, as in the case of UC molecular complexes.54 Such UC thin films can offer some unique features over the NPs, such as conformality and improved adhesion to the substrate in practical devices.
An elegant way to fabricate state-of-the-art inorganic–organic thin films from gaseous precursors is to employ the currently strongly emerging atomic/molecular layer deposition (ALD/MLD) technique.55–57 This technique is derived from the well-known industry-feasible ALD (atomic layer deposition) technique for ultrathin conformal inorganic thin films, and its less exploited counterpart for purely organic thin films, i.e. molecular layer deposition (MLD). Most importantly, the combined ALD/MLD technique allows the production of unique inorganic–organic hybrid materials with atomic/molecular level accuracy.
5 Examples of Ln-organic UC materials
5.1 Core-only nanoparticles
Zou et al. pioneered in the use of dye-sensitization to improve the NIR absorption and photon upconversion property of β-NaYF4:Yb,Er NPs (average diameter 16 nm); they utilized cyanine dye, IR-780, as the starting material and replaced its central chlorine atom with a carboxylic acid group by nucleophilic substitution reaction.24 After the carboxylation step, the absorption spectrum of the dye peaked at 806 nm (in CHCl3), and thus the derivatized product was named as IR-806 dye. The derivatization of the IR-780 dye was crucial to facilitate the binding of dye molecules to the surface of oleyl-amine capped β-NaYF4:Yb,Er NPs. The extinction coefficient of IR-806 at 806 nm was ca. 5 × 106 times higher than that of the β-NaYF4:Yb,Er NPs at 980 nm. The emission spectrum of IR-806 and the absorption spectrum of β-NaYF4:Yb,Er NPs shared an overlapping region at 900–1000 nm, which allowed Förster-type energy transfer, at 800 nm laser excitation, from the IR-806 dye to the Yb3+ ions on the surface of NPs. A subsequent energy transfer from Yb3+ to the Er3+ ions led to an efficient UC emission at the characteristic Er3+ emission bands of 510–530, 530–570 and 630–680 nm due to the 2H11/2 → 4I15/2, 4S3/2 → 4I15/2 and 4F9/2 → 4I15/2 transitions, respectively. When compared to the bare NPs, the dye-coated NPs exhibited ca. 3300 times brighter UC response within the broad spectral range of 720–1000 nm (Fig. 6A). As an extension of the aforementioned work, Dupuy et al. combined different organic dyes with varying infrared absorption and emission bands to NPs of different compositions (e.g. NaYF4:Er3+ and NaYF4:Yb3+,Ln3+ (Ln = Tm3+, Ho3+, Er3+)) that had differing activation wavelengths and UC emission bands.58 In this way, it was possible to observe unique visible emission associated with different bands of IR excitation. Also, a set of different NIR dyes with distinct absorption wavelengths can be combined with a specific type of UCNPs (e.g. β-NaYF4:Yb,Er NPs) to enable programming of the excitation bands.59 The NPs decorated with mixed dyes of distinct absorbing bands (783, 845 nm) allow an expanded excitation wavelength range (790–850 nm) that otherwise cannot be realized using a single dye.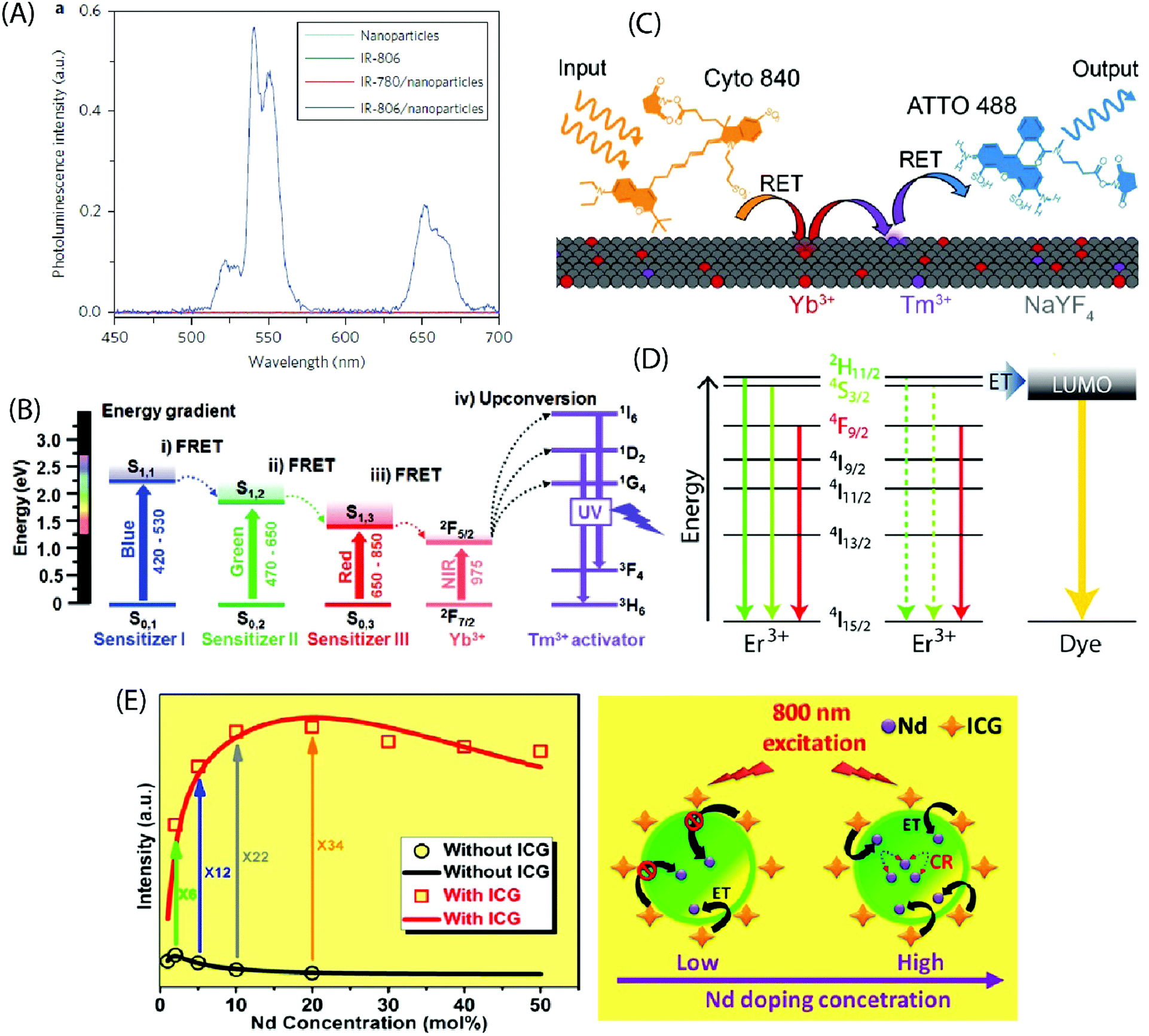 | ||
| Fig. 6 (A) Steady-state emission spectra of NaYF4:Yb,Er NPs (cyan line), IR-806 (green line), NaYF4:Yb,Er NPs/IR-780 (red line) and NaYF4:Yb,Er NPs/IR-806 (blue line) in CHCl3 excited by a 2 mW, 800 nm c.w. laser. (Reproduced from ref. 24, with permission from Springer Nature.)24 (B) Energy diagram of three sensitizers (sensitizer I: BODIPY-FL, sensitizer II: Cy3.5, sensitizer III: IR-806) and Ln3+ ions, showing concurrent sensitization of NaYF4:Yb,Tm NPs. (Reproduced from ref. 60, with permission from Wiley.)60 (C) Schematic illustrating the composition and operation of the RET relay. Cyto 840 is excited at 810 nm and transfers its energy to the Ln3+ dopants near the surface of the UCNPs. After multiple transfer events, Tm3+ reaches a highly excited state and transfers this energy to ATTO 488, which can then fluoresce. (Reproduced from ref. 61, with permission from Wiley.)61 (D) UCNPs are decorated with the fluorescent dye ATTO 542. The LUMO of the dye is aligned with the 2H11/2 and 4S3/2 states in Er3+, enabling efficient energy transfer to the dye molecules. (Reproduced from ref. 62, with permission from American Chemical Society.)62 (E) Experimental results (black circle and red square) and theoretical modelling (black and red curves) of integrated UC intensities of a set of NaYF4:Nd UCNPs with and without ICG sensitization. The energy transfer mechanism from ICG to NaYF4:Nd NPs, considering both low and high Nd3+ doping levels with ICG sensitization, is also shown. (Reproduced from ref. 63, with permission from American Chemical Society.)63 | ||
An appropriate combination of organic dyes can enable ultra-wideband excitation of photon-upconverting NPs for a Vis/NIR-to-UV UC emission. For example, combining dyes that absorb in the blue, green and red regions of visible light with the NIR-absorbing β-NaYF4:20%Yb3+,0.5%Tm3+ NPs containing UV-emitting Tm3+ ions, the absorption band can be extended to the entire visible and NIR range (450–975 nm).60 This expansion of the excitation window allows the utilization of diverse excitation sources for different applications (Fig. 6B). LaBoda et al. used a combination of two organic chromophores (Cyto 840, ATTO 488) and NaYF4:Tb3+,Tm3+ NPs to design fluorescent resonance energy transfer (FRET) networks.61 Cyto 840 absorbs in the NIR region and its emission spectrum sufficiently overlaps with the excitation spectrum of the UCNPs employed. ATTO 448 possesses a significant excitation overlap with the emission spectrum of NaYF4:Tb3+,Tm3+ NPs and a high quantum yield of 80% that increases the possibility of energy transfer process. Upon excitation at 810 nm, Cyto 840 transfers its energy to the lanthanide dopants near the surface of the NPs. After multiple transfer events, Tm3+ ions reach the highly excited states 1G4 and 1D2 and transfer the energy to ATTO 488, which then undergoes emission in the visible range (Fig. 6C). Such a dye-assisted UC process provides a promising way to achieve intensity enhanced UC emission.
Lanthanide UC relies on parity forbidden radiative transitions that exhibit very long radiative lifetimes (microseconds). The occurrence of faster non-radiative decay pathways, such as phonon coupling and surface quenching effects, cause a loss of the input energy that translates into poor UC emission. Wisser et al. solved this issue by anchoring ATTO 542 dye molecules to the surface of Na(Y/Gd/Lu)0.8F4:Yb0.18Er0.02 NPs.62 The absorption spectrum of ATTO 542 overlaps with the green emission peaks of Er3+. The gap between the lowest unoccupied molecular orbital (LUMO) and the highest occupied molecular orbital (HOMO) of ATTO 542 is very similar in energy to that separating the 2H11/2 and 4S3/2 excited states from the 4I15/2 ground state in Er3+, which can facilitate efficient energy transfer from Er3+ to the dye (Fig. 6D). The Er3+ to ATTO 542 energy transfer significantly enhanced the UC emission from dye molecules that was peaked at 580 nm.
The decoration of inorganic UCNPs with organic dyes also offers a powerful solution to alleviate the concentration quenching effect in highly doped materials. For example, for uncoated NaYF4:Nd NPs, the optimal concentration of Nd3+ ions is around 2%, which is similar to other activators as well, such as Er3+, Tm3+ and Ho3+. Higher levels of Nd3+ dopant ions give rise to cross-relaxation process between two Nd3+ ions, resulting in the quenching of the UC emission. In contrast, when highly doped (ca. 20%) NaYF4:Nd NPs were sensitized with indocyanine green dye, the UC emission was strongly enhanced.63 The indocyanine dye has ca. 30![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 times larger absorption cross-section than Nd3+ ions, and it facilitates efficient Förster-type energy transfer to the surface Nd3+ ions. As the Nd3+ dopant concentration increases, the number of surface Nd3+ ions also increases, introducing more channels for dye sensitization for every single NaYF4:Nd NP. This enhancement effect competes with the concentration induced quenching rate and yields an overall improvement in the UC luminescence (Fig. 6E). Jiao et al. demonstrated reduced surface-induced luminescence quenching for Phen, TTA modified NaYbF4:Tb,Eu NPs, exhibiting both photon-upconversion and down-shifting under IR and UV irradiation, respectively.64 The antenna effect of organic ligands was operational only under UV light for the down-shifting process. The enhancement of the UC luminescence was attributed to the passivation of the NPs by the organic ligands that reduced the quenching effect originating from surface defects.
000 times larger absorption cross-section than Nd3+ ions, and it facilitates efficient Förster-type energy transfer to the surface Nd3+ ions. As the Nd3+ dopant concentration increases, the number of surface Nd3+ ions also increases, introducing more channels for dye sensitization for every single NaYF4:Nd NP. This enhancement effect competes with the concentration induced quenching rate and yields an overall improvement in the UC luminescence (Fig. 6E). Jiao et al. demonstrated reduced surface-induced luminescence quenching for Phen, TTA modified NaYbF4:Tb,Eu NPs, exhibiting both photon-upconversion and down-shifting under IR and UV irradiation, respectively.64 The antenna effect of organic ligands was operational only under UV light for the down-shifting process. The enhancement of the UC luminescence was attributed to the passivation of the NPs by the organic ligands that reduced the quenching effect originating from surface defects.
A critical analysis of the afore-mentioned UC systems indicates a limited understanding of the dye-NP interactions and a possibility of the involvement of processes that may limit efficiency of UCL even after dye-sensitization. To elaborate on this, consider that the absorption cross-section of NIR absorbing dyes is significantly greater than the Ln ions, and each UCNP contains several hundreds of dye molecules on its surface. This should boost UC by several hundreds of times, but that is not the case. Recently, Garfield et al. proposed new design rules for constructing highly efficient UC nanosystems by understanding the dye-NP interactions during the energy transfer process.65 The spin-triplet states in the dye molecules are predominantly responsible for the sensitization of UCNPs and the Ln ions situated on the NP surface can enhance singlet to triplet intersystem crossing (ISC) within the dye by spin–orbit coupling (Fig. 7). By designing dyes whose excited-state wavefunctions are located closer to the NP surface, it is possible to boost the energy transfer efficiency due to an improved spin–orbit coupling and Dexter electron transfer processes. In addition, the doping of the UCNPs with heavier Ln ions can further improve the UC emission due to the heavy atom effect that increases ISC within the dye. It is also important to consider that the UCNP-to-dye energy back transfer and the Ln–Ln energy migration losses are among the probable factors that may potentially restrict the theoretically anticipated UC luminescence intensity.77
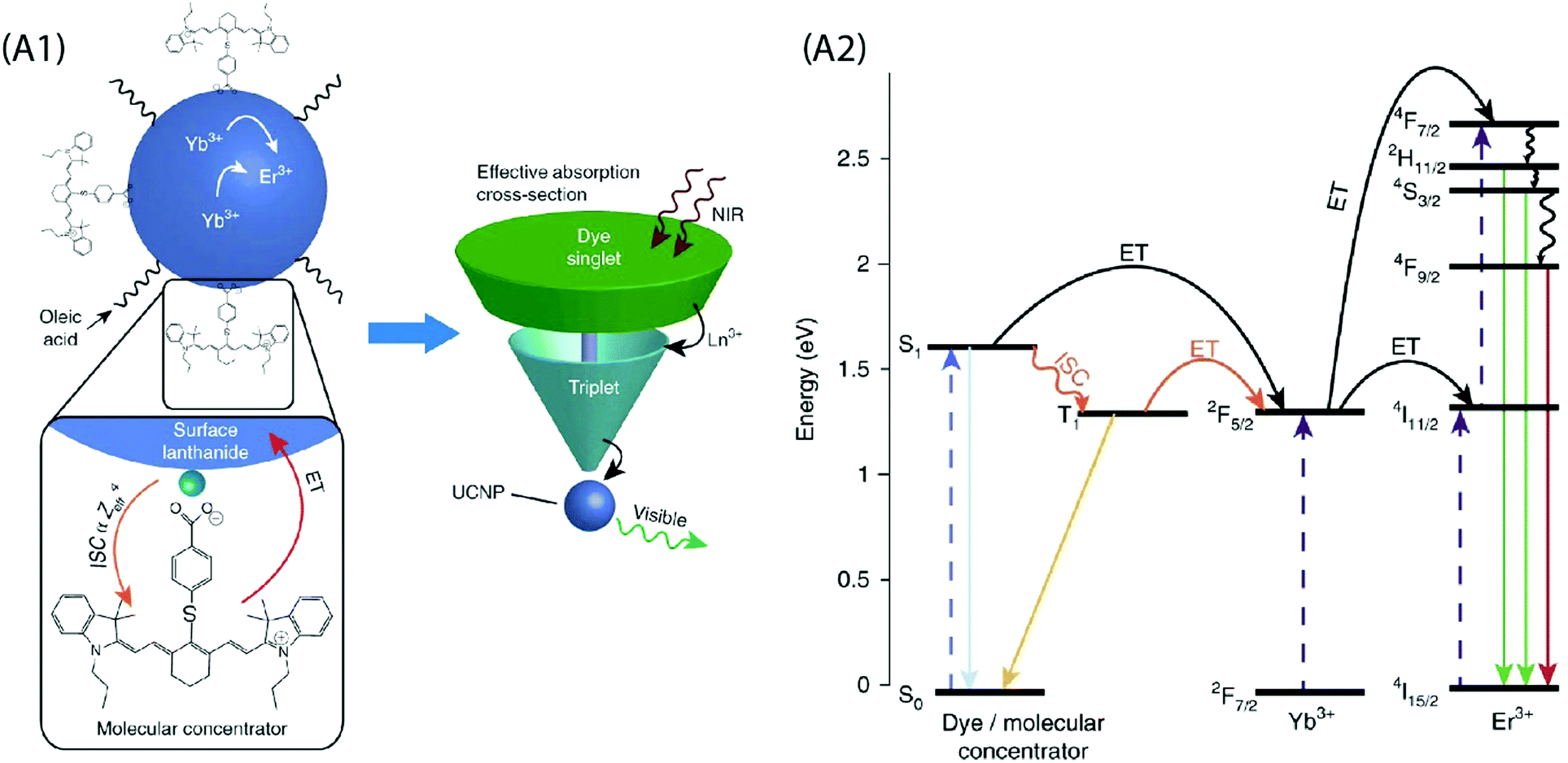 | ||
| Fig. 7 (A1) Schematic of the dye-sensitized UCNP system, showing IR806 bound to the UCNP surface, and an upconversion event inside the UCNP where two excited Yb3+ non-radiatively and sequentially excite an Er3+ into a higher energetic state. Bottom left is the magnified illustration of the interactions between IR-806 and a surface lanthanide—the heavy nucleus of the lanthanide aids in ISC from IR806 S1 → T1 states, allowing much more efficient T1 sensitization of the Ln3+ atoms within the UCNP. Right, Depiction of the antenna-like nature of IR806 in sensitizing the NP upconversion, conveying the much larger absorption cross-section of IR-806 relative to the UCNP, as well as S1 → T1 ISC enhancement by Ln3+. (A2) Diagram of proposed energy transfer landscape, showing dye ISC to the triplet state T1 before transfer to UCNP lanthanides. (Reproduced from ref. 65, with permission from Nature Publishing Group.)65 | ||
5.2 Core/shell nanoparticles
In the core-only NP configuration, the large size of organic ligands may limit the full surface passivation. The core/shell NPs efficiently address this issue as they take benefit of an epitaxially grown inorganic shell to passivate the core structure. The UC quenching may be suppressed by a factor of two to three orders of magnitude for the core/shell NPs.83 Absorption sensitization of these core/shell NPs with organic ligands can result in bright UC luminescence due to combination of the antenna effect and the suppressed surface-induced quenching. Judicious choice of dopants in the core/shell structure can improve spectral overlap between the emission spectrum of the dye and absorption spectrum of the sensitizer ions in the core/shell structure.Chen et al. sensitized NaYbF4:Tm3+/NaYF4:Nd3+ based core/shell NPs using the IR-808 dye to demonstrate a multistep energy transfer from the antenna to the activator ions within the core of the NPs.44 A strong overlap of the IR-808 emission spectrum with the absorption bands of Nd3+ in the shell resulted in an efficient energy transfer from the dye molecules to the Nd3+ ions, which subsequently sensitized Yb3+ in the core. An intracore energy transfer from Yb3+ to Tm3+ ions produced efficient multiphoton UC emission (Fig. 8A). The emission peaks were located at 340 nm (1I6 → 3F4, five photon luminescence); 350, 450 and 510 nm (1D2 → 3H6, 3F4 and 3H5, four photon luminescence) and 480, 650 nm (1G4 → 3H6 and 3F4, three photon luminescence). An increase in the dye:NP ratio first increased the luminescence intensity due to the overall enhancement of the energy absorption. However, at a further increased dye:NP ratio; an increased interaction among the dye molecules resulted in quenching of the luminescence intensity. The optimal dye:NP ratio was determined to be ∼830, which refers to ca. 830 dye molecules bound to the surface of each NaYbF4:Tm3+/NaYF4:Nd3+ core/shell NP. The nonradiative energy transfer efficiency from the dye to the Nd3+ ion across the organic/inorganic interface and sequentially the Yb3+ ions in the core was determined to be 82% and 80%, respectively, and enabled an UC efficiency as high as 19%.
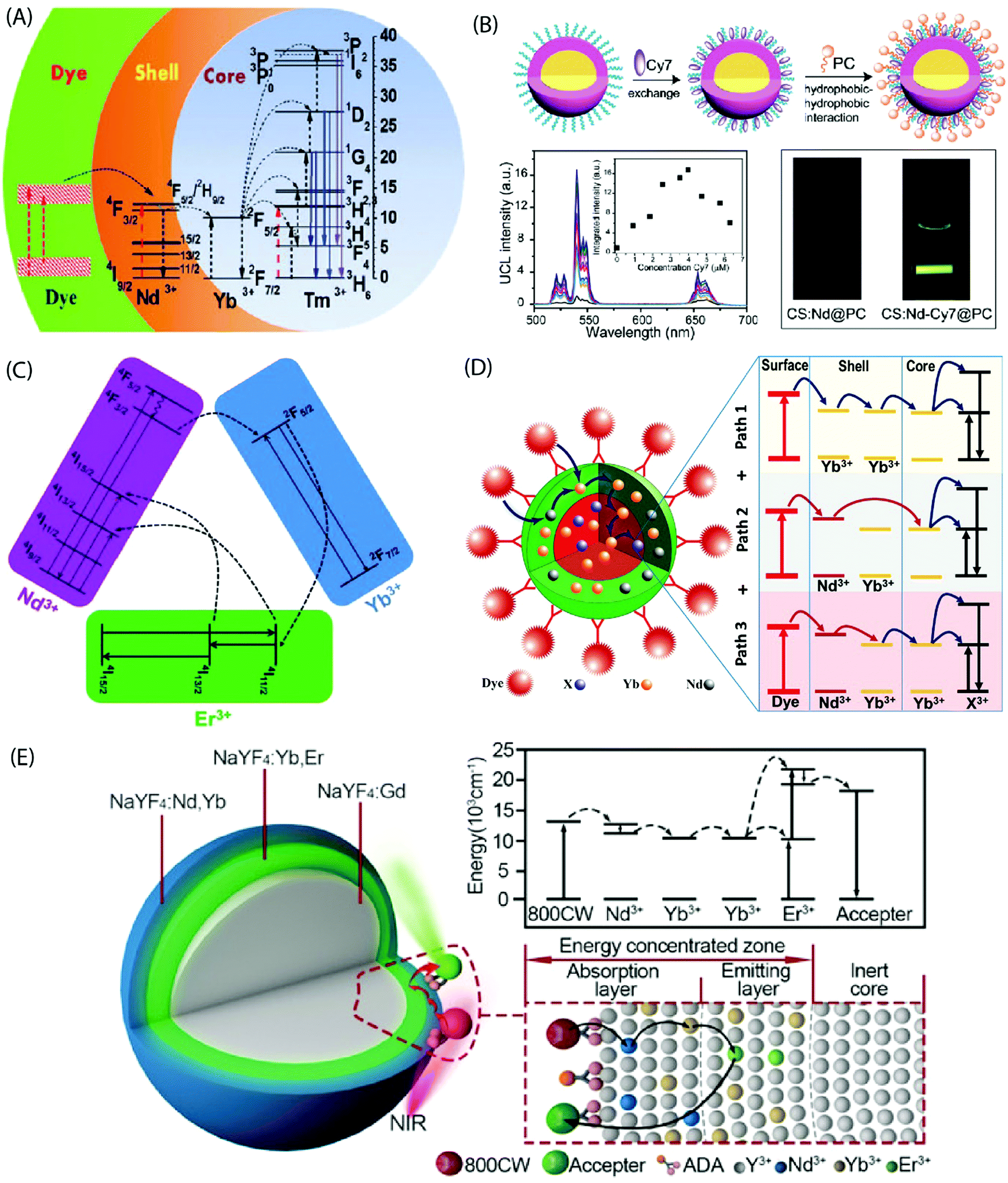 | ||
| Fig. 8 (A) Energy transfer pathways in IR-808-NaYbF4:Tm3+/NaYF4:Nd3+ core/shell NPs. The dye molecules on the core/shell NP surface absorb photons and transfer excitation energy to the Nd3+ ions in the shell, which in turn sensitize Yb3+ ions in the core. Subsequently, intracore energy transfer from Yb3+ to Tm3+ excites the Tm3+ ions from the ground state to the 1G4, 1D2, and the 1I6 states. (Reproduced from ref. 44, with permission from American Chemical Society.)44 (B) Synthesis of a water-dispersible dye-sensitized UC nanocomposite. UC of UCNPs loaded with different concentrations of Cy7 in water and emission intensity integrated in the range of 500–675 nm as a function of the Cy7 content (inset) excited using an 808 nm c.w. laser. Photographic images of the UCNPs without and with Cy7 antenna dye, excited at 808 nm. (Reproduced from ref. 66, with permission from Royal Society of Chemistry.)66 (C) Simplified energy-level diagram depicting the energy transfer from Nd3+ to Yb3+, Yb3+ to Er3+, and energy back transfer from Er3+ to Nd3+ upon NIR excitation. (Reproduced from ref. 68, with permission from Royal Society of Chemistry.)68 (D) Multi-dimensional energy transfer pathways from the NIR dye molecules on the surface of NaYF4:Yb3+,X3+/NaYF4:Nd3+/Yb3+ NPs to the lanthanide ions in the core. (Reproduced from ref. 74, with permission from Wiley.)74 (E) Structure of the UCNPs with an energy-concentrating zone to boost energy transfer efficiency and a simplified energy level diagram. (Reproduced from ref. 80, with permission from Wiley.)80 | ||
The dye-sensitized UCNPs are mostly produced as dispersions in organic solvents. The fluorescence efficiency of NIR dyes in aqueous environment is typically much less than in the organic phase. However, water-based UC systems are strongly desired for several practical applications. Zou et al. made Cy7 dye-sensitized NaYF4:Yb,Nd,Er@NaYF4:Nd core/shell NPs that were subsequently coated with phosphatidylcholine through hydrophobic–hydrophobic self-assembly to achieve the water-dispersibility.66 The optimal Cy7 dye:NP ratio was ca. 84 and it yielded 17 times greater UC luminescence at 808 nm irradiation than the control samples without a dye-sensitization (Fig. 8B).
A tailoring of the structure and composition of the core/shell NPs can offer additional benefits. For instance, the core/shell/shell structure can suppress surface-related quenching of the core nanocrystal by spatially isolating it from the environment. Additionally, it offers a directional energy flow from the surface-bound organic to the core of NPs for an enhanced emission. Shao et al. introduced an efficient downshifting system composed of ICG-sensitized NaYF4:Yb3+,X3+/NaYbF4/NaYF4:Nd3+ (X = none, Er, Ho, Tm or Pr) core/shell/shell design.67 Under 800 nm excitation, this system was able to produce a set of narrow band emissions with a large Stokes shift (>200 nm) in the 1000–1700 nm range. Yang's group adopted a similar design concept to produce upconverting IR-808-sensitized NaGdF4:Yb,Er/NaGdF4:Yb/NaNdF4:Yb NPs.68 Later, NaLuF4:Gd,Yb,Tm/NaLuF4:Gd,Yb/NaNdF4:Yb NPs sensitized with IR-820 were reported.69 In these studies, Nd3+ was incorporated into the outermost inorganic shell and the activator ions were doped in the inner core. The intermediate layer between the core and the outermost shell served as a shield that spatially isolated the Nd3+ ions from the activator ions embedded in the core, and consequently prevented the cross-relaxation induced quenching effects. An efficient energy transfer from dye to the Nd3+ ions and subsequently to the core could still be achieved through the Yb3+ mediated core/shell/shell interface (Fig. 8C). The second shell can also be made of an inert material for certain applications, such as drug loading.70
It is also possible to dope the shell with Yb3+ ions instead of typically used Nd3+ ions, as was done for IR-806 sensitized NaYF4:Yb3+,Er3+/NaYF4:Yb3+ and IR-820 bound NaLuF4:Gd,Yb,Er/NaLuF4:Yb,Pr core/shell NPs.71–73 These single sensitizer-doped NPs utilize a single exclusive energy transfer pathway for the transfer of absorbed energy from the dye molecules, across the inorganic shell and subsequently to the ions in the core. An availability of multiple energy transfer pathways across the shell holds a possibility to boost the UC efficiency. This can be achieved by doping the shell with more than one type of sensitizer ions. Chen et al. combined Nd3+ and Yb3+ ions in the shell of UCNPs to take benefit of three exclusive energy transfer pathways from the surface-bound ICG dye to the Yb3+ ions in the core.74 The overlap between the emission spectrum of ICG and the absorption spectrum of Nd3+ and Yb3+ ions enabled a Förster-type energy transfer from the excited ICG molecules to both ions embedded in the shell.
The three underlying energy transfer pathways can be summarized as: (1) ICG → Nd3+ (shell) → core; (2) ICG → Yb3+ (shell) → core; and (3) ICG → Nd3+ (shell) → Yb3+ (shell) → core. The resulting UCL intensities were about 2–3 times higher than observed from the single energy transfer pathway in NPs with only Nd3+ or Yb3+ ions in the shell (Fig. 8D). A similar improvement in UC emission was recorded by Shao et al. using IR-806 functionalized NaYF4:Yb3+,Er3+/NaYF4:Yb3+,Nd3+ core/shell NPs.75
Another approach to improve energy transfer efficiency in multi-layered UCNPs is to use an inert core and an energy-concentrating zone to confine the energy transfer process in a narrow region near the particle surface.80 The inert core blocks the energy transfer to deep interior of the UCNPs and spatially concentrates it more in the proximity of the UCNP surface to enhance the local excitation energy density. The synergistic effect of outer surface of maximal energy absorption, combined with effect of the energy-concentrating zone, holds a remarkable promise to achieve high UC emission intensity in various applications (Fig. 8E).
5.3 Metal–organic network-type structures
Coordination polymers (CPs) are single-phase metal–organic networks of repeating units of metal ions and organic ligands that possess a unique structural and functional diversity; these materials are commonly called metal–organic frameworks (MOFs) when they possess high degree of porosity. A majority of the CP and MOF materials highlighted are highly crystalline, but the concept of amorphous MOFs (aMOFs) has also gained increasing interest in recent years.84 Upconverting lanthanide-based MOF structures (Ln-MOFs) can offer exciting features for several practical applications, such as for imaging-monitored drug delivery, etc. Various functional Ln-MOFs have already been developed for several applications.85–87 Most of these Ln-MOFs require excitation by UV light that may pose limitations for their use in certain applications. Thus, development of NIR excitable Ln-MOFs is of significant interest.Li et al. synthesized a series of doped Er-MOF and Y-MOF materials with biphenyl-2,5,2′,5′-tetracarboxylate and 1,4-dicarboxybenzene ligands,38 and observed the strongest UC emission from Er-MOFs when 980 nm excitation was used; by doping Yb3+ ions into Er-MOF, the emission intensity increased with increasing Yb3+ substitution level. The larger Ln⋯Ln distance (6.63 Å) in Ln-MOFs as compared to typical NaYF4:Yb,Er UCNPs (3.5 Å) was suspected to be the cause of inefficient energy transfer between Yb3+ and Er3+ ions. This is an important finding and must be considered while designing NIR-to-Vis UC Ln-MOFs. Later, Yuan et al. produced several Zr-based MOFs that were in situ decorated with ligand-free NaYF4:Yb,Er UCNPs using electrostatically driven self-assembly.81 The nanocomposites exhibited characteristic Er3+ UC emission under 980 nm laser excitation (Fig. 9A).
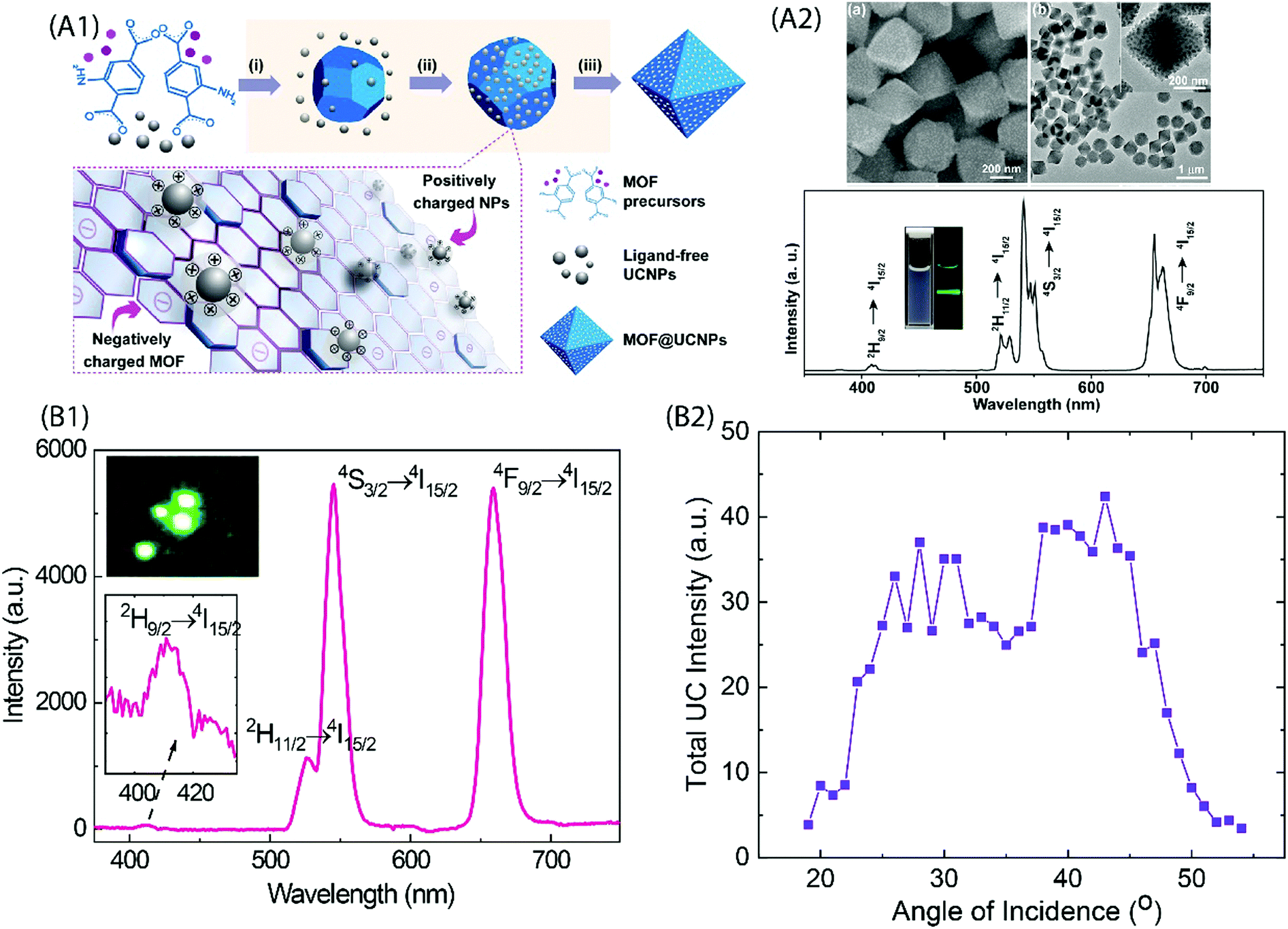 | ||
| Fig. 9 (A1) Fabrication of UCNP and MOF nanocomposites. Precursors of MOF and ligand-free UCNP are mixed directly for the in situ formation of the nanocomposite. Yellow shaded steps show the proposed formation mechanisms: (i) MOF nucleation, (ii) attachment of nanoparticles onto MOFs through electrostatic interaction, and (iii) nanocomposite formation. (A2) Top, SEM and TEM images of the UiO-66-NH2@NaYF4:Yb,Er nanocomposites; Bottom, UC emission spectrum of UiO-66-NH2@NaYF4:Yb,Er nanocomposites dispersed in DMF under 980 nm excitation. The inset shows photographs of UiO-66-NH2@NaYF4:Yb/Er nanocomposites dispersed in DMF under daylight (left) and the excitation of a 980 nm laser (right). (Reproduced from ref. 81, with permission from American Chemical Society.)81 (B1) UC emission spectrum for (Y,Yb,Er)–pyrazine thin film grown on the Si substrate. Inset is a composite image of the film excited at 974 nm. (B2) Dependence of the UC emission intensity on the incident angle of excitation.37 | ||
In recent years, advances in the ALD/MLD thin-film technique48–50 has made it possible to deposit directly from gaseous/evaporated precursors hybrid metal–organic materials with atomic/molecular level accuracy, both in situ crystalline MOF-like and amorphous aMOF-like thin-film materials.88–90 It is worth noting that the ALD/MLD approach also enables the growth of novel hybrid materials with unique molecular networks that cannot be readily realized using any other synthesis route.91–93 Recently, our group reported the NIR-to-Vis UC emission for amorphous ALD/MLD-fabricated (Y,Yb,Er)–pyrazine thin films.37 The films were deposited using mixed (Y,Yb,Er)(thd)3 and 2,3-pyrazinedicarboxylic acid as the metal and organic precursors, respectively. The precursors were separately vaporized and sequentially pulsed inside the reactor to form hybrid thin films. Upon exposure to 974 nm NIR laser light, the films exhibited all the three red-green-blue colours that were a result of two- and three-photon NIR-to-Vis excitation processes. Interestingly, the films produced UC emission only when the incident angles were between 20 and 50°. Such a narrow angular range is otherwise seen in the case of surface plasmon-coupled emissions (Fig. 9B).94
More recently, ALD/MLD was used to combine Yb3+ and Er3+ ions with the IR-806 dye molecules via Yb–O and/or Yb–N bonds, with the aim of improving the NIR absorption of the films.82 Upon excitation with 974 nm NIR laser, characteristic green and red UC emission was observed from Er3+ ions. The underlying two-photon stacking process followed the following possible schemes: (i) Energy transfer up conversion (ETU): Yb3+ absorbed the first excitation photon through the 2F7/2 → 2F5/2 transition and the energy was transferred to Er3+ which excited to the 4I11/2 state. Next, this excited state absorbed a second photon promoting Er3+ to the 4F7/2 state. (ii) Ground state absorption followed by excited state absorption (GSA + ESA): The first photon was absorbed directly by Er3+ through the 4I15/2 → 4I11/2 transition. Then, this excited state absorbed a second photon promoting Er3+ to the 4F7/2 state. In both cases, the 4F7/2 state nonradiatively relaxed to the 2H11/2, 4S3/2 and/or 4F9/2 states, which finally relaxed to 4I15/2 yielding the green and/or red-light emission. Interestingly, no UC emission was observed upon excitation of the (Yb,Er)-IR-806 films under 800 nm, which is the absorption wavelength for IR-806. This generally means that either no dye-to-Ln3+ energy transfer took place, or that the lifetime of the excited IR-806 was not long enough to allow the stacking of the photons. It was speculated that one reason for this could be self-quenching caused by a too high concentration of the IR-806 molecules. This assumption was based on the earlier report that the ideal IR-806 distance for high upconversion intensity when acting as antennas for NaYF4:Yb,Er was only 3.4 nm.95 In the films, the distance was expected to be much shorter. A partial fragmentation of the dye molecules at evaporation/deposition temperature was also thought to be possible. Thus, a careful selection of the organic molecules is critical for growth of UC thin films to realize their role in the energy absorption and transfer steps. The use of larger organic molecules, such as IR absorbing dyes can greatly improve absorption of low-energy photons, but these are prone to fragmentation at higher temperature. Moreover, the use of large molecules in metal–organic network type structures can result in larger Ln⋯Ln interionic distances that can reduce the efficiency of non-radiative energy transfer between the sensitizer and activator ions. Despite the foreseeable challenges, such MOF/aMOF type structures prepared by vapour-phase growth methods hold several benefits compared to UCNPs, for instance, solvent-free synthesis for solid-state devices, compatibility with scalable large-area growth on various substrates, etc.
6 Examples of applications
6.1 Bioimaging
The commonly used clinical fluorophores for bioimaging are organic molecules that emit Vis-light upon excitation by UV or blue photons. However, photobleaching of organic probes, autofluorescence of the biological specimen, phototoxicity under UV irradiation and limited penetration depth into/out of the sample are some of the major problems. Hence, the use of NIR-light for cellular and deep-tissue imaging is highly advantageous because the so-called NIR biological windows provide a transparency towards the biological materials (Fig. 10).96 Inorganic UCNPs have already been used for several biomedical applications in recent years. Their superior features compared to quantum dots and organic fluorophores, such as sharp emission peaks, low toxicity, photochemical stability, long emission lifetimes and a resistance to photobleaching, make them potential candidates for use as contrast agents in biomedical imaging and sensing applications. However, the inorganic UCNPs, which predominantly utilize Yb3+ ions as the absorbers, require 980 nm laser excitation. In this spectral region, water molecules tend to strongly absorb the IR photons. Because water molecules are an essential constituent of biological samples, the absorption causes both an attenuation of the excitation signal and possible overheating of biological specimens that can cause cell death and tissue damage.97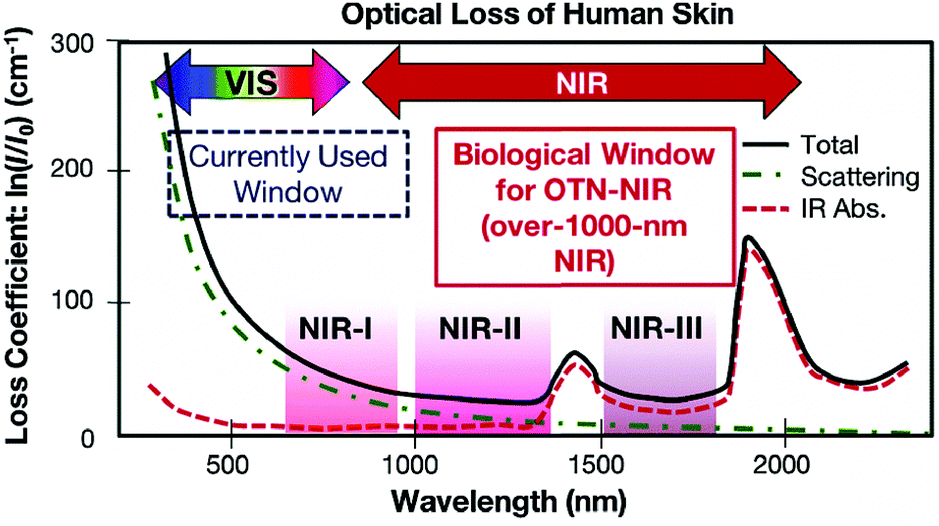 | ||
| Fig. 10 Absorption spectrum of human skin showing the first (NIR-I), second (NIR-II) and third (NIR-III) biological windows. (Reproduced from ref. 96, with permission from Royal Society of Chemistry.)96 | ||
The inorganic–organic hybrid UCNPs provide a plausible solution against this issue, because the organic antenna molecules can be carefully designed to exhibit absorption at wavelengths of interest. Most of the organic dyes used as sensitizers absorb at ∼800 nm, which is a spectral region minimally attenuated by the biological samples. Thus, a choice of excitation wavelength from this region not only improves the penetration depth, but also reduces tissue-overheating problem.
Chen et al. encapsulated ICG-sensitized core/shell UCNPs into amphiphilic micelles to render them dispersible in an aqueous phase for in vitro imaging of cancer cells.74 However, the UC luminescence decreased by a factor of 5 after the phase transfer from DMF to water, which can be possibly associated with the quenching due to water molecules. As NIR light at 800 nm can penetrate transcranially to a depth of several cm through the human skull, meninges, scalp, and brain; the UCNPs can be used for deep-tissue imaging and optogenetics. Certain light-sensitive proteins (opsins), control neuronal activity upon exposure to certain wavelengths of light.
Wu et al. demonstrated neuron activation by IR-806-sensitized core/shell NPs embedded in poly(methyl methacrylate) film, under 800 nm excitation.71 The film was placed directly beneath a glass coverslip containing cultured hippocampal neurons. Red light sensitive channel rhodopsin revealed consistent depolarization and firing in response to 800 nm light, in an intensity and pulse-pattern dependent manner. A subcutaneous administration of water solubilized UCNPs into the mouse was used for in vivo imaging. However, this system also suffered from significant loss in UC emission in aqueous environment due to the quenching by water molecules. A relatively more stable upconverting system was obtained when phosphatidylcholine (PC) coating was applied to Cy7-sensitized core/shell UCNPs for cellular and lymphatic imaging.66 The UC intensity of this upconverting nanocomposite system remained above 70% of the initial intensity even after seven days as compared to the initial value, which reflects improved protection of the Cy7-sensitized UCNPs by the PC molecules against aqueous environment (Fig. 11A).
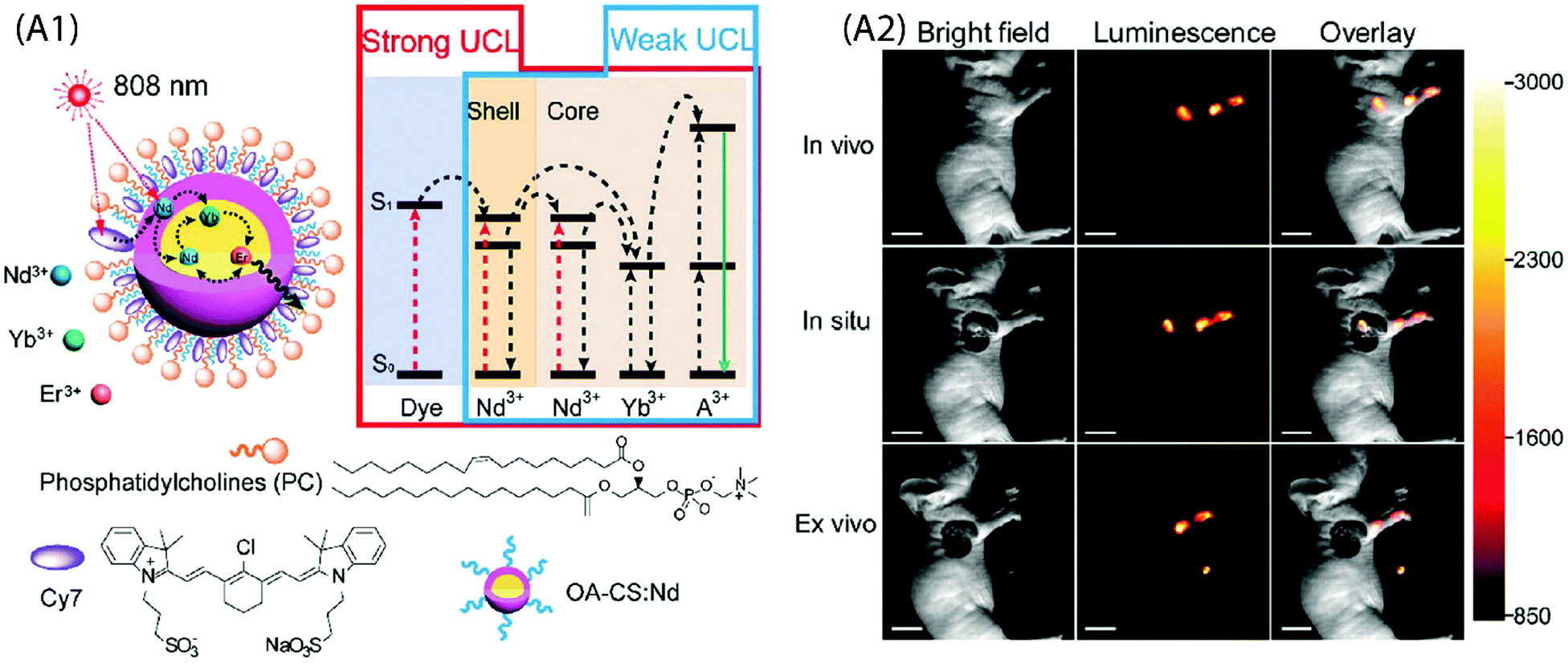 | ||
| Fig. 11 (A1) Schematic design and simplified energy level diagram of a water-dispersible PC-Cy7-UCNPs. (A2) UCL lymphatic imaging at 30 min post injection of PC-Cy7-UCNPs under irradiation at 808 nm (λUCL = 540 ± 12 nm, scale bar = 10 mm). (Reproduced from ref. 66, with permission from Royal Society of Chemistry.)66 | ||
6.2 Photodynamic therapy
Another potential biomedical application of inorganic–organic UCNPs is for the photodynamic therapy and simultaneous multimodal bioimaging of cancer cells and infectious pathogens. Photodynamic therapy (PDT) involves the use of photosensitizers, visible light and ambient oxygen to generate reactive oxygen species (ROS) that trigger cancer cell death.2 Because visible light suffers from limited penetration depth, its use in PDT is restricted to diseased tissues closely underneath the skin or near organ lining. Xu et al. developed IR-808-sensitized NaGdF4:Yb,Er/NaGdF4:Nd,Yb UCNPs coated with mesoporous SiO2 shell that housed green- and red-light sensitive dual-photosensitizers.70 Upon excitation by 808 nm NIR photons, the energy was absorbed by IR-808 and transferred to Nd,Yb ions in the shell and further to the Yb,Er ions in the core to radiate green and red photons. These visible-light photons activated dual-photosensitizers to generate a burst of ROS for an impressive in vivo tumour inhibition efficacy. Moreover, in addition to UC imaging, the presence of Gd and Yb ions in the UCNPs allowed for in vitro magnetic resonance imaging and computerized tomography. Such multimodal imaging capability holds a promise for imaging-guided PDT.6.3 Detection of (bio)chemicals
Multiplexed, sensitive (bio)molecular detection of target molecules in clinical diagnostics requires sophisticated analytical approaches due to the heterogeneous and complex nature of specimens. Conventional bioanalytical methods based on PCR gene chips, electrophoresis and mass spectrometry are not directly suited for the analysis of such heterogeneous samples. Moreover, these methods require destructive sample preparation and cannot be applied to live cells/tissues. The use of quantum dots or fluorescent dyes for the analysis of protein biomarkers has emerged as a powerful approach, but it also has several limitations, such as background autofluorescence, photobleaching, etc. Hence, more suitable multiplexed biomarker detection technologies are required. One of the possibilities towards this end is to use narrow line width, single-band (sb) UC emission that can increase the number of distinct emission bands and enable multiplexed analysis of analytes. For instance, Zhou et al. have used sb-UCNPs for the quantitative and simultaneous in situ profiling of multiple biomarkers in intact breast cancer cells and tissues.46 The sb emission in the blue (480nm), green (550nm) and red (650nm) regions was achieved by coating UCNPs with selected organic dyes to function as optical filters that possessed an absorption band overlapping with one of the UC emission bands. The antibody-conjugated sb-UCNPs were successfully applied to simultaneously detect and quantify multiple tumour biomarkers (Fig. 12A).
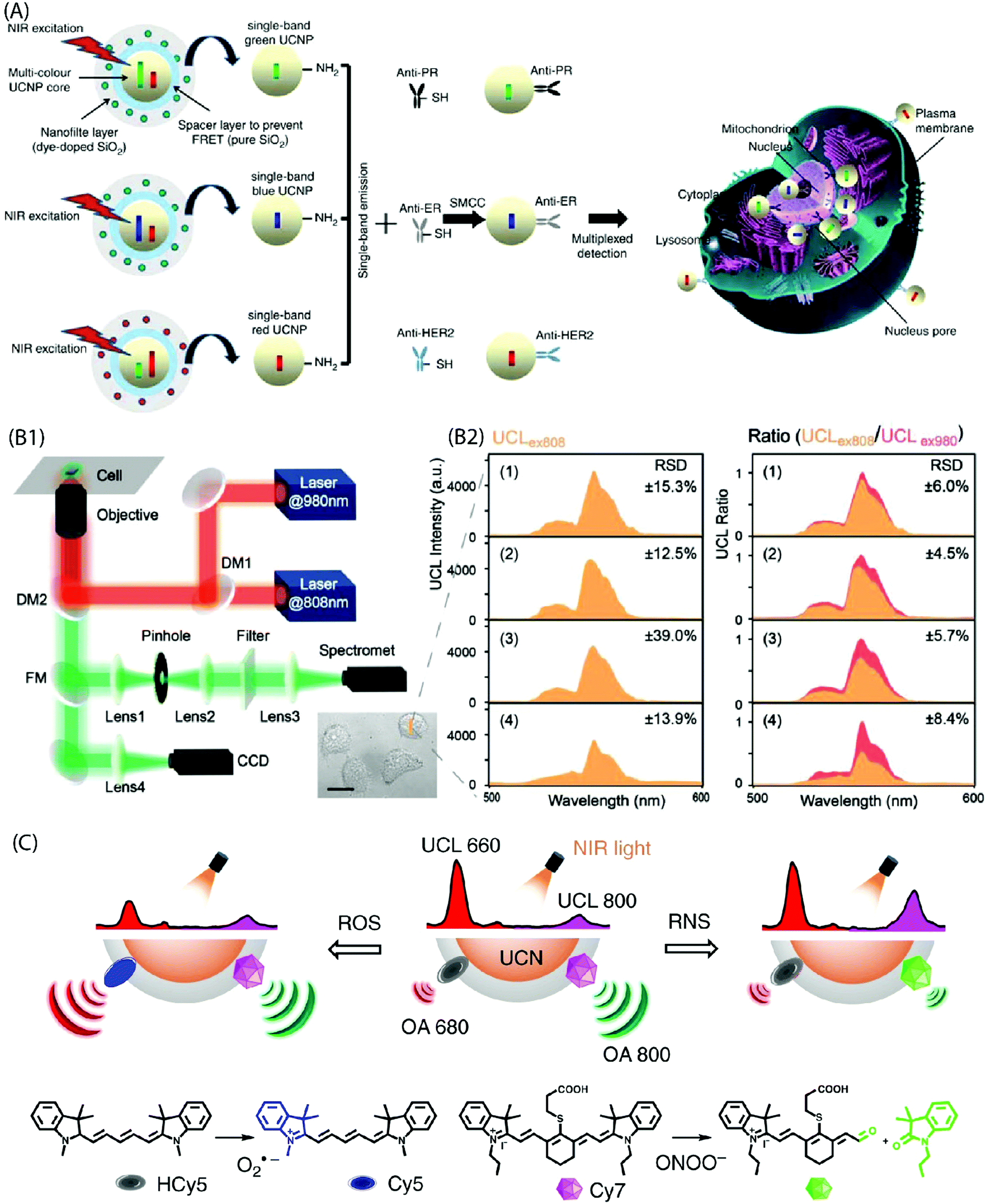 | ||
| Fig. 12 (A) Design of the multi-layer structure of green, blue and red sb-UCNPs and their antibody – for multiplexed breast cancer biomarker mapping.46 (B1) Optical layout of the purpose-built NIR dual laser confocal microscope system (DM: dichroic mirror, FM: flip mirror, CCD: charge coupled device). (B2) UC spectra collected at 500–600 nm under 808 nm excitation (UCLex808) in probe-loaded cells. Panels (1–4) show different cell groups upon addition of NaClO. Also shown is the UCL ratio (UCLex808/UCLex980) derived by normalizing the UCLex808 intensities to their corresponding 540 nm emission under 980 nm excitation. A significant reduction in the relative standard deviation (RSD) is evident from the plots after the ratiometric normalization (n = 6 cells).45 (C) Design of NIR light-mediated UCNPs by comprising reactive oxygen species-responsive HCy5 and reactive nitrogen species-responsive Cy7 dyes. Upon radical stimulation, HCy5 and Cy7 underwent a structural regeneration and degradation, respectively, leading to ratiometric UCL and optoacoustic signal variations that enabled screening of diverse endogenous redox biomarkers.79 | ||
The use of inorganic–organic hybrid UCNPs is becoming more popular also for ratiometric intracellular detection. In a ratiometric analysis, the signal output from two or more excitation or emission sources is compared to deduce changes in an intracellular environment. For instance, Ke et al. reported recently nanomolar hypochlorite (ClO−) quantification in live cancer cells using IR-808-sensitized NaGdF4:Yb,Er/NaGdF4:Yb UCNPs and a purpose-built confocal microscope equipped with 808 nm and 980 nm dual-laser excitation source (Fig. 12B).45 The C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond at the secondary amine substitution of IR-808 can be reduced by the ClO− molecules to interrupt energy transfer pathway and thus markedly suppress 540 nm UC emission under 808 nm excitation. However, as different intracellular environments can give rise to variations in the emission intensity even in the case of cells from the same group; the authors normalized 540 nm emission under 808 nm excitation to the corresponding 540 nm emission under 980 nm excitation. As a result, accurate quantification of intracellular intrinsic and exogenous ClO− was made possible due to the minimal interference from other biomolecular constituents, pH perturbation and inhomogeneous probe distribution within the cellular environment. Liu et al. also developed a ratiometric strategy for the selective detection of peroxynitrite (ONOO−) as the hepatitis indicator for in vivo and in vitro application.78 Moreover, specific radical-sensitive NIR cyanine fluorophores were combined with UCNPs for simultaneous visualization of endogenous redox biomarkers with excellent spatiotemporal resolution in living conditions (Fig. 12C).79
C bond at the secondary amine substitution of IR-808 can be reduced by the ClO− molecules to interrupt energy transfer pathway and thus markedly suppress 540 nm UC emission under 808 nm excitation. However, as different intracellular environments can give rise to variations in the emission intensity even in the case of cells from the same group; the authors normalized 540 nm emission under 808 nm excitation to the corresponding 540 nm emission under 980 nm excitation. As a result, accurate quantification of intracellular intrinsic and exogenous ClO− was made possible due to the minimal interference from other biomolecular constituents, pH perturbation and inhomogeneous probe distribution within the cellular environment. Liu et al. also developed a ratiometric strategy for the selective detection of peroxynitrite (ONOO−) as the hepatitis indicator for in vivo and in vitro application.78 Moreover, specific radical-sensitive NIR cyanine fluorophores were combined with UCNPs for simultaneous visualization of endogenous redox biomarkers with excellent spatiotemporal resolution in living conditions (Fig. 12C).79
Liang et al. developed NaYF4:20%Yb,2%Er/NaYF4:20%Nd UCNPs using chemically modified heptamethine cyanine dye as a target-modulated sensitizing switch for the detection of glutathione (GSH); a cancer biomarker.76 The modified dye contained a nitroazo group to serve a dual purpose, i.e. the recognition of GSH molecules, and to annihilate the emission from the dye itself by photoinduced electron transfer process. In the presence of GSH, the nitroazo group from the dye was replaced, thereby resuming a strongly emissive nature of it. Since the sensitization effect for UCL was triggered exclusively by the interaction of target molecules with the dye-decorated NPs, it allowed a promising way for quantitative detection of GSH in live cells.
6.4 Solar cells
The standard spectrum of sunlight (280–2500 nm) is composed of about 3% UV, 44% Vis and 53% IR light (Fig. 1B). The UV photons are only a fraction of the solar spectrum and there is a high variation in its irradiance due to atmospheric factors (ozone, clouds, etc.). On the other hand, IR light is very abundant, but the photon energies are too low to be absorbed by semiconducting materials commonly used in solar cells due to their wide bandgap. Hence, most of the solar cell technologies rely on Vis-portion of the solar spectrum because the energy of Vis-photons matches the bandgap of the semiconductor used. The loss of sub-bandgap photons is one of the major limitations that confront further improvement of solar cell efficiencies, particularly in single-junction solar cells.The absorption of only those photons with energies higher than the semiconductor bandgap contribute to an electric current. The widely applicable solar cell technologies utilize only Vis-light; whereas the UV and IR light is lost to thermalization and transmission losses, respectively.98 As a result, the solar cell efficiencies are severely limited close to the Schockley–Queisser limit (e.g. 30% for c-Si solar cells). Earlier computational studies have shown that the performance of c-Si PV cells can be enhanced up to 63% and 47.6% for concentrated and nonconcentrated sunlight, respectively, by IR-to-Vis UC.99 Other solar cell technologies (e.g. a-Si, perovskites, dye-sensitized solar cells (DSSC), organic solar cells, etc.) can benefit even better from the IR-to-Vis UC process as these materials severely fail to absorb photons at longer wavelengths. UC can improve solar light harvesting ability of these materials without altering the design and merits of the existing solar cells. For example, NaYF4:10%Yb3+,2%Er3+/NaYF4:30%Nd3+ NPs functionalized with IR-783 dye were incorporated onto the TiO2 photoanode of a dye-sensitized solar cell to harvest energy from NIR photons that is otherwise lost due to transmission losses.43 The solar cell efficiency improved from 7.57 to 8.57% under direct AM1.5 G simulated sunlight irradiation (100 mW cm−2), which corresponds to more than 13% improvement compared to the control. The UC contribution from harvesting NIR sunlight was quantified to be 7.1%, along with the scattering contribution of ∼6%. By selecting a range of suitable organic dyes, a significant portion of the IR spectrum can be harvested and converted into visible light for considerable increase in the overall efficiency of a solar cell. Jiao and co-workers achieved both UC and DS for polymer solar cells by using NaYbF4:Tb,Eu NPs decorated with Phen and TTA ligands.64 Here, the antenna ligands were sensitive to UV photons (372 nm excitation) only and the energy transfer took place from Phen/TTA → Tb → Eu. Under 980 nm excitation, Yb was populated to 2F5/2 level to non-radiatively transfer the energy to Tb by cooperative sensitization, which was then followed by energy transfer to Eu for visible-light emission. The authors reported a 10.2% increase in power conversion efficiency when these UC and DS NPs were incorporated into the polymer solar cells. These recent reports indicate a considerable promise that inorganic–organic UC materials offer to solar cell technologies. More research focus is required to design efficient UC materials and combine them with different solar cell devices to study improvement in power conversion efficiency under solar irradiation.
7 Summary and outlook
In this review, we introduced the lanthanide-based upconversion phenomenon, the underlying energy transfer processes, sensitization strategies taking advantage of organic linker/ligand molecules, the emerging hybrid Ln-organic UC materials and their design concepts, and finally the prominent applications of such materials in a broader context. The hybrid UC materials have emerged as promising candidates for a diverse range of applications that can benefit from IR photons’ good biological penetrability or high availability in sunlight. Whether it be the larger absorption cross-section of the organic sensitizers, emission sensitization, emission band tuning or their use in metal–organic framework type structures, the organic molecules offer a breadth of unique features that otherwise cannot be realized using purely inorganic UC materials.However, despite the recent remarkable developments, there is a lot of room for further improvement. For instance, the existing UC materials require laser excitation for UC process to take place and the quantum efficiencies are still very low. The conversion of IR solar spectrum into Vis light would greatly benefit solar cell technologies and solar-driven photocatalysis, but the efficiency of most Ln-based UC materials is still poor under normal solar irradiance (ca. 1000 W m−2). The organic sensitizers attached to UC materials can provide a potential route to improve the absorption of sub-bandgap photons. This feature can be combined with novel design principles to construct UC materials with strong emission characteristics.
The MOF-type UC materials may find applications for sensing or detection of chemicals. A modulation of the luminescence property upon the absorption of guest molecules in porous UC metal–organic structures can open new ways of chemical sensing. However, the use of UCNPs and MOF-type materials can face some serious challenges. They can be difficult to apply onto solid-state devices in an appropriate form, such as thin films. Furthermore, the incorporation of additional solvent impurities may degrade the performance or lifetime of the devices. The currently strongly emerging ALD/MLD technique can provide an attractive approach for the integration of hybrid UC materials in e.g. solar cells or photocatalysts.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge the funding from European Research Council under the European Union's Seventh Framework Programme (FP/2007-2013)/ERC Advanced Grant Agreement (339478) and Academy of Finland (296299).Notes and references
- T. M. Liu, J. Conde, T. Lipiński, A. Bednarkiewicz and C. C. Huang, NPG Asia Mater., 2016, 8, 1–25 Search PubMed.
- M. R. Hamblin, Dalton Trans., 2018, 47, 8571–8580 RSC.
- D. Li, H. Ågren and G. Chen, Dalton Trans., 2018, 47, 8526–8537 RSC.
- W. Guan, W. Zhou, J. Lu and C. Lu, Chem. Soc. Rev., 2015, 44, 6981–7009 RSC.
- X. Wang, O. S. Wolfbeis and R. J. Meier, Chem. Soc. Rev., 2013, 42, 7834 RSC.
- Y. Wang, K. Zheng, S. Song, D. Fan, H. Zhang and X. Liu, Chem. Soc. Rev., 2018, 47, 6473–6485 RSC.
- G. Tian, Z. Gu, L. Zhou, W. Yin, X. Liu, L. Yan, S. Jin, W. Ren, G. Xing, S. Li and Y. Zhao, Adv. Mater., 2012, 24, 1226–1231 CrossRef CAS PubMed.
- M. Pawlicki, H. A. Collins, R. G. Denning and H. L. Anderson, Angew. Chem., Int. Ed., 2009, 48, 3244–3266 CrossRef CAS PubMed.
- M. Drobizhev, A. Karotki, M. Kruk, A. Krivokapic, H. L. Anderson and A. Rebane, Chem. Phys. Lett., 2003, 370, 690–699 CrossRef CAS.
- F. Auzel, Chem. Rev., 2004, 104, 139–173 CrossRef CAS PubMed.
- D. Bloor, G. Cross, P. K. Sharma, J. A. Elliott and G. Rumbles, J. Chem. Soc., Faraday Trans., 1993, 89, 4013–4015 RSC.
- J. Liu, A. M. Kaczmarek and R. Van Deun, Chem. Soc. Rev., 2018, 47, 7225–7238 RSC.
- F. Zhang, Photon upconversion nanomaterials, Springer-Verlag, Berlin, 2015 Search PubMed.
- Q. Liu, B. Yin, T. Yang, Y. Yang, Z. Shen, P. Yao and F. Li, J. Am. Chem. Soc., 2013, 135, 5029–5037 CrossRef CAS PubMed.
- X. Wang, R. R. Valiev, T. Y. Ohulchanskyy, H. Ågren, C. Yang and G. Chen, Chem. Soc. Rev., 2017, 46, 4150–4167 RSC.
- S. Wen, J. Zhou, P. J. Schuck, Y. D. Suh, T. W. Schmidt and D. Jin, Nat. Photonics, 2019, 13, 828–838 CrossRef CAS.
- D. Yin, K. Song, Y. Ou, C. Wang, B. Liu and M. Wu, J. Nanosci. Nanotechnol., 2013, 13, 4162–4167 CrossRef CAS PubMed.
- F. Huang, X. Liu, Y. Ma, S. Kang, L. Hu and D. Chen, Sci. Rep., 2015, 5, 8233 CrossRef CAS PubMed.
- C. Jiang and P. Song, in Nonlinear-Emission Photonic Glass Fiber and Waveguide Devices, ed. C. Jiang and P. Song, Cambridge University Press, Cambridge, 2019, pp. 65–73 Search PubMed.
- K. Lingeshwar Reddy, R. Balaji, A. Kumar and V. Krishnan, Small, 2018, 14, 1–27 CrossRef PubMed.
- F. Wang and X. Liu, Chem. Soc. Rev., 2009, 38, 976–989 RSC.
- X. Xie, N. Gao, R. Deng, Q. Sun, Q. H. Xu and X. Liu, J. Am. Chem. Soc., 2013, 135, 12608–12611 CrossRef CAS PubMed.
- Y. Zhong, G. Tian, Z. Gu, Y. Yang, L. Gu, Y. Zhao, Y. Ma and J. Yao, Adv. Mater., 2014, 26, 2831–2837 CrossRef CAS PubMed.
- W. Zou, C. Visser, J. A. Maduro, M. S. Pshenichnikov and J. C. Hummelen, Nat. Photonics, 2012, 6, 560–564 CrossRef CAS.
- J. Schmidt and A. Penzkofer, J. Chem. Phys., 1989, 91, 1403–1409 CrossRef CAS.
- G. Chen, J. Damasco, H. Qiu, W. Shao, T. Y. Ohulchanskyy, R. R. Valiev, X. Wu, G. Han, Y. Wang, C. Yang, H. Ågren and P. N. Prasad, Nano Lett., 2015, 15, 7400–7407 CrossRef CAS PubMed.
- Y. Suffren, B. Golesorkhi, D. Zare, L. Guénée, H. Nozary, S. V. Eliseeva, S. Petoud, A. Hauser and C. Piguet, Inorg. Chem., 2016, 55, 9964–9972 CrossRef CAS PubMed.
- C. Reinhard and H. U. Güdel, Inorg. Chem., 2002, 41, 1048–1055 CrossRef CAS PubMed.
- D. L. Dexter, J. Chem. Phys., 1953, 21, 836–850 CrossRef CAS.
- T. Förster, Radiat. Res., Suppl., 1960, 2, 326–339 CrossRef.
- A. Nadort, J. Zhao and E. M. Goldys, Nanoscale, 2016, 8, 13099–13130 RSC.
- P. A. Tanner, L. Zhou, C. Duan and K. L. Wong, Chem. Soc. Rev., 2018, 47, 5234–5265 RSC.
- R. E. Joseph, C. Jiménez, D. Hudry, G. Gao, D. Busko, D. Biner, A. Turshatov, K. Krämer, B. S. Richards and I. A. Howard, J. Phys. Chem. A, 2019, 123, 6799–6811 CrossRef CAS PubMed.
- A. L. Pellegrino, S. La Manna, A. Bartasyte, P. Cortelletti, G. Lucchini, A. Speghini and G. Malandrino, J. Mater. Chem. C, 2020, 8, 3865–3877 RSC.
- S. Fan, G. Gao, S. Sun, S. Fan, H. Sun and L. Hu, J. Mater. Chem. C, 2018, 6, 5453–5461 RSC.
- M. Pollnau, D. Gamelin, S. Lüthi, H. Güdel and M. Hehlen, Phys. Rev. B: Condens. Matter Mater. Phys., 2000, 61, 3337–3346 CrossRef CAS.
- Z. Giedraityte, M. Tuomisto, M. Lastusaari and M. Karppinen, ACS Appl. Mater. Interfaces, 2018, 10, 8845–8852 CrossRef CAS PubMed.
- M. Li, S. Gul, D. Tian, E. Zhou, Y. Wang, Y. Han, L. Yin and L. Huang, Dalton Trans., 2018, 47, 12868–12872 RSC.
- J. Rissler, Chem. Phys. Lett., 2004, 395, 92–96 CrossRef CAS.
- G. Patonay, J. Salon, J. Sowell and L. Strekowski, Molecules, 2004, 9, 40–49 CrossRef CAS PubMed.
- J. Fabian, H. Nakazumi and M. Matsuoka, Chem. Rev., 1992, 92, 1197–1226 CrossRef CAS.
- M. Matsuoka, Infrared Absorbing Dyes, Springer, US, Boston, MA, 1990, pp. 19–33 Search PubMed.
- S. Hao, Y. Shang, D. Li, H. Ågren, C. Yang and G. Chen, Nanoscale, 2017, 9, 6711–6715 RSC.
- G. Chen, J. Damasco, H. Qiu, W. Shao, T. Y. Ohulchanskyy, R. R. Valiev, X. Wu, G. Han, Y. Wang, C. Yang, H. Ågren and P. N. Prasad, Nano Lett., 2015, 15, 7400–7407 CrossRef CAS PubMed.
- J. Ke, S. Lu, X. Shang, Y. Liu, H. Guo, W. You, X. Li, J. Xu, R. Li, Z. Chen and X. Chen, Adv. Sci., 2019, 6, 1901874 CrossRef CAS PubMed.
- L. Zhou, R. Wang, C. Yao, X. Li, C. Wang, X. Zhang, C. Xu, A. Zeng, D. Zhao and F. Zhang, Nat. Commun., 2015, 6, 1–10 Search PubMed.
- F. Wang, J. Wang and X. Liu, Angew. Chem., Int. Ed., 2010, 49, 7456–7460 CrossRef CAS PubMed.
- J. C. Boyer, M. P. Manseau, J. I. Murray and F. C. J. M. V. Veggel, Langmuir, 2010, 26, 1157–1164 CrossRef CAS PubMed.
- R. Arppe, I. Hyppänen, N. Perälä, R. Peltomaa, M. Kaiser, C. Würth, S. Christ, U. Resch-Genger, M. Schäferling and T. Soukka, Nanoscale, 2015, 7, 11746–11757 RSC.
- F. T. Rabouw, P. T. Prins, P. Villanueva-Delgado, M. Castelijns, R. G. Geitenbeek and A. Meijerink, ACS Nano, 2018, 12, 4812–4823 CrossRef CAS PubMed.
- H. X. Mai, Y. W. Zhang, L. D. Sun and C. H. Yan, J. Phys. Chem. C, 2007, 111, 13721–13729 CrossRef CAS.
- H. S. Qian and Y. Zhang, Langmuir, 2008, 24, 12123–12125 CrossRef CAS PubMed.
- J. Shan, J. Chen, J. Meng, J. Collins, W. Soboyejo, J. S. Friedberg and Y. Ju, J. Appl. Phys., 2008, 104, 094308 CrossRef.
- I. Hyppänen, S. Lahtinen, T. Ääritalo, J. Mäkelä, J. Kankare and T. Soukka, ACS Photonics, 2014, 1, 394–397 CrossRef.
- X. Meng, J. Mater. Chem. A, 2017, 5, 18326–18378 RSC.
- S. M. George, A. A. Dameron and B. Yoon, Acc. Chem. Res., 2009, 42, 498–508 CrossRef CAS PubMed.
- H. Van Bui, F. Grillo and J. R. V. Ommen, Chem. Commun., 2017, 53, 45–71 RSC.
- C. G. Dupuy, T. L. Allen, G. M. Williams and D. Schut, J. Nanotechnol., 2014, 1–13 Search PubMed.
- X. Wu, H. Lee, O. Bilsel, Y. Zhang, Z. Li, T. Chen, Y. Liu, C. Duan, J. Shen, A. Punjabi and G. Han, Nanoscale, 2015, 7, 18424–18428 RSC.
- J. Lee, B. Yoo, H. Lee, G. D. Cha, H.-S. Lee, Y. Cho, S. Y. Kim, H. Seo, W. Lee, D. Son, M. Kang, H. M. Kim, Y. Il Park, T. Hyeon and D.-H. Kim, Adv. Mater., 2017, 29, 1603169 CrossRef PubMed.
- C. D. Laboda and C. L. Dwyer, Adv. Funct. Mater., 2016, 26, 2866–2874 CrossRef CAS.
- M. D. Wisser, S. Fischer, C. Siefe, A. P. Alivisatos, A. Salleo and J. A. Dionne, Nano Lett., 2018, 18, 2689–2695 CrossRef CAS.
- W. Wei, G. Chen, A. Baev, G. S. He, W. Shao, J. Damasco and P. N. Prasad, J. Am. Chem. Soc., 2016, 138, 15130–15133 CrossRef CAS.
- J. Jiao, S. Gai, Y. Li, W. Shen, J. Tang, Y. Wang, L. Huang, J. Liu, W. Wang and L. A. Belfiore, Electrochim. Acta, 2018, 260, 959–964 CrossRef CAS.
- D. J. Garfield, N. J. Borys, S. M. Hamed, N. A. Torquato, C. A. Tajon, B. Tian, B. Shevitski, E. S. Barnard, Y. D. Suh, S. Aloni, J. B. Neaton, E. M. Chan, B. E. Cohen and P. J. Schuck, Nat. Photonics, 2018, 12, 402–407 CrossRef CAS.
- X. Zou, M. Xu, W. Yuan, Q. Wang, Y. Shi, W. Feng and F. Li, Chem. Commun., 2016, 52, 13389–13392 RSC.
- W. Shao, G. Chen, A. Kuzmin, H. L. Kutscher, A. Pliss, T. Y. Ohulchanskyy and P. N. Prasad, J. Am. Chem. Soc., 2016, 138, 16192–16195 CrossRef CAS PubMed.
- J. Xu, M. Sun, Y. Kuang, H. Bi, B. Liu, D. Yang, R. Lv, S. Gai, F. He and P. Yang, Dalton Trans., 2017, 46, 1495–1501 RSC.
- F. Zhao, D. Yin, C. Wu, B. Liu, T. Chen, M. Guo, K. Huang, Z. Chen and Y. Zhang, Dalton Trans., 2017, 46, 16180–16189 RSC.
- J. Xu, P. Yang, M. Sun, H. Bi, B. Liu, D. Yang, S. Gai, F. He and J. Lin, ACS Nano, 2017, 11, 4133–4144 CrossRef CAS PubMed.
- X. Wu, Y. Zhang, K. Takle, O. Bilsel, Z. Li, H. Lee, Z. Zhang, D. Li, W. Fan, C. Duan, E. M. Chan, C. Lois, Y. Xiang and G. Han, ACS Nano, 2016, 10, 1060–1066 CrossRef CAS PubMed.
- D. Yin, Y. Liu, J. Tang, F. Zhao, Z. Chen, T. Zhang, X. Zhang, N. Chang, C. Wu, D. Chen and M. Wu, Dalton Trans., 2016, 45, 13392–13398 RSC.
- S. Alyatkin, E. Urena-Horno, B. Chen, O. L. Muskens, A. G. Kanaras and P. G. Lagoudakis, J. Phys. Chem. C, 2018, 122, 18177–18184 CrossRef CAS.
- G. Chen, W. Shao, R. R. Valiev, T. Y. Ohulchanskyy, G. S. He, H. Ågren and P. N. Prasad, Adv. Opt. Mater., 2016, 4, 1760–1766 CrossRef CAS.
- Q. Shao, X. Li, P. Hua, G. Zhang, Y. Dong and J. Jiang, J. Colloid Interface Sci., 2017, 486, 121–127 CrossRef CAS PubMed.
- T. Liang, Z. Li, P. Wang, F. Zhao, J. Liu and Z. Liu, J. Am. Chem. Soc., 2018, 140, 14696–14703 CrossRef CAS PubMed.
- B. Xue, D. Wang, L. Tu, D. Sun, P. Jing, Y. Chang, Y. Zhang, X. Liu, J. Zuo, J. Song, J. Qu, E. J. Meijer, H. Zhang and X. Kong, J. Phys. Chem. Lett., 2018, 9, 4625–4631 CrossRef CAS PubMed.
- X. Liu, H. Lai, J. Peng, D. Cheng, X. Zhang and L. Yuan, Small, 2019, 15, 1902737 CrossRef CAS PubMed.
- X. Ai, Z. Wang, H. Cheong, Y. Wang, R. Zhang, J. Lin, Y. Zheng, M. Gao and B. Xing, Nat. Commun., 2019, 10, 1–11 CrossRef CAS PubMed.
- X. Zhang, W. Chen, X. Xie, Y. Li, D. Chen, Z. Chao, C. Liu, H. Ma, Y. Liu and H. Ju, Angew. Chem., Int. Ed., 2019, 58, 12117–12122 CrossRef CAS PubMed.
- Z. Yuan, L. Zhang, S. Li, W. Zhang, M. Lu, Y. Pan, X. Xie, L. Huang and W. Huang, J. Am. Chem. Soc., 2018, 140, 15507–15515 CrossRef CAS PubMed.
- M. Tuomisto, Z. Giedraityte, L. Mai, A. Devi, V. Boiko, K. Grzeszkiewicz, D. Hreniak, M. Karppinen and M. Lastusaari, J. Lumin., 2019, 213, 310–315 CrossRef CAS.
- G. Chen, H. Ågren, T. Y. Ohulchanskyy and P. N. Prasad, Chem. Soc. Rev., 2015, 44, 1680–1713 RSC.
- T. D. Bennett, A. L. Goodwin, M. T. Dove, D. A. Keen, M. G. Tucker, E. R. Barney, A. K. Soper, E. G. Bithell, J. C. Tan and A. K. Cheetham, Phys. Rev. Lett., 2010, 104, 115503 CrossRef PubMed.
- Y. Su, J. Yu, Y. Li, S. F. Z. Phua, G. Liu, W. Q. Lim, X. Yang, R. Ganguly, C. Dang, C. Yang and Y. Zhao, Chem. Commun., 2018, 1, 1–13 CrossRef CAS.
- J. N. Hao and B. Yan, Chem. Commun., 2015, 51, 7737–7740 RSC.
- X. Rao, T. Song, J. Gao, Y. Cui, Y. Yang, C. Wu, B. Chen and G. Qian, J. Am. Chem. Soc., 2013, 135, 15559–15564 CrossRef CAS PubMed.
- E. Ahvenniemi and M. Karppinen, Chem. Commun., 2016, 52, 1139–1142 RSC.
- E. Ahvenniemi and M. Karppinen, Chem. Mater., 2016, 28, 6260–6265 CrossRef CAS.
- A. Ghazy, M. Safdar, M. Lastusaari and M. Karppinen, Chem. Commun., 2020, 56, 241–244 RSC.
- M. Nisula, J. Linnera, A. J. Karttunen and M. Karppinen, Chem. – Eur. J., 2017, 23, 2988–2992 CrossRef CAS PubMed.
- J. Penttinen, M. Nisula and M. Karppinen, Chem. – Eur. J., 2017, 23, 18225–18231 CrossRef CAS PubMed.
- A. Khayyami, A. Philip and M. Karppinen, Angew. Chem., Int. Ed., 2019, 58, 13400–13404 CrossRef CAS PubMed.
- S.-H. Cao, W.-P. Cai, Q. Liu and Y.-Q. Li, Annu. Rev. Anal. Chem., 2012, 5, 317–336 CrossRef CAS PubMed.
- W. Zou, PhD thesis, University of Groningen, 2015.
- E. Hemmer, A. Benayas, F. Légaré and F. Vetrone, Nanoscale Horiz., 2016, 1, 168–184 RSC.
- Y. F. Wang, G. Y. Liu, L. D. Sun, J. W. Xiao, J. C. Zhou and C. H. Yan, ACS Nano, 2013, 7, 7200–7206 CrossRef CAS PubMed.
- J. C. Goldschmidt and S. Fischer, Adv. Opt. Mater., 2015, 3, 510–535 CrossRef CAS.
- T. Trupke, M. A. Green and P. Würfel, J. Appl. Phys., 2002, 92, 4117–4122 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2020 |