
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Dual-triggered nanoaggregates of cucurbit[7]uril and gold nanoparticles for multi-spectroscopic quantification of creatinine in urinalysis†
Weng-I Katherine
Chio
 ab,
Suresh
Moorthy
c,
Jayakumar
Perumal
b,
Dinish
U. S.
b,
Ivan P.
Parkin
a,
Malini
Olivo
b and
Tung-Chun
Lee
*ac
ab,
Suresh
Moorthy
c,
Jayakumar
Perumal
b,
Dinish
U. S.
b,
Ivan P.
Parkin
a,
Malini
Olivo
b and
Tung-Chun
Lee
*ac
aDepartment of Chemistry, University College London (UCL), London WC1H 0AJ, UK. E-mail: tungchun.lee@ucl.ac.uk
bLaboratory of Bio-Optical Imaging, Singapore Bioimaging Consortium (SBIC), Agency for Science Technology and Research (A*STAR), Singapore 138667, Singapore
cInstitute for Materials Discovery, University College London (UCL), London WC1H 0AJ, UK
First published on 7th April 2020
Abstract
Plasmonic nanocomposites of cucurbit[7]uril (CB7) and gold nanoparticles (Au NPs) have been optimised and applied to rapidly detect and quantify creatinine (CRN) of clinically relevant levels in urinalysis. The in situ formation of plasmonic nanocomposites via aqueous self-assembly is mediated by a combination of the portal binding of CB7 and the electrostatic effects of CRN molecules, allowing independent spectral signatures to be extracted from the same sample solution using surface-enhanced Raman spectroscopy (SERS) and UV-Visible spectroscopy. Meanwhile the formation of host–guest complexes between CB7 and CRN allows quantification of CRN in highly diluted synthetic urine by localising CRN at or in close proximity to the plasmonic hotspots within the Au NP:CB7 nanoaggregates, which enables highly reproducible SERS signals (within 5% error) with a sub-μM detection limit of 12.5 ng mL−1 (111 nM) and has tolerance against the presence of proteins and other biomolecules in a complex matrix. Our nanobiosensing material platform demonstrates the potential to be extended to other in-field applications.
Introduction
Urine is one of the major body fluids from which information on metabolism of the body and renal function can be extracted.1,2 It is readily available and can be obtained in a non-invasive way as opposed to blood serum. Creatinine (CRN), which is the end product of muscle metabolism, is a very important biomarker in clinical diagnosis, for example, it can be used to monitor the kidney filtration function in renal clearance tests.3,4 As the excretion rate of CRN in urine is almost constant, it is used as the internal standard to normalise the variation of water and thus other analytes in urine.5,6 Early diagnosis of urine biomarkers can help to improve the efficiency of medical treatment and to prevent the progression from renal diseases to kidney failure, which requires dialysis or kidney transplant to maintain life. In addition, elevated CRN levels are related to diseases such as muscular dystrophy, hyperthyroidism and poliomyelitis.7–9The standard clinical method for CRN quantification is based on the Jaffe reaction first developed more than a century ago.10,11 The major drawback of the Jaffe reaction is, however, its non-specificity as picric acid forms a reddish-orange complex with many other biomarkers besides CRN in basic solution.6,12–14 Other clinical methods include enzymatic methods,15 capillary electrophoresis,16 high-performance liquid chromatography17 and isotope dilution mass spectrometry.18 Nevertheless, not all of these methods allow real-time point-of-care diagnosis with high sensitivity and specificity.
Analytical techniques such as Raman spectroscopy and surface-enhanced Raman spectroscopy (SERS) are promising for diagnostic applications as they offer advantages including rapid response, high-sensitivity, cost-effectiveness, ease of use, minimal sample preparation and wider information window of the analyte molecules via their vibrational fingerprints.19–21 SERS can dramatically amplify the Raman signals of the analyte molecules located in close proximity to the plasmonic nanostructures (e.g. gold nanoparticles, Au NPs, and silver nanoparticles, Ag NPs) by 4 to 10 orders of magnitude via strong electromagnetic field enhancement.22
Despite the strong merits of using colloidal Au NPs or Ag NPs as SERS substrates for CRN detection in urinalysis, application has been limited due to the relatively low reproducibility, particularly in quantitative measurements. Clarke et al.8 demonstrated the feasibility of using Raman and SERS for quantitative detection of CRN in synthetic urine (SU) with Au NPs in the range of 1.25 mg dL−1 – 5 mg dL−1 and Chiang et al.4 later reported SERS sensing of CRN in diluted human urine with linear range from 2.56 to 6.4 mg dL−1. Chen et al.23 showed the SERS detection of CRN in water with colloidal Ag NPs with a detection limit of 10 mg dL−1. Nanostructured SERS substrates such as Ag-coated parylene,24 Jaffe complex-Ag film,16 nanoporous Au disk,25 boron nitride/Au nanocomposites,26 superhydrophobic Ag film,27 and Au NP-deposited blu-ray DVD28 were then developed, aiming to improve the sensitivity and signal reproducibility for CRN sensing. However, the fabrication of substrates is time-consuming and often involves complicated procedures.
Recently, it has been demonstrated that the issues of poor reproducibility and weak signal intensity resulted from uncontrolled aggregations of colloidal Au NPs can be addressed by attaching macrocyclic molecules, cucurbit[n]urils (CBn where n = 5–8 and 10), onto the Au NP surface to form precise plasmonic nanojunctions with a controlled interparticle spacing.29–35 The aggregation of Au NPs showing SERS signals can be checked directly by dark- and bright-field microscopy.36 Furthermore, CBs, which are highly symmetric, rigid and Raman-active, can form host–guest complexes with small analyte molecules via encapsulation inside their hydrophobic cavity.37 The analyte molecules are hence localised right at the centre or in close proximity of the plasmonic hotspots with enhanced SERS sensitivity.38 While there has been previous work on Au NP-CB SERS sensing, applications of this promising system in biosensing remain relatively limited in the literature.33,35 For instance, there is only one illustrative example on using this promising system for multiplexed sensing of neurotransmitters in SU, which has left plenty of room for more application-oriented studies.33
Herein, we report the host–guest complexation between CB7 and an important biomarker, CRN, and the quantitative detection of CRN in water and in diluted SU using Au NP-CB nanoaggregates for the first time. In contrast to precedent examples of Au NP-CB systems, however, the aggregation of Au NPs is mediated by both the portal binding of CB7 and the neutralisation of surface charges on the citrate-protected Au NP surface by CRN and its CB7 complexes, which exist in their protonated forms [CRN-H]+ and [CB7-CRN-H]+ under our experimental conditions. The plasmonic nanosubstrates formed by a combination of CB-mediated aggregation and CRN-mediated electrostatic self-assembly allow CRN to be detected and quantified via a facile multi-spectroscopic approach, i.e. SERS and UV-Vis spectroscopy. Our novel detection scheme can rapidly cross-validate the quantification results, where a single sample solution can simultaneously exhibit two independent spectral signatures as revealed by the far-field and the near-field effects of the plasmonic nanosubstrates (Fig. 1). Notably, the SERS signals of CRN in urinalysis using our approach are highly reproducible (within 5% error) and tolerant to matrix effects with a sub-μM detection limit of 12.5 ng mL−1 (111 nM), which is the lowest among all current state-of-the-art methods to the best of our knowledge (Table S1, ESI†). Our macrocycle-functionalised nanosubstrates have the potential for multiplexed biosensing in a complex matrix and are promising for other in-field monitoring outside biological and medical applications.
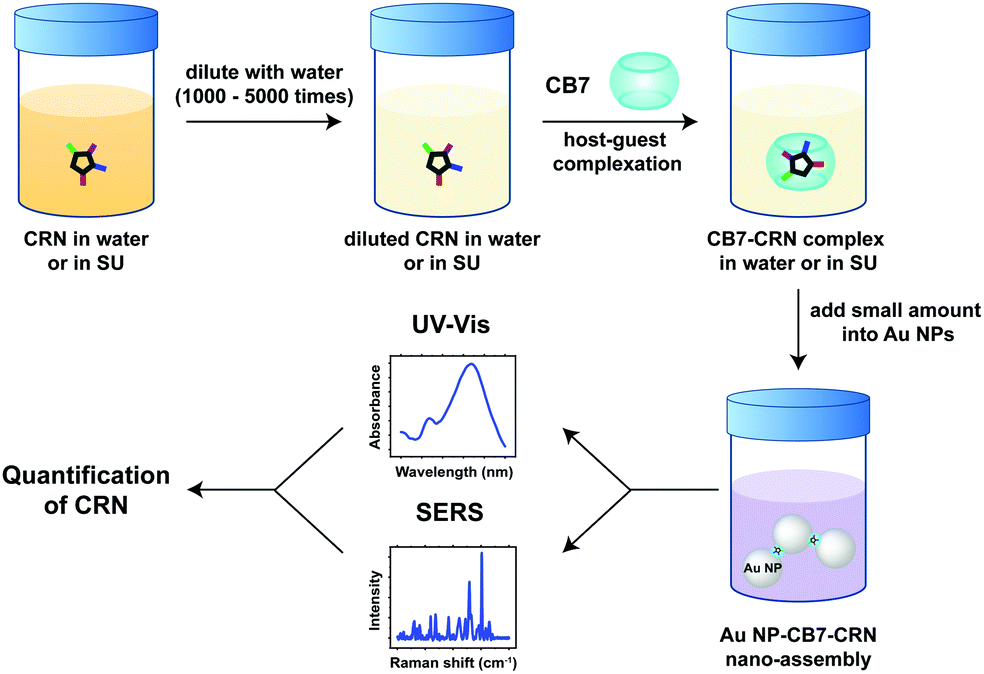 | ||
| Fig. 1 Schematic illustration of our dual UV-Vis and SERS detection scheme triggered by the formation of host–guest complexes between CB7 and CRN in highly diluted SU and the self-assembly of Au NPs mediated by portal binding of CB7 and electrostatic interactions with CRN. | ||
Materials and methods
Materials
40 nm citrate-capped Au NPs were purchased from nanoComposix. Paraformaldehyde, HCl, creatinine, urea, uric acid, critic acid, sodium phosphate monobasic and albumin from human serum were purchased from Sigma-Aldrich. Methanol, ethanol and KCl were purchased from VWR. Glycoluril was purchased from Acros Organics. NaCl was purchased from Kanto Chemical. CB7 was synthesised according to the literature.39 All chemicals were used as received without further purification. 18.2 MΩ Milli-Q water was used in all experiments.Preparation of synthetic urine
Synthetic urine mimicking a person with normal and excess CRN levels was prepared according to the literature with modifications.40 For 100 mL of synthetic urine, 0.9 g of urea, 16 mg of uric acid, 0.5 g of NaCl, 0.45 g of KCl, 40 mg of citric acid, 0.48 g of sodium phosphate, 5 mg of albumin and 0 to 300 mg of CRN were dissolved in 90 mL of water. The prepared SU was then diluted with water for UV-Vis and SERS measurements.Simulations
Density functional theory (DFT) calculations were performed using Gaussian and force-field calculations were performed using Chem3D. Geometry optimisation was performed using MMFF94, followed by full optimisation using a dispersion-corrected functional (wB97XD/6-31G* and CPCM/wB97XD/6-31G*). Restricted (close-shell) models were used in all DFT calculations. The binding energy of the [CB7–CRN-H]+ inclusion complex was calculated from the energy difference between the [CB7–CRN-H]+ complex and the total energies of CB7 and [CRN-H]+ optimised and calculated at the same level of theory. Raman spectra were simulated at the CPCM/wB97XD/6-31G* level of theory.Nuclear magnetic resonance (NMR) spectroscopy
A 4 mM CB7–CRN solution was prepared in D2O with a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 molar ratio. 1H NMR spectra were measured using a Bruker Avance III 400 spectrometer. Chemical shifts (in ppm) were referenced to D2O with δ = 4.79 ppm for 1H.
:1 molar ratio. 1H NMR spectra were measured using a Bruker Avance III 400 spectrometer. Chemical shifts (in ppm) were referenced to D2O with δ = 4.79 ppm for 1H.
UV-Visible spectroscopy
UV-Vis measurements were performed using a Boeco S-200 Vis spectrophotometer and a Shimadzu UV-3600 UV-Vis-NIR spectrophotometer. For CB7–CRN binding studies, a small volume of 5 mM CB7 solution was added to 50 μM CRN solution in a cuvette with 1 cm optical path length sequentially up to 2 equivalents. The concentration of CB7 was increased gradually, while that of CRN was kept approximately constant. For CRN sensing and quantification experiments, 20 μL of the pre-mixed stock solution of CB7 and CRN in water (or diluted SU) was added to 180 μL of Au NP solution in a 0.5 mL Eppendorf tube. The sample solution was vortexed for 30 s before loading 100 μL into the cuvette. Controls were prepared in the absence of CB7. Five spectra were measured for each sample and an averaged value was taken.Dynamic light scattering (DLS) and zeta potential measurements
500 μL of a Au NP–CRN or Au NP–CB7–CRN solution was added to a disposable semi-micro cuvette to measure average size, while 750 μL of the same solution was added to a folded capillary zeta cell to measure zeta potential using a Malvern ZEN 3600 instrument. Five measurements were taken for each sample and an average was taken.Raman and surface-enhanced Raman spectroscopy (SERS)
Raman and SERS spectra were acquired using a Reinshaw Raman InVia Microscope with a 633 nm He–Ne laser (9.3 mW). The laser was focused onto the sample via a 50× objective lens (N.A. = 0.75) and the grating used was 1800 lines mm−1, which gave a spectral resolution of 1 cm−1. All spectra were calibrated with respect to Si and measured at room temperature. The sample solution was prepared as above for UV-Vis spectroscopy and 15 μL was loaded onto a custom-made cell for SERS measurements. Five measurements were taken across different regions of interest per sample and three accumulations of 30 s scans were acquired on each measurement. The spectra were averaged and baseline corrected using an asymmetric least squares (ALS) plugin in Origin.Results and discussions
Host–guest chemistry of CB7 and CRN
Creatinine (CRN) exists in various molecular forms in water due to tautomerism. The most stable form of CRN under standard conditions is (amino tautomer, 2-amino-1-methyl-2-imidazoline-4-one).41 The pKa of the conjugated acid of CRN [CRN-H]+ is 9.2,42 implying that CRN exists in the protonated form in water and in synthetic urine (SU) where the pH is between 5 and 7.Upon addition of cucurbit[7]uril (CB7), CRN readily forms host–guest complexes mainly due to the release of high-energy water from the CB7 cavity,43 together with ion–dipole interactions and hydrogen bonding between the protonated site on CRN and the electron-rich carbonyl portal of CB7. This host–guest complexation has been verified by the characteristic upfield shifts of the CRN proton signals in 1H NMR spectra (Fig. S1, ESI†). The change in chemical shift (Δδ) for Hb (Δδb = −0.15 ppm) is much greater than for Ha (Δδa = −0.02 ppm) after host–guest complexation of CB7 and CRN. This suggests that Hb (methyl group) is deep inside the CB7 cavity, while Ha (CH2 group) stays closer to the carbonyl rim of CB7. A binding constant for the CB7–CRN complex of (1.17 ± 0.03) × 104 M−1 was also obtained from UV-Vis titration by fitting a 1:1 binding model (Fig. S2, ESI†). The relatively low binding energy is probably due to the small molecular size and the high solubility of CRN in water.
Energy-minimised molecular models of the complex in the gas phase (MMFF94 and wB97XD/6-31G*) and in water (CPCM/wB97XD/6-31G*) both support the NMR-derived host–guest binding geometry. The binding energy can be estimated by the energy of the complex [CB7–CRN-H]+ minus those of the individual [CRN-H]+ and CB7. The binding energy in the gas phase and in water is calculated to be −36.12 kcal mol−1 and −85.00 kcal mol−1, respectively (see ESI† for details), consistent with a previous report on similar host–guest complexes.44 As CRN is fully encapsulated by CB7, both of the CB7 portals should be available to bind to the surface of the Au NPs. Hence, CRN can be localised at the centre of the plasmonic hotspots, which is particularly important for the subsequent SERS sensing.
Colourimetric sensing of Au NP–CB7–CRN via UV-Vis spectroscopy
Quantitative detection of CRN was first investigated using UV-Vis spectroscopy via CB7-mediated aggregation of citrate-stabilised Au NPs. Clinically relevant CRN concentrations in urine (300–3000 μg mL−1, from normal to excess level)2 were prepared in this study. UV-Vis titrations of CRN were performed in water and in diluted SU (2000× and 5000×) with a constant concentration of 10 μM CB7 to ensure the formation of reproducible nanoaggregates (Fig. 2 and Fig. S3, S5, ESI†). It should be noted that 5 μM CB7 can also aggregate Au NPs but takes a longer time, while 20 μM CB7 results in faster aggregation but shorter measurement windows due to faster precipitation. The aggregation of Au NPs by CB7 was evidenced by the characteristic colour change of the solution from red to purple and the appearance of a new localised surface plasmon resonance (LSPR) peak between 690–760 nm in the UV-Vis spectrum. This peak arises from the longitudinal plasmonic resonance, which suggests the formation of chain-like Au nanostructures in the sample solution and the double-capping of CB7 by Au NPs.29,38 | ||
| Fig. 2 (a and b) UV-Vis spectra of CRN in (a) water (see Fig. S5 for all concentrations) and (b) SU after 2000× dilution with concentrations from 0 to 1.50 μg mL−1 (13.3 μM) with aggregation of Au NPs triggered by 10 μM CB7, with arrows indicating the shifting of the LSPR peaks. (c and d) Corresponding plots of λLSPR from (a and b) against CRN concentration, respectively. Logarithmic fittings were performed to reveal the correlation between λLSPR and CRN concentrations. (Note: 1 μg mL−1 in measurement samples corresponds to 2000 μg mL−1 of the actual CRN concentration in undiluted patient samples.) | ||
Notably, a high linearity of the LSPR peak wavelength (λLSPR) and CRN concentration was found in water (R2 ∼ 0.93), as shown in Fig. 2a and c, and in diluted SU (R2 ∼ 0.98), as shown in Fig. 2b and d, with a detection limit as low as 0.06 μg mL−1 (0.53 μM). The low detection limit allows CRN to be quantified in highly diluted SU samples, which could minimise the signal perturbation from the complex matrix (i.e. matrix effects, see latter subsection). The optimal dilution factor of SU samples was found to be 2000× (Fig. 2b and d), while reasonable correlation (R2 ∼ 0.81) can still be observed with a dilution factor up to 5000× (Fig. S3, ESI†). As a control experiment, the same titration experiments were repeated in the absence of CB7 (Fig. S6, ESI†). Interestingly, we found that CRN alone can also trigger aggregation of Au NPs in water to give red-shifted LSPR signals that correlate to the CRN concentration. Nevertheless, the correlation was found to be less robust and no aggregation can be observed for <0.30 μg mL−1 CRN in water. In contrast, CRN alone cannot aggregate Au NPs in SU, probably due to the preferential surface crowding by proteins and other biomolecules present in the matrix,25 thus illustrating the key role of CB7 in mediating aggregation of Au NPs in a controlled fashion.
While CB7 is known to mediate aggregation of Au NPs via portal binding,29,30,38 our results show that CB7–CRN complexes can further speed up the aggregation of Au NPs. The enhancement in aggregation speed can be attributed to the neutralisation of negative surface charges of the citrate-protected Au NPs upon attachment of the [CB7–CRN-H]+ complexes, which subsequently leads to a decrease in the long-range electrostatic repulsion between Au NPs, lowering the colloidal stability. This hypothesis can be verified by the decrease in the magnitude of zeta potential (i.e. less negative) and increase in hydrodynamic size of the nanoaggregates for the same concentration of CRN in the presence of CB7 (Fig. S4, ESI†). In the absence of CB7, [CRN-H]+ can also trigger aggregation of Au NPs via a similar electrostatic self-assembly mechanism, upon electrostatic interactions or chemisorption with the surface of Au NPs via its nitrogen containing groups, as supported by our UV-Vis, dynamic light scattering and zeta potential measurements.
While UV-Vis spectroscopy can potentially be used for CRN sensing in urine via CB-mediated aggregation of Au NPs and formation of host–guest complexes, there is no direct evidence to show whether the shift in λLSPR is caused by CRN or other molecules in the matrix; for instance, the λLSPR of 0.60 μg mL−1 CRN is at 738 nm in 2000× diluted SU but at 731 nm in 5000× diluted SU. Nevertheless, colourimetric sensing via UV-Vis spectroscopy can be used as a rapid test to complement and cross-validate the quantitative SERS sensing in the next section.
SERS sensing of CRN via CB-mediated Au nanoaggregates in water
Quantitative detection of CRN in water and in diluted SU was also investigated using SERS in a similar manner to the UV-Vis spectroscopy study. Unlike colourimetric sensing, SERS is highly specific as it can identify CRN via its vibrational fingerprints. The Raman spectrum of CRN powder is characterised by multiple peaks at 412, 577, 608, 674, 839, 904, 1246, 1416, 1478, 1657 and 1712 cm−1 (Fig. S7a, ESI†). The Raman spectrum of CB7 powder is characterised by two major peaks at 444 cm−1 and 833 cm−1, which are attributed to ring scissor and ring deformation modes, respectively (Fig. S7b, ESI†). It was challenging to measure the Raman signals of CB7, CRN and CB7–CRN in water, even at high concentration, because of the intrinsically low Raman scattering cross-sections.SERS signals were then measured by adding CB7, CRN and pre-mixed CB7–CRN solutions into a citrate-capped Au NP solution, respectively. The SERS peaks of CB7 and CRN solutions are broadened and slightly shifted in position when compared to the Raman spectra of CB7 and CRN powder (Fig. S8b, ESI†), owing to solution effect and molecular interaction of CB7 with Au NPs.45 The experimental SERS data show a very good match with the Raman spectra of CB7, CRN and CB7–CRN in water modelled at the CPCM/wB97XD/6-31G* level of theory (Fig. S8, ESI†). As the SERS signals of the complex are approximately equal to the sum of those of CB7 and CRN, we can deduce that the major tautomerism of CRN in aqueous solution stays the same upon binding to CB7.
SERS titration of CRN was first performed in water by adding different concentrations of CRN from 0 to 1.50 μg mL−1 (13.3 μM), as in the UV-Vis titration, at a constant CB7 concentration of 10 μM to ensure the reproducibility of the SERS nanosubstrates (Fig. 3a and b, see full spectra in Fig. S9, ESI†). CB7 defines the precise spacing between Au NPs by capping them via their electron-rich carbonyl portals, leading to the formation of strong and localised plasmonic hotspots. Although most of the CRN molecules are encapsulated within CB7, it is possible that a tiny amount of uncomplexed CRN could be trapped within the Au NP:CB7 nanoaggregates, when the concentration of CRN is relatively high (ratio of CRN:CB7 ≥ 1). The SERS signals of CRN can be observed clearly at 0.06 μg mL−1 (0.53 μM) with a good signal-to-noise ratio (SNR) of 12.8, which implies that the actual detection limit should be 4.69 ng mL−1 (41.5 nM). The SNR is calculated by the intensity of the major CRN peak at 1434 cm−1 against the background signal at 1800–2000 cm−1. Good linearity (R2 ∼ 0.78–0.91) between the SERS intensity and log concentration of CRN was obtained for multiple characteristic peaks at 721, 877, 947, 1435 and 1461 cm−1, respectively (Fig. 3c). The slight drop in linearity from 1.05 μg mL−1 (9.28 μM) could be due to the LSPR peak shifting away from 633 nm excitation.38 It should be noted that SERS signals of CRN could be observed in the absence of CB7 but a correlation between intensity and concentration of CRN is lacking (Fig. S10a, ESI†).
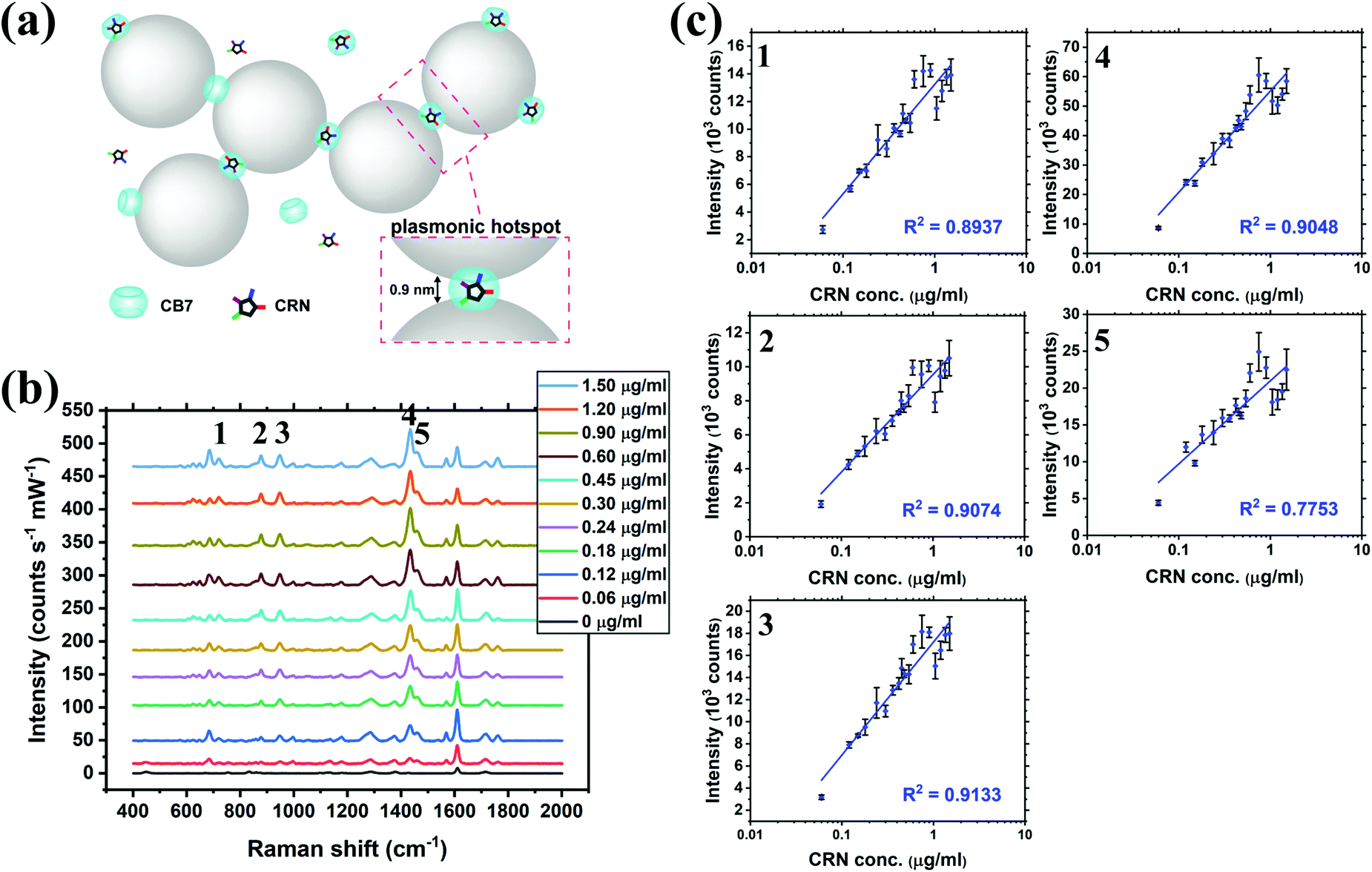 | ||
| Fig. 3 (a) Schematic illustration of the precise plasmonic hotspots within dynamic aggregates of Au NPs formed by 10 μM CB7 for CRN detection in water (not to scale). (b) SERS spectra of CRN in water with concentrations from 0 to 1.50 μg mL−1 (13.3 μM) with aggregation of Au NPs triggered by 10 μM CB7 (see Fig. S9 (ESI†) for all concentrations). Spectra were baseline corrected and offset for clarity. (c) Corresponding plots of SERS intensity of the five characteristic CRN peaks in (b) against CRN concentration. Logarithmic fittings were performed to reveal correlation between SERS intensity and CRN concentration. | ||
Quantitative SERS sensing of CRN in SU
The practical application of CRN detection in SU by SERS was investigated, in a similar manner to the UV-Vis spectroscopy study. The Raman signals in the prepared SU were attributed to urea and phosphate, while the CRN signals were too weak to be observed using normal Raman spectroscopy (Fig. S10c, ESI†). SU was diluted with a high dilution factor (1000×–5000×) for SERS measurements to minimise the matrix effect. It should be noted that only a very small amount of sample is needed for sensing. This has important implications for point-of-care monitoring of patients in critical conditions who often excrete a very small amount of urine. Since the sensing scheme is highly sensitive, dilution is needed to bring the concentration to a suitable range. Meanwhile dilution will also allow us to work on a larger sample volume in subsequent steps, therefore decreasing error in determining CRN concentration due to evaporation of water, as well as reducing loss of sample due to liquid transfer, together with other benefits such as minimising the matrix effect. No SERS signals of CRN can be observed in diluted SU in the absence of CB7 due to preferential surface crowding by other molecules as aforementioned (Fig. S10b, ESI†). Good general linearity was observed (R2 ∼ 0.81–0.94) in 1000× diluted SU but the error bars are relatively large for samples with high CRN concentrations (Fig. S11, ESI†). We found that the presence of excess [CRN-H]+ in the sample with a constant CB7 concentration of 10 μM caused a shift in the aggregation kinetics, resulting in faster aggregation and a narrower measurement window.SU was then diluted to 2000× to overcome the problem of a high CRN to CB7 ratio in the samples. Strong correlations between the SERS intensity and log concentration of CRN (R2 > 0.95) were found for all of the five characteristic CRN peaks with very small error bars (Fig. 4b and c), indicating the high reproducibility of the SERS signals. Herein, our high-performance SERS sensing platform based on Au NP:CB7 nanoaggregates is able to detect a particular analyte (i.e. CRN) in a complex matrix (i.e. SU) via the formation of host–guest complexes and is tolerant to protein fouling (Fig. 4a). The lowest CRN concentration measured in this study was 0.06 μg mL−1 (0.53 μM) in 5000× diluted SU with a decent SNR of 4.8, which indicates that the actual detection limit is 12.5 ng mL−1 (111 nM) (Fig. S12, ESI†). Notably, the sub-μM CRN concentration measured by our SERS system is lower than those of all the previous reports (Table S1, ESI†).
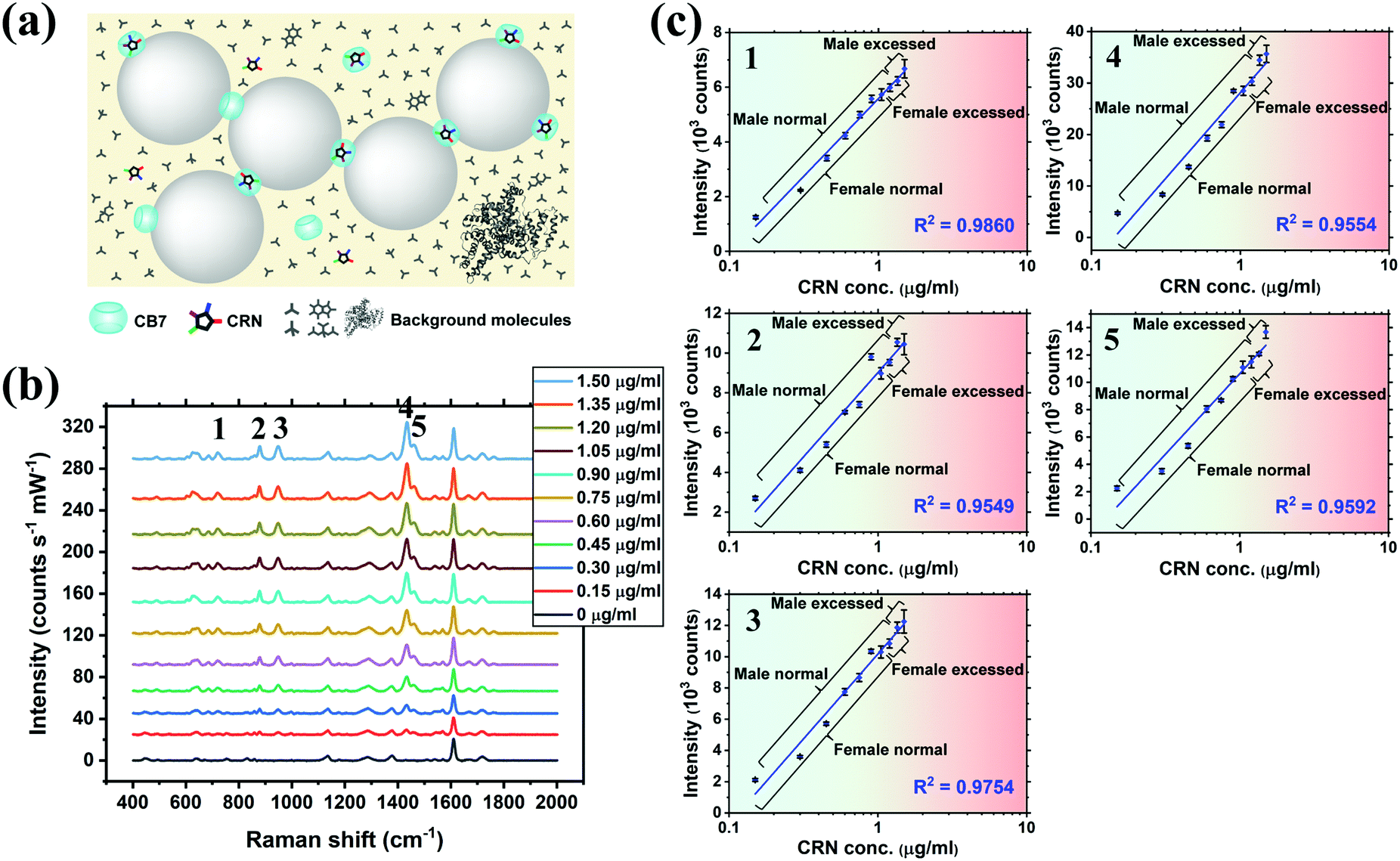 | ||
| Fig. 4 (a) Schematic illustration of the precise plasmonic hotspots within dynamic aggregates of Au NPs formed by 10 μM CB7 for CRN detection in SU after 2000× dilution with water (not to scale). (b) SERS spectra of CRN in SU with concentrations from 0 to 1.50 μg mL−1 (13.3 μM) with aggregation of Au NPs triggered by 10 μM CB7. Spectra were baseline corrected and offset for clarity. (c) Corresponding plots of SERS intensity of the five characteristic CRN peaks in (b) against CRN concentration. Logarithmic fittings were performed to reveal correlation between SERS intensity and CRN concentration. (Note: 1 μg mL−1 in measurement samples corresponds to 2000 μg mL−1 of the actual CRN concentration in undiluted patient samples.) | ||
Matrix effect on SERS signals of CRN
The SERS signals of CRN characteristic peaks in 2000× diluted SU are 34.0–52.6% lower than those in water at the same concentration (Fig. 5). This could be due to competitive binding between CRN and other biomolecules in the matrix to CB7 and Au NPs.40 For instance, uric acid can form a host–guest complex with CB7, while phosphate may bind to the Au NP surface. The matrix effect on the SERS signals of CRN can be reduced by increasing the dilution factor and thus recovering the CRN signals in water and increasing the SNR. Our SNRs are generally higher than the previously reported values.25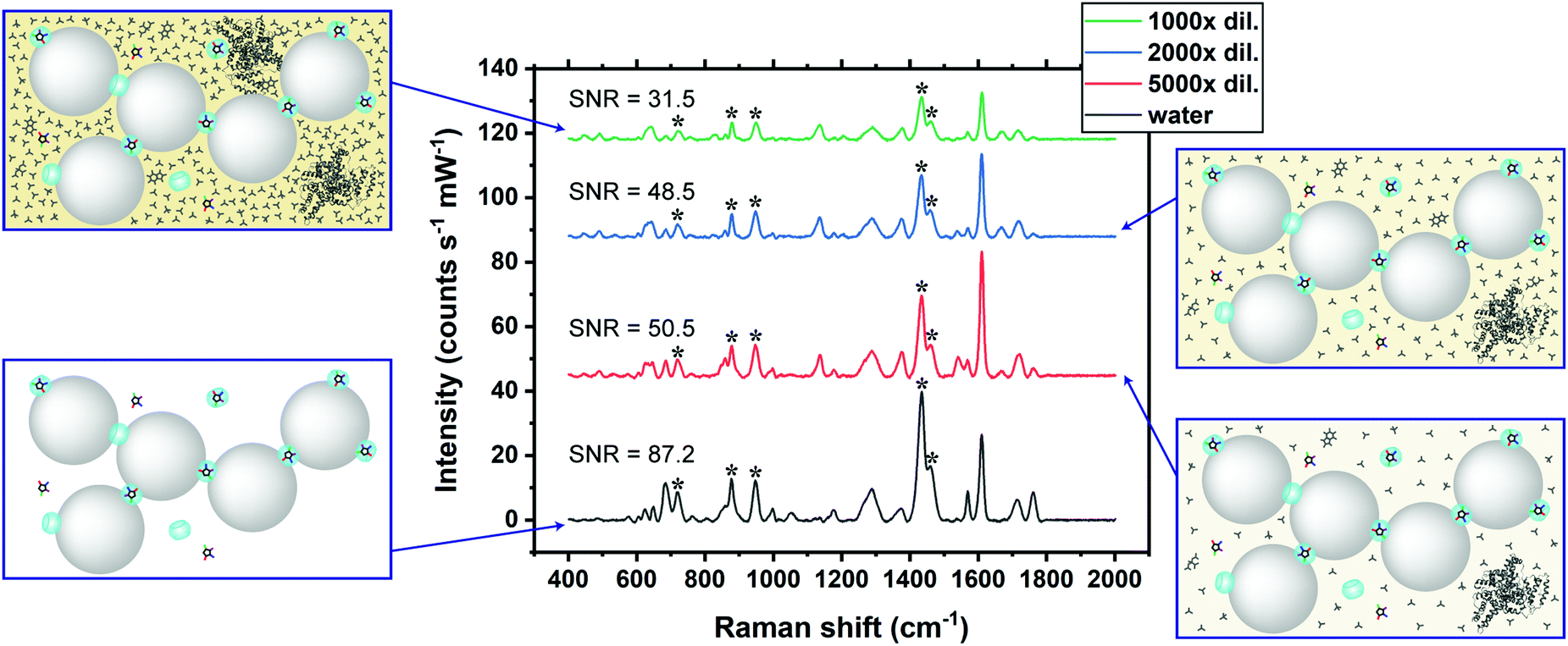 | ||
| Fig. 5 SERS spectra of 0.60 μg mL−1 CRN in water and SU after subsequent dilutions, showing the matrix effect on the SERS signals of CRN. The CRN signals (marked by asterisks) in water are gradually recovered and the SNR is higher as the concentration of SU is lowered. | ||
Conclusions
In this work, we report a novel multi-spectroscopic nanosensing platform based on SERS and UV-Vis spectroscopy for an important biomarker, creatinine (CRN), in urinalysis. Our approach achieved quantification of CRN of clinically relevant concentrations with a rapid response, high sensitivity (detection limit down to 111 nM) and reproducibility (within 5% error). The dual-triggered self-assembly of the plasmonic nanosubstrates allows CRN quantification via independent spectral signatures from a single sample solution, which enhances data consistency and measurement throughput. Specific identification of CRN can be achieved using the SERS signals, which are selectively enhanced via novel host–guest complexation between cucurbit[7]uril (CB7) and CRN. Meanwhile, the bright SERS hotspots from precise sub-nm plasmonic junctions mediated by the rigid CB7 molecular spacer allow CRN to be detected in highly diluted synthetic urine (SU), minimising matrix effects. The tolerance of our system against the presence of a large variety of molecules and proteins in SU makes it promising for point-of-care and clinical applications. Given the wide range of small-molecular biomarkers that exist in their protonated forms in physiological conditions, we expect this nanobiosensing approach to be readily extended for detection of other types of molecules in complex media for in-field applications.Conflicts of interest
There are no conflicts to declare.Acknowledgements
SM and TCL are grateful to the Research Project Grant (RPG-2016-393) funded by the Leverhulme Trust. WIKC, IPP, MO and TCL are grateful to the Studentship funded by the A*STAR-UCL Research Attachment Programme through the EPSRC M3S CDT (EP/L015862/1). WIKC, JK, DUS and MO thank SBIC LBOI lab for use of facilities and resources. WIKC thanks IMRE for use of facilities.Notes and references
- H. M. Heise, G. Voigt, P. Lampen, L. Küpper, S. Rudloff and G. Werner, Appl. Spectrosc., 2001, 5, 434 Search PubMed.
- C. J. Saatkamp, M. L. de Almeida, J. A. M. Bispo, A. L. Pinheiro, A. B. Fernandes and L. Silveria Jr, J. Biomed. Opt., 2016, 21, 37001 CrossRef PubMed.
- T. W. Bell, Z. Hou, Y. Luo, M. G. Drew, E. Chapoteau, B. P. Czech and A. Kumar, Science, 1995, 269, 671 CrossRef CAS PubMed.
- T.-L. Wang, H. K. Chiang, H.-H. Lu and F.-Y. Peng, Opt. Quantum Electron., 2005, 37, 1415 CrossRef CAS.
- J. W. McMurdy III and A. J. Berger, Appl. Spectrosc., 2003, 57, 522 Search PubMed.
- U. Lad, S. Khokhar and G. M. Kale, Anal. Chem., 2008, 80, 7910 CrossRef CAS PubMed.
- S.-L. Yan, P.-Z. Lin and M.-W. Hsiao, J. Chromatogr. Sci., 1999, 37, 45 CAS.
- W. R. Premasiri, R. H. Clarke and M. E. Womble, Lasers Surg. Med., 2001, 28, 330 CrossRef CAS PubMed.
- J.-C. Chen, A. S. Kumar, H.-H. Chung, S.-H. Chien, M.-C. Kuo and J.-M. Zen, Sens. Actuators, B, 2006, 115, 473 CrossRef CAS.
- M. Z. Jaffe, Z. Physiol. Chem., 1886, 10, 391 Search PubMed.
- O. Folin, J. Biol. Chem., 1914, 17, 469 Search PubMed.
- R. W. Bonsnes and H. H. Taussky, J. Biol. Chem., 1945, 158, 581 CAS.
- H. Du, R. Chen, J. Du, J. Fan and X. Peng, Ind. Eng. Chem. Res., 2016, 55, 12334 CrossRef CAS.
- D. Gangopadhyay, P. Sharma, R. Nandi, M. Das, S. Ghosh and R. K. Singh, RSC Adv., 2016, 6, 112562 RSC.
- J. A. Weber and A. P. van Zanten, Clin. Chem., 1991, 37, 695 CrossRef CAS.
- E. Liotta, R. Gottardo, L. Bonizzato, J. P. Pascali, A. Bertaso and F. Tagliaro, Clin. Chem. Acta., 2009, 409, 52 CrossRef CAS PubMed.
- Y. Zuo, C. Wang, J. Zhou, A. Sachdeva and V. C. Ruelos, Anal. Sci., 2008, 24, 1589 CrossRef CAS PubMed.
- X. Li, X. Fang, Z. Yu, G. Sheng, M. Wu, J. Fu and H. Chen, Anal. Chim. Acta, 2012, 748, 53 CrossRef CAS PubMed.
- A. Bonifacio, S. Cervo and V. Sergo, Anal. Bioanal. Chem., 2015, 407, 8265 CrossRef CAS PubMed.
- B. Yu, M. Ge, P. Li, Q. Xie and L. Yang, Talanta, 2019, 191, 1 CrossRef CAS PubMed.
- K. Kong, G. Kendall, N. Stone and I. Notingher, Adv. Drug Delivery Rev., 2015, 89, 121 CrossRef CAS PubMed.
- Y. Fang, N.-H. Seong and D. D. Dlott, Science, 2008, 321, 288 CrossRef PubMed.
- Y. Wang, J. Chen, Y. Wu, Y. Chen, J. Pan, J. Lei, Y. Chen, L. Sun, S. Feng and R. Chen, Proc. SPIE, 2011, 8329, 83290K CrossRef.
- H. Wang, N. Malvadkar, S. Koytek, J. Bylander, W. B. Reeves and M. C. Demirel, Biomed. Opt., 2010, 15, 027004 Search PubMed.
- M. Li, Y. Du, F. Zhao, J. Zeng, C. Mohan and W.-C. Shin, Biomed. Opt. Express, 2015, 6, 849 Search PubMed.
- H. Zhang, G. Li, S. Li, L. Xu, Y. Tian, A. Jiao and X. Liu, Appl. Surf. Sci., 2018, 457, 684 CrossRef CAS.
- Y. Lu, C. Wu, R. You, Y. Wu, H. Shen, L. Zhu and S. Feng, Biomed. Opt. Express, 2018, 9, 4988 CrossRef CAS PubMed.
- N. Chamuah, A. Saikia, A. M. Joseph and P. Nath, Sens. Actuators, B, 2019, 285, 108 CrossRef CAS.
- T.-C. Lee and O. A. Scherman, Chem. Commun., 2010, 46, 2438 RSC.
- T.-C. Lee and O. A. Scherman, Chem. – Eur. J., 2012, 18, 1628 CrossRef CAS PubMed.
- S. Kasera, F. Biedermann, J. J. Baumberg, O. A. Sherman and S. Mahajan, Nano Lett., 2012, 12, 5924 CrossRef CAS PubMed.
- R. W. Taylor, R. J. Coulston, F. Biedermann, S. Mahajan, J. J. Baumberg and O. A. Scherman, Nano Lett., 2013, 13, 5985 CrossRef CAS PubMed.
- S. Kasera, L. O. Herrmann, J. D. Barrio, J. J. Baumberg and O. A. Scherman, Sci. Rep., 2015, 4, 6785 CrossRef PubMed.
- W.-I. K. Chio, W. J. Peveler, K. I. Assaf, S. Moorthy, W. M. Nau, I. P. Parkin, M. Olivo and T.-C. Lee, J. Phys. Chem. C, 2019, 123, 15769 CrossRef CAS PubMed.
- B. de Nijs, C. Carnegie, I. Szabó, D.-B. Grys, R. Chikkaraddy, M. Kamp, S. J. Barrow, C. A. Readman, M.-E. Kleemann, O. A. Scherman, E. Rosta and J. J. Baumberg, ACS Sens., 2019, 4, 2988 CrossRef CAS PubMed.
- T. Itoh, Y. S. Yamamoto and T. Okamoto, Phys. Rev. B, 2019, 99, 235409 CrossRef CAS.
- W. M. Nau, M. Florea and K. I. Assaf, Isr. J. Chem., 2011, 51, 559 CrossRef CAS.
- R. W. Taylor, T.-C. Lee, O. A. Scherman, R. Esteban, J. Aizpurua, F. M. Huang, J. J. Baumberg and S. Mahajan, ACS Nano, 2011, 5, 3878 CrossRef CAS.
- J. Lagona, P. Mukhopadhyay, S. Chakrabarti and L. Issacs, Angew. Chem., Int. Ed., 2005, 44, 4844 CrossRef CAS PubMed.
- J. E. L. Villa and R. J. Poppi, Analyst, 2016, 141, 1966 RSC.
- Y. Valadbeigi, V. Ilbeigi and M. Tabrizchi, Comput. Theor. Chem., 2015, 1061, 27 CrossRef CAS.
- C. Eisner, R. Faulhaber-Walter, Y. Wang, A. Leelahavanichkul, P. S. T. Yuen, D. Mizel, R. A. Star, J. P. Briggs, M. Levine and J. Schnermann, Kidney Int., 2010, 77, 519 CrossRef CAS PubMed.
- F. Biedermann, V. D. Uzunova, O. A. Scherman, W. M. Nau and A. De Simone, J. Am. Chem. Soc., 2012, 134, 15318 CrossRef CAS PubMed.
- T.-C. Lee, E. Kalenius, A. I. Lazar, K. I. Assaf, N. Kuhnert, C. H. Grün, J. Jänis, O. A. Scherman and W. M. Nau, Nat. Chem., 2013, 5, 376 CrossRef CAS PubMed.
- S. Mahajan, T.-C. Lee, F. Biedermann, J. T. Hugall, J. J. Baumberg and O. A. Scherman, Phys. Chem. Chem. Phys., 2010, 12, 10429 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Detection limit, computational details, NMR, UV-Vis, DLS, zeta potential, Raman and SERS. See DOI: 10.1039/d0tc00931h |
| This journal is © The Royal Society of Chemistry 2020 |