
Electrochemical reduction of europium(iii) using tetra-n-octyl diglycolamide functionalized ordered mesoporous carbon microelectrodes†
 a
Nolan C.
Kovach,
a
Brian G.
Trewyn,
ab
Mark R.
Antonio
c
and
Jenifer C.
Shafer
*ad
a
Nolan C.
Kovach,
a
Brian G.
Trewyn,
ab
Mark R.
Antonio
c
and
Jenifer C.
Shafer
*ad
Abstract
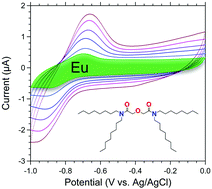
This work investigates the one-electron reduction of Eu(III) to Eu(II) with ordered mesoporous carbon (OMC) in cavity microelectrode (CME) systems. OMC materials with and without tetra-n-octyl diglycolamide (TODGA) functionalization were subjected to voltammetric measurements and compared with commercial carbon black Vulcan® XC-72. The electrochemical reduction of solution Eu(III) with unfunctionalized OMC, XC-72, and TODGA-functionalized OMC—both within the electrode matrix and on the electrode surface—is reported. The complexation of Eu(III) by TODGA-functionalized OMC prior to electrode preparation incorporates Eu(III) as part of the bulk electrode matrix. Under these conditions, the high capacitance obscures the Eu(III)/Eu(II) redox couple. A signal emerges above the background (capacitive) currents when 2-octanol is added to the TODGA-functionalized OMC as a wetting agent. In contrast, surface Eu(III)–TODGA complexation, when Eu(III) contacts the electrode surface exclusively after electrode preparation, provides a strong response. The addition of 2-octanol to TODGA reduces the capacitance of the electrode and narrows the Eu(III)/Eu(II) redox peak widths. The desorption by reductive stripping of Eu(II) was demonstrated using a 2-octanol modified TODGA OMC CME, opening the possibility for selective separation of Eu from adjacent trivalent lanthanides.
 Please wait while we load your content...
Please wait while we load your content...

