Bright and efficient red emitting electroluminescent devices fabricated from ternary europium complexes†
Received
13th February 2020
, Accepted 20th March 2020
First published on 23rd March 2020
Abstract
The photophysical properties of two previously synthesized, highly efficient, bright red emitting complexes [Eu(btfa)3DPEPO] (CIE, 0.672; 0.326) (Eu-1) and [Eu(nta)3DPEPO] (CIE, 0.673; 0.326) (Eu-2) (btfa = 4,4,4-trifluoro-1-phenyl-1,3-butanedione, nta = 4,4,4-trifluoro-1-(2-naphthyl)-1,3-butanedione and DPEPO = bis(2-(diphenylphosphino)phenyl)ether oxide) are reported. The complexes have been used as dopants to fabricate several single and double emitting layer (EML) electroluminescent (EL) devices. At the optimum doping concentration of 4 wt%, the single EML device of Eu-1 exhibited a pure red color with an external quantum efficiency (EQE) of 2.9% and a maximum brightness (B) = 1320 cd m−2, maximum current efficiency (ηc) = 4.08 cd A−1, maximum power efficiency (ηp) = 3.37 lm W−1 with a very low turn-on voltage (Vturn-on) = 3.8 V. Importantly, the single EML device of Eu-2 at the optimum doping concentration exhibited pure red EL with EQE of 6.0% with the best EL performance: B = 2108 cd m−2, ηc = 8.45 cd A−1, and ηp = 6.98 lm W−1 at a very low Vturn-on = 3.5 V. The performances of this device are among the best reported for devices incorporating a europium complex as a red emitter. Furthermore, the EL performances were achieved at the current density of 10 mA cm−2, thereby alleviating a serious efficiency roll-off as a major obstacle to the development of Eu-complex based EL devices.
1. Introduction
Efficient photoluminescent materials with desirable properties are becoming increasingly crucial for the development of organic light-emitting devices (OLEDs), luminescence thermometer (LMT), bioassay, etc.1 A plethora of reports on photoluminescent materials comprising either organic or metal complexes have appeared and many of the materials have fascinating applications. Among the materials in this category trivalent lanthanide [Ln(III)] complexes, especially Eu(III) coordination complexes (Eu-CCs) have the shown potential to be used in a range of applications such as OLEDs,2 sensors,3 sensitizers to improve the electroluminescence (EL) performance of red-emitting Ir(III) complex,1a,4 and laser materials.5 This is because of their intriguing photophysical properties, e.g., highly monochromatic red emission (full width at half maxima (FWHM) less than 10 nm, CIEx,y = 0.67, 0.32),5b,c,6 high photoluminescence quantum yield (PLQY),7 large energy shift between absorbed and emitted radiation, millisecond (ms) to microsecond (μs) long luminescence lifetimes. However, according to the Laporte rule8 for the optical absorption, the redistribution of electrons within a given subshell is forbidden. This limitation can be overcome by the introduction of appropriate organic chromophore(s) (referred to as “antenna” ligands) with high absorption coefficients (ε) in ultraviolet (UV) or visible (Vis) region of the spectrum to harvest the absorbed energy to the Ln(III) emitting state, typically known as the “antenna effect”.9
Among the many strongly absorbing chelating multidentate organic antenna ligands, acetylacetone (Hacac) and its derivatives, known as β-diketones (β-diket) have dominated lanthanide coordination chemistry.10 This is primarily because of their ease of synthesis, high ε values, and intense red or green emission, which is caused by the high energy transfer (ET) from the β-diket to the Eu(III) or Tb(III) ion emitting states. Most importantly, they form strongly coordinated neutral and stable lanthanide coordination complexes (Ln-CCs). However, water or alcohol molecule(s) always complement the unsaturated coordination sphere affecting the overall photophysical properties due to the vibronic quenching by high energy oscillators. This issue can be simply resolved by utilizing co-ligands. One of the co-ligands of choice is rigid “bis(2-(diphenylphosphino)phenyl)ether oxide (DPEPO)” because of its intriguing properties. It serves as a hard Lewis base forming very stable complexes with hard Lewis acid [Ln(III)] ions and as an excellent photosensitizer for the Ln(III) ions generating high PLQY (up to 80%) with 97% sensitization efficiency (ηsen).7a It provides a large bite angle that imposes a distortion to the complex symmetry, leading to additional enhancement of PLQY. Furthermore, the coordination of DPEPO increases the thermal- and photostability of the complexes, thus complexes formed would be ideal candidates for the fabrication of OLEDs via a thermal evaporation process. An additional advantage of DPEPO lies in its capacity to be used as a host material for the EML in OLEDs.2a,11 In continuation of our on-going research to improve the EL performance of the red-emitting Eu(III) complexes, herein, we report systematic and comparative photophysical analyses of two complexes [Eu(btfa)3DPEPO] (CIE, 0.672; 0.326) (Eu-1) and [Eu(nta)3DPEPO] (CIE, 0.673; 0.326) (Eu-2) (Chart 1) first reported by Hasegawa et al.12 Our experimental and theoretical studies include determination of Judd–Ofelt (J–O) intensity parameter (Ω2 and Ω4), radiative (AR) and non-radiative (ANR) rate constants, radiative lifetime (τrad), forward (WET) and backward (WBT) ET rates and intrinsic quantum yield (QEuEu). Since the pioneering works of Kido et al.,13 on the EL of lanthanide complexes, research on OLEDs is growing because of their potential use in the next-generation flat panel displays and solid-state lighting (SSL) applications. Xu et al.,14 used bidentate DPEPO ligand to synthesize a [Eu(tta)3DPEPO] complex (tta = 2-thenoyltrifluoroacetonate) and utilized it to fabricate red OLEDs. The fabricated device displayed impressive brightness (B) of 632 cd m−2 at 25 V with maximum EQE of 2.89% and ηc and ηp of 4.58 cd A−1 and 2.05 lm W−1 were achieved at 7 V. Later, the same group reported15 EL devices of modified monodentate phosphine oxide ligands and synthesized [Eu(tta)3(tapo)2], [Eu(tta)3(nadapo)2] and [Eu(tta)3(cppo)2] (tapo = 4-diphe-nylaminophenyl)diphenylphosphineoxide, (nadapo = naphthalen-1-yl-phe-nylaminophenyl)diphenylphosphineoxide and cppo = [4-((diphenyl-phosphinoyl)phenyl]-9H-carbazole). Inspiring EL performances were obtained from the devices using these complexes as an EML with B = 1195 cd m−2 at 19.6 V and EQE = 3.2%, for [Eu(tta)3(tapo)2], B = 1195 cd m−2 at 18.0 V and EQE = 3.71% for [Eu(tta)3(nadapo)2] and B = 852 cd m−2 at 22.2 V and EQE = 2.08%, respectively. However, these devices utilized very high operating voltages to achieve these EL performances and are therefore not suitable for real-life applications. The objective of producing low Vturn-on, Eu-CCs based red-emitting OLEDs is important and needs to be investigated further. Based on this idea, Zhao et al.16 utilized exciplex of 4,4′,4′′-tri(N-carbazolyl)-triphenylamine:4,7-diphenyl-1,10-phenanthroline (TCTA:Bphen) as a host to sensitize the [Eu(DBM)3Phen] complex to achieve a high purity red color OLED. At the optimum doping concentration of 10 wt%, the device exhibited ηc and EQE of 3.85 cd A−1 and 2.12% at Vturn-on = 2.9 V. Recently, we have reported a light red (CIEx,y, 0.574; 0.275) EL device of [Eu(tfac)3DPEPO],2a (tfac = trifluoroacetylacetonate) with an impressive B = 1274 cd m−2 at very low Vturn-on = 3.4 V. Motivated by these findings, we further employed the Eu-1 and Eu-2 complexes as EML to fabricate single- and double-layer OLEDs. We found that at the optimum doping concentration of 4 wt% Eu-2, single-layer devices outperformed many of the Eu-CCs and displayed the best EL performances, B, ηc, ηp, and EQE of 2108 cd m−2, 8.45 cd A−1, 6.98 lm W−1 and 6.0%, respectively, at very low Vturn-on = 3.4 V.
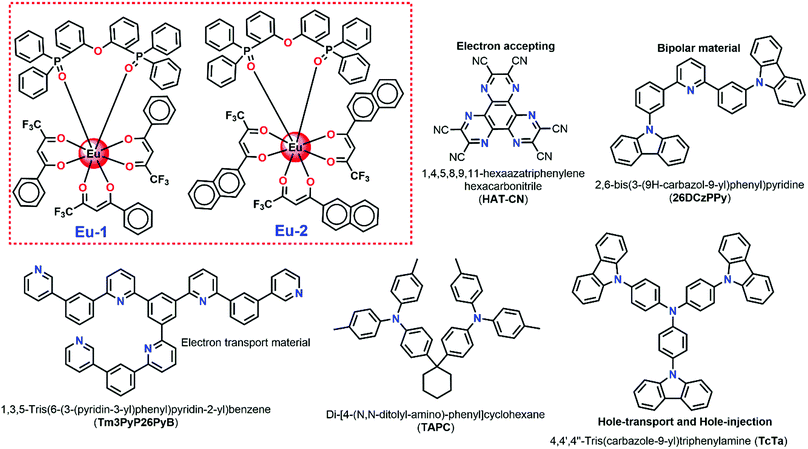 |
| | Chart 1 Structural representation of Eu complexes and chemicals used in EL device fabrication. | |
2. Photophysical parameters
Details of the syntheses and photophysical measurements of Eu-1 and Eu-2 (Chart 1) are given in the ESI.† Important experimental photophysical parameters such as J–O parameters (Ω2 and Ω4), radiative (AR) and non-radiative (ANR) decay rates, radiative lifetime (τrad) and intrinsic quantum yield (QEuEu) were calculated using following eqn (1)–(7) and details are reported elsewhere.1a,17| | 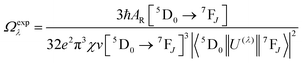 | (1) |
| | 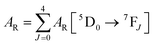 | (2) |
| | 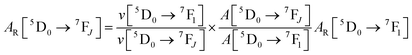 | (3) |
| | 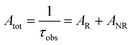 | (4) |
| | 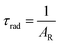 | (5) |
| | 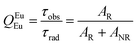 | (6) |
| | 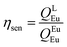 | (7) |
2.1. Details of theoretical calculations
2.1.1. Ground state geometry and singlet (S) and triplet (T) energy levels.
Structural modeling of Eu-1 and Eu-2 was performed using a reference crystal structure of an analogous europium complex (CSD CODE 1942486) reported by us.2a The structures were optimized without any symmetry restrictions using the RM1 model18 implemented in the MOPAC2016 program.19 A search in the Cambridge Crystallographic Database (CSD)20 for [Ln(β-diket)3DPEPO] complexes showed that β-diketonate ligands with different substituents adopt different relative positions with respect to the orientation of the same substituent. As a result, eight different starting structures were considered for Eu-1 and Eu-2. Fig. S1 and S2, ESI† show the geometries optimized with RM1 for all structures considered for both complexes. Vibrational frequency calculations using the RM1 model showed that all the optimized structures for both complexes were at a minimum of energy.
Additionally, density functional theory (DFT) calculations with the B3LYP/SVP level of theory, implemented into ORCA 4.2.1,21 were also performed by using as a starting structure with the lowest energy RM1 geometry for both complexes. The SVP basis function treats the electronic structure of hydrogen (H) atom as (4s1p)/[2s1p], carbon (C), oxygen (O) and fluorine (F) atoms as (7s4p1d)/[3s2p1d], and phosphorus (P) as (10s7p1d)/[4s3p1d]. The electronic structure of Eu was represented by the MWB52 effective core potential,22 which represents the core by 46 + 4f6, with the valence orbitals being treated as (7s6p5d)/[5s4p3d]. The advantage of using a small optimized basis set is that it reduces the computational cost compared to a calculation involving all electrons. Since Eu-1 and Eu-2 have 133 and 151 atoms, respectively, the use of a basis set containing even more components is prohibitive, so the cost-effective SVP is an appropriate basis set.23
Singlet and triplet states of the coordinated ligands were determined from the B3LYP/SVP/MWB52 geometries. Therefore, the Time Dependent-Density Functional Theory (TD-DFT) approach to the CAM-B3LYP/SVP/MWB52 level of theory implemented in ORCA4.2.1 was applied. The effect of the dichloromethane (DCM) solvent was implicitly considered in the excited state calculations using the Conductor-like Polarizable Continuum Model approximation (CPCM). In addition to the TD-DFT approach, INDO/S-CIS semiempirical model, in which the Ln(III) ion is replaced by a 3e+ point charge, has also been applied for comparative purposes. For the INDO configuration interaction single (CIS) excitation approach, 20 occupied molecular orbitals (MOs) and 20 virtual MOs were considered. The absorption spectra were fitted to a Lorentzian function with a half-width at half maximum (HWHM) of 15 nm for all calculations. Details of the theoretical J–O intensity parameters (Ωλ, λ = 2, 4 and 6) and eqn (S1)–(S9) are included in ESI.†
2.1.2. Energy transfer (ET) rates.
According to Malta's model,24 if the direct coulombic interaction is only taken into account, the ligand–metal ET rate is calculated by:| | 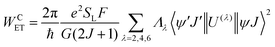 | (8) |
| | 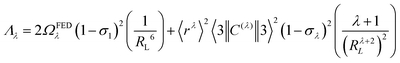 | (9) |
The reduced matrix element 〈Ψ′J′‖U(![[small script l]](https://www.rsc.org/images/entities/char_e146.gif) )‖ΨJ〉2 provides the selection rule for total angular momentum J and is given by |ΔJ| ≤ 6 with |ΔJ| = 2, 4, 6 (J = J′ = 0 excluded). As a result, this mechanism is operative to promote the electronic excitation, for instance, involving the states 5D4 ← 7F0, 5G6 ← 7F0, and 5L6 ← 7F0 for the Eu(III) ion. The values of 〈α′J′‖U()‖αJ〉2 is taken from the work of Carnall et al.,25 and are implemented into LUMPAC26 and considered for calculation of ET rates for Eu-1 and Eu-2. The ref. 27 describes all quantities of the eqn (8) and (9) in detail.
)‖ΨJ〉2 provides the selection rule for total angular momentum J and is given by |ΔJ| ≤ 6 with |ΔJ| = 2, 4, 6 (J = J′ = 0 excluded). As a result, this mechanism is operative to promote the electronic excitation, for instance, involving the states 5D4 ← 7F0, 5G6 ← 7F0, and 5L6 ← 7F0 for the Eu(III) ion. The values of 〈α′J′‖U()‖αJ〉2 is taken from the work of Carnall et al.,25 and are implemented into LUMPAC26 and considered for calculation of ET rates for Eu-1 and Eu-2. The ref. 27 describes all quantities of the eqn (8) and (9) in detail.
The contribution regarding the exchange (Ex) mechanism for the ligand–metal ET rate is given by:
| | 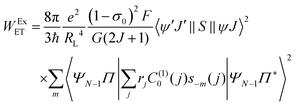 | (10) |
From the matrix elements 〈
ψ′
J′‖
S‖
ψJ〉
2 the selection rule is derived for the Ex mechanism (ExM), which is given by |Δ
J| = 0, ±1 (
J =
J′ = 0 excluded) and Δ
S = 0 for the Ln(
III) ion, the latter can be relaxed by the spin–orbit coupling. In this way, the ExM is operative for the excitations
5D
1 ←
7F
0 and
5D
0 ←
7F
1 for the Eu(
III) ion. For more details of the terms in the
eqn (10)ref. 27 should be consulted.
The calculation of the factor F present in eqn (8) and (10) can be approximated by the following expression:
| | 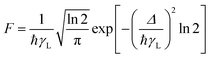 | (11) |
The expression assumes that ligand bandwidth at half-height,
γL (in s
−1), is much larger than the widths of the 4f–4f transitions
γLn of Ln(
III) ions.
Δ is the energy difference between the donor (excited singlet and triplet states of the organic part of the complex in question and acceptor excited states of the lanthanide ion) involved in the ET process.
The term (1 − σ0) in eqn (10), recently revisited27 by the group led by Malta is given by eqn (12). Utilizing eqn (12) the new version of the equation for calculating ET rate via ExM is formed and implemented in LUMPAC software26
| | 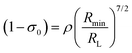 | (12) |
| | 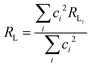 | (13) |
where
ρ is the overlap integral (
ca. 0.05) between the valence shells of the Ln(
III) ion and the ligating atom and
Rmin is the smallest distance in the first coordination sphere.
RL is the distance from the Ln(
III) ion nucleus to the electronic barycenter of the ligand donor (or acceptor) state,
ci is the molecular orbital coefficient of atom-
i at the ligand donor (or acceptor) state and
RLi is the distance of atom-
i to the Ln(
III) ion.
Back ET rate is obtained simply by multiplying the direct ET rate by the Boltzmann factor 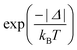 , where T is the environment temperature and kB stands for the Boltzmann constant. The theoretical quantum yields of the complexes were also determined and the details are presented in the ESI† (eqn (S10) and (S11)).
, where T is the environment temperature and kB stands for the Boltzmann constant. The theoretical quantum yields of the complexes were also determined and the details are presented in the ESI† (eqn (S10) and (S11)).
2.2. Fabrication of EL devices
The molecular structure of the materials used in the fabrication processes, together with the complexes Eu-1 and Eu-2 are shown in Chart 1. All the organic materials used in the fabrication processes were obtained commercially and used as received without further purification. The device fabrication details are given in the ESI.†
3. Results and discussion
3.1. Synthesis and ground state geometry of the europium complexes
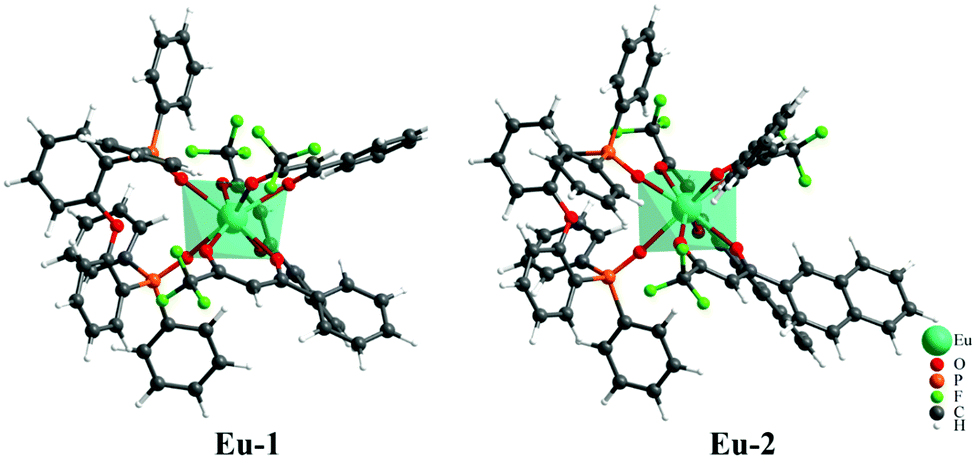
Mixed ligand europium complexes Eu-1 and Eu-2 were synthesized as previously reported12 and were characterized by the elemental analysis, mass spectrometry and FTIR spectroscopy (Fig. S3–S6, ESI†) which are in agreement with the proposed chemical structures and stoichiometry, as shown in Chart 1. Repeated attempts to obtain single crystals of the complexes failed to produce suitable single crystals for X-ray analysis. As a matter of fact, for predicting the photophysical properties of the complex, one needs to have the theoretical structure of the complex, even though the experimental X-ray structure is available. In view of this, the structure of both Eu-1 and Eu-2 were first optimized at the semi-empirical and DFT levels. The root-mean-square-deviation (RMSD) between the geometries optimized by RM1 and B3LYP/SVP/MWB52 are 0.9882 Å and 0.5340 Å for the Eu-1 and Eu-2 complexes, respectively (Fig. S7, ESI†). Although the different methods describe the ligand orientation slightly differently for both complexes, the first coordination sphere remains in good agreement for both the methods. Fig. 1 shows the geometry of Eu-1 and Eu-2 optimized with B3LYP/SVP/MWB52. The information on the polyhedral point groups for the complexes was obtained from a brute force symmetry analyzer implemented into C++ code (https://github.com/nquesada/symmetry). The coordination polyhedron of the B3LYP/SVP/MWB52 model for Eu-1 and Eu-2 (Table S1, ESI†) showed a low symmetry C1 point group. Even by flexing specific parameters of the code, the C1 symmetry remains unaltered. The coordination geometry around the metal approximates most closely to a trigonal dodecahedron for both the complexes. The three β-diketonate ligands coordinate to the Eu ion with Eu–O bond length ranging from 2.4187 to 2.4830 Å and from 2.4152 to 2.5030 Å, for Eu-1 and Eu-2, respectively. The chelating DPEPO ligand coordinates to Eu ion with the Eu–O bond lengths of 2.3937 and 2.4169 Å and 2.3872 and 2.4321 Å, respectively, for the Eu-1 and Eu-2 complexes.
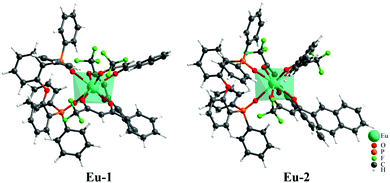 |
| | Fig. 1 Ground state geometry for complexes Eu-1 and Eu-2 calculated by the B3LYP/SVP/MWB52 level of theory. | |
The Eu–O(DPEPO) bond distances found in the literature for related eight coordinate Eu(III) phosphine oxide complexes are in the range of 2.345–2.446 Å and are in agreement with those observed in the literature.28 The Eu–O bond distances are longer in Eu-2 compared to Eu-1. This could be attributed to the bulkier (napthayl vs. phenyl) nature of the β-diketone in the former, forcing the ligating O atoms to adopt longer distances to reduce the steric crowding around the metal center.
3.2. Experimental and theoretical determination of photophysical properties
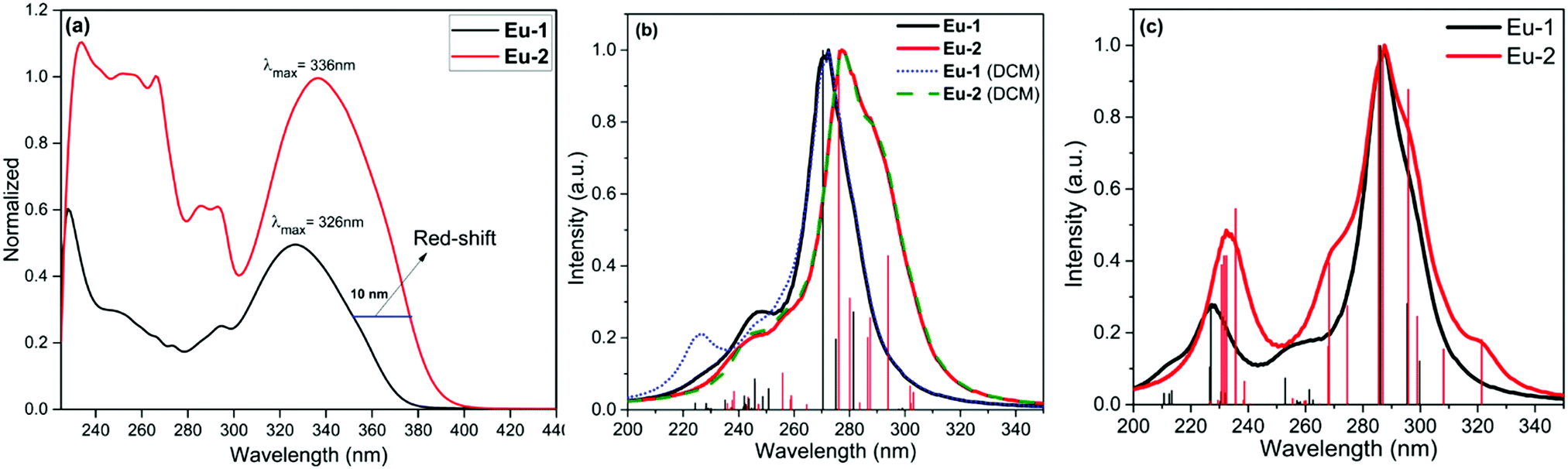
The optical absorption spectra of the complexes were measured in DCM solution, as shown in Fig. 2a. The uncoordinated DPEPO displays two major absorption peaks at 228 nm (ε = 48![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 506 M−1 cm−1) and 290 nm (ε = 8863 M−1 cm−1).2a The absorption spectra of the complexes exhibit combined absorption of the primary β-diketone and ancillary DPEPO ligand in DCM solution and the band shapes are similar with λmax = 326 nm (30674 cm−1) and 336 nm (29761 cm−1) for Eu-1 and Eu-2, respectively. The absorption band-edge of the π–π* transition of Eu-1 is at around 382 nm (26187 cm−1) while this is at 404 nm (24752 cm−1) for Eu-2 showing a red shift of 22 nm. The redshift could be attributed to the enhanced conjugation when a terminal phenyl is substituted with the naphthyl group in the case of Eu-2, thus resulting in less photobleaching. Fig. 2b and c show the absorption spectra calculated using both the TD-DFT, CAM-B3LYP/SVP/MWB52 approach and the INDO/S-CIS semiempirical model from the B3LYP/SVP/MWB52 geometries. While the TD-DFT spectra effectively reproduced the redshift (Fig. 2b), this was not the case in the spectra obtained with the INDO/S-CIS model (Fig. 2c). By considering the effect of the DCM solvent implicitly in TD-DFT calculations, this effect does not cause a shift of the more intense absorption.
506 M−1 cm−1) and 290 nm (ε = 8863 M−1 cm−1).2a The absorption spectra of the complexes exhibit combined absorption of the primary β-diketone and ancillary DPEPO ligand in DCM solution and the band shapes are similar with λmax = 326 nm (30674 cm−1) and 336 nm (29761 cm−1) for Eu-1 and Eu-2, respectively. The absorption band-edge of the π–π* transition of Eu-1 is at around 382 nm (26187 cm−1) while this is at 404 nm (24752 cm−1) for Eu-2 showing a red shift of 22 nm. The redshift could be attributed to the enhanced conjugation when a terminal phenyl is substituted with the naphthyl group in the case of Eu-2, thus resulting in less photobleaching. Fig. 2b and c show the absorption spectra calculated using both the TD-DFT, CAM-B3LYP/SVP/MWB52 approach and the INDO/S-CIS semiempirical model from the B3LYP/SVP/MWB52 geometries. While the TD-DFT spectra effectively reproduced the redshift (Fig. 2b), this was not the case in the spectra obtained with the INDO/S-CIS model (Fig. 2c). By considering the effect of the DCM solvent implicitly in TD-DFT calculations, this effect does not cause a shift of the more intense absorption.
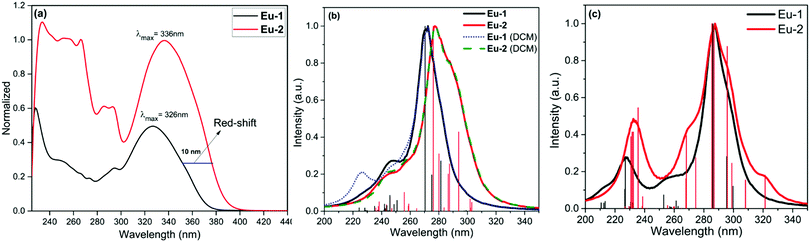 |
| | Fig. 2 (a) Electronic spectra of Eu-1 and Eu-2 in DCM at RT. Estimated electronic spectra by using different levels of theory for the complexes under investigation (b) CAM-B3LYP/SVP/MWB52//B3LYP/SVP/MWB52 with and without the effect of the DCM solvent, and (c) INDO/S-CIS//B3LYP/SVP/MWB52. | |
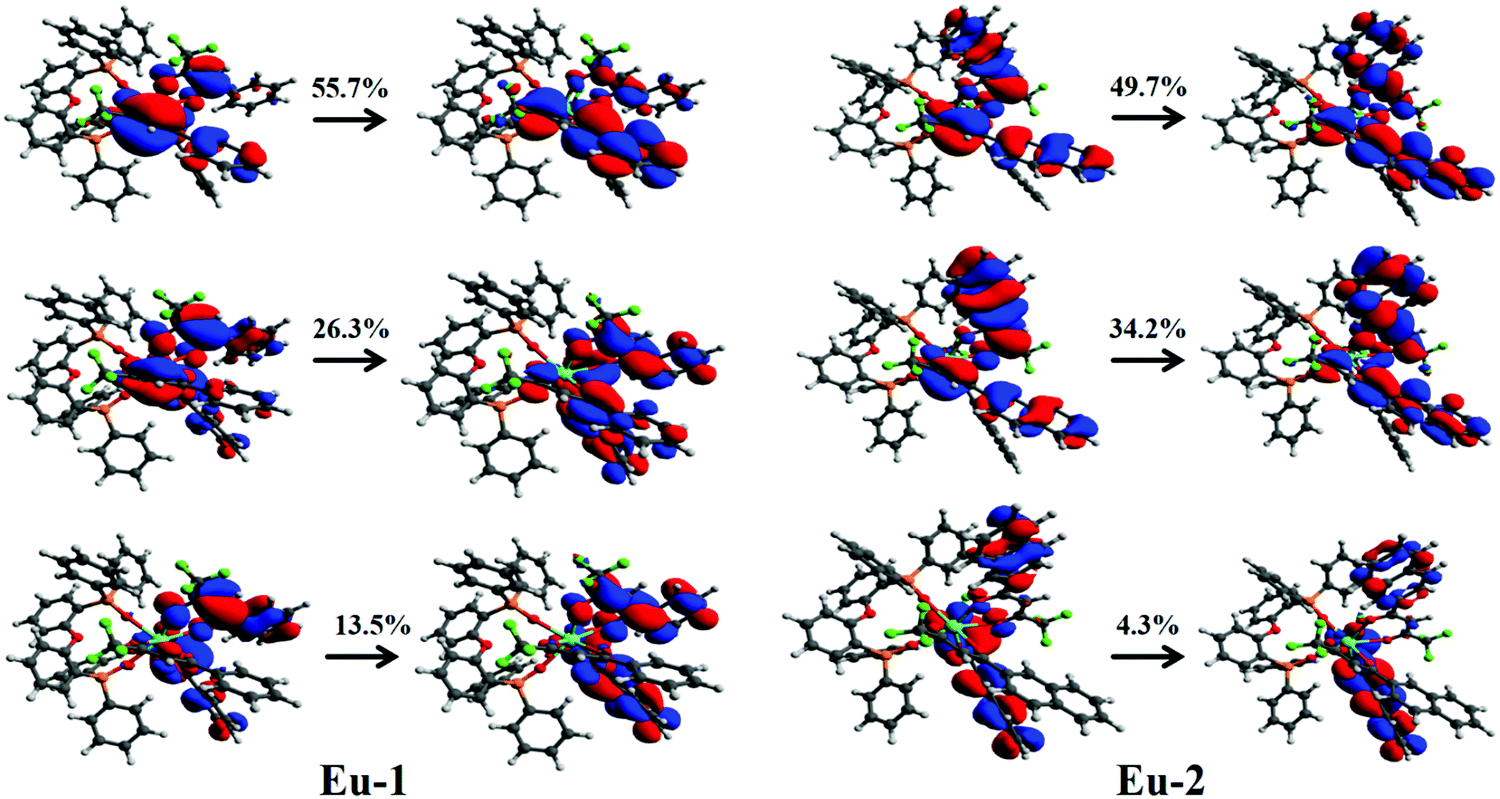
Table S2, ESI,† shows the highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO) energy levels and ΔE(HOMO–LUMO), respectively for the Eu-1 and Eu-2 calculated by different levels of theory. While the RM1 energies were calculated for the optimized geometry at the same computational level, the CAM-B3LYP energies were calculated by considering the optimized geometry at the B3LYP/SVP/MWB52 level of theory. At the same level of theory, the Eu-2 complex has a higher ΔE(HOMO–LUMO) than the one calculated for Eu-1. As suggested by Nüesch et al.,29 the hole and electron trapping ability of dopants depend on the HOMO and LUMO energy levels of dopant and host molecules in the EL devices. Thus, these results indicate that both the complexes could be good candidates for trapping the charge in the EL devices. In the case of Eu-2, for instance, the singlet excited state with the higher oscillator strength relies on the contribution of 18 electronic transitions, making it difficult to determine the qualitative nature of the excited state. In order to elucidate the nature of the orbitals involved in the UV electronic absorption transitions regarding the singlet excited state of interest (higher oscillator strength) for Eu-1 and Eu-2, calculations of the natural transition orbitals (NTOs) were employed (Fig. 3). The NTO analysis offers a simple representation of the transition density between the ground and the excited state. It is possible to observe from Fig. 3 that the most important electronic transitions are π → π* centered on the different β-diketone ligands. These transitions involve MOs energetically close to the HOMO and LUMO frontier MOs for both complexes. It is important to emphasize that the MO center on the DPEPO does not contribute to the most intense absorption band (Fig. 3). However, the DPEPO plays an important role in completing the coordination sphere of the Ln(III) ion and thus preventing the coordination of solvent molecule(s), which may enable the luminescence suppression via non-radiative decay. Simultaneously, imposing a highly asymmetric coordination geometry such as trigonal dodecahedral in our case is an important factor for enhancing the PLQY with large radiative rate constants.30
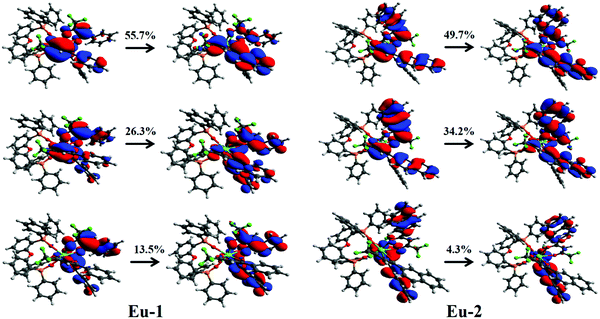 |
| | Fig. 3 Pictures of the NTOs mainly contributing to the singlet excited state with higher oscillator strength for Eu-1 (around 270.4 nm) and Eu-2 (around 276.1 nm). For each transition, the contribution percentages of the main NTOs are also included. | |
The PL properties of both the Eu-1 and Eu-2 were determined in DCM solution at RT (Fig. 4). In order to evaluate the detailed photophysical properties of the complexes and to maximize the steady-state emission, we first obtained the excitation spectra (Fig. S8, ESI†) for both the complexes by monitoring the 5D0 → 7F2 emission transition. The excitation spectra of both complexes in DCM solution are similar and displayed a broadband in the region between 350 nm and 440 nm with a very faint intra-configurational f–f transition at 7F0 → 5D2 (21505 cm−1; 465 nm), suggesting that the antenna mechanism as noted in similar [Eu(tfac)3DPEPO] and other related ternary β-diketonate complexes is functioning here.2a,5c,17 Moreover, the excitation spectra of Eu-2 exhibited a bathochromic shift, as noted in the electronic spectra of the complexes at RT and could be caused by the same effect.
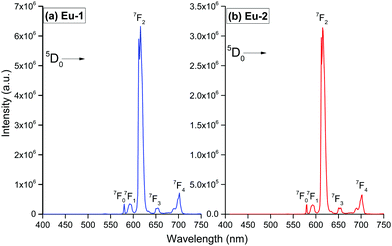 |
| | Fig. 4 Emission spectra of the Eu(III) complexes in DCM solution at RT (a) λmaxEx = 380 nm and (b) λmaxEx = 393 nm. | |
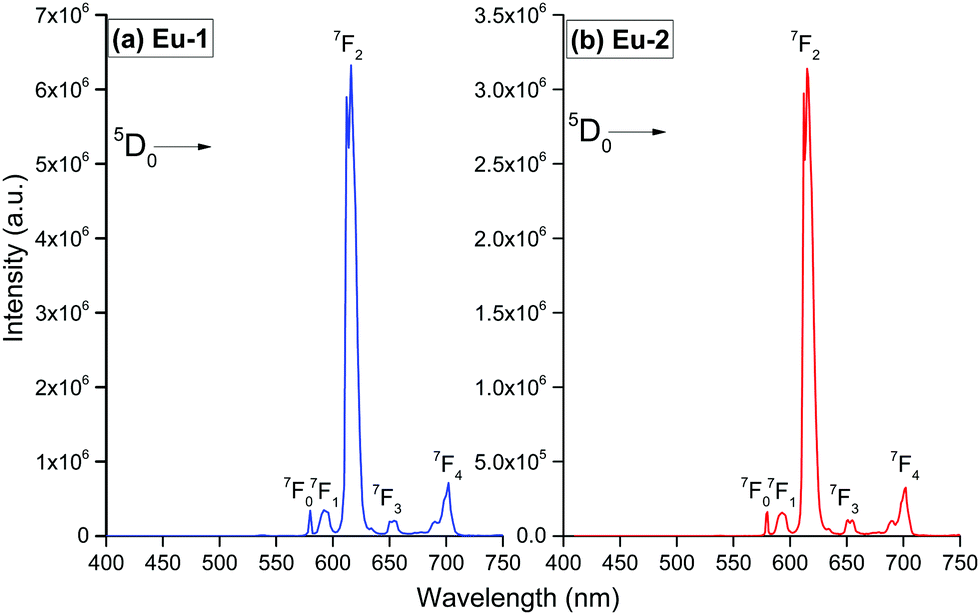
The PL spectra shown in Fig. 4 for the complexes were obtained by exciting them at λExmax mentioned in the figure caption. The spectra displayed essentially five well-resolved emission transitions emanating from the 5D0 state to 7FJ (J = 0, 1, 2, 3 and 4). The data obtained for the parameters such as intensity, barycenter, and % contribution of each transition is gathered in Table S3, ESI.† A well-resolved and faint (% contribution of 0.23–0.24%) emission transition 5D0 → 7F0 with FWHM of 1.17 nm and 1.55 nm (Table 1) for Eu-1 and Eu-2, respectively, can be seen in Fig. 4, which suggests a single chemical environment around the Eu(III) ion1a,2a,5c,17 in both complexes. The fourth emission transition 5D0 → 7F3 follows the same mechanism as the 5D0 → 7F0 transition, i.e., is not allowed by either mechanism, the magnetic-dipole (MD) and electric-dipole (ED) and contribute only 2.21–2.49% to the total integral emission output. The second MD transition 5D0 → 7F1 and its intensity is independent of the Eu(III) coordination sphere; however, it varies with the change in the refractive index of the medium.5c,31 Thus it is used as a standard for the dipole strength and allows (will be discussed later) the determination of the natural radiative lifetime (τrad) for a given Eu-CC. The third transition 5D0 → 7F2 in the series, ED is “hypersensitive” in nature, and so its intensity will change with the subtle change in the environment around the Eu(III) ion. The PL spectra are dominated by this narrow emission transition with FWHM of (10.31 nm)Eu-1 and (9.66 nm)Eu-2 (Table 1) of the total integral of the intensity (82.77%)Eu-1 and (83.44%)Eu-2 (Table S3, ESI†) suggesting highly monochromatic bright red emission with CIE color coordinates (0.672; 0.326)Eu-1 and (0.673; 0.326)Eu-2 (Table 1, Fig. S9 and S10, ESI†). The calculated CIE color coordinates obtained from the PL spectra are very similar to the National Television System Committee (NTSC: x – 0.67; y – 0.33), suggesting that these Eu-CCs could be a potential candidates as red components for the fabrication of EL devices. The higher total integral intensity of ED 5D0 → 7F2 (% contribution of 82–84%) transition compared to MD 5D0 → 7F1 transition indicates that forced ED and the dynamic coupling (DC) mechanism are dominant over MD. Furthermore, the PL spectra of both the complexes exhibit 5D1 → 7F1 transition (Fig. S11 and S12, ESI†). The PL spectrum of Eu(III) ion is quite informative because of the presence of MD 5D0 → 7F1 transition and a simple analysis and calculations can give plenty of information together with experimentally observed lifetime (τobs). For example, the intensity ratio (R21) is a measure of asymmetric environment around Eu(III) ion and a value higher than 0.7 could be attributed to the non-centrosymmetric and low symmetric environment. The high value of R21 = (18.89)Eu-1 and (20.42)Eu-2, respectively (Table 1) suggests that the complexes are non-centrosymmetric with C1 point group as determined by the ground state geometry of the complexes.
Table 1 Experimental and theoretical photophysical parameters of Eu-1 and Eu-2 at RT in DCM
|
|
Ω
2
|
Ω
4
|
FWHM (nm) |
τ
obs
|
τ
rad
|
A
R
|
A
NR
|
Q
EuEu
|
Q
LEu
|
R
21
|
η
sen
|
CIE(x,y) |
| ×10−20 cm2 |
(μs) |
(s−1) |
(%) |
|
Calculated using eqn (1).
Calculated using eqn (5).
Calculated using eqn (2)–(4).
Calculated using eqn (2)–(4).
Calculated using eqn (6).
|
|
Eu-1
|
33.29a |
8.18a |
1.17; 10.31 |
875.36 ± 8.06 |
923b |
989.03c |
153.30d |
86.58 |
80.1 |
18.89 |
92.51 |
0.672; 0.326 |
| Theoretical |
33.28 |
8.16 |
— |
— |
|
999.41 |
142.93 |
87.5 |
86.6 |
— |
98.98 |
— |
|
Eu-2
|
35.74a |
8.53a |
1.55; 9.66 |
779.15 ± 2.86 |
901b |
1056.16c |
227.20d |
82.30 |
79.2 |
20.42 |
96.23 |
0.673; 0.326 |
| Theoretical |
35.74 |
8.52 |
— |
— |
|
1066.89 |
216.48 |
83.13 |
82.3 |
— |
99.00 |
— |
3.3. J–O parameters, excited-state lifetime, and rate constants
In order to get more information regarding the photophysical properties, the PL lifetime (τobs) of the 5D0 excited state was determined in solution by exciting it at λmaxEx and fitting the decay curves (Fig. S13 and S14, ESI†) for Eu-1 and Eu-2 (Table 1). The fitting of the decay curves is in good agreement with monoexponential behavior (χ2 = 1.00–1.01) suggesting the single dominant emitting species and attests the results of steady-state measurement where only one line is observed for the 5D0 → 7F0 emission transition. The τobs value of the present complexes display τobs = 875.36 ± 8.06 and 779.15 ± 2.86 μs for Eu-1 and Eu-2 (Table 1), respectively, typical for ternary europium complexes of β-diketone. As mentioned above, we further utilized the steady-state emission spectra and τobs to get the information of radiative (AR) and non-radiative (ANR) decay constants with the help of eqn (2)–(5). The data obtained are gathered in Table 1. It is very clear that the Eu-2 complex displayed a marginally larger value of the ANR ≈ 227.20 s−1 compared to Eu-1, ANR ≈ 153.30 s−1 which could be due to the presence of the higher number of C–H oscillators in the case of Eu-2. The PLQY of the complexes in solution exhibited very large values of 80.1 and 79.2% and sensitization (ηsen) efficiency of 92.51% and 96.23% for Eu-1 and Eu-2, respectively (Table 1). Finally, Ω2 and Ω4 were calculated (Table 1), the high values of Ω2 = 33.29–35.74 × 10−20 cm2 suggest that the Eu(III) ion in both the complexes is surrounded by a highly polarizable environment.1a The parameter Ω4 is related to the rigidity and long-range effects such as hydrogen bonding, π–π stacking and substantially large value (Ω4 ≈ 8.18 × 10−20 cm2)Eu-1 and (Ω4 ≈ 8.53 × 10−20 cm2)Eu-2 is an indication of the presence of these effects.5c Zero Differential Overlap (ZDO) electronic densities (q) and electrophilic superdelocalizabilities (SE) calculated with RM1, by considering the B3LYP/SVP/MWB52 geometry of each atom directly coordinated to the Eu(III) ion, in Eu-1 and Eu-2, together with the corresponding charge factors g and polarizabilities α from the fitting (Table S4, ESI†). Moreover, through a series of tests using crystallographically-related structures, it has been shown that the ratio between adjustable D and C parameters has a value greater than unity where the experimental intensity parameters were appropriately adjusted.32 Furthermore, the forced ED contribution to the intensity parameters (ΩFEDλ) was calculated using the Q, D, and C values, and ΩFEDλ are important for the calculation of the ET rates related to the MM mechanism (eqn (9)).
3.4. Intermolecular energy transfer (IET) process
For an efficient sensitization of Ln(III) ion emissions in organo-lanthanide complexes, an appropriate energy difference between the triplet state of organic ligands and the emitting levels of Ln(III) ions must exist, which is >2500 cm−1 for Eu(III) ion.1a,2a,33 This criterion is satisfied by both complexes. The energy difference corresponds to the Δ parameter in the eqn (11), and this equation contains the dependence of the ET rate on the triplet and singlet energy of the coordinated ligands. Table 2 shows the lowest triplet energy for both complexes as well as the electron transition configurations mainly contributing to the state. Although structurally different, the Eu-1 and Eu-2 have only a marginal difference in energy for the lowest triplet state.
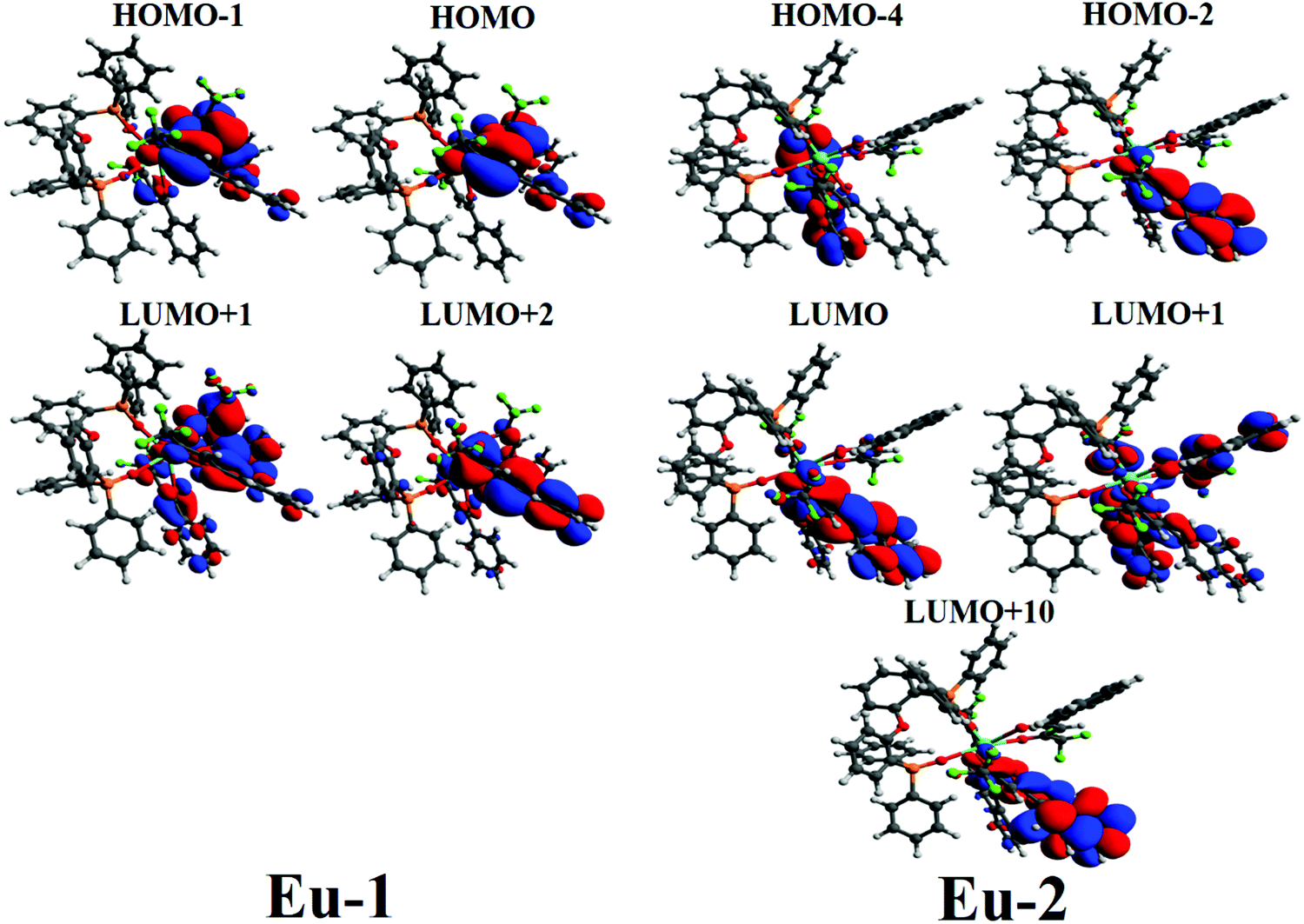
Table 2 Electron transition configurations for the lowest triplet state (T1) of Eu-1 and Eu-2
|
|
Major contribution |
|
Major contribution |
|
Eu-1 22413.7 cm−1 |
HOMO−1 → LUMO+2 (16.6%) |
Eu-2 22280.4 cm−1 |
HOMO−4 → LUMO (14.9%) |
| HOMO → LUMO+1 (7.2%) |
HOMO−4 → LUMO+10 (7.0%) |
| HOMO → LUMO+2 (54.6%) |
HOMO−2 → LUMO (46.2%) |
|
|
HOMO−2 → LUMO+1 (5.2%) |
Fig. 5 shows the greater involvement of the MOs localized on the β-diketonate ligands and thus clearly indicates that the β-diketonate ligands are of great importance for the lowest triplet state in both complexes. Additionally, it also indicates that the PO-ligand does not play a crucial role in the luminescence sensitization. This observation is further supported by the TD-DFT work of Congiu et al.,34 on the [Eu(hfaa)3DPEPO] complex (hfaa = hexafluoroacetylacetone) where they reported similar behavior i.e., the excited singlet–triplet is located on the β-diketonate ligand.
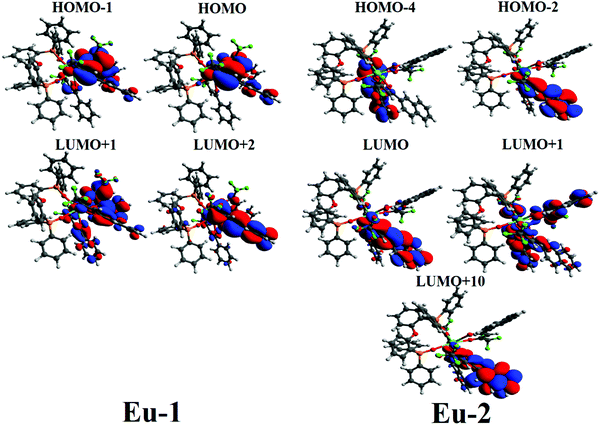 |
| | Fig. 5 CAM-B3LYP/SVP/MWB52 molecular orbitals mainly contributing to the lowest triplet state of Eu-1 and Eu-2. | |
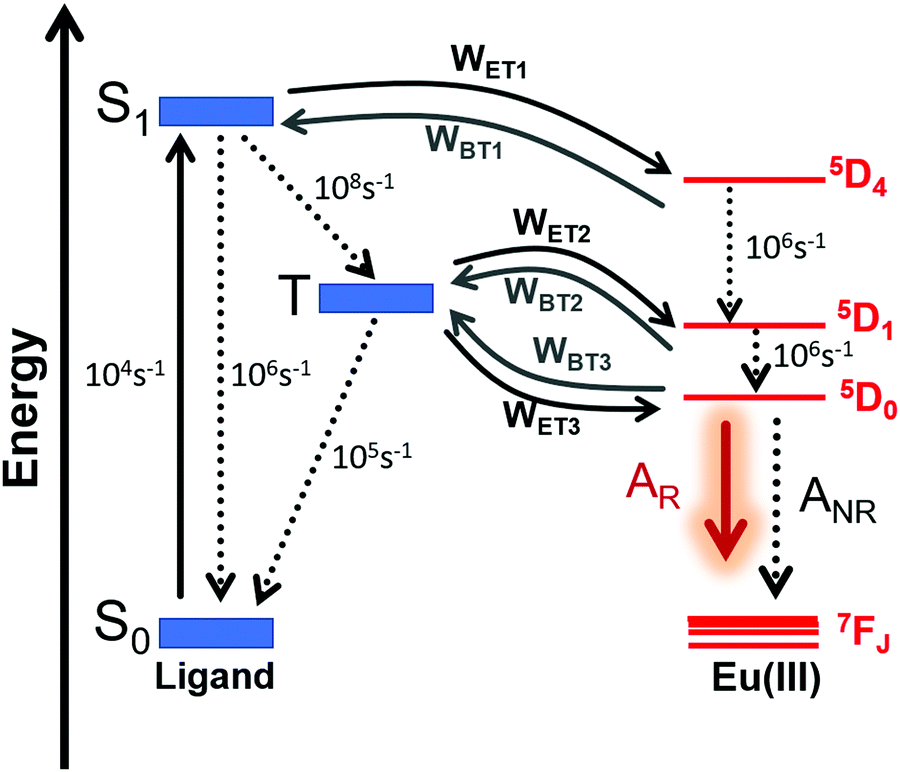
The ET rates were calculated using the values obtained employing the LUMPAC software package26 and are summarized in Table 3. Eu-1 was chosen as a representative for the discussion. The main accepting levels for the Eu(III) ion 5D4 (∼27586 cm−1), 5D1 (∼19027 cm−1), and 5D0 (∼17293 cm−1) are well above the singlet and the triplet state (Table 3). The selection rules state that ET from singlet to 5D4 (5D4 ← 7F0) excitation in Eu(III) ion occurs through a multipolar mechanism (MM) and the lower value suggests that MM mechanism is not the dominant ET process (Table 3). The ET involving the channels, triplet → 5D1 and triplet → 5D0, are related to the 5D1 ← 7F0 and 5D0 ← 7F1 excitations respectively, considering that the thermally populated 7F1 state proceeds through an ExM (Dexter mechanism).2b A representative Jablonski energy diagram representing the most probable ET channels is displayed in Fig. 6. The higher values of WET rate of T1 → 5D1 ≈ 1.2 × 1010 s−1 and T1 → 5D0 ≈ 8.5 × 109 s−1 suggest that the direct ligand-to-metal ET is dominated by the ExM. Interestingly, Eu-2 showed marginally smaller value of WET rate of T1 → 5D1 ≈ 1.9 × 108 s−1 and T1 → 5D0 ≈ 1.4 × 108 s−1 compared to Eu-1. A plausible explanation for this observation could be attributed to the longer RL value (ca. 5 Å) for Eu-2, which is due to the extended conjugation of the substituent (naphthyl) on Eu-2. Thus, the electronic density involved in the ligand–metal ET is further away from the energy acceptor center in Eu-2 compared to Eu-1 (Fig. 5). Furthermore, since the enthalpy of formation difference (ΔHF) among some of the different geometries calculated by RM1 model are very similar, it is worthwhile to evaluating the effect of the geometry on the excited state energies and their corresponding RL distances. We applied the INDO/S-CIS model and the obtain data are summarized in Table S5, ESI,† it shows small variation for the different geometries and therefore the main results regarding the ET process remain unaltered in the complexes. Moreover, the solvent effect provides a very modest difference in the most relevant quantities for the ET process (RL,sing, Esing, RL,trip, and Etrip) in comparison with the same quantities without solvent (Table S6, ESI†). Because the ligand triplet state is well above the 5D0 and 5D1 levels of the Eu(III) ion, the direct ET is favored and a small energy difference is not enough for significantly changing the ET.
Table 3 ET rates calculated by the Malta's model for the Eu-1 and Eu-2. The ligand state energies considered were those calculated with the CAM-B3LYP/SVP/MWB52//B3LYP/SVP/MWB52 method
| Complex |
Ligand state (cm−1) |
|
4f state (cm−1) |
ET rate (WET) (s−1) |
Energy back-transfer (WBT) rate (s−1) |
Quantum yield (%) |
|
Eu-1
|
Singlet (36977.2) |
→ |
5D4 (27586) |
W
ET1 = 9.1 × 103 |
W
BT1 = 0 |
86.6 |
| Triplet (22413.7) |
→ |
5D1 (19027) |
W
ET2 = 1.2 × 1010 |
W
BT2 = 1.0 × 103 |
| Triplet (22413.7) |
→ |
5D0 (17293) |
W
ET3 = 8.5 × 109 |
W
BT3 = 1.8 × 10−1 |
|
|
|
Eu-2
|
Singlet (36220.4) |
→ |
5D4 (27586) |
W
ET1 = 1.9 × 102 |
W
BT1 = 0 |
82.3 |
| Triplet (22280.4) |
→ |
5D1 (19027) |
W
ET2 = 1.9 × 108 |
W
BT2 = 3.2 × 10 |
| Triplet (22280.4) |
→ |
5D0 (17293) |
W
ET3 = 1.4 × 108 |
W
BT3 = 5.8 × 10−3 |
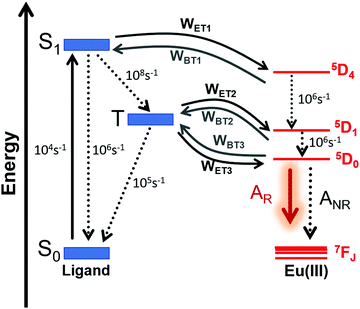 |
| | Fig. 6 Schematic energy-level diagram, ET processes, and transfer rates considered for both complexes. The rates shown are typical experimental values for the non-radiative rates in coordination compounds35 and are implemented into LUMPAC.26 Please see Table 1 for AR and ANR, and Table 3 for WET and WBT values for both complexes. | |
The theoretical PLQY for Eu-1 and Eu-2 are shown in Table 3. Theoretical PLQY depends on the ET rates, AR and ANR decay rates, in addition to the other non-radiative rates, whose typical experimental values are presented in Fig. 6. The normalized populations of the ligand and metal ion states considered in the PL process are calculated by using all rates and, in particular, the most important populations are those associated with states S0 and 5D0 (eqn (S10), ESI†). One reason of the direct transfer rate for the two complexes to be greatly favored, such as indicated by the theoretical ET modeling, the sensitization efficiency for each complex is large considering either experimental or theoretical value (Table 1). In this way, the most relevant ET channels were considered in the theoretical ET modeling.
3.5. EL performance of Eu-1 and Eu-2
The impressive photophysical properties of the two europium complexes such as narrow ED 5D0 → 7F2 (FWHM = 9.66–10.31 nm) emission transition with the total integral contribution (82.00–83.44%) and color coordinates (CIE; 0.673; 0.326) suggest that the complexes could be utilized as efficient emitting materials in EL devices. To characterize the EL properties, Eu-1 and Eu-2 were used as emitters to fabricate single- and double EML devices with the following general device configuration (please see the ESI† for further details).
Single-EML device.
ITO/HAT-CN (6 nm)/HAT-CN (0.2 wt%): TAPC (50 nm)/Eu-1 or Eu-2 (x wt%): 26DCzPPy (10 nm)/Tm3PyP26PyB (60 nm)/LiF (1 nm)/Al (100 nm).
Double-EML device.
ITO/HAT-CN (6 nm)/HAT-CN (0.2 wt%): TAPC (50 nm)/Eu-1 or Eu-2 (x wt%): TcTa (10 nm)/Eu-1 or Eu-2 (x wt%): 26DCzPPy (10 nm)/Tm3PyP26PyB (60 nm)/LiF (1 nm)/Al (100 nm).
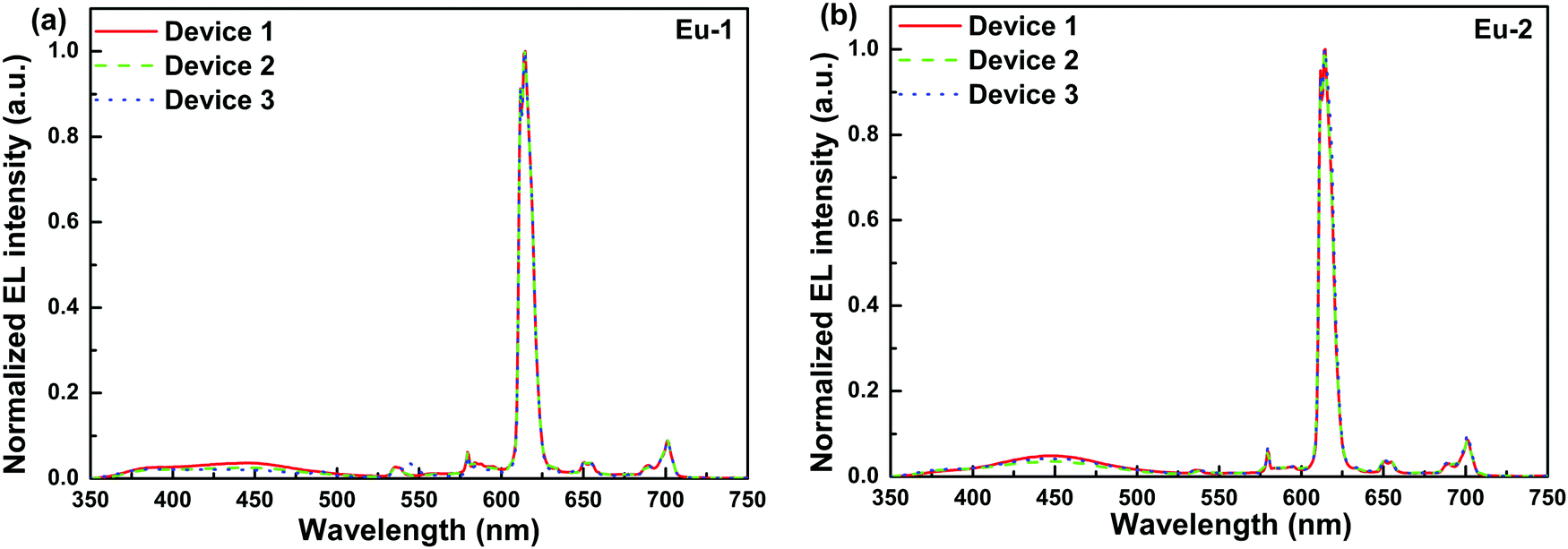
EL devices based on Eu-1 and Eu-2 were fabricated by thermal evaporation under vacuum, which was facilitated by high thermal stability of the complexes (318–320 °C).12 The doping concentration for europium complexes was modulated to be 3.0 wt%, 4.0 wt% and 5.0 wt%, respectively, As the doping concentration increases, the evaporation temperature increases gradually from 140 to 151 °C and 147 to 159 °C for Eu-1 and Eu-2, respectively. The low thermal evaporation temperature of the complexes thus ensures negligible decomposition of the complexes during the thermal evaporation process. The normalized EL spectra of Eu-1 and Eu-2 based single and double EML devices are shown in Fig. 7 and Fig. S15 and S16, ESI.†
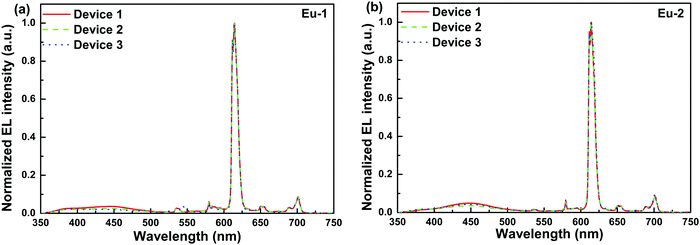 |
| | Fig. 7 Normalized EL spectra of the single-EML devices 1, 2 and 3 of (a) Eu-1 and (b) Eu-2 at 10 mA cm−2. | |
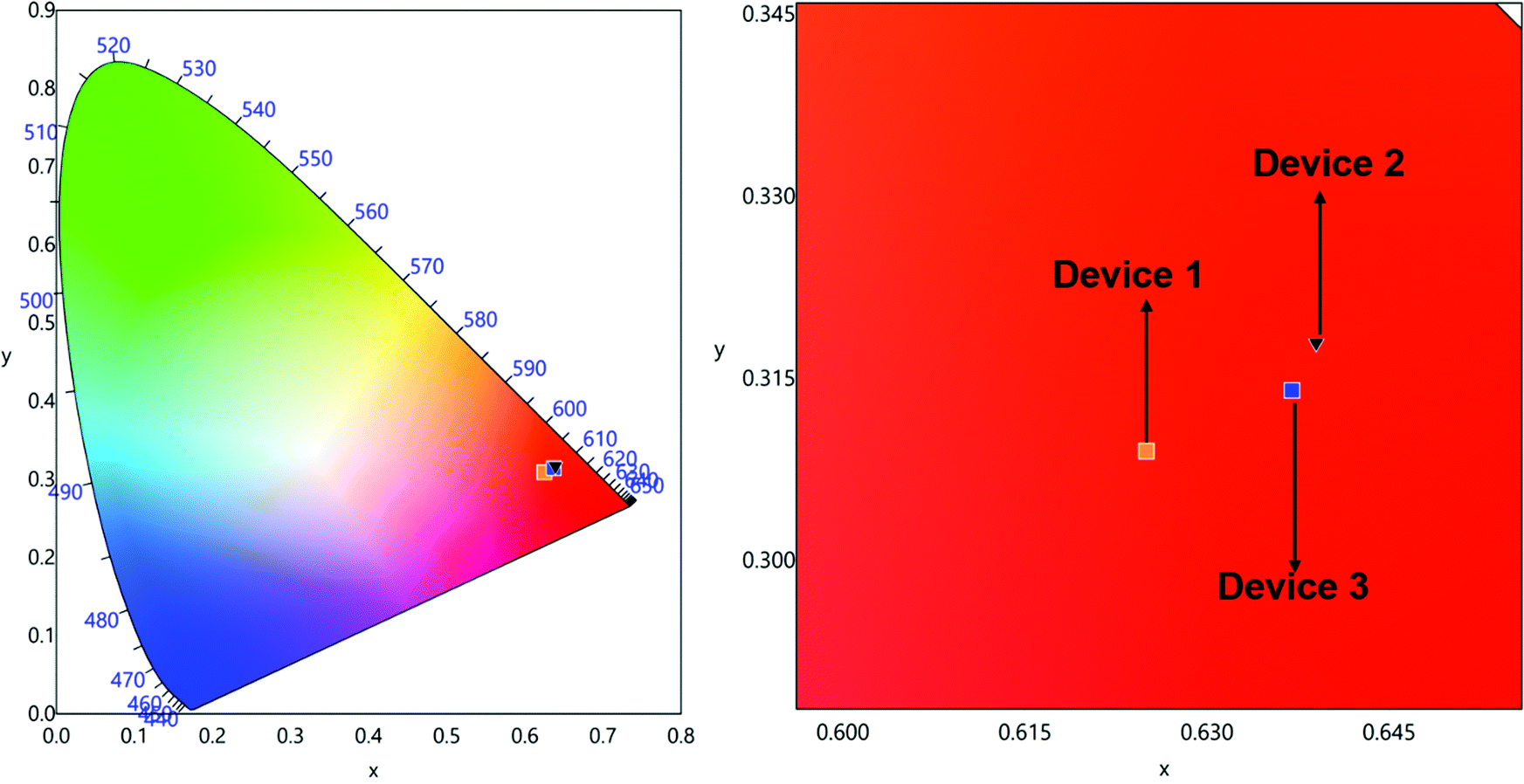
The spectra displayed extremely sharp EL similar to the PL spectra for both the complexes and is dominated by ED transition at 615 nm characteristic for the Eu(III) ion as observed in the steady-state emission analysis. Moreover, they displayed minor high-lying emissions in the range between 400 and 500 nm of host emission, which decreases as the doping concentration increases. The devices emitted pure red emission in spite of the presence of the very faint host emission with (CIE)x,y color coordinates of (x = 0.622, y = 0.306)–(x = 0.648, y = 0.319) (Table 4, Fig. 8 and Fig. S17–S19, ESI†). In the device luminescence process for the present complexes, we speculate that both carriers’ trapping and Förster ET may co-exist simultaneously36 since the EL spectra (Fig. 7) in the range of 500–750 nm resembles well the PL spectra (Fig. 4) except the presence of a faint broadband in the range between 350 and 450 nm due to the incomplete ET from the host material.
Table 4 Key properties of single EML EL device 1, 2 and 3 for Eu-1 and Eu-2
| Device |
V
turn-on (V) |
B
(cd m−2) |
η
c
(cd A−1) |
η
p
(lm W−1) |
EQEd (%) |
CIEx,ye |
| The values in square brackets are EL performances of the double layer devices 4, 5 and 6 of Eu-1 and Eu-2 complexes. The data for maximum brightness (B). Maximum current efficiency (ηc). Maximum power efficiency (ηp). Maximum external quantum efficiency (EQE). CIEx,y at 10 mA cm−2. |
|
Eu-1
|
| 1 |
3.4 [3.7] |
1402 [1213] |
4.20 [3.98] |
3.47 [3.13] |
3.0 [2.8] |
0.625, 0.309 [0.636, 0.310] |
| 2 |
3.8 [3.9] |
1320 [1300] |
4.08 [3.66] |
3.37 [2.82] |
2.9 [2.6] |
0.637, 0.314 [0.640, 0.311] |
| 3 |
4.0 [4.2] |
1237 [1185] |
3.09 [3.58] |
2.33 [2.56] |
2.2 [2.5] |
0.639, 0.318 [0.635, 0.315] |
|
|
|
Eu-2
|
| 1 |
3.3 [3.5] |
1707 (1852) |
6.00 [7.30] |
5.39 [6.03] |
4.3 [5.2] |
0.615, 0.302 [0.624, 0.310] |
| 2 |
3.5 [3.5] |
2108 (1890) |
8.45 [8.92] |
6.98 [7.18] |
6.0 [6.3] |
0.628, 0.308 [0.639, 0.314] |
| 3 |
3.4 [3.5] |
1633 (1739) |
7.03 [7.33] |
6.02 [6.06] |
5.0 [5.2] |
0.622, 0.306 [0.648, 0.319] |
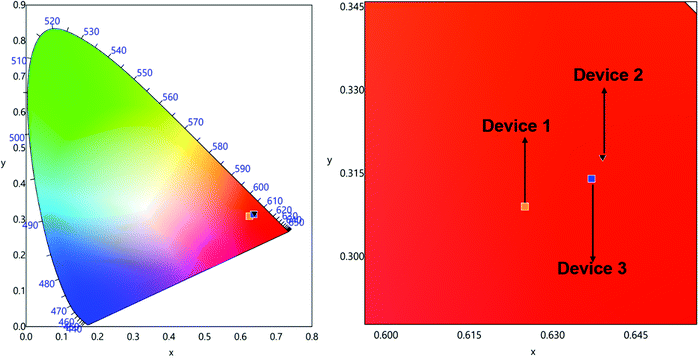 |
| | Fig. 8 CIE 1931 chromaticity diagrams of single EML devices 1, 2 and 3 of Eu-1 with a magnified view at 10 mA cm−2. | |
The EL efficiency and current density curves together with the voltage (V)–brightness (B) and current density curves as an inset are shown in Fig. 9. The detailed EL performance parameters such as B, ηc, ηp and EQE of single- and double-EML devices are gathered in Table 4. For Eu-1, at the optimum doping concentration of 4.0 wt%, the single EML displayed, B = 1320 cd m−2, ηc = 4.08 cd A−1, ηp = 3.37 lm W−1 with an impressive EQE = 2.9% at very low Vturn-on = 3.8 V with (CIE)x,y = 0.637, 0.314 in the pure red region. The double-EML devices at the same doping concentration do not exhibit any significant change except for a marginal change in the brightness. For Eu-2 at the similar optimum concentration, the EL performance of the single EML device showed outstanding results with B = 2108 cd m−2, ηc = 8.45 cd A−1, ηp = 6.98 lm W−1 with an impressive EQE = 6.0% at very low Vturn-on = 3.5 V with (CIE)x,y = 0.628, 0.308. As suggested by Xu et al.,37 the Vturn-on is relative to the carrier injection and transport ability of each layer. The low Vturn-on and high EL efficiency of Eu-2 could be caused by the barrier-free carrier injection, balanced carrier transport and recombination coupled with simultaneous high ET efficiency from the exciplex host to Eu-2. Interestingly the device based on Eu-2 outperformed many of the ternary europium complexes (Chart 2) reported14,15,37,38 in the literature as can be seen in a comparative Table 5. In fact, this is the best performance to the best of our knowledge.
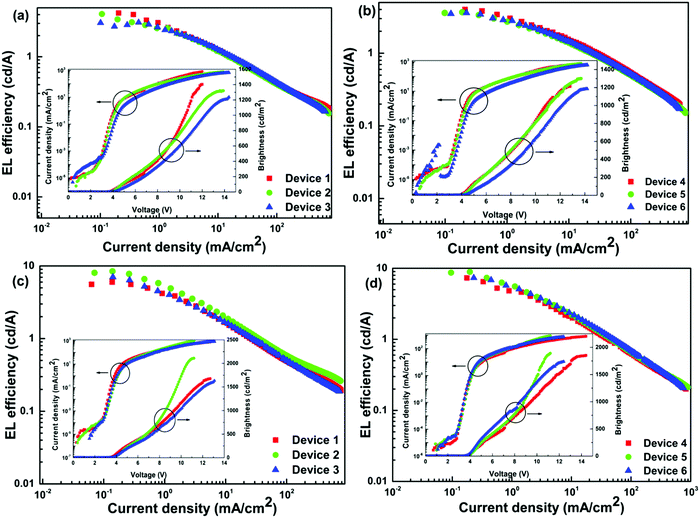 |
| | Fig. 9 EL efficiency–current density characteristics of devices. Inset: Current density–brightness–voltage characteristics of their respective devices. (a and b) Single and double EML devices of Eu-1 while (c and d) are single and double EML devices of Eu-2 operating at 10 mA cm−2. | |
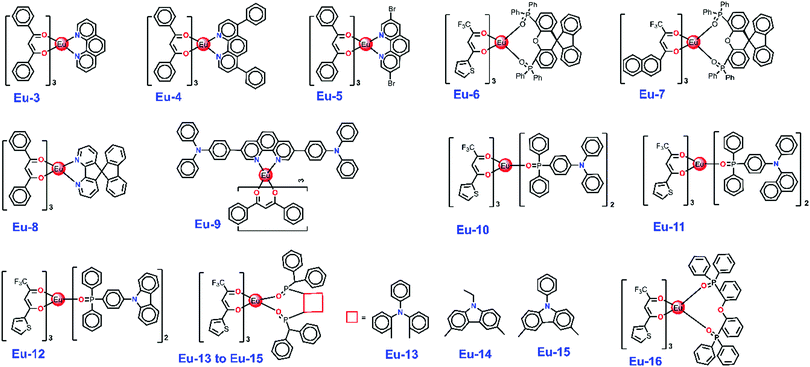 |
| | Chart 2 Chemical structure of the reported efficient ternary Eu-CCs. | |
Table 5 Comparative EL performances of reported Eu-CCs (their chemical structures are shown in Chart 2)
| Emitting layer |
V
turn-on (V) |
B
(cd m−2) |
η
c
(cd A−1) |
EQEd (%) |
η
p
(lm W−1) |
CIEx,ye |
Ref. |
| Where, BPhen = 4,7-diphenyl-1,10-phenanthroline; PBD = 2-(tert-butylphenyl)-5-biphenylyl-1,3,4-oxadiazole; CBP = 4,4′-N,N′-dicarbazole-biphenyl. The data for maximum brightness (B). Maximum current efficiency (ηc). Maximum power efficiency (ηp). Maximum external quantum efficiency (EQE). CIEx,y at 10 mA cm−2. |
|
Eu-1 (3 wt%): 26DCzPPy (10 nm) |
4.0 |
1237 |
3.09 |
2.2 |
2.33 |
0.639, 0.318 |
This work |
|
Eu-2 (3 wt%): 26DCzPPy (10 nm) |
3.5 |
2108 |
8.45 |
6.0 |
6.98 |
0.628, 0.308 |
This work |
|
Eu-3 (10 wt%): TCTA:Bphen |
2.9 |
— |
3.85 |
2.12 |
2.26 |
— |
16
|
|
Eu-4 (5 wt%): PBD (90 nm) |
— |
— |
10.0 |
5.30 |
— |
— |
38
|
|
Eu-5 (90 nm): CBP: PBD |
12 |
278 |
— |
1.45 |
— |
— |
38
|
|
Eu-6 (90 nm): CBP: PBD |
— |
— |
3.2 |
2.40 |
0.6 |
0.65, 0.32 |
38
|
|
Eu-7 (90 nm): CBP: PBD |
— |
— |
5.1 |
3.70 |
1.0 |
0.66, 0.33 |
38
|
|
Eu-8 (8 wt%): CBP (30 nm, 8%) |
19.8 |
1365 |
3.31 |
1.80 |
— |
— |
38
|
|
Eu-9 (80 nm): PBD |
19.6 |
1271 |
5.71 |
3.60 |
2.35 |
— |
38
|
|
Eu-10 (40 nm) |
5.7 |
— |
5.07 |
3.20 |
3.62 |
— |
15
|
|
Eu-11 (40 nm) |
5.0 |
— |
5.88 |
3.71 |
3.69 |
— |
15
|
|
Eu-12 (40 nm) |
8.4 |
— |
3.25 |
2.08 |
1.23 |
— |
15
|
|
Eu-13: CBP (40 nm, 10%) |
6.0 |
945 |
— |
2.96 |
— |
— |
37
|
|
Eu-14: CBP (40 nm, 10%) |
7.6 |
1276 |
— |
3.54 |
— |
— |
37
|
|
Eu-15: CBP (40 nm, 10%) |
7.2 |
1163 |
N/A |
3.20 |
— |
— |
37
|
|
Eu-16: CBP (30 nm, 10%) |
7 |
632 |
4.58 |
2.89 |
2.89 |
— |
14
|
4. Conclusion
In conclusion, we have synthesized two highly efficient red emitting complexes Eu-1 and Eu-2 with PLQY values as large as 80%. The EL performance of the single EML device of Eu-2 exhibited bright red EL with the EQE of 6.0% and B = 2108 cd m−2, ηc = 8.45 cd A−1, ηp = 6.98 lm W−1 at very low Vturn-on = 3.5 V which are the best reported for devices with Eu-CC as emitters. Moreover, it is important to note that doping concentrations as low as 4 wt% generated the best EL performance compared to other red-emitting europium-based OLEDs reported in the literature (Chart 2). To the best of our knowledge, this is the best EL performance shown by a Eu-CC. Furthermore, the single EML device makes the fabrication of EL devices easy and decreases the manufacturing cost as well as the time taken to fabricate them, making these complexes important EL materials for real-life applications. Work on new phospho-oxide type ligand with large bite angle to impose asymmetric coordination geometry around Eu(III) and optimization of the device structures is in progress in our laboratory to further improve the device efficiency of the red emitting OLEDs.
Conflicts of interest
The authors declare no conflicts of interest.
Acknowledgements
MSK acknowledges His Majesty's Trust Fund for Strategic Research (Grant No. SR/SQU/SCI/CHEM/16/02) for funding. RI thanks HM's Trust Fund for a postdoctoral fellowship. WYW thanks the Hong Kong Polytechnic University (1-ZE1C) and the Endowed Professorship in Energy from Ms Clarea Au (847S) for the financial support. WFO thanks PIBIC/CNPq/UFS (02/2019 COPES/POSGRAP/UFS) for a scientific initiation fellowship. LZ is grateful to the financial aid from National Natural Science Foundation of China (21771172), Youth Innovation Promotion Association of Chinese Academy of Sciences (2013150). PRR is grateful to the Engineering and Physical Sciences Research Council (EPSRC) for continued funding (Grant EP/K004956/1).
References
-
(a) A. Haque, L. Xu, R. A. Al-Balushi, M. K. Al-Suti, R. Ilmi, Z. Guo, M. S. Khan, W. Y. Wong and P. R. Raithby, Chem. Soc. Rev., 2019, 48, 5547–5563 RSC;
(b) R. Ilmi, E. Tseriotou, P. Stylianou, Y. A. Christou, I. Ttofi, N. Dietis, C. Pitris, A. D. Odysseos and S. N. Georgiades, Mol. Pharmaceutics, 2019, 16, 4260–4273 CrossRef CAS PubMed;
(c) R. Ilmi, I. Juma Al-busaidi, A. Haque and M. S. Khan, J. Coord. Chem., 2018, 71, 3045–3076 CrossRef CAS;
(d)
C. D. S. Brites, A. Millán and L. D. Carlos, in Handbook on the Physics and Chemistry of Rare Earths, ed. B. Jean-Claude and P. K. Vitalij, Elsevier, 2016, vol. 49, pp. 339–427 Search PubMed;
(e) C.-L. Ho and W.-Y. Wong, Coord. Chem. Rev., 2013, 257, 1614–1649 CrossRef CAS;
(f) H. Xu, R. Chen, Q. Sun, W. Lai, Q. Su, W. Huang and X. Liu, Chem. Soc. Rev., 2014, 43, 3259–3302 RSC.
-
(a) R. Ilmi, M. S. Khan, W. Sun, L. Zhou, W.-Y. Wong and P. R. Raithby, J. Mater. Chem. C, 2019, 7, 13966–13975 RSC;
(b) R. Ilmi, M. S. Khan, Z. Li, L. Zhou, W.-Y. Wong, F. Marken and P. R. Raithby, Inorg. Chem., 2019, 58, 8316–8331 CrossRef CAS PubMed;
(c) B. Francis, M. M. Nolasco, P. Brandão, R. A. S. Ferreira, R. S. Carvalho, M. Cremona and L. D. Carlos, Eur. J. Inorg. Chem., 2020 DOI:10.1002/ejic.202000027.
-
(a) A. N. Al-Khalili, I. J. Al-Busaidi, R. Ilmi, M. Al-Mandhary, M. S. Khan and N. K. Al-Rasbi, Inorg. Chim. Acta, 2020, 501, 119226 CrossRef;
(b) Y. Liu, J. Ma, C. Xu, Y. Yang, M. Xia, H. Jiang and W. Liu, Dalton Trans., 2018, 47, 13543–13549 RSC;
(c) W. Liu, X. Huang, C. Chen, C. Xu, J. Ma, L. Yang, W. Wang, W. Dou and W. Liu, Chem. – Eur. J., 2019, 25, 1090–1097 CrossRef CAS PubMed.
- L. Zhou, L. Li, Y. Jiang, R. Cui, Y. Li, X. Zhao and H. Zhang, ACS Appl. Mater. Interfaces, 2015, 7, 16046–16053 CrossRef CAS PubMed.
-
(a) P. K. Shahi, A. K. Singh, S. K. Singh, S. B. Rai and B. Ullrich, ACS Appl. Mater. Interfaces, 2015, 7, 18231–18239 CrossRef CAS PubMed;
(b) K. Nakamura, Y. Hasegawa, H. Kawai, N. Yasuda, N. Kanehisa, Y. Kai, T. Nagamura, S. Yanagida and Y. Wada, J. Phys. Chem. A, 2007, 111, 3029–3037 CrossRef CAS PubMed;
(c) R. Ilmi, S. Anjum, A. Haque and M. S. Khan, J. Photochem. Photobiol., A, 2019, 383, 111968 CrossRef CAS;
(d) K. Nakamura, Y. Hasegawa, H. Kawai, N. Yasuda, Y. Wada and S. Yanagida, J. Alloys Compd., 2006, 408–412, 771–775 CrossRef CAS.
- R. Ilmi, A. Haque, I. J. Al-Busaidi, N. K. Al Rasbi and M. S. Khan, Dyes Pigm., 2019, 162, 59–66 CrossRef CAS.
-
(a) O. Moudam, B. C. Rowan, M. Alamiry, P. Richardson, B. S. Richards, A. C. Jones and N. Robertson, Chem. Commun., 2009, 6649–6651, 10.1039/B914978C;
(b) A. S. Kalyakina, V. V. Utochnikova, M. Zimmer, F. Dietrich, A. M. Kaczmarek, R. Van Deun, A. A. Vashchenko, A. S. Goloveshkin, M. Nieger, M. Gerhards, U. Schepers and S. Brase, Chem. Commun., 2018, 54, 5221–5224 RSC.
- O. Laporte and W. F. Meggers, J. Opt. Soc. Am., 1925, 11, 459–463 CrossRef CAS.
-
(a)
N. Sabbatini, M. Guardigli and I. Manet, in Handbook on the Physics and Chemistry of Rare Earths, ed. K. A. Gschneidner Jr. and E. LeRoy, Elsevier, 1996, vol. 23, pp. 69–119 Search PubMed;
(b) S. I. Weissman, J. Chem. Phys., 1942, 10, 214–217 CrossRef CAS.
-
(a)
K. Binnemans, in Handbook on the Physics and Chemistry of Rare Earths, ed. J. C. B. K. A. Gschneidner and V. K. Pecharsky, Elsevier, 2005, vol. 35, pp. 107–272 Search PubMed;
(b) R. Ilmi, S. Kansız, N. K. Al Rasbi, N. Dege, P. R. Raithby and M. S. Khan, New J. Chem., 2020 10.1039/c9nj06287d;
(c) R. Ilmi and K. Iftikhar, Polyhedron, 2017, 127, 191–202 CrossRef CAS.
-
(a) E. Busby, J. Xia, Q. Wu, J. Z. Low, R. Song, J. R. Miller, X. Y. Zhu, L. M. Campos and M. Y. Sfeir, Nat. Mater., 2015, 14, 426 CrossRef CAS PubMed;
(b) Q. Zhang, B. Li, S. Huang, H. Nomura, H. Tanaka and C. Adachi, Nat. Photonics, 2014, 8, 326 CrossRef CAS.
- T. Koizuka, M. Yamamoto, Y. Kitagawa, T. Nakanishi, K. Fushimi and Y. Hasegawa, Bull. Chem. Soc. Jpn., 2017, 90, 1287–1292 CrossRef CAS.
-
(a) J. Kido, K. Nagai and Y. Ohashi, Chem. Lett., 1990, 657–660 CrossRef CAS;
(b) J. Kido, K. Nagai, Y. Okamoto and T. Skotheim, Chem. Lett., 1991, 1267–1270 CrossRef CAS;
(c) J. Kido, K. Nagai and Y. Okamoto, J. Alloys Compd., 1993, 192, 30–33 CrossRef CAS.
- H. Xu, L.-H. Wang, X.-H. Zhu, K. Yin, G.-Y. Zhong, X.-Y. Hou and W. Huang, J. Phys. Chem. B, 2006, 110, 3023–3029 CrossRef CAS PubMed.
- H. Xu, K. Yin and W. Huang, Chem. – Eur. J., 2007, 13, 10281–10293 CrossRef CAS PubMed.
- B. Zhao, H. Zhang, Y. Miao, Z. Wang, L. Gao, H. Wang, Y. Hao, B. Xu and W. Li, J. Mater. Chem. C, 2017, 5, 12182–12188 RSC.
- R. Ilmi, S. Kansız, N. Dege and M. S. Khan, J. Photochem. Photobiol., A, 2019, 377, 268–281 CrossRef CAS.
- M. A. Filho, J. D. L. Dutra, H. L. Cavalcanti, G. B. Rocha, A. M. Simas and R. O. Freire, J. Chem. Theory Comput., 2014, 10, 3031–3037 CrossRef CAS PubMed.
-
J. J. P. Stewart, MOPAC2016, Stewart Computational Chemistry, CO, Colorado Springs, USA, 2016 Search PubMed.
- C. R. Groom, I. J. Bruno, M. P. Lightfoot and S. C. Ward, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 2016, 72, 171–179 CrossRef CAS PubMed.
- F. Neese, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2018, 8, e1327 Search PubMed.
- M. Dolg, H. Stoll and H. Preuss, J. Chem. Phys., 1989, 90, 1730–1734 CrossRef CAS.
- S. M. Ali, S. Pahan, A. Bhattacharyya and P. K. Mohapatra, Phys. Chem. Chem. Phys., 2016, 18, 9816–9828 RSC.
-
(a) O. L. Malta, J. Lumin., 1997, 71, 229–236 CrossRef CAS;
(b) F. R. G. e Silva and O. L. Malta, J. Alloys Compd., 1997, 250, 427–430 CrossRef.
-
W. T. Carnall, H. Crosswhite and H. M. Crosswhite, Energy level structure and transition probabilities in the spectra of the trivalent lanthanides in LaF3, United States, 1978 Search PubMed.
- J. D. Dutra, T. D. Bispo and R. O. Freire, J. Comput. Chem., 2014, 35, 772–775 CrossRef CAS PubMed.
-
A. N. Carneiro Neto, E. E. S. Teotonio, G. F. de Sá, H. F. Brito, J. Legendziewicz, L. D. Carlos, M. C. F. C. Felinto, P. Gawryszewska, R. T. Moura, R. L. Longo, W. M. Faustino and O. L. Malta, in Handbook on the Physics and Chemistry of Rare Earths, ed. J.-C. G. Bünzli and V. K. Pecharsky, Elsevier, 2019, vol. 56, pp. 55–162 Search PubMed.
-
(a) K. L. Nash, R. D. Rogers, J. Ferraro and J. Zhang, Inorg. Chim. Acta, 1998, 269, 211–223 CrossRef CAS;
(b) U. Englert, B. Ganter, T. Wagner and W. Kläui, Z. Anorg. Allg. Chem., 1998, 624, 970–974 CrossRef CAS.
- F. Nüesch, D. Berner, E. Tutiš, M. Schaer, C. Ma, X. Wang, B. Zhang and L. Zuppiroli, Adv. Funct. Mater., 2005, 15, 323–330 CrossRef.
- K. Miyata, T. Nakagawa, R. Kawakami, Y. Kita, K. Sugimoto, T. Nakashima, T. Harada, T. Kawai and Y. Hasegawa, Chem. – Eur. J., 2011, 17, 521–528 CrossRef CAS PubMed.
- R. Ilmi, A. Haque and M. S. Khan, J. Photochem. Photobiol., A, 2019, 370, 135–144 CrossRef CAS.
- J. D. L. Dutra, N. B. D. Lima, R. O. Freire and A. M. Simas, Sci. Rep., 2015, 5, 13695 CrossRef PubMed.
- M. Latva, H. Takalo, V. M. Mukkala, C. Matachescu, J. C. RodriguezUbis and J. Kankare, J. Lumin., 1997, 75, 149–169 CrossRef CAS.
- M. Congiu, M. Alamiry, O. Moudam, S. Ciorba, P. R. Richardson, L. Maron, A. C. Jones, B. S. Richards and N. Robertson, Dalton Trans., 2013, 42, 13537–13545 RSC.
- O. L. Malta, F. R. G. E. Silva and R. Longo, Chem. Phys. Lett., 1999, 307, 518–526 CrossRef CAS.
-
(a) P.-P. Sun, J.-P. Duan, H.-T. Shih and C.-H. Cheng, Appl. Phys. Lett., 2002, 81, 792–794 CrossRef CAS;
(b) F. Liang, Q. Zhou, Y. Cheng, L. Wang, D. Ma, X. Jing and F. Wang, Chem. Mater., 2003, 15, 1935–1937 CrossRef CAS.
- H. Xu, K. Yin and W. Huang, J. Phys. Chem. C, 2010, 114, 1674–1683 CrossRef CAS.
-
(a) S. Zhang, G. A. Turnbull and I. D. W. Samuel, Org. Electron., 2012, 13, 3091–3096 CrossRef CAS;
(b) Y. Liu, J. Li, C. Li, J. Song, Y. Zhang, J. Peng, X. Wang, M. Zhu, Y. Cao and W. Zhu, Chem. Phys. Lett., 2007, 433, 331–334 CrossRef CAS;
(c) M. Pietraszkiewicz, M. Maciejczyk, I. D. W. Samuel and S. Zhang, J. Mater. Chem. C, 2013, 1, 8028–8032 RSC;
(d) S. Wang, J. Zhang, Y. Hou, C. Du and Y. Wu, J. Mater. Chem., 2011, 21, 7559–7561 RSC;
(e) Y. Liu, Y. Wang, H. Guo, M. Zhu, C. Li, J. Peng, W. Zhu and Y. Cao, J. Phys. Chem. C, 2011, 115, 4209–4216 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0tc00749h |
|
| This journal is © The Royal Society of Chemistry 2020 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  *a,
Rashid
Ilmi
*a,
Rashid
Ilmi

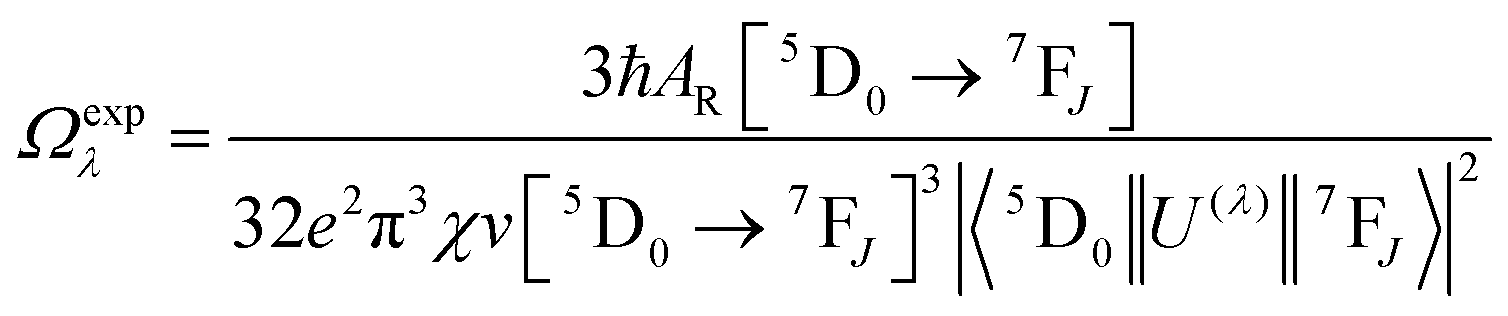
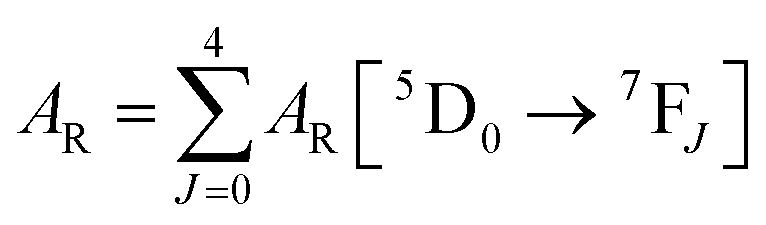


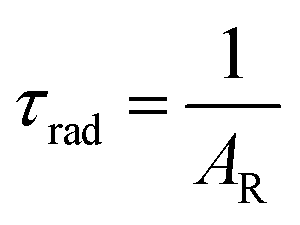
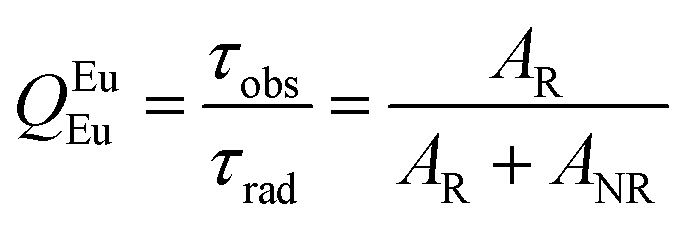
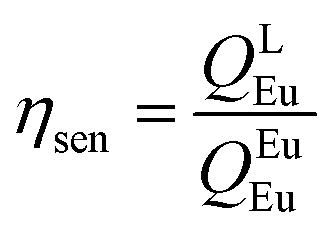


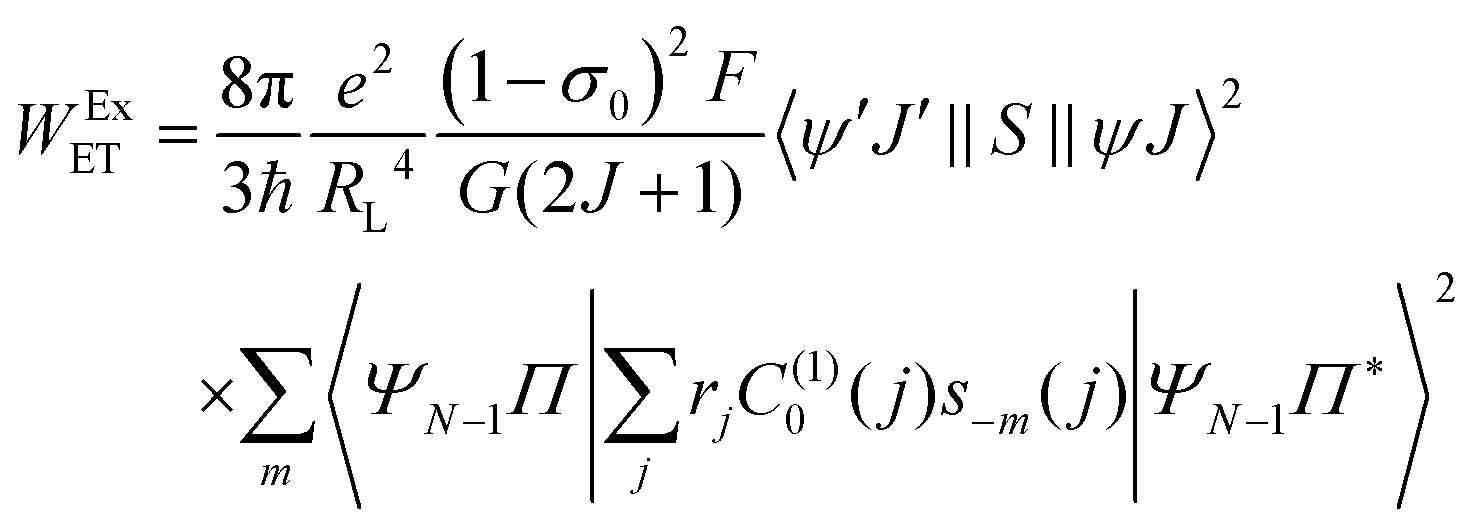

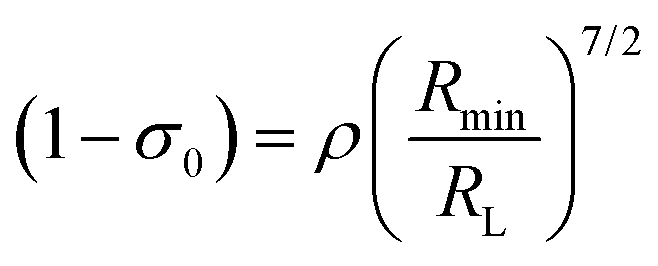

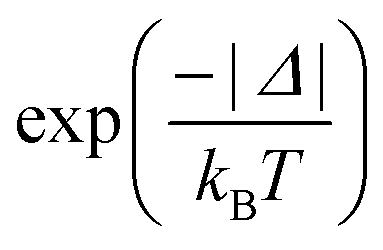 , where T is the environment temperature and kB stands for the Boltzmann constant. The theoretical quantum yields of the complexes were also determined and the details are presented in the ESI† (eqn (S10) and (S11)).
, where T is the environment temperature and kB stands for the Boltzmann constant. The theoretical quantum yields of the complexes were also determined and the details are presented in the ESI† (eqn (S10) and (S11)).