
S,N-Codoped oil-soluble fluorescent carbon dots for a high color-rendering WLED†

Abstract
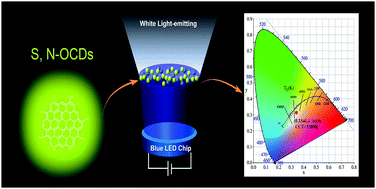
Carbon dots (CDs) have attracted widespread attention in light-emitting-diode (LED) applications owing to their environmental friendliness, easy processing and unique optical properties. For the practical use of CDs in LEDs, the controllable synthesis of high performance oil-soluble carbon dots is still in urgent demand. In this study, S,N-codoped oil-soluble carbon dots (S,N-OCDs) with an average size of 3.38 nm were prepared by solvothermal reaction of acetone, dimethyl trithiocarbonate (DMTTC) and nitric acid. Characterization results reveal that both the conjugated aromatic π systems from the aldol condensation and highly active free radicals from DMTTC essentially dominate the development of these yellow-green emitting S,N-OCDs, which possess high carbonization, quantum yield (21.08%) and oil-solubility. Furthermore, a white light-emitting-diode (WLED) was fabricated through mixing the obtained S,N-OCDs and epoxy and drop-casting the mixture on the surface of gallium nitride (GaN)-based blue chips. The as-prepared WLED showed excellent color rendering properties (CCT of 5389 K, CIE coordinates of (0.33, 0.30), and CRI of 88.38).
 Please wait while we load your content...
Please wait while we load your content...

