
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Correction: Towards predicting the power conversion efficiencies of organic solar cells from donor and acceptor molecule structures
Yecheng
Zhou
*a,
Guankui
Long
b,
Ailin
Li
c,
Angus
Gray-Weale
a,
Yongsheng
Chen
b and
Tianying
Yan
c
aSchool of Chemistry, The University of Melbourne, Parkville, VIC 3010, Australia. E-mail: zhouych87@gmail.com
bKey Laboratory of Functional Polymer Materials, College of Chemistry, Nankai University, Tianjin 300071, China
cCollege of Materials Science and Engineering, National Institute of Advanced Materials, Nankai University, Tianjin 300071, China
First published on 16th January 2020
Abstract
Correction for ‘Towards predicting the power conversion efficiencies of organic solar cells from donor and acceptor molecule structures’ by Yecheng Zhou et al., J. Mater. Chem. C, 2018, 6, 3276–3287.
There are errors in the equations on page 3279 of the article, under the heading ‘2.4 DOS determination.’ The correct equations and related text are below.
DOSs are essential for the numerical simulations of CT in solar cells. However, we haven’t found a theoretical method to estimate them for OSCs without any empirical or experimental parameters. Here, we put forward a method to estimate the DOS in OSCs. We assumed that each orbital can be occupied by two electrons, then DOS(E) can be calculated by 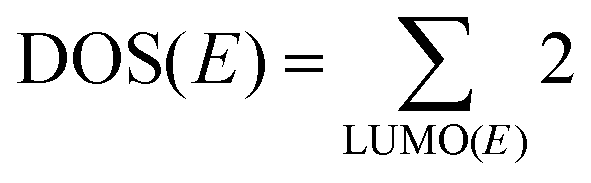 , which is the summation over all LUMOs whose energies are E. The DOS(E) should be Gaussian-like due to the Gaussian distribution of LUMOs. Assuming the charge occupation in the semiconductor obeys the Fermi–Dirac distribution, then, the charge density of the OSCs is given by:
, which is the summation over all LUMOs whose energies are E. The DOS(E) should be Gaussian-like due to the Gaussian distribution of LUMOs. Assuming the charge occupation in the semiconductor obeys the Fermi–Dirac distribution, then, the charge density of the OSCs is given by:
 | (8) |
 . Therefore, the DOS of the conduction band can be expressed as:
. Therefore, the DOS of the conduction band can be expressed as: | (9) |
The changes in the equations do not affect the interpretation of the results or the conclusions. The authors apologise for this oversight and for any confusion that it may have caused.
The Royal Society of Chemistry apologises for these errors and any consequent inconvenience to authors and readers.
| This journal is © The Royal Society of Chemistry 2020 |