
The structure optimization of phenanthroimidazole based isomers with external quantum efficiency approaching 7% in non-doped deep-blue OLEDs†

Abstract
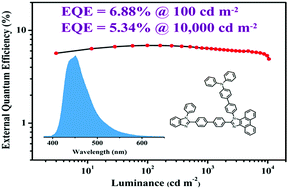
In this work, four phenanthroimidazole (PI) based isomers TPA-PPI-PBI, TPA-PPI-NPBI, PBI-PPI-TPA and NPBI-PPI-TPA for high-efficiency deep-blue organic light-emitting diodes (OLEDs) have been designed and synthesized. The structure–property relationship is systematically studied. Devices based on TPA-PPI-PBI, TPA-PPI-NPBI, PBI-PPI-TPA and NPBI-PPI-TPA achieved deep-blue emissions with Commission Internationale de L'Eclairage (CIE) coordinates of (0.15, 0.07), (0.15, 0.07), (0.15, 0.09) and (0.15, 0.05) and high external quantum efficiencies (EQEmax) of 4.12%, 4.66%, 6.88% and 5.59%, respectively. The PBI-PPI-TPA based device exhibited negligible efficiency roll-off with an EQE of 6.48% at practical 1000 cd m−2. Moreover, the EQE is still above 5% even at a high brightness of 10 000 cd m−2. Comparing the four isomers, we found that the substituent at the C2 position of the PI core has a significant influence on the emission wavelength and CIE coordinates. This work provides a rational design strategy where modifying an electron acceptor (A) at the C2 position and an electron donor (D) at the N1 position of the PI core will be an effective way to fabricate high-performance PI-based bipolar emitters.
 Please wait while we load your content...
Please wait while we load your content...

