
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Designing highly fluorescent, arylated poly(phenylene vinylene)s of intrinsic microporosity†
Ana Clara B.
Rodrigues‡
 a,
Isabell S.
Geisler‡
b,
Patrick
Klein
b,
João
Pina
a,
Fabian J. H.
Neuhaus
b,
Elke
Dreher
c,
Christian W.
Lehmann
c,
Ullrich
Scherf
*b and
J. Sérgio
Seixas de Melo
*a
a,
Isabell S.
Geisler‡
b,
Patrick
Klein
b,
João
Pina
a,
Fabian J. H.
Neuhaus
b,
Elke
Dreher
c,
Christian W.
Lehmann
c,
Ullrich
Scherf
*b and
J. Sérgio
Seixas de Melo
*a
aCQC, Department of Chemistry, University of Coimbra, P3004-535 Coimbra, Portugal. E-mail: sseixas@ci.uc.pt
bBergische Universitat Wuppertal, Macromolecular Chemistry Group (buwmakro) and Institute for Polymer Technology, Gauss-Str. 20, D-42119 Wuppertal, Germany. E-mail: scherf@uni-wuppertal.de
cMax-Planck-Institut für Kohlenforschung, Kaiser-Wilhelm-Platz 1, D-45470 Mülheim an der Ruhr, Germany
First published on 6th December 2019
Abstract
Three new polymers containing tetraphenylethylene and diphenyl-dinaphthylethylene cores and their corresponding monomeric model compounds were synthesized and fully characterized aiming to investigate their photoluminescence efficiency, microporosity and Brunauer–Emmett–Teller-derived surface areas (SBET). Comprehensive photophysical characterization was undertaken in the solid state (powder and thin films), in tetrahydrofuran (THF) solution and in mixtures of “good” and “poor” solvent to induce aggregation (THF:water mixtures). Aggregation induced emission (AIE) was found for the tert-butyl-TPE monomer and polymer and diphenyl-dinaphthylethylene monomer with the increase of the water amount in THF:water mixtures and in the solid state. The tert-butyl substituted TPE derivatives display the highest fluorescence quantum yield (ϕF) values: 0.14 to 0.30 (in powder) and 0.46 to 0.64 in thin films. In contrast, with the diphenyl-dinaphthylethylene (meta and para-phenylene) polymers aggregation caused quenching (ACQ) occurs in THF:water mixtures (ϕF ≤ 0.011) and in the solid state (ϕF ≤ 0.012). The microporosity of the soluble conjugated polymers as potential conjugated polymers of intrinsic microporosity (cPIMs) was further investigated. The SBET of the polymers were related to their optical properties. The polymers show an attractive combination of high SBET surface area (417 m2 g−1) and the occurrence of distinct AIE effects for the tert-butyl-TPE polymer while the diphenyl-dinaphthylethylene polymers do not exhibit microporosity (SBET ≤ 17 m2 g−1) and show ACQ behavior.
Introduction
Aggregation induced emission (AIE, or even aggregation induced enhanced emission, AIEE) corresponds to the description of the luminescence phenomenon in which the luminescence efficiency of a luminogen is increased upon its aggregation.1 The most accepted mechanism for the occurrence of AIE is through a restriction of intramolecular motions (RIM), including rotations and vibrations, that would deactivate non-radiatively the excited state of the luminogens.2 This phenomenon occurs in some luminogens in contrast to aggregation caused quenching (ACQ) commonly described for traditional fluorophores in which the π–π-stacking of the molecules at high concentration or in the solid state leads to the formation of H-aggregates, which decreases the emission efficiency.3 Typically, low molecular weight AIE chromophores possess a propeller-shaped structure with rotatable peripheral phenyl rings (rotors),4 such as 1,1,2,3,4,5-hexaphenylsilole (HPS),5 2,3,3-triphenylacrylonitrile (TPAN)6 and 1,1,2,2,-tetraphenylethylene (TPE).7TPE derivatives have demonstrated a wide range of applications in optoelectronic devices,8–10 bioimaging,11 biosensors,12etc. due to their AIE properties.TPE-based conjugated polymers – poly(arylene-diphenylvinylenes) – have been known since the 1960s.13 Soluble, high molecular weight poly-TPEs (Mn 10–40 kDa) can be made in reductive polyolefinations of aromatic, bisgeminal tetrachlorides as diketone derivatives with Cr2ac4 or Co2(CO)8 as reducing agents14–16 or in carbonyl polyolefinations after McMurry with TiCl3/LiAlH4.17 Some of them show distinct AIE properties leading to high solid state photoluminescence quantum yields of >70% for some of the poly-TPEs.4,18 Since 2011,19,20 the easily functionalized four phenyl rings of the TPE unit were combined with a variety of reactive groups in construction of TPE-based polymers with inherent porosity.21 The development of microporous organic polymers (MOPs) with very high surface area is an active field of current research,22 with high potential in a variety of applications4,23–26 such as energy storage, light harvesting, catalysis and sensing of hazardous chemicals and explosives. Several classes of MOPs, such as amorphous hyper-cross-linked polymers (HCPs), polymers of intrinsic microporosity (PIMs), and conjugated microporous polymers (CMPs), have been reported.27 CMPs are also potential fluorescent materials due to their extended conjugated frameworks and tunable optoelectronic properties.28
In this work, two types of poly(1,4-phenylene-diarylvinylene)s with 4-tert-butylphenyl or naphthyl as aryl substituents at the vinylene units as well as the corresponding poly(1,3-phenylene-dinaphthylvinylene) are investigated for their photophysical properties in tetrahydrofuran (THF) and THF:water mixtures (a mixture of a “good solvent”, THF, and water, a “poor solvent” to induce the formation of aggregates) as well as in the solid state (powder and films) for studying the effect of aggregation on the photoluminescence efficiency. The results are further rationalized with investigations on monomeric model monomer compounds and with the parent TPE molecule under the same conditions to also elucidate the effect of the substituent and polymerization on the photophysical properties of these compounds. Furthermore, the porosity of these new TPE-based polymers is also investigated.
Experimental section
Materials
Most chemicals were purchased from Sigma-Aldrich, Fischer Scientific or TCI and used without further purification, unless described otherwise. For the photophysical studies, the solutions were prepared with solvents of spectroscopic grade or equivalent: tetrahydrofuran (THF, Uvasol Merck), analytical grade chloroform (Fischer Chemical) or deionized water (18.2 MΩ cm at 25 °C, Milli-Q, Millipore).Synthesis and structural characterization of the model compounds and polymers
All reactions were carried out under an argon atmosphere using flame dried glassware. The detailed synthetic procedure and structural characterization are provided in the ESI.† NMR spectra were recorded on a Bruker AVANCE 400 or AVANCE III 600. APCI (Atmospheric Pressure Chemical Ionization) and ESI (electrospray ionisation) mass spectra were obtained on a Bruker Daltronik micrOTOF system. Gel permeation chromatography (GPC) measurements were carried out on a PSS/Agilent SECurity GPC system equipped with polystyrene gel columns using chloroform or THF as the eluent. Nitrogen adsorption–desorption isotherms were recorded on a BEL Japan Inc Belsorp-max system at 77 K. The surface areas were calculated using the BET model in the pressure range p/p0 from 0.05–0.25. All samples were degassed offline at >100 °C for 16 hours under a vacuum. Single crystal X-ray structures were obtained on a Bruker-AXS Kappa Mach3 APEX-II-diffractometer with a FR591 rotating anode, equipped with graded multilayer optics emitting copper radiation (1.54178 Å), and measured at 100 K. For structure solution and refinement, the SHELX-package29,30 was used as integrated in Olex2.31Solutions and film preparation
An appropriate amount of powder of each compound was diluted in THF to prepare a stock solution with an optical density of 1.0 at the excitation wavelength used for the experiments. Then, 100 μL of the stock solution was then diluted with the proper amount of THF or THF:water mixture to obtain the desired water fraction (fW = 0–95%, v/v) in 2 mL of final volume.Thin films from the compounds were obtained with a desktop precision spin-coating system, model P6700 series from Speedline Technologies, as described elsewhere.32 Briefly, thin films from the samples were obtained by deposition of ca. 50 μL from a solution of the compounds onto a circular sapphire substrate (10 mm diameter) followed by spin-coating (2500 rpm) in a nitrogen-saturated atmosphere (2 psi). The solutions for spin-coating were prepared by adding 2 mg of the samples to 200 μL of chloroform solution, with stirring, at environment temperature, overnight. 15 mg of Zeonex® was added to the chloroform solution of the model compounds, TPE, Mono-t-Bu and Mono-Np, as a polymeric matrix to obtain thin films of these samples.
Steady state and time-resolved fluorescence measurements
The absorption spectra were recorded using Shimadzu UV-2450 or Agilent Cary 5000 UV-vis-NIR spectrometers. Absorption spectra of the transparent thin films were obtained in absorption mode using a clean sapphire substrate as the reference sample. The absorption spectra of the amorphous powder samples were recorded by collecting diffuse reflectance using a Cary 5000 DRA (an integrating sphere accessory with detection in the 200–2500 nm range). Background correction was performed by collecting the baseline with 100% and 0% reflectance (using a polytetrafluoroethylene, PTFE, reference sample and a blocked beam, respectively) prior to the determination of the spectra of the solid samples. Conversion to absorption was performed assuming the Kubelka–Munk function, F(R).33Fluorescence spectroscopic studies were performed using a Horiba-Jobin-Yvon Fluorolog 3-22 spectrofluorimeter. The fluorescence quantum yields (ϕF) of all compounds, in solution or in the solid state, were measured using the absolute method with a Hamamatsu Quantaurus QY absolute photoluminescence quantum yield spectrometer, model C11347 (integrating sphere). A clean sapphire substrate was used as a reference for the ϕF measurements of solid-state thin films.
Fluorescence decays were measured using a home-built Time-Correlated Single Photon Counting (TCSPC) apparatus described previously.32 An IBH nanoLED (339 nm, 1.0 kHz) was used as the excitation source. The fluorescence decays and the instrumental response function (IRF) were collected using 1024 channels on a time scale of up to 48.8 ps per channel; alternate measurements (500 counts) of the pulse profile at the excitation wavelength and the sample emission were performed until 3000 counts at the maximum were reached. Deconvolution of the fluorescence decay curves was performed using the modulation function method in the SAND program, as previously described.34
Results and discussion
Synthesis and physical characterization of the polymers and monomeric model compounds
The structures and acronyms of the investigated compounds are depicted in Scheme 1.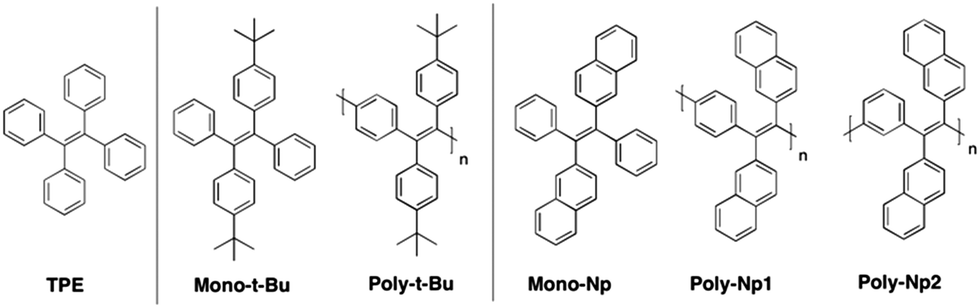 | ||
| Scheme 1 Representative structures and acronyms of the investigated polymers, and TPE and tert-butylbenzyl and naphthyl monomeric model compounds. | ||
Three new polymers comprising tetraphenylethylene and diphenyl-dinaphthylethylene backbones together with their corresponding monomeric analogues were synthesized according to the general strategic methodology described in Scheme 2. These polymers were inspired by the well-known aggregation induced emission, AIE, luminogen, 1,1,2,2-tetraphenylethylene (TPE).1,7,24,35–37
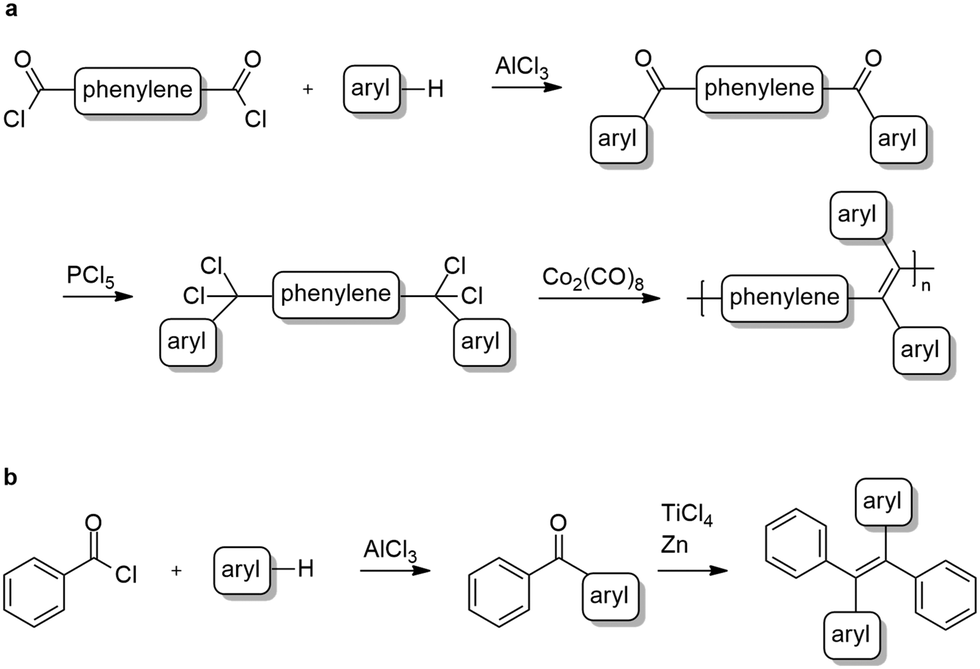 | ||
| Scheme 2 General synthesis of polymers (a) and phenyl-terminated monomers (b). | ||
All polymers were synthesized via reductive polyolefinations of aromatic, bisgeminal tetrachlorides following a procedure initially described by Hörhold and co-workers in the 1970s.14,38,39 In contrast to the original procedure dicobalt octacarbonyl was used as a condensing agent, a variation of Hörhold's method developed by us in the 1990s.15 The required tetrachlorides were generated starting from the commercially available tere- or isophthaloyl chlorides in two steps (Scheme 2a). In the first step diketones were synthesized following a modified Friedel–Crafts-acylation procedure using tere-/isophthaloyl chloride and tert-butylbenzene or naphthalene.40,41 Next, the bisgeminal tetrachlorides were obtained by reaction of the diketones with phosphorous pentachloride as described by Hörhold et al.14,38
The monomeric model compounds were prepared in a two-step synthesis. First benzoyl chloride and naphthalene or tert-butylbenzene were converted to the corresponding diarylketones in a Friedel–Crafts-acylation protocol42 (Scheme 2b). In the case of naphthalene, the resulting mixture of 1-benzoylnaphthalene and 2-benzoylnaphtalene was separated using flash column chromatography. In the second step the desired tetraarylethylenes were obtained by reductive coupling of the ketones using titanium tetrachloride and zinc.43 The 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixtures of E/Z-isomers in the products (as determined from the 1H-NMR spectrum) were separated by recrystallization.
:1 mixtures of E/Z-isomers in the products (as determined from the 1H-NMR spectrum) were separated by recrystallization.
The average molecular weight, ![[M with combining macron]](https://www.rsc.org/images/entities/i_char_004d_0304.gif) n, weight average molecular weight, w, and polydispersity (PD) values of the polymers are listed in Table 1. Based on the n values the degrees of polymerization (DP) have been calculated. The two naphthyl-substituted polymers, Poly-Np1 and Poly-Np2, can be assigned to the same model compound, Mono-Np; Poly-Np1 incorporates para-phenylene main chain linker units, while Poly-Np2 contains meta-phenylene main chain units (Scheme 1). Poly-Np1 and Poly-Np2 display reduced molecular weights, most probably caused by the increased steric demand coupled with decreased polydispersity (PD) values. Table 1 also lists the BET-based surface areas (SBET) of the solid polymer powders extracted from nitrogen sorption/desorption isotherms (Fig. SI1 and SI2, ESI†). Obviously, only Poly-t-Bu exhibits intrinsic microporosity with a high SBET surface area of up to 417 m2 g−1 as a so-called conjugated polymer of intrinsic microporosity (cPIM).23
n, weight average molecular weight, w, and polydispersity (PD) values of the polymers are listed in Table 1. Based on the n values the degrees of polymerization (DP) have been calculated. The two naphthyl-substituted polymers, Poly-Np1 and Poly-Np2, can be assigned to the same model compound, Mono-Np; Poly-Np1 incorporates para-phenylene main chain linker units, while Poly-Np2 contains meta-phenylene main chain units (Scheme 1). Poly-Np1 and Poly-Np2 display reduced molecular weights, most probably caused by the increased steric demand coupled with decreased polydispersity (PD) values. Table 1 also lists the BET-based surface areas (SBET) of the solid polymer powders extracted from nitrogen sorption/desorption isotherms (Fig. SI1 and SI2, ESI†). Obviously, only Poly-t-Bu exhibits intrinsic microporosity with a high SBET surface area of up to 417 m2 g−1 as a so-called conjugated polymer of intrinsic microporosity (cPIM).23
| Compound |
n (g mol−1) |
w (g mol−1) |
PD | DP | S BET (m2 g−1) |
|---|---|---|---|---|---|
|
a Mean and weight average molecular weights (n and w), polydispersity (PD) and the degree of polymerization (DP) calculated based on the average molecular weight (n) and BET-derived surface area (SBET).
|
|||||
| Poly-t-Bu | 29300 |
164000 |
5.62 | 80 | 417 |
| Poly-Np1 | 11600 |
17800 |
1.54 | 33 | 14 |
| Poly-Np2 | 5300 | 9400 | 1.77 | 15 | 17 |
The investigated phenyl-terminated monomeric model compounds, Mono-t-Bu and Mono-Np, are both of E-configuration as determined by single crystal X-ray crystallography (see Fig. 1). In the crystalline state, Mono-t-Bu adopts a propeller like conformation with dihedral angles between the plane subtended by the central double bond and the phenyl rings of 52.3° and 55.9°. The ring planes of the two tert-butylphenyl groups are rotated slightly less, by 38.8° and 41.1°. The molecular conformation of Mono-Np is similar to that of Mono-t-Bu. The molecule is located on a crystallographic twofold axis and consequently only one independent dihedral angle between the two phenyl rings and the olefinic bond exists. This amounts to 56.2°. The same symmetry restriction holds for the naphthyl rings, which are rotated by 53.3° against the plane of the double bond. While the molecular conformations of the two propeller shaped molecules are similar, a marked difference is observed regarding the intermolecular interactions. Mono-t-Bu does not form π–π interactions, either between the tert-butyl rings or the phenyl rings. In contrast, Mono-Np exhibits π-stacking between the naphthyl rings. The shortest C–C contact between the rings is 3.32 Å and slightly shorter than in graphite (Fig. 2). The distance between the centroids of the overlapping halves of the naphthyl rings is 4.8 Å, indicating ring slippage and reduced overlap of the π-systems. However, the π-stacking is not limited to pairs of molecules but extends infinitely through the crystal along the c-axis. Also, the observed differences in the surface area values of the polymers should result from the different packing behaviour of the 4-tert-butylphenyl vs. naphthyl side groups. Indeed, while the naphthyl side groups tend to aggregate (π-stack) with neighbouring side groups in the solid state, the 4-tert-butylphenyl groups with their bulky tert-butyl substituents create free volume and microporosity (please see also the discussion of the X-ray crystal structures of the monomers).
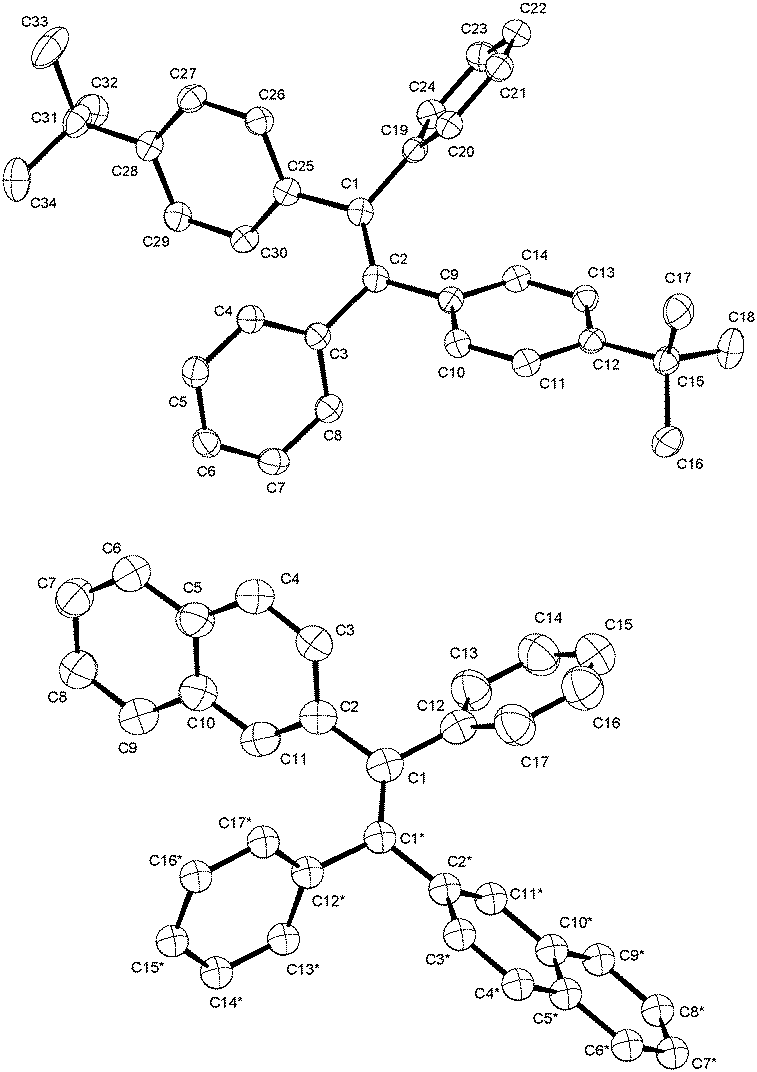 | ||
| Fig. 1 Molecular structures of the phenyl-terminated monomeric model compounds, Mono-t-Bu and Mono-Np, determined by single crystal X-ray crystallography. | ||
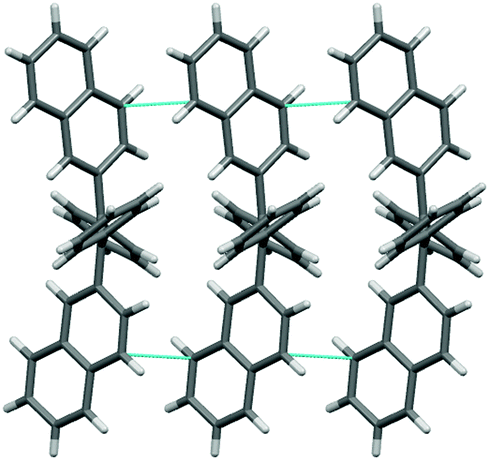 | ||
| Fig. 2 View of the crystal packing of Mono-Np, showing short Cπ–Cπ interactions (in cyan) parallel to the c-axis. | ||
Electronic spectral data
The photophysical properties of the polymers and model compounds, TPE and phenyl-terminated monomers, were investigated in the solid state (amorphous powders and thin films), in tetrahydrofuran (THF) solution and in mixtures of good and poor solvent (THF:water mixtures) to evaluate the effect of aggregation on the fluorescence emission efficiency.Fig. 3 presents the absorption and fluorescence emission spectra of the polymers and model compounds in the solid-state (powder and thin films) and in THF solutions. The spectroscopic data in Fig. 3 are designed and structured in order to compare the polymer with the respective monomeric model compound plus the model AIGen, TPE.
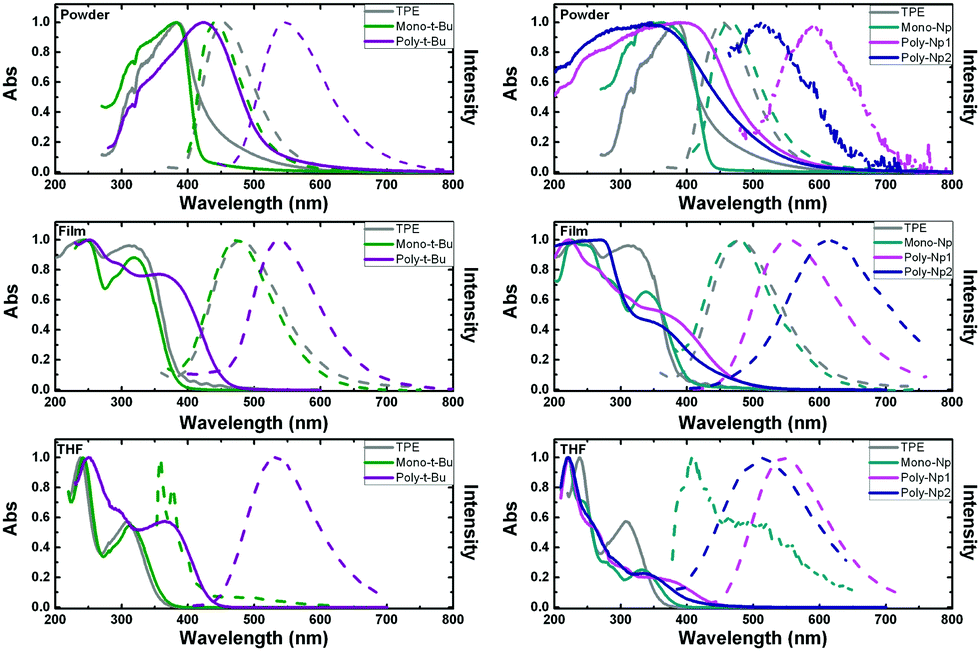 | ||
| Fig. 3 Normalized absorption and emission spectra for the compounds in powder and thin films (i.e., in a solid environment) and in THF solution. For better comparison the polymers together with the corresponding monomer are presented the same graph. Solid line: normalized absorption spectra; dashed line: normalized emission spectra. For comparison, the TPE absorption and emission spectra are shown (grey lines). | ||
The same wavelength scale is presented for all the compounds, which further allows a direct comparison between the two types of polymers.
The absorption and emission spectra of the investigated compounds in the amorphous powders are red-shifted and broader than in THF solution, indicating an extent of conjugation through intermolecular packing in the solid state (see Fig. 3 and Table 2). Additional observation of the spectral behavior shows that there is a good match between the solution absorption band profiles and those obtained in thin films for the polymers and monomers (although with a different maximum, Table 2).
| Compound | Medium | λ abs (nm) | λ em (nm) | Δ SS (cm−1) |
|---|---|---|---|---|
| a Data from ref. 44. b Prepared using Zeonex® as a polymeric matrix. | ||||
| TPE | Powdera | 381 | 454 | 4220 |
| Filmb | 243/312 | 482 | 11304 |
|
| THFa | 238/308 | — | — | |
| Mono-t-Bu | Powder | 382 | 441 | 3052 |
| Filmb | 245/320 | 473 | 10108 |
|
| THF | 242/314 | 358, 490 | 3914 | |
| Poly-t-Bu | Powder | 421 | 546 | 5438 |
| Film | 252/355 | 537 | 9547 | |
| THF | 250/366 | 530 | 8454 | |
| Mono-Np | Powder | 362 | 468 | 6257 |
| Filmb | 226/337 | 479 | 8797 | |
| THF | 220/334 | 407, 490 (sh) | 5370 | |
| Poly-Np1 | Powder | 391 | 590 | 8626 |
| Film | 222/351 | 553 | 10407 |
|
| THF | 222/343 | 545 | 10806 |
|
| Poly-Np2 | Powder | 346 | 511 | 9332 |
| Film | 268/348 | 611 | 12369 |
|
| THF | 221/335 | 518 | 10546 |
|
Observation of Fig. 3 and Table 2 shows that the emission spectra of the polymers are red-shifted relative to the monomeric model compounds, emitting in the visible green region of the light spectrum: in solution, the emission maxima correspond to 530 nm, for Poly-t-Bu, and 545 nm and 518 nm for Poly-Np1 and Poly-Np2, respectively. In films, Poly-Np2 presents the highest emission wavelength found for the studied compound, with a maximum at 611 nm (Table 2). Generally, the polymers also have higher Stokes shifts (ΔSS) when compared to their respective monomers, in the same medium. Since charge transfer bands cannot occur in these polymers this behavior points to the adoption of different conformations in the ground- and excited-state of the polymers (probably adopting a more planar conformation in the excited-state, which can lead to an extended degree of conjugation). The more extended nature of the π-conjugation in Poly-Np1 compared to Poly-Np2 in THF solution may be explained by the fact that the para-phenylene units in Poly-Np1 formally allow extended π-conjugation along the main chain (not much due to its strongly distorted conformation), while in Poly-Np2, the meta-phenylene main chain units act now as effective conjugation barriers. This is further reflected in the differences observed in the solution absorption and emission (solvent:THF) spectra (Fig. 3, bottom right hand panel). In contrast with these results, a different behavior is found in thin films (Fig. 3, middle right hand panel) where the fluorescence spectrum of Poly-Np2 is red-shifted when compared to Poly-Np1. While the THF solution and thin film emission maxima of Poly-Np1 are quite similar (545 nm in THF solution vs. 553 nm in the thin film), Poly-Np2 shows a distinctly red-shifted PL maximum when compared to the solution value (611 nm in the thin film vs. 518 nm for the THF solution). This finding may be explained by increased side chain aggregation (and AQC) in Poly-Np2 with the aggregate emission as the dominating feature. This explanation is supported by the reduced thin film ϕF for Poly-Np2 (PLQY: 0.02 for Poly-Np1vs. 0.001 for Poly-Np2, which will be further discussed in the next section on Table 3).
| THF | 90W:10 THF |
Powder | Film | |
|---|---|---|---|---|
| a Data from ref. 44. b Prepared using Zeonex® as a polymeric matrix. c ϕ F determined by comparison with the emission area of the Poly-Np1 film, with the same absorption at the excitation wavelength. | ||||
| TPE | 0.003 | 0.25 | 0.23a | 0.26b |
| Mono-t-Bu | 0.024 | 0.20 | 0.14 | 0.46b |
| Poly-t-Bu | 0.046 | 0.62 | 0.30 | 0.64 |
| Mono-Np | 0.003 | 0.043 | 0.14 | 0.12b |
| Poly-Np1 | 0.028 | 0.011 | 0.012 | 0.010 |
| Poly-Np2 | 0.008 | 0.003 | 0.004 | 0.002c |
Upon going from the monomeric model compound, Mono-t-Bu, to the polymer Poly-t-Bu a significant red shift of the absorption spectra is observed (39 nm in powder, 35 nm in film and 52 nm in THF). The same is not found for the Poly-Np polymers in THF and thin films where a small red shift is observed when compared to Mono-Np (see Table 2). This shows that the chromophoric unit is in these polymers limited to the monomeric counterpart.
In the case of Mono-Np the two bands at maxima 407 nm and 498 nm (Fig. 3, right hand bottom panel) indicate the coexistence of monomer and aggregate species. This is further complemented with a concentration dependence study in Fig. SI3 (ESI†).
It is also worth noting that in the solid state (powder and thin films) the investigated phenyl-terminated monomers retain the electronic spectral absorption features and maxima found for TPE (Fig. 3). On going to solution (THF), although red-shifted (∼6 nm for Mono-t-Bu and ∼26 nm for Mono-Np, see Table 2), similar broad absorption bands are observed for the monomeric model compounds when compared with TPE. The higher bathochromic shift found for Mono-Np can be attributed to the increase in the conjugation segment promoted by the naphthyl moieties when compared with the phenyl units in TPE.37
Clearly contrasting with the behavior found for TPE in good solvents, where fluorescence emission is almost negligible (with ϕF values of 0.002 or 0.0024 and of 0.003 in Table 3),37,45,46 the phenyl-terminated monomeric model compounds, Mono-t-Bu and Mono-Np, display fluorescence both in solution and in the solid state (Fig. 3 and Table 3). Indeed, TPE itself fluoresces when aggregated in solution37,47 or in the solid state (Fig. 3), i.e., when restriction of intramolecular rotation (RIR) occurs, thus hindering the excited state radiationless processes.
In THF solution, when TPE is compared with the tert-butyl-TPE (Mono-t-Bu) derivative, the absorption spectra almost totally match and the emission is absent in TPE. However, in the case of Mono-t-Bu, the emission spectrum presents two bands: a vibronically resolved band with a maximum at ∼358 nm (corresponding to the monomer) and a second broad band with a maximum at ∼490 nm corresponding to the emission of the aggregate. This last band increases and red-shifts with the addition of water (Fig. 4). Moreover the ϕF value of Mono-t-Bu in THF is one order of magnitude higher than that of TPE (Table 3).
 | ||
| Fig. 4 Room temperature fluorescence emission (photoluminescence, PL) spectra for the model compounds (TPE, Mono-t-Bu and Mono-Np) and polymers (Poly-t-Bu, Poly-Np1 and Poly-Np2) in THF:water mixtures and their respective correlation of the fluorescence quantum yield and emission maxima with increasing water fraction, fW = 0–95%. The lines in the right hand panels are just meant to be a guide to the eye. | ||
Although the emission spectra of the model compounds Mono-t-Bu and Mono-Np in films quite overlap with that of TPE, in powder the emission of Mono-t-Bu is blue-shifted when compared to TPE, with emission maxima of 441 nm and 454 nm for Mono-t-Bu and TPE, respectively (Fig. 3 and Table 2). In the case of Mono-Np and again compared with TPE, there is now a red-shift of the emission maximum of 14 nm (Table 2). For the latter monomer the observed red-shift together with the increase in the fluorescence quantum yield on going from solution to the solid state gives support to the formation of J-aggregates.48
Aggregation induced emission (AIE) studies
To further explore the occurrence of AIE in the investigated compounds, the emission behavior was studied in THF:water mixtures (Fig. 4). TPE blue emission is only visible in high water content, either in acetonitrile:water7 or in THF:water mixtures.49 The broad emission band associated with this emissive aggregate, centered at ca. 470 nm, becomes detectable at a water fraction (fW) ≥ 85%.Enhancing the steric effect is an efficient means to activate the restriction of intramolecular motion (RIM) process,47 which is considered one of the possible mechanisms to explain the AIE phenomenon.50 The phenyl rings in TPE have been substituted with multiple methyl groups at the o-positions to examine how the intramolecular steric effects affect its photophysical properties and AIE properties.46 It was shown that the sterically crowded TPE derivative with four methyl groups loses its AIE activity, directly validating the RIM hypothesis.51 However, it is expected that the t-Bu groups substituted in the p-position of the benzyl rings of TPE should enhance the steric effect and not affect the rotation of the aromatic rotors; therefore, the Mono-t-Bu monomer should present AIE properties. To verify this hypothesis, the emission spectra and fluorescence quantum yields (ϕF) for Mono-t-Bu were obtained in THF:water mixtures. Naphthalene-substituted ethenes, such as Mono-Np, are AIE-active luminogens.37
The spectral behavior of the investigated model compounds and polymers in THF:water mixtures, aiming to observe the presence of and to quantify the AIE effect, is shown in Fig. 4 and Table 3. From Fig. 4, it can be seen that Mono-t-Bu presents a structured emission spectrum in THF:water mixtures up to fW = 60% (fW: water:THF fraction, v/v), increasing in intensity with the addition of water. From fW ≥ 70% on, the emission maximum for Mono-t-Bu red-shifts and the ϕF value increases with the addition of water, up to ϕF = 0.2 for fW = 90%, i.e., ∼1 order of magnitude higher than in pure THF (Table 3).
Mono-Np shows a similar behavior (to that found for Mono-t-Bu), i.e., a vibronically resolved spectrum up to fW = 60% and a red-shifted emission band, followed by an increase in the ϕF values for fW ≥ 70%. This observation is likely due to the formation of J-aggregates in the two phenyl-terminated monomers. Indeed, although the presence of the naphthalene rings could favor π–π interactions, leading to the formation of H-aggregates and consequently to aggregation caused quenching (ACQ), the opposite behavior was found.
The ϕF values for TPE and Mono-Np in films have been previously reported as 0.49 and 0.30, respectively.37 Our values in Table 3 are 0.24 (TPE) and 0.46 (Mono-t-Bu), while for TPE in a THF:water mixture ϕF = 0.14 for fW = 95% was previously reported46 in comparison with our ϕF of 0.25 (for fW = 90%). The values obtained in this study are slightly different from those reported previously in films. This is likely because the photophysical properties are intrinsically dependent on the morphological properties of the films. Indeed, and exemplifying this, the ϕF of TPE in powder was described as 24.1%,52 found to be in good agreement with the value previously obtained by us (23%)44 with the same equipment that was used to perform all the ϕF measurements.
Usually, when AIEgens, such as TPE and its derivatives, are chemically incorporated into polymeric structures, polymers with AIE characteristics could be obtained.32,36,53 Thus, it was anticipated that Poly-t-Bu and Poly-Np1 and Poly-Np2, whose monomeric model compounds are AIE active, would also maintain AIE properties. Although Poly-t-Bu presents an increase of fluorescence quantum yield upon the increase of the water fraction, the fluorescence of the naphthyl polymers is quenched with the addition of water in THF:water mixtures. Indeed, as can be seen from Fig. 2Poly-Np1 and Poly-Np2 present ACQ. Moreover, as shown in Table 3, the ϕF of Poly-Np2 is smaller than Poly-Np1 in solution and the solid state and when compared at the same fW in THF:water mixtures. The ACQ behaviour of Poly-Np1,2 may be caused by the stacking tendency of the naphthyl groups, leading to the formation of H-type aggregates. This is supported by the observed intermolecular π-interactions of the naphthyl substituents in the single crystal structure of Mono-Np.
In the solid state, the highest photoluminescence efficiency was obtained for Poly-t-Bu, with ϕF = 64% in film – an increase of ca. 14× when compared to THF and higher than TPE and Mono-t-Bu – and when aggregated in THF:water mixtures it shows a value of ϕF = 62% for fW = 90%.
The combination of TPE and carbazole groups also generated porous organic polymers with higher SBET area, varying from 472 to 2200 m2 g−1,24,25,54 with fluorescence quantum yields of up to 40% in films55 (see also Table SI5, ESI,† for a literature review of SBET and fluorescence quantum yield values for porous TPE based polymers). In our set of TPE derived polymers, a new compound with moderate surface area (412 m2 g−1), but higher fluorescence quantum yields (64% in spin coated films), is described.
Time resolved fluorescence decays were also obtained in order to gain further insight into the aggregate formation, i.e., AIE in Poly-t-Bu. The fluorescence decays of Poly-t-Bu in THF and THF:water mixtures were found to be well fitted with a bi-exponential decay law (Fig. SI4, ESI†), thus indicating the presence of two emitting species in the aggregate region (λem = 525 nm). This possibly indicates that the emissive aggregate results from two main conformers with different contributions. Indeed, the fractional contribution (Ci), in Table 4, of each species is given by the following equation56
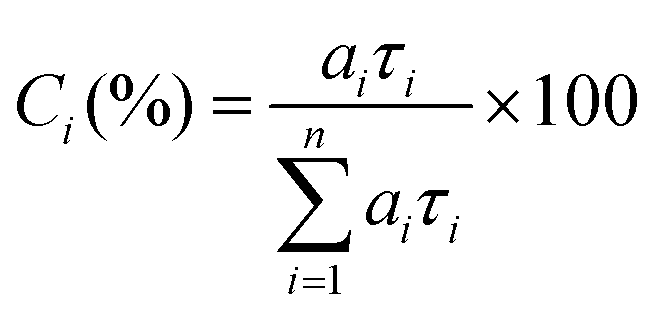 | (1) |
| Solvent | a 1 | τ 1 (ns) | a 2 | τ 2 (ns) | χ 2 | ϕ F | %C1 | %C2 | k F (ns−1) | k NR (ns−1) |
|---|---|---|---|---|---|---|---|---|---|---|
a Experimental conditions: nanoLed λex = 339 nm; λem = 525 nm; 48.1 ps per channel, 3k counts.
b
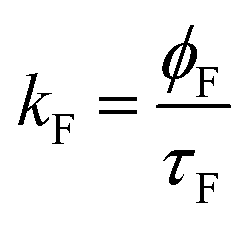 .
c .
c
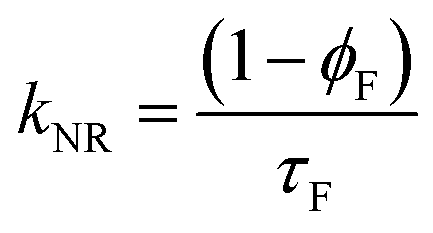 . .
|
||||||||||
| 100% THF:0% water |
0.947 | 0.23 | 0.053 | 1.45 | 1.01 | 0.046 | 73.9 | 26.1 | 0.032 | 0.658 |
| 80% THF:20% water |
0.768 | 0.5 | 0.232 | 1.87 | 1.29 | 0.161 | 47.0 | 53.0 | 0.086 | 0.449 |
| 50% THF:50% water |
0.692 | 0.78 | 0.308 | 2.36 | 1.27 | 0.332 | 42.6 | 57.4 | 0.141 | 0.283 |
| 30% THF:70% water |
0.627 | 0.98 | 0.373 | 2.74 | 1.35 | 0.485 | 37.5 | 62.5 | 0.177 | 0.188 |
| 10% THF:90% water |
0.601 | 1.23 | 0.399 | 3.3 | 1.05 | 0.612 | 36.0 | 64.0 | 0.185 | 0.118 |
The decay time values together with the fractional contribution of each species (Ci) at the aggregate emission wavelength (525 nm) change with the increase of the water fraction in the mixture (Fig. 5). For Poly-t-Bu in THF a fast decay component, τ1, in the 0.23–1.2 ns range and a longer decay time, τ2, ranging from 1.5 ns to 3.3 ns were found on going from 0% to 90% water content (Table 4). As shown in Fig. 5 the fluorescence quantum yields and the fluorescence lifetimes follow a similar trend, i.e., a concomitant increase of these parameters with the increase of the water fraction. In addition, the determination of the radiative (kF) and radiationless (kNR) rate constants clearly shows a decrease of kNR and an increase of kF with fW. This is valid for the two decay components, but in Table 4 the data are only shown for the longer (τ2) component. The radiationless decay is therefore dominant up to fW = 70%, and from there on the radiative deactivation begins to dominate.
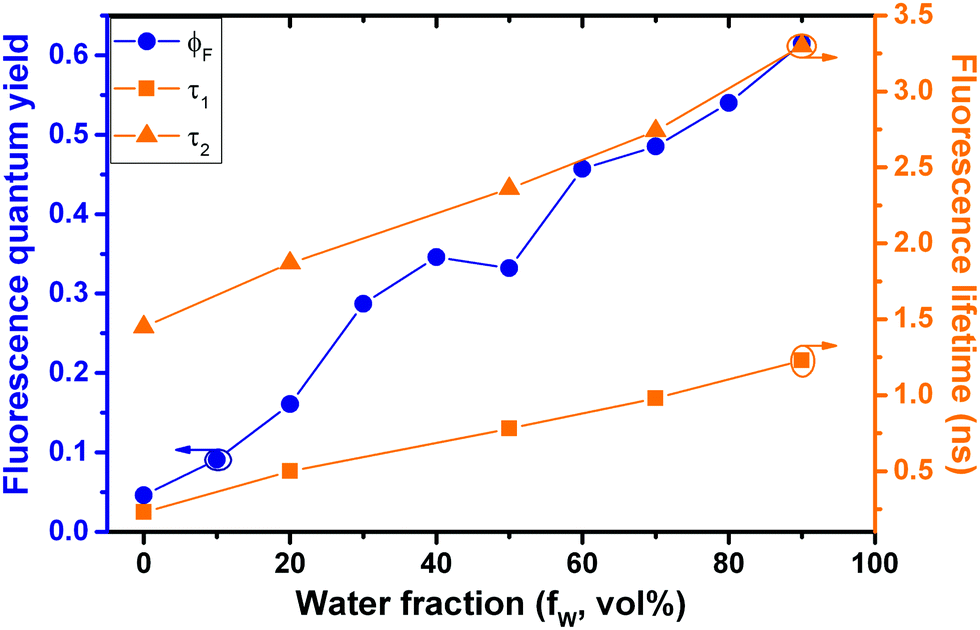 | ||
| Fig. 5 Correlation of fluorescence quantum yields and fluorescence decay times for Poly-t-Bu in THF:water mixtures with increasing water fraction (fW). | ||
Conclusions
In summary, in this work we have synthesized propeller-shaped structures derived from tetraphenylethylene (TPE). The optical and PL properties of these compounds were studied in the solid state (powder and films) and in solution (THF and THF:water mixtures). Their properties were compared to the model compound TPE in similar experimental conditions. Mono-t-Bu and Mono-Np, monomeric model compounds with para-position tert-butyl substituents and naphthalene-substituted ethenes, respectively, showed AIE active properties in the solid state and in THF:water mixtures. This behavior is attributed to RIR in the solid state and when the compounds aggregate with the addition of the “poor solvent” (water), which hinders the excited state radiationless channels. The polymer Poly-t-Bu was found to be more fluorescent than its monomer Mono-t-Bu and to retain its AIE active properties. The ϕF value of Poly-t-Bu is found to be one order of magnitude higher in the solid state than in solution (ϕF = 64% in film vs. 4.6% in THF). This is accompanied by distinct microporosity in the solid state with a high SBET surface area of 417 m2 g−1, probably driven by the steric demand of the tert-butyl groups. Poly-t-Bu, therefore, represents a so-called conjugated polymer of intrinsic microporosity (cPIM) with an attractive combination of intrinsic microporosity and occurrence of distinct AIE effects, thus predestining further experiments into stimuli-responsive properties in contact with suited analytes. In contrast, the diphenyl-dinaphthylethylene derivative polymers, Poly-Np1 and Poly-Np2, are less emissive than Mono-Np and presented ACQ properties. These properties are assigned to the occurrence of strong π–π interactions in these polymers, leading to the formation of non-fluorescent H-aggregates. The occurrence of π–π interactions may also lead to tight packing of Poly-Np1 and Poly-Np2, since these polymers do not show intrinsic microporosity (SBET < 17 m2 g−1).Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by Project “Hylight” (no. 031625) 02/SAICT/2017 which is funded by the Portuguese Science Foundation and Compete Centro 2020. We acknowledge funding from Fundo Europeu de Desenvolvimento Regional (FEDER) through Programa Operacional Factores de Competitividade (COMPETE) and project ROTEIRO/0152/2013. The Coimbra Chemistry Centre is supported by the Fundacão para a Ciência e a Tecnologia (FCT), Portuguese Agency for Scientific Research, through the Project PEst-OE/QUI/UI0313/2014. The research leading to these results has received funding from Laserlab-Europe (grant agreement no. 284464, EC's Seventh Framework Programme).Notes and references
- Y. Tang and B. Z. Tang, Principles and Applications of Aggregation-Induced Emission, Springer International Publishing, Cham, 2019 Search PubMed.
- Y. Hong, J. W. Lam and B. Z. Tang, Chem. Commun., 2009, 4332–4353, 10.1039/b904665h.
- J. B. Birks, Photophysics of Aromatic Molecules, Wiley- Interscience, London, 1970 Search PubMed.
- S. Baysec, E. Preis, S. Allard and U. Scherf, Macromol. Rapid Commun., 2016, 37, 1802–1806 CrossRef CAS PubMed.
- J. Chen, C. C. W. Law, J. W. Y. Lam, Y. Dong, S. M. F. Lo, I. D. Williams, D. Zhu and B. Z. Tang, Chem. Mater., 2003, 15, 1535–1546 CrossRef CAS.
- W. Z. Yuan, Y. Gong, S. Chen, X. Y. Shen, J. W. Y. Lam, P. Lu, Y. Lu, Z. Wang, R. Hu, N. Xie, H. S. Kwok, Y. Zhang, J. Z. Sun and B. Z. Tang, Chem. Mater., 2012, 24, 1518–1528 CrossRef CAS.
- Y. Dong, J. W. Y. Lam, A. Qin, J. Liu, Z. Li, B. Z. Tang, J. Sun and H. S. Kwok, Appl. Phys. Lett., 2007, 91, 011111 CrossRef.
- X. Du, J. Qi, Z. Zhang, D. Ma and Z. Y. Wang, Chem. Mater., 2012, 24, 2178–2185 CrossRef CAS.
- S. Biswas, D. Jana, G. S. Kumar, S. Maji, P. Kundu, U. K. Ghorai, R. P. Giri, B. Das, N. Chattopadhyay, B. K. Ghorai and S. Acharya, ACS Appl. Mater. Interfaces, 2018, 10, 17409–17418 CrossRef CAS PubMed.
- J. Huang, Y. Jiang, J. Yang, R. Tang, N. Xie, Q. Li, H. S. Kwok, B. Z. Tang and Z. Li, J. Mater. Chem. C, 2014, 2, 2028–2036 RSC.
- G. Liang, J. W. Y. Lam, W. Qin, J. Li, N. Xie and B. Z. Tang, Chem. Commun., 2014, 50, 1725–1727 RSC.
- M. H. Chua, H. Zhou, T. T. Lin, J. Wu and J. Xu, ACS Omega, 2018, 3, 16424–16435 CrossRef CAS.
- J. M. Hoyt and C. E. Frank, US Pat., 3345332, Apr. 2, 1964, patented Oct. 3, 1967 Search PubMed.
- H. H. Hörhold and D. Raabe, Acta Polym., 1979, 30, 86–92 CrossRef.
- H. Reisch, U. Wiesler, U. Scherf and N. Tuytuylkov, Macromolecules, 1996, 29, 8204–8210 CrossRef CAS.
- E. Preis and U. Scherf, Macromol. Rapid Commun., 2006, 27, 1105–1109 CrossRef CAS.
- W. J. Feast and I. S. Millichamp, Polym. Commun., 1983, 24, 102–103 CAS.
- C. Belton, D. F. O'Brien, W. J. Blau, A. J. Cadby, P. A. Lane, D. D. C. Bradley, H. J. Byrne, R. Stockmann and H. H. Hörhold, Appl. Phys. Lett., 2001, 78, 1059–1061 CrossRef CAS.
- Q. Chen, J.-X. Wang, F. Yang, D. Zhou, N. Bian, X.-J. Zhang, C.-G. Yan and B.-H. Han, J. Mater. Chem., 2011, 21, 13554–13560 RSC.
- Y. Xu, L. Chen, Z. Guo, A. Nagai and D. Jiang, J. Am. Chem. Soc., 2011, 133, 17622–17625 CrossRef CAS PubMed.
- S. Dalapati, C. Gu and D. Jiang, Small, 2016, 12, 6513–6527 CrossRef CAS PubMed.
- J. C. Jansen, E. Esposito, A. Fuoco and M. Carta, Polymers, 2019, 11, 844, DOI:10.3390/polym11050844 CrossRef PubMed.
- G. Cheng, B. Bonillo, R. S. Sprick, D. J. Adams, T. Hasell and A. I. Cooper, Adv. Funct. Mater., 2014, 24, 5219–5224 CrossRef CAS.
- A. Palma-Cando, D. Woitassek, G. Brunklaus and U. Scherf, Mater. Chem. Front., 2017, 1, 1118–1124 RSC.
- V. S. Mothika, A. Räupke, K. O. Brinkmann, T. Riedl, G. Brunklaus and U. Scherf, ACS Appl. Nano Mater., 2018, 1, 6483–6492 CrossRef CAS.
- Y. Xu, S. Jin, H. Xu, A. Nagai and D. Jiang, Chem. Soc. Rev., 2013, 42, 8012–8031 RSC.
- A. Patra and U. Scherf, Chem. – Eur. J., 2012, 18, 10074–10080 CrossRef CAS PubMed.
- D. Chen, C. Liu, J. Tang, L. Luo and G. Yu, Polym. Chem., 2019, 10, 1168–1181 RSC.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 3–8 CrossRef PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
- J. Pina, J. S. D. Melo, H. D. Burrows, A. Bilge, T. Farrell, M. Forster and U. Scherf, J. Phys. Chem. B, 2006, 110, 15100–15106 CrossRef CAS PubMed.
- D. P. Law, A. B. Blakeney and R. Tkachuk, J. Near Infrared Spectrosc., 1996, 4, 189–193 CrossRef CAS.
- G. Striker, V. Subramaniam, C. A. M. Seidel and A. Volkmer, J. Phys. Chem. B, 2002, 103, 8612–8617 CrossRef.
- C. Gao, J. Y. Seow, B. Zhang, C. R. Hall, A. J. Tilley, J. M. White, T. A. Smith and W. W. H. Wong, ChemPlusChem, 2019, 746–753, DOI:10.1002/cplu.201900100.
- F. De Nisi, R. Francischello, A. Battisti, A. Panniello, E. Fanizza, M. Striccoli, X. Gu, N. L. C. Leung, B. Z. Tang and A. Pucci, Mater. Chem. Front., 2017, 1, 1406–1412 RSC.
- J. Zhou, Z. Chang, Y. Jiang, B. He, M. Du, P. Lu, Y. Hong, H. S. Kwok, A. Qin, H. Qiu, Z. Zhao and B. Z. Tang, Chem. Commun., 2013, 49, 2491–2493 RSC.
- H.-H. Hörhold, J. Gottschaldt and J. Opfermann, J. Prakt. Chem., 1977, 319, 611–621 CrossRef.
- H.-H. Hörhold, M. Helbig, D. Raabe, J. Opfermann, U. Scherf, R. Stockmann and D. Weiß, Z. Chem., 1987, 27, 126–137 CrossRef.
- S. Gonzalez, R. Pelaez, F. Sanz, M. B. Jimenez, J. R. Moran and M. C. Caballero, Org. Lett., 2006, 8, 4679–4682 CrossRef CAS PubMed.
- D. Sek, Polym. J., 1981, 13, 13–22 CrossRef CAS.
- C. Friedel and J. Crafts, C. R. Chim., 1877, 84, 1392–1396 Search PubMed.
- T. Mukaiyama, T. Sato and J. Hanna, Chem. Lett., 1973, 1041–1044 CrossRef CAS.
- A. C. B. Rodrigues, J. Pina, W. Dong, M. Forster, U. Scherf and J. S. S. D. Melo, Macromolecules, 2018, 51, 8501–8512 CrossRef CAS.
- W. Tian, T. Lin, H. Chen and W. Wang, ACS Appl. Mater. Interfaces, 2019, 11, 6302–6314 CrossRef CAS PubMed.
- G. F. Zhang, Z. Q. Chen, M. P. Aldred, Z. Hu, T. Chen, Z. Huang, X. Meng and M. Q. Zhu, Chem. Commun., 2014, 50, 12058–12060 RSC.
- J. Mei, Y. Hong, J. W. Y. Lam, A. Qin, Y. Tang and B. Z. Tang, Adv. Mater., 2014, 26, 5429–5479 CrossRef CAS PubMed.
- B.-K. An, S.-K. Kwon, S.-D. Jung and S. Y. Park, J. Am. Chem. Soc., 2002, 124, 14410–14415 CrossRef CAS PubMed.
- H. Zhang, J. Liu, L. Du, C. Ma, N. L. C. Leung, Y. Niu, A. Qin, J. Sun, Q. Peng, H. H. Y. Sung, I. D. Williams, R. T. K. Kwok, J. W. Y. Lam, K. S. Wong, D. L. Phillips and B. Z. Tang, Mater. Chem. Front., 2019, 1143–1150, 10.1039/c9qm00156e,.
- G. Tian, D. Sun, Y. Zhang and X. Yu, Angew. Chem., Int. Ed., 2019, 58, 5951–5955 CrossRef CAS PubMed.
- J. Mei, N. L. C. Leung, R. T. K. Kwok, J. W. Y. Lam and B. Z. Tang, Chem. Rev., 2015, 115, 11718–11940 CrossRef CAS PubMed.
- Y. Cai, L. Du, K. Samedov, X. Gu, F. Qi, H. H. Y. Sung, B. O. Patrick, Z. Yan, X. Jiang, H. Zhang, J. W. Y. Lam, I. D. Williams, D. Lee Phillips, A. Qin and B. Z. Tang, Chem. Sci., 2018, 9, 4662–4670 RSC.
- Z. Qiu, X. Liu, J. W. Y. Lam and B. Z. Tang, Macromol. Rapid Commun., 2019, 40, 1–15 CrossRef PubMed.
- E. Preis, W. Dong, G. Brunklaus and U. Scherf, J. Mater. Chem. C, 2015, 3, 1582–1587 RSC.
- C. Gu, N. Huang, Y. Wu, H. Xu and D. Jiang, Angew. Chem., Int. Ed., 2015, 54, 11540–11544 CrossRef CAS PubMed.
- C. S. de Castro, T. F. G. G. Cova, A. C. C. Pais, D. Pinheiro, C. Nuñez, C. Lodeiro and J. S. Seixas de Melo, Dyes Pigm., 2016, 134, 601–612 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Detailed synthesis and structural characterization of model compounds and polymers; crystal data and structure refinement of monomers; nitrogen gas adsorption and desorption isotherms of polymers and their BET surface area fits and room temperature fluorescence decays for Poly-t-Bu. CCDC 1961126 and 1961127. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9tc06028f |
| ‡ These authors contributed equally to the work. |
| This journal is © The Royal Society of Chemistry 2020 |