Nitrogen doped carbon aerogel composites with TiO2 and ZnO prepared by atomic layer deposition†
Received
31st October 2019
, Accepted 4th April 2020
First published on 16th April 2020
Abstract
A nitrogen doped carbon aerogel was used as a substrate for the atomic layer deposition of TiO2 and ZnO layers in various thicknesses. The bare and composite products were analyzed using Raman spectroscopy, XRD, N2 adsorption, SEM-EDX, TEM, XPS, and ICP-OES and their photocatalytic activity was investigated in decomposing methyl orange dye under UV light irradiation. We investigated the effect of the different metal oxides and their thickness dependence on the photocatalytic activity.
1. Introduction
Carbon aerogels have been widely investigated nanostructured materials since Pekala et al. reported a synthesis method for them from resorcinol and formaldehyde in 1989. Because of their high specific surface area, porous structure and very low density, they have potential applications in many fields, including waste water treatment, heat insulation, electrochemistry and catalysis.1–6 By doping the carbon matrix of the aerogel with nitrogen, the electron distribution can be significantly altered through introducing more active sites and defects.5,7
TiO2 and ZnO are widely researched photocatalysts to decompose organic pollutants in the environment, since they are chemically stable, non-toxic, and possess high reactivity. As photo-excited charge carriers (electrons and holes) recombine rapidly, a co-catalyst can be helpful to inhibit this phenomenon in order to increase the photocatalytic activity.8–11 Carbon aerogels can be good candidates for this purpose via their electron acceptor nature, and their ability to increase the lifetime of the separated electron–hole pairs.12,13 Moreover, the presence of carbon may lead to a band gap narrowing effect in the semiconductor oxides, as it acts as a sensitizer to visible light, which is beneficial in applications utilizing solar radiation (though visible light activity is not the topic of this present study).14,15 Furthermore, carbon materials possess significant photocatalytic activity even by themselves, as it was reported for activated carbon, graphene oxide and carbon nanotubes, which is not a widely researched phenomenon.16–20
Carbon aerogel composites with semiconductor oxides can be prepared by numerous methods, e.g. by incorporating a titania precursor during the sol–gel process of the aerogel synthesis, or depositing an oxide with atomic layer deposition (ALD) on carbon aerogels.21,22 Among the various synthesis methods, ALD is unique as it allows the coating of the surface of nanostructures in a homogeneous way, and can be used for many substrates with different morphologies, e.g. nanospheres, nanotubes, fullerenes, nanofibers and nanoparticles equally.23–29
The purpose of our research was to synthesize a nitrogen doped carbon aerogel (NCA) and to use it as a substrate for the atomic layer deposition of titanium dioxide and zinc oxide. First, a polymer hydrogel was prepared by the sol–gel process, using formaldehyde, resorcinol and melamine. The latter was used to introduce nitrogen into the sample. The resulting gel was dried with supercritical carbon dioxide, which produced a polymer aerogel, and it was annealed in a nitrogen atmosphere to get the nitrogen doped carbon aerogel. Four NCA–metal oxide composites were made using atomic layer deposition (ALD), with TiO2 and ZnO, each with two different thicknesses. The NCA and the resulting composites were characterized with Raman spectroscopy, X-ray diffraction (XRD), low temperature N2 adsorption, scanning electron microscopy-energy-dispersive X-ray spectroscopy (SEM-EDX), transmission electron microscopy (TEM), X-ray photoelectron spectroscopy (XPS), and inductively coupled plasma-optical emission spectrometry (ICP-OES) and their photocatalytic activity was investigated by studying the decomposition of methyl orange dye under UV light irradiation. Special attention was given to the nitrogen content of the aerogel, as our group have already investigated carbon aerogel/titania composites where the aerogel was devoid of nitrogen.22
2. Experimental
2.1. Synthesis of the nitrogen doped carbon aerogel
1.5990 g resorcinol, 0.7326 g melamine, and 0.0208 g Na2CO3 in 48 cm3 distilled water were stirred for 15 minutes, until the melamine dissolved. After that, 6.5 cm3 aq. formaldehyde (36.5%) was added to the solution and stirred for an additional 5 minutes. Then it was poured into glass vials, sealed and kept in an oven at 85 °C for one week. The polymer hydrogels were removed from the tubes and soaked in acetone to replace the water in the gel with a less polar solvent, which is important for the supercritical drying with CO2. The drying was performed under 100 bar and at 42 °C for 80 minutes, yielding nitrogen doped polymer aerogels. To obtain nitrogen doped carbon aerogels (NCA), the polymer aerogels were carbonized in a rotating quartz tube under a dry nitrogen atmosphere (25 cm3 min−1) at 900 °C for 60 min.5,30,31
2.2. Atomic layer deposition
For the deposition of TiO2 and ZnO on the carbon aerogels, a Beneq TFS-200-186 flow ALD reactor was used in thermal mode, at 1 mbar pressure in the reaction chamber. The parameters of the ALD are shown in Table 1. One deposition cycle consisted of a 0.3 s pulse of the metallic precursor (TiCl4 and (C2H5)2Zn, respectively), a 3 s nitrogen purge, a 0.3 s pulse of H2O and a 3 s nitrogen purge, which was repeated for the given number of cycles. Our previous work on the thermal behavior of the NCA proved its stability at the applied ALD temperatures.30
Table 1 Sample names and ALD parameters
| Sample name |
Deposition cycles |
Deposited oxide |
Temperature [°C] |
| NCA–TiO2 60 |
60 |
TiO2 |
300 |
| NCA–TiO2 600 |
600 |
| NCA–ZnO 60 |
60 |
ZnO |
200 |
| NCA–ZnO 600 |
600 |
| NCA |
No deposition |
2.3. Characterization methods
Raman measurements were carried out by using a Jobin Yvon Labram Raman instrument equipped with an Olympus BX41 microscope by using a green (532 nm) Nd-YAG laser. X-ray diffraction (XRD) analysis was performed on a PANanalytical X’Pert Pro MPD X-ray diffractometer with Cu Kα radiation.
Nitrogen adsorption–desorption isotherms were recorded on a NOVA 2000e automated volumetric nitrogen gas adsorption instrument at −196 °C. The apparent surface area (SBET) was calculated with the Brunauer–Emmett–Teller (BET) model.32 The total pore volume (Vtot) was derived from the amount of nitrogen adsorbed at relative pressure p/p0 → 1 presuming that the pores are filled with liquid nitrogen. The micropore volume (W0) was calculated from the Dubinin–Radushkevich (DR) plot.33 As no kernel files are available for the atomic layer deposited systems necessary for DFT methods the pore size distribution was calculated from the desorption branch of the isotherms, using the Barrett–Joyner–Halenda (BJH) method.34
A LEO 1540 XB scanning electron microscope (SEM) was used to observe the surface morphology of the samples, in high vacuum mode with a secondary electron detector. Adhesive carbon tape was used to fasten the samples on a copper sample holder. To prevent the samples from charging, they were coated with Au/Pd for imaging. TEM images were taken with a JEOL JEM-ARM200F transmission electron microscope operating at 200 kV. The as-prepared samples were dispersed in ethanol with an ultrasonic bath for 5 minutes, and then a few drops of suspension were spread on Cu grids for TEM investigation.
EDX spectra were taken on a JEOL JSM-5500LV scanning electron microscope, and the approximate composition was calculated from three different spots on each sample. The oxidation states (and the atomic ratio of the elements) were studied by X-ray photoelectron spectroscopy with a SPECS instrument equipped with a Phoibos 150 MCD-9 analyzer. The Al Kα X-ray source was operated at 14 kV and 10.8 mA (150 W) and the analyzer was used in FAT mode with a pass energy of 20 eV in the case of high resolution spectra. CasaXPS software was used for data evaluation. The binding energy was correlated to the adventitious carbon C1s peak at 284.8 eV. ICP-OES measurements were performed to get the metal content of the composites on a Labtest Plasmalab ICP-OES spectrometer. Solutions were prepared by microwave-assisted digestion of the samples in a HCl–HNO3–HF mixture, and then the excess HF was neutralized with boric acid.
2.4. Photocatalysis
Analysis of the photocatalytic activity was carried out by adding 1.0 mg of the samples into 3 ml aqueous solution (concentration: 4 × 10−5 M) of methyl orange dye in quartz cuvettes. They were left in the dark for 24 hours for the adsorption equilibrium to occur, and then they were placed between two parallel Osram 18 W UV lamps (see the spectrum in Fig. S1, ESI†), 5 cm from each, and the decomposition of the methyl orange was followed by measuring the absorption of its most intense peak at 464 nm every half hour with a Jasco V-550 UV-vis spectrometer for four hours at ambient temperature. For the investigation of the cyclic reusability, after one photocatalytic cycle, the old methyl orange solution was replaced with fresh in the cuvettes, and the samples were put in the dark for one day, and then the UV illumination was started, and their activity was measured. This process was repeated, with three parallel measurements.
3. Results and discussion
3.1. Raman spectroscopy
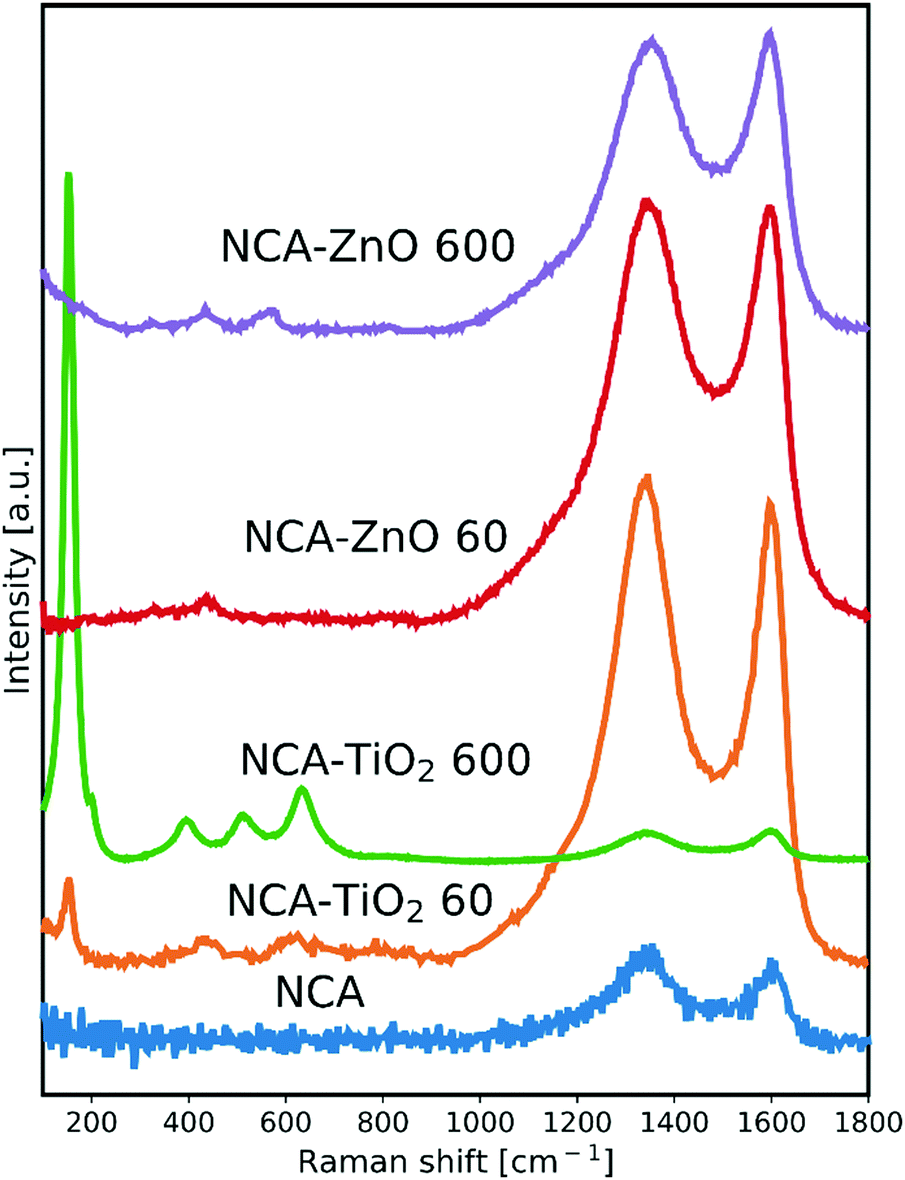
The Raman spectra are shown in Fig. 1. The NCA has only the typical D (disordered) and G (graphitic) peaks of carbon, which are visible in the spectrum of each sample. The ratio of their intensity (ID/IG) provides an indication of the level of graphitization (Table 2); its value of 1.20 for the bare aerogel points to the presence of structural defects. The utilized temperature and the exothermic nature of the reactions during the deposition caused this value to decrease, meaning that the carbon matrix became more ordered for the composites, particularly with 600 cycles as these samples spent the most time in the ALD reactor. Besides the D and G peaks, the presence of the metal oxide peaks in the spectra is apparent. In the NCA–TiO2 60 and NCA–TiO2 600 samples, the specific peaks for anatase TiO2 are present at 144, 400, 507 and 640 cm−1 with much higher intensity in the case of the thicker coating.35 The NCA–ZnO 60 and NCA–ZnO 600 samples show three bands of ZnO, at 332, 440 and 583 cm−1, but their intensity is much lower compared to the peaks of TiO2.36
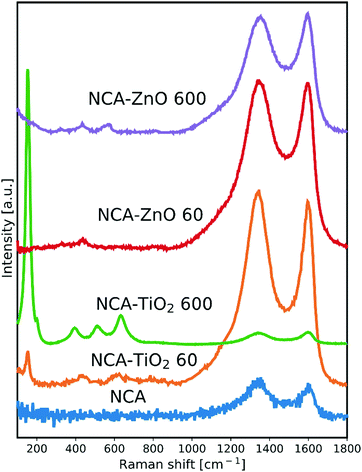 |
| | Fig. 1 Raman spectra of the samples, scaled for the visibility of the peaks. | |
Table 2 Position and intensity ratio of the D and G peaks of the carbon in the samples
|
|
NCA |
NCA–TiO2 60 |
NCA–TiO2 600 |
NCA–ZnO 60 |
NCA–ZnO 600 |
| D peak [cm−1] |
1355 |
1344 |
1343 |
1340 |
1353 |
| G peak [cm−1] |
1597 |
1596 |
1597 |
1595 |
1595 |
|
I
D/IG |
1.20 |
1.06 |
0.94 |
1.02 |
0.98 |
3.2. Powder XRD
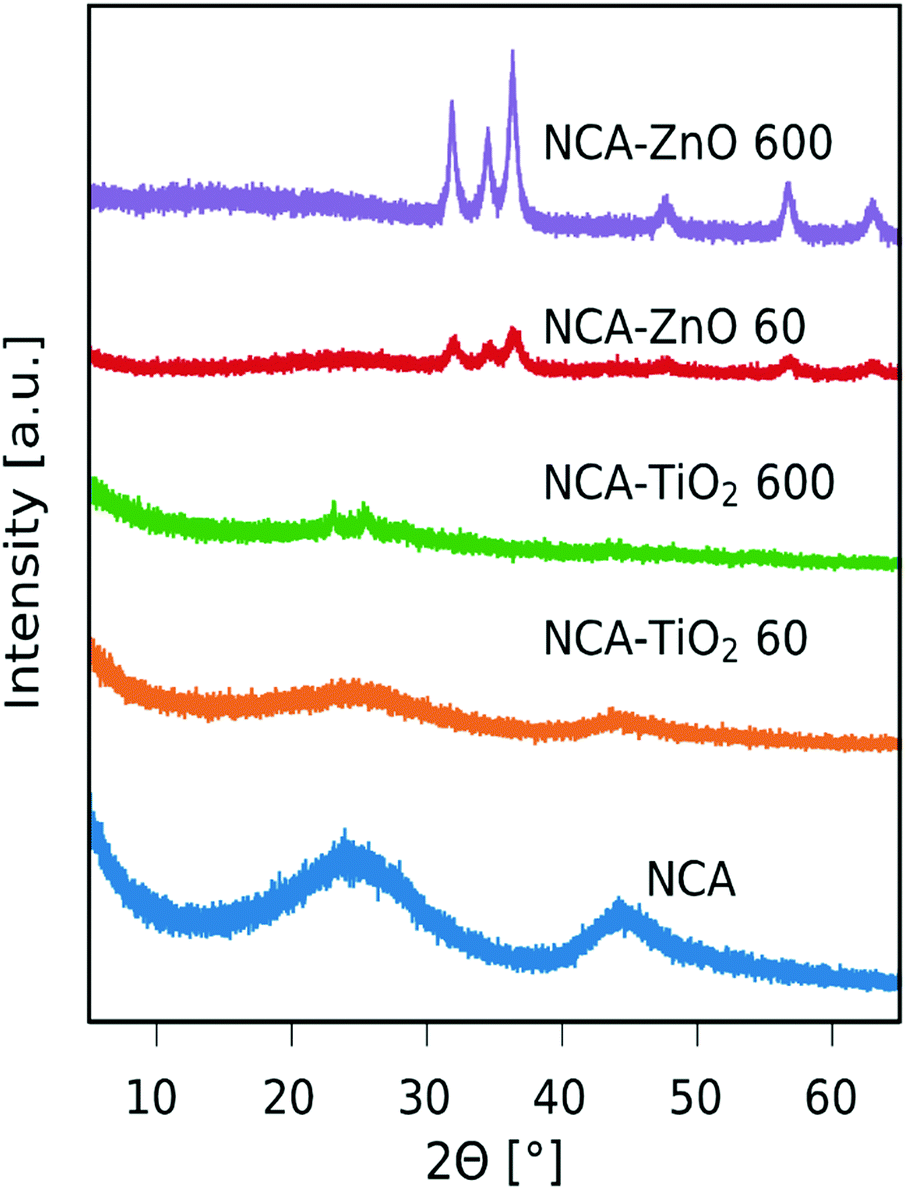
The recorded XRD patterns are shown in Fig. 2. The NCA is amorphous, only showing some reflections belonging to disordered carbon.37 The peaks corresponding to TiO2 are hardly visible, only an anatase peak is at around 2θ = 25° (ICDD card number: 01-075-2546) in the sample with thicker layers, from which the estimated crystallite size using the Scherrer equation is 7.1 nm. The anatase peak is not visible in the sample with the thinner coating. The peaks of ZnO were identified as hexagonal zinc oxide (ICDD card number: 01-080-4199) and have much higher intensity in the case of both composites compared to the TiO2 containing ones, mainly because the particles are larger in general and the coverage of the aerogel is better as seen in SEM and TEM images (Fig. 5 and 6). The approximate crystallite size for NCA–ZnO 60 was around 9.7 nm and it was 17.0 nm for NCA–ZnO 600; a thicker ZnO coating resulted in larger particles, and they caused higher intensity XRD peaks.
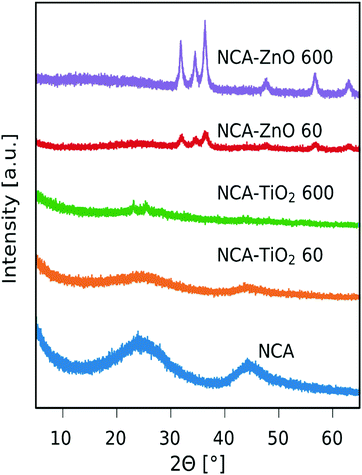 |
| | Fig. 2 XRD diffractograms of the samples. | |
3.3. Nitrogen adsorption
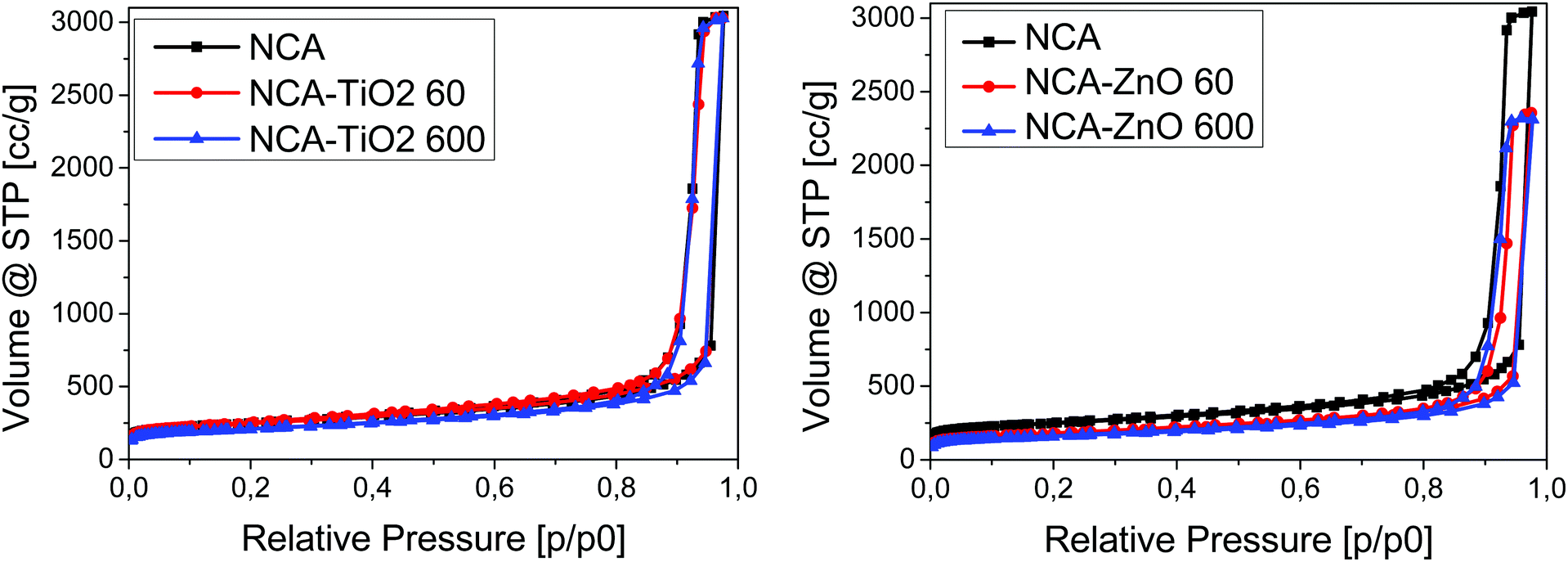
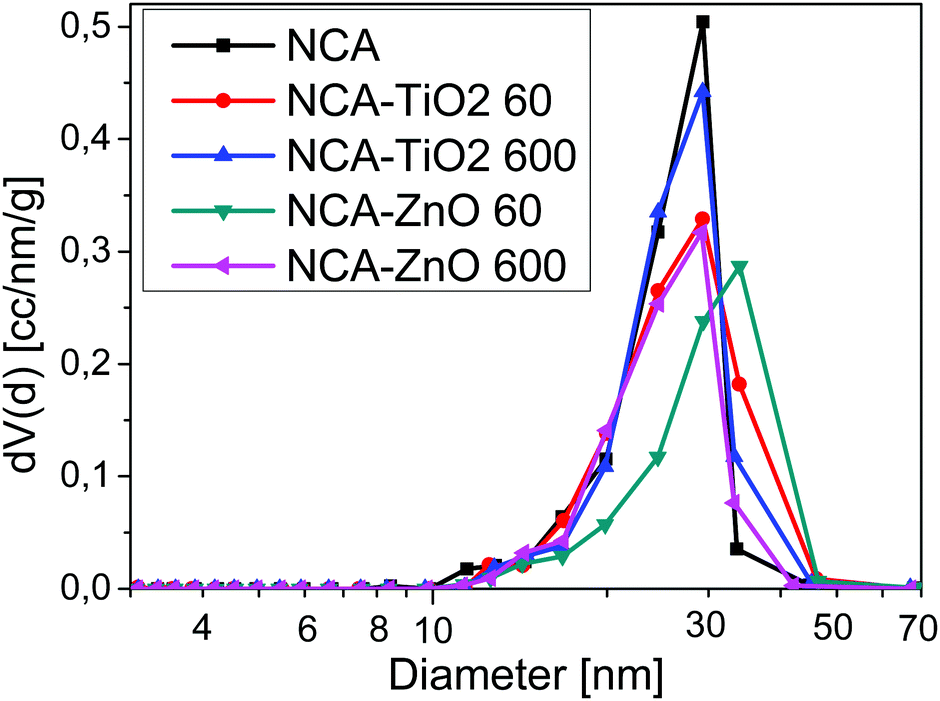
Low temperature nitrogen adsorption/desorption isotherms as well as SEM and TEM imaging were used to characterize the morphological consequences of the ALD treatments. The expected effect of the coating may be twofold. On one hand, it is related to the increased overall density of the samples: the density of the metal oxides (∼4.2 g cm−3 for TiO2 and ∼5.6 g cm−3 for ZnO) is substantially higher than that of the carbon matrix (∼2.2 g cm−3). On the other hand, the ALD layers may block and/or narrow the pores in a different way, limited by the diameter of the TiCl4 (0.64 nm) and [C2H5]2Zn molecules (around 0.7 nm).38,39 The isotherms, the pore size distribution curves and the parameters calculated from the isotherms are shown in Fig. 3 and 4, Fig. S2 (ESI†) and Table 3, respectively. Each sample has a significant specific surface area even after the deposition. The effect of the TiO2 and ZnO depositions, however, is different. The isotherms of the untreated and both TiO2 treated samples practically overlap. The 60-cycle TiO2 ALD coating has no effect on any of the nitrogen adsorption related parameters. The longer treatment results in the partial blocking of the micropores, reflected by the ca. 85% concomitant drop of the micropore volume and the apparent surface area. The outcome of the treatment is more pronounced in the SEM images (Fig. 5B and D), revealing the filled large macropores non-detectable by the gas adsorption method. The total pore volume of NCA–ZnO 60 and NCA–ZnO 600 decreased significantly, because the deposited ZnO forms thicker layers than TiO2 as seen in the SEM, EDX, XPS and ICP-OES results. Therefore, many pores became filled with ZnO and, consequently, this reduced the available surface area and pore volume.
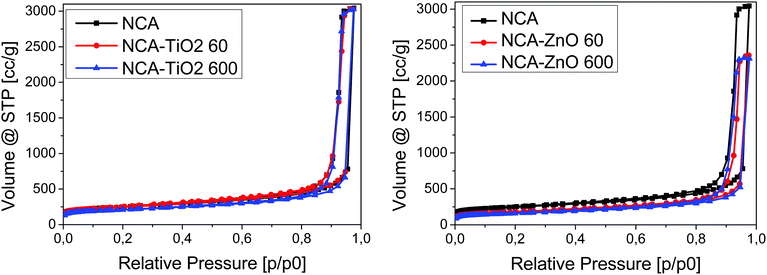 |
| | Fig. 3 Low temperature nitrogen adsorption–desorption isotherms. | |
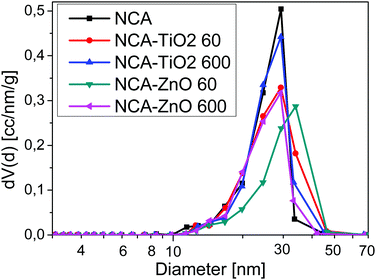 |
| | Fig. 4 Differential pore size distribution from the BJH model. | |
Table 3 Parameters calculated from the nitrogen adsorption–desorption isotherms
| Property |
NCA |
NCA–TiO2 60 |
NCA–TiO2 600 |
NCA–ZnO 60 |
NCA–ZnO 600 |
|
S
BET [m2 g−1] |
890 |
890 |
770 |
636 |
569 |
|
V
tot [cm3 g−1] |
4.70 |
4.68 |
4.68 |
3.65 |
3.58 |
|
W
0 [cm3 g−1] |
0.36 |
0.36 |
0.31 |
0.26 |
0.23 |
 |
| | Fig. 5 SEM pictures of the samples at 200k× magnification (A: NCA, B: NCA–TiO2 60, C: NCA–ZnO 60, D: NCA–TiO2 600, E: NCA–ZnO 600). | |
The effect of the ZnO deposition on the morphology is more obvious already from the adsorption isotherms: there is an increasing discrepancy in the N2 uptake. That is, not only the micropores but also the mesopores are blocked by the deposition already after 60 cycles. Interestingly, the drop of the micropore volume and the surface area is also very similar, 71 and 64% after 60 and 600 ZnO cycles, respectively. The total pore volume, however, shows an almost identical drop of 77%. The NCA–ZnO 60 sample shows a significant widening in the pore size distribution, but it shifts back to the untreated range in NCA–ZnO 600. It is very probable that the deposited ZnO further blocks the narrow pores, but by covering the walls of the wider pores previously “unseen” by nitrogen they shift into the measurable range. This is confirmed by the SEM image (Fig. 5E).
3.4. SEM
The SEM images in Fig. 5 show the morphology of the specimens. The porous surface and globular structure of the NCA are displayed clearly with its uniform texture. The metal oxides grew on the globular clusters. In particular, TiO2 coated the aerogels in spherical layers. Its presence is apparent when deposited in thicker layers, but barely visible in NCA–TiO2 60. The effective thickness of ZnO was greater with the same number of ALD cycles than TiO2 because of its higher growth rate. For NCA–ZnO 600, the aerogel was covered to the highest degree, and the globular morphology disappeared. The SEM images confirm the results of the surface area and pore volume measurements.
3.5. TEM
Fig. 6 shows the TEM images of the samples. The globular structure of the aerogel is present here as well. After ALD, the crystal lattice of the deposited metal oxide particles becomes visible on the amorphous carbon aerogel. The particles are crystalline with clearly visible lattice fringes, as can be seen in D and H for the TiO2 coated composites, and in F and J for the samples with ZnO. The approximate size of the metal oxide particles from TEM is 4–5 nm for NCA–TiO2 60, 7–8 nm for NCA–ZnO 60, 9–10 nm for NCA–TiO2 600 and 12–13 nm for NCA–ZnO 600, which is similar to the XRD results.
 |
| | Fig. 6 TEM images of the samples (A, B: NCA, C, D: NCA–TiO2 60, E, F: NCA–ZnO 60, G, H: NCA–TiO2 600, I, J: NCA–ZnO 600). | |
3.6. Elemental composition
The elemental composition from the EDX and XPS spectra in atomic% and metal content in weight% from EDX, XPS and ICP-OES measurements are displayed in Table 4. The values are different for EDX and XPS, because of the different sample preparation and depth of information provided by the two methods, and EDX is not that accurate to measure light (C, N, and O) elements precisely. For carbon, the trend in the EDX and XPS results aligns with other measurements; for NCA–TiO2 60, its amount decreased only slightly as it was the thinnest coating, for NCA–TiO2 600 and NCA–ZnO 60, it decreased further, and it become the smallest for NCA–ZnO 600. The amount of nitrogen present was the same after deposition from XPS, and varied greatly in EDX. The oxygen content increased with the presence of more deposited metal oxide. For the precise metal content, ICP-OES is the most accurate measurement method used, as it gives information about the bulk of the materials, not just locally from the surface. Its results align with the XPS in most samples, but the trend is the same in all cases for the metals in EDX as well. Less TiO2 was detected than ZnO with the same number of cycles, because the ALD growth rate per cycle is less for the TiO2, and the amount of metal increased when using 600 cycles instead of 60 as expected, as seen in the SEM images as well. Compared to our previous work on nitrogen-free carbon aerogel–TiO2 composites, where 200 ALD cycles were used to deposit TiO2 at 250 °C, the atomic% of Ti per ALD cycle was higher now according to EDX, even with 60 ALD cycles, as the presence of nitrogen could provide further active sites for the initial nucleation sites for the ALD reactions.22
Table 4 Elemental composition from EDX and XPS in atomic%, and metal content in weight% from EDX, XPS and ICP-OES
| Sample |
From EDX [at%] |
From XPS [at%] |
Ti or Zn [wt%] |
| C |
N |
O |
Ti |
Zn |
C |
N |
O |
Ti |
Zn |
EDX |
XPS |
ICP-OES |
| NCA |
96.7 |
0.9 |
2.4 |
|
|
93.9 |
1.5 |
4.7 |
|
|
|
|
|
| NCA–TiO2 60 |
90.9 |
0.6 |
7.6 |
1.0 |
|
93.3 |
1.5 |
5.1 |
0.1 |
|
3.8 |
0.4 |
0.244 |
| NCA–TiO2 600 |
75.3 |
2.2 |
15.1 |
7.2 |
|
89.2 |
1.5 |
8.4 |
1.0 |
|
22.7 |
3.8 |
4.01 |
| NCA–ZnO 60 |
82.9 |
0.2 |
11.0 |
|
5.9 |
89.5 |
1.4 |
7.1 |
|
2.0 |
24.7 |
9.8 |
10.3 |
| NCA–ZnO 600 |
11.3 |
0.1 |
22.4 |
|
66.2 |
78.3 |
1.1 |
15.8 |
|
4.9 |
89.7 |
21.0 |
35.7 |
3.7. XPS
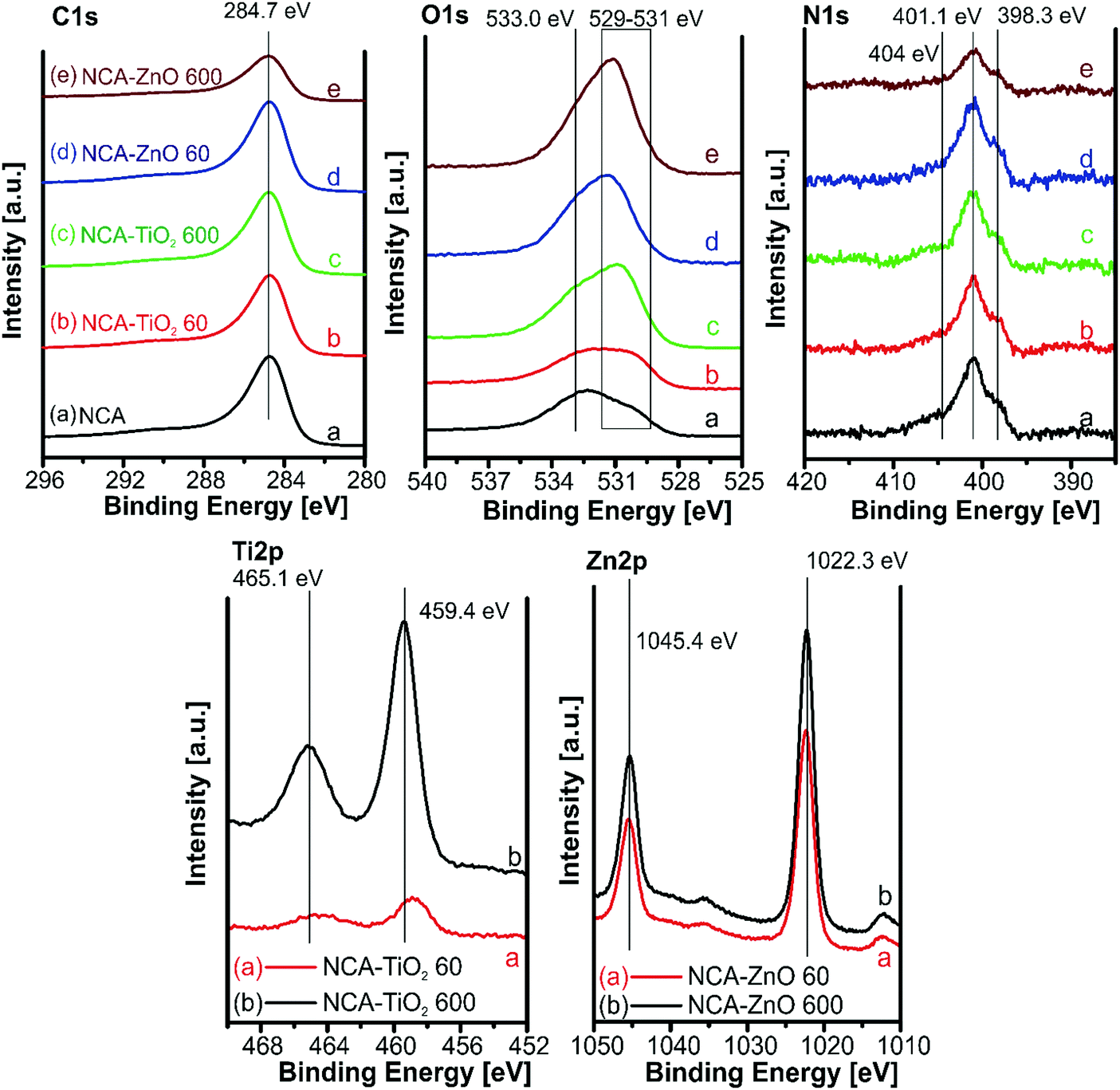
The XPS spectra for the samples are shown in Fig. 7. The amounts of atoms in different chemical states were calculated from the deconvoluted C, O and N peaks (Fig. S3–S5, ESI†), and are in Table 5. The carbon 1s peak was resolved into four components; their relative amount was similar in all samples. Oxygen has three overlapping peaks in the composites: one at 533 eV for the oxygen in the C–O–H bond, and two between 531.5 and 530 eV for the surface carbonate/carboxyl groups and the lattice oxygen; the latter was not present in the bare aerogel. All samples contained the same types of nitrogen, of which four types can be distinguished: pyridinic nitrogen in the lattice of the carbon structure at 398.4 eV, pyrollic at 400.4 eV, N–H bonds on the surface at 401.3 eV, and N–Ox around 404.7 eV. XPS further confirmed the presence of the metal oxides: the Ti2p region with a binding energy of 459 eV indicates that Ti is present as TiO2. The 1 eV shift of the Ti(IV) peaks to higher binding energies signals an electron deficient Ti environment and improper conductivity of the TiO2 particles in the composite, which also appeared in the lattice oxygen region. In the Zn2p region it is difficult to differentiate between Zn metal and ZnO, but the Zn Auger position is at 987 eV indicating that it is ZnO (Zn LMM peak in Fig. S6, ESI†). In the case of ZnO, no difference was observed between the peak positions obtained from the thinner and the thicker coating, as both coatings were relatively thick compared to TiO2.22,40,41
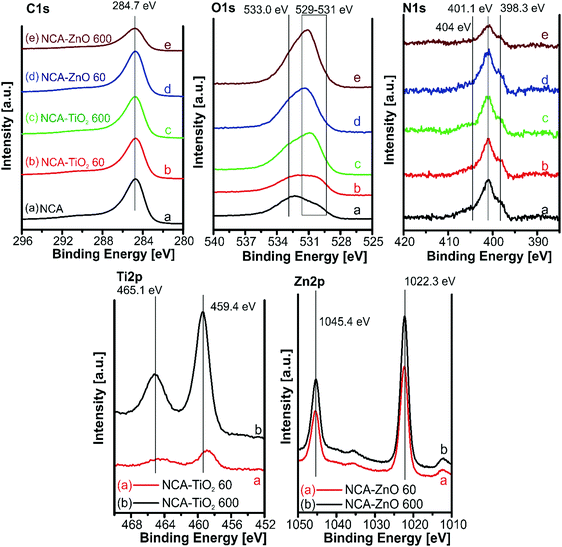 |
| | Fig. 7 XPS spectra for the samples in the different binding energy regions. | |
Table 5 Deconvolution of the C, O and N peaks of the XPS spectra
| C 1s peak [at%] |
C–C, C–H/graphitic 284.6 eV |
sp2 C–OH, C–O–C, C–N 285.8 eV |
sp3 C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, O–CO, C–N 287.4 eV O, O–CO, C–N 287.4 eV |
Loss feature 289.9 eV |
| NCA |
59.0 |
20.1 |
7.4 |
13.6 |
| NCA–TiO2 60 |
60.8 |
18.6 |
6.9 |
13.8 |
| NCA–TiO2 600 |
59.9 |
20.1 |
6.8 |
13.2 |
| NCA–ZnO 60 |
61.3 |
17.7 |
6.7 |
14.3 |
| NCA–ZnO 600 |
58.8 |
21.3 |
6.8 |
13.2 |
| O 1s peak [at%] |
CO 530–531.5 eV |
C–O– 533 eV |
Lattice oxygen 530–531.5 eV |
| NCA |
23.0 |
77.0 |
|
| NCA–TiO2 60 |
29.3 |
35.0 |
35.7 |
| NCA–TiO2 600 |
42.7 |
36.5 |
20.8 |
| NCA–ZnO 60 |
38.9 |
38.3 |
22.8 |
| NCA–ZnO 600 |
31.3 |
39.2 |
29.5 |
| N 1s peak [at%] |
Pyridinic N 398.4 eV |
Pyrrolic 400.4 eV |
Quaternary 401.3 eV |
N–Ox 404.7 eV |
| NCA |
21.1 |
15.9 |
41.7 |
21.4 |
| NCA–TiO2 60 |
20.3 |
18.1 |
39.3 |
22.8 |
| NCA–TiO2 600 |
16.2 |
16.8 |
44.9 |
22.1 |
| NCA–ZnO 60 |
24.6 |
12.9 |
41.7 |
20.8 |
| NCA–ZnO 600 |
23.5 |
12.6 |
46.9 |
17.0 |
3.8. Photocatalysis
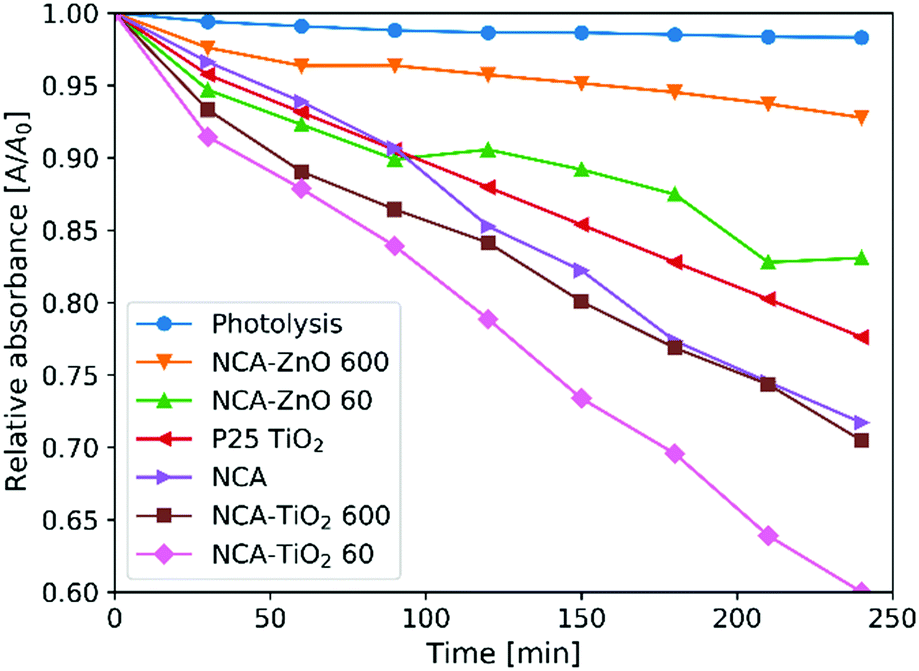
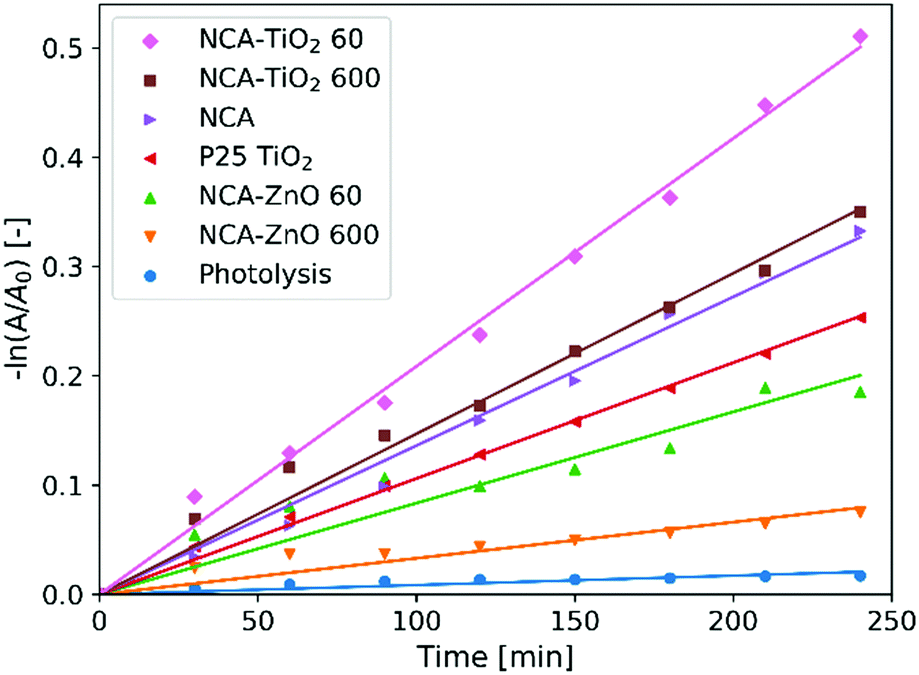
Fig. 8 shows the measurements of the photocatalytic activity. The apparent rate constants (kapp) of the decomposition (Table 6) were calculated from pseudo first order reaction kinetics (Fig. 9), by assuming the Langmuir–Hinshelwood mechanism.42 The coefficient of determination (R2) was above 0.96 in the case of all samples, making the assumption correct. The photolysis of the methyl orange dye was miniscule without a catalyst. P25 TiO2 powder was used for a reference material, as it is widely used and represents one of the most active forms of titania regarding the photocatalytic performance. The optical band gaps of the pure anatase TiO2 (3.2 eV) and ZnO (3.3 eV) are similar. The utilized UV source was radiating mainly at these energies (Fig. S1, ESI†).26 For our samples, the band gap was not possible to measure since all of them are dark materials with absorption in the whole wavelength range. In the case of the ZnO coated samples, the thinner layer showed considerably better activity, because of the 11% higher specific surface area and smaller oxide crystallite size. NCA–ZnO 600 was the worst of all the specimens, for its crystallites were the largest and the coverage of the NCA was the most complete. The bare NCA and the TiO2 containing samples exhibited better performance than P25 TiO2. The high activity of the NCA can be attributed to the presence of the nitrogen and oxygen containing surface groups on the carbon surface. The activity was similar to the NCA–TiO2 600 sample, while the latter had about 15% lower surface area compared to that of the NCA.43 The sample NCA–TiO2 60 showed the highest photocatalytic efficiency, decomposing about 40% of methyl orange dye after 240 minutes. The specific surface area and pore volumes were the same as for the bare NCA. The origin of this high activity is likely due to synergistic effects, improved charge separation and sensitization through the presence of C–TiO2 bonds and to the particle size of TiO2, which was the smallest of all in this case. The recombination of the photo-generated electron–hole pairs was inhibited as the low electrical resistance of the carbon aerogel makes it a suitable electron acceptor, so the charge carriers are removed from each other's vicinity.14,22,44 Another source of this enhancement effect may be similar to the one proposed by Zhang et al, where TiO2 nanoparticles were loaded on activated carbon. The adsorption of the dye onto the carbon material increases its concentration near the TiO2 particles, but when using only bare TiO2, the dye molecules have to collide with the TiO2 particles, which have limited surface area and thus fewer adsorption sites compared to a nanostructured carbon matrix, and this phenomenon was the most pronounced in the case of sample NCA–TiO2 60, which had the lowest coverage among all samples.45 The results of the cyclic reusability in Fig. S7 (ESI†) show that the activities decrease with repeated use, with significant standard deviations. The most pronounced activity decrease was observed for the TiO2 covered samples.
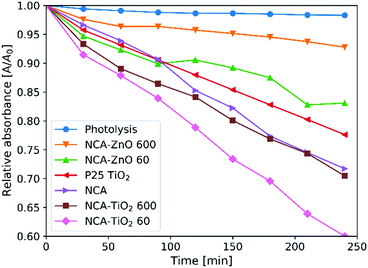 |
| | Fig. 8 Photocatalytic activity of the samples. | |
Table 6 Results of the photocatalytic measurements
| Sample |
Decomposition [%] |
k
app [10−4 min−1] |
R
2
|
| Photolysis |
1.7 |
0.9 |
0.9485 |
| P25 TiO2 |
22.4 |
11.0 |
0.9989 |
| NCA |
28.3 |
13.6 |
0.9962 |
| NCA–TiO2 60 |
40.0 |
21.0 |
0.9982 |
| NCA–TiO2 600 |
29.5 |
15.7 |
0.9956 |
| NCA–ZnO 60 |
16.9 |
8.4 |
0.9729 |
| NCA–ZnO 600 |
7.2 |
3.3 |
0.9699 |
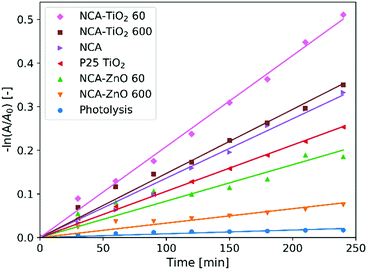 |
| | Fig. 9 Pseudo first order linear fitting for the photocatalysis. | |
Compared to previous work by our group, where the carbon aerogel was prepared without nitrogen, and the TiO2 coating was made with 200 ALD cycles at 250 °C, the N-free bare aerogel showed much better performance than the composite, which is in contrast to this case where the thin TiO2 coating improved the activity, and the NCA is similar to NCA–TiO2 600 in photocatalysis. This implies that the presence of nitrogen is beneficial for the photocatalytic activity of the composites.22 Regarding the initial adsorption of the methyl orange dye on the N-free and N-doped bare aerogels after 24 h, the amount was considerably higher when nitrogen was not present in the carbon matrix (Table S1, ESI†).
4. Conclusions
In this work, we prepared a nitrogen doped carbon aerogel, and deposited TiO2 and ZnO layers via ALD, with two different thicknesses for each metal oxide. The coating with the metal oxides was successful, as Raman spectroscopy showed the presence of anatase TiO2 and ZnO on the samples. XRD measurements identified the deposited ZnO as the wurtzite phase. The samples had significant specific surface area, total pore volume and micropore volume, which decreased in the composites, especially when depositing thicker layers. SEM and TEM images showed the globular morphology of the bare aerogel and the large porosity of the structures. According to the EDX and XPS results, nitrogen doping was successful, and the relative amount of the oxides showed that more ZnO was deposited than TiO2 under the same number of ALD cycles, as also observed in the SEM images. Photocatalytic decomposition of methyl orange dye was investigated, and all samples showed photocatalytic activity. The bare carbon aerogel and the samples coated with TiO2 were the most active ones, which showed even higher efficiency than that of P25 TiO2. The highest photocatalytic efficiency was observed for the one coated with thinner layers of TiO2.
Author contributions
The manuscript was written through the contributions of all authors. All authors have given approval to the final version of the manuscript.
Conflicts of interest
The authors declare no competing financial interest.
Acknowledgements
I. M. Szilágyi is thankful for a János Bolyai Research Fellowship of the Hungarian Academy of Sciences. The ÚNKP-18-4-BME-238 New National Excellence Program of the Ministry of Human Capacities, Hungary, and GINOP-2.2.1-15-2017-00084, GINOP-2.3.2-15-2016-00041, NRDI K 124212 and NRDI TNN_16 123631 grants are acknowledged. The work performed within project VEKOP-2.3.2-16-2017-00013 was supported by the European Union and the State of Hungary, co-financed by the European Regional Development Fund. The research reported in this paper was supported by the Higher Education Institutional Excellence Program of the Ministry of Human Capacities in the frame of Nanotechnology and Materials Science research area of Budapest University of Technology (BME FIKP-NAT) and the Energetics thematic programme of the University of Debrecen (NKFIH-1150-6/2019). J.W. Seo and C. Zhou are grateful to FWO (Research project G0B8915N) and Flemish Hercules Stichting (AKUL/13/19). The authors are thankful to György Bosznai (Department of Physical Chemistry and Materials Science, Budapest University of Technology and Economics) for his help in the synthesis of the aerogel and nitrogen adsorption measurements, Tamás Igricz (Department of Organic Chemistry and Technology, Budapest University of Technology and Economics) for the Raman measurements and László Bezur for the ICP-OES measurements (Department of Inorganic and Analytical Chemistry, Budapest University of Technology and Economics).
References
- R. W. Pekala, J. Mater. Sci., 1989, 24, 3221–3227 CrossRef CAS.
- C. Moreno-Castilla and F. J. Maldonado-Hódar, Carbon, 2005, 43, 455–465 CrossRef CAS.
- K. McEnaney, L. Weinstein, D. Kraemer, H. Ghasemi and G. Chen, Nano Energy, 2017, 40, 180–186 CrossRef CAS.
- C. Byrne, G. Subramanian and S. C. Pillai, J. Environ. Chem. Eng., 2018, 6, 3531–3555 CrossRef CAS.
- B. Nagy, S. Villar-Rodil, J. M. D. Tascón, I. Bakos and K. László, Microporous Mesoporous Mater., 2016, 230, 135–144 CrossRef CAS.
- J. Li, X. Wang, Q. Huang, S. Gamboa and P. J. Sebastian, J. Power Sources, 2006, 158, 784–788 CrossRef CAS.
- D. Long, J. Zhang, J. Yang, Z. Hu, G. Cheng, X. Liu, R. Zhang, L. Zhan, W. Qiao and L. Ling, Carbon, 2008, 46, 1259–1262 CrossRef CAS.
- D. Sudha and P. Sivakumar, Chem. Eng. Process., 2015, 97, 112–133 CrossRef CAS.
- S. Kansal, N. Kaur and S. Singh, Nanoscale Res. Lett., 2009, 4, 709–716 CrossRef CAS PubMed.
- K. Woan, G. Pyrgiotakis and W. Sigmund, Adv. Mater., 2009, 21, 2233–2239 CrossRef CAS.
- T. Xu, L. Zhang, H. Cheng and Y. Zhu, Appl. Catal., B, 2011, 101, 382–387 CrossRef CAS.
- H. Dong, G. Zeng, L. Tang, C. Fan, C. Zhang, X. He and Y. He, Water Res., 2015, 79, 128–146 CrossRef CAS PubMed.
- A. Di Mauro, M. E. Fragalà, V. Privitera and G. Impellizzeri, Mater. Sci. Semicond. Process., 2017, 69, 44–51 CrossRef CAS.
- R. Leary and A. Westwood, Carbon, 2011, 49, 741–772 CrossRef CAS.
- A. S. Alshammari, L. Chi, X. Chen, A. Bagabas, D. Kramer, A. Alromaeh and Z. Jiang, RSC Adv., 2015, 5, 27690–27698 RSC.
- L. F. Velasco, J. B. Parra and C. O. Ania, Appl. Surf. Sci., 2010, 256, 5254–5258 CrossRef CAS.
- L. F. Velasco, I. M. Fonseca, J. B. Parra, J. C. Lima and C. O. Ania, Carbon, 2012, 50, 249–258 CrossRef CAS.
- N. Justh, B. Berke, K. László, L. P. Bakos, A. Szabó, K. Hernádi and I. M. Szilágyi, Appl. Surf. Sci., 2018, 453, 245–251 CrossRef CAS.
- Y. Luo, Y. Heng, X. Dai, W. Chen and J. Li, J. Solid State Chem., 2009, 182, 2521–2525 CrossRef CAS.
- I. Velo-Gala, J. J. López-Peñalver, M. Sánchez-Polo and J. Rivera-Utrilla, Appl. Catal., B, 2017, 207, 412–423 CrossRef CAS.
- C. Moreno-Castilla, F. J. Maldonado-Hódar, F. Carrasco-Marín and E. Rodríguez-Castellón, Langmuir, 2002, 18, 2295–2299 CrossRef CAS.
- N. Justh, G. J. Mikula, L. P. Bakos, B. Nagy, K. László, B. Parditka, Z. Erdélyi, V. Takáts, J. Mizsei and I. M. Szilágyi, Carbon, 2019, 147, 476–482 CrossRef CAS.
- S. M. George, Chem. Rev., 2010, 110, 111–131 CrossRef CAS PubMed.
- J. Y. Park, S. W. Choi, J. W. Lee, C. Lee and S. S. Kim, J. Am. Ceram. Soc., 2009, 92, 2551–2554 CrossRef CAS.
- X. Meng, Y. Zhong, Y. Sun, M. N. Banis, R. Li and X. Sun, Carbon, 2011, 49, 1133–1144 CrossRef CAS.
- N. Justh, L. P. Bakos, K. Hernádi, G. Kiss, B. Réti, Z. Erdélyi, B. Parditka and I. M. Szilágyi, Sci. Rep., 2017, 7, 4337 CrossRef PubMed.
- N. Justh, T. Firkala, K. László, J. Lábár and I. M. Szilágyi, Appl. Surf. Sci., 2017, 419, 497–502 CrossRef CAS.
- H. A. Borbón-Nuñez, D. Dominguez, F. Muñoz-Muñoz, J. Lopez, J. Romo-Herrera, G. Soto and H. Tiznado, Powder Technol., 2017, 308, 249–257 CrossRef.
- H. Kim, H.-B.-R. Lee and W.-J. Maeng, Thin Solid Films, 2009, 517, 2563–2580 CrossRef CAS.
- L. P. Bakos, J. Mensah, K. László, T. Igricz and I. M. Szilágyi, J. Therm. Anal. Calorim., 2018, 134, 933–939 CrossRef CAS.
- O. Czakkel, E. Székely, B. Koczka, E. Geissler and K. László, Microporous Mesoporous Mater., 2012, 148, 34–42 CrossRef CAS.
- S. Brunauer, P. H. Emmett and E. Teller, J. Am. Chem. Soc., 1938, 60, 309–319 CrossRef CAS.
- M. M. Dubinin and L. V. Radushkevich, Proc. Natl. Acad. Sci. U. S. A., 1947, 55, 331–333 Search PubMed.
- H. Jin, H. Zhang, H. Zhong, J. Zhang, R. Mukundan, N. Garland, D. Myers, M. Wilson, F. Garzon, D. Wood, P. Zelenay, K. More, K. Stroh, T. Zawodzinski, J. Boncella, J. E. McGrath, M. Inaba, K. Miyatake, M. Hori, K. Ota, Z. Ogumi, S. Miyata, A. Nishikata, Z. Siroma, Y. Uchimoto, K. Yasuda, K. I. Kimijima and N. Iwashita, Energy Environ. Sci., 2011, 4, 3389 RSC.
- M. J. Šćepanović, M. Grujić-Brojčin, Z. D. Dohčević-Mitrović and Z. V. Popović, Sci. Sintering, 2009, 41, 67–73 CrossRef.
- M. Marie, S. Mandal and O. Manasreh, Sensors, 2015, 15, 18714–18723 CrossRef CAS PubMed.
- C. Macias, G. Rasines, T. García, M. Zafra, P. Lavela, J. Tirado and C. Ania, Gels, 2016, 2, 4 CrossRef PubMed.
- J. Aarik, A. Aidla, H. Mändar and T. Uustare, Appl. Surf. Sci., 2001, 172, 148–158 CrossRef CAS.
- J. Bacsa, F. Hanke, S. Hindley, R. Odedra, G. R. Darling, A. C. Jones and A. Steiner, Angew. Chem., Int. Ed., 2011, 50, 11685–11687 CrossRef CAS PubMed.
- T. Jia, F. Fu, D. Yu, J. Cao and G. Sun, Appl. Surf. Sci., 2018, 430, 438–447 CrossRef CAS.
- F. Kayaci, S. Vempati, C. Ozgit-Akgun, I. Donmez, N. Biyikli and T. Uyar, Nanoscale, 2014, 6, 5557–6188 RSC.
- Y. Yu, J. C. Yu, J.-G. Yu, Y.-C. Kwok, Y.-K. Che, J.-C. Zhao, L. Ding, W.-K. Ge and P.-K. Wong, Appl. Catal., A, 2005, 289, 186–196 CrossRef CAS.
- A. Gomis-Berenguer, L. F. Velasco, I. Velo-Gala and C. O. Ania, J. Colloid Interface Sci., 2017, 490, 879–901 CrossRef CAS PubMed.
- R. W. Pekala, J. C. Farmer, C. T. Alviso, T. D. Tran, S. T. Mayer, J. M. Miller and B. Dunn, J. Non-Cryst. Solids, 1998, 225, 74–80 CrossRef CAS.
- X. Zhang, M. Zhou and L. Lei, Carbon, 2005, 43, 1700–1708 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Spectrum of the UV lamp; integral pore size distribution; XPS survey spectra; deconvoluted XPS peaks; cyclic investigation of photocatalysis; methyl orange adsorption on bare aerogels. See DOI: 10.1039/c9tc05953a |
|
| This journal is © The Royal Society of Chemistry 2020 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  *a,
Joshua
Mensah
a,
Krisztina
László
*a,
Joshua
Mensah
a,
Krisztina
László