
Molecular conducting magnetic heterostructures†
 a
Shenqiang
Ren
*a
a
Shenqiang
Ren
*a
Abstract
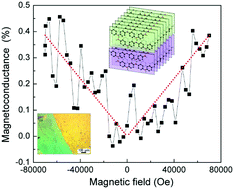
Building molecular conducting magnets has been of considerable interest due to their metallic transport and magnetic properties. Conjugated aniline anions and transition-metal cations are recognized to synthesize such π–d interaction frameworks. Here, an interfacial assembly of quasi-two-dimensional aniline heterojunction frameworks is developed, which consists of a proton-aniline conducting layer (conductivity of 9.2 × 102 S cm−1) and a transition metal–aniline magnetic layer. The two-dimensional aniline coordination framework with pronounced π–d interactions is indispensable for obtaining ordered spin states and metallic transport conductivity. We apply optical and Raman spectroscopy to study the interactions between the metal cations and nitrogen atoms in the aniline chains. The bilayer heterostructure maintains a high conductivity of 3.5 × 102 S cm−1 and increased magnetization, while the coupling of metallic conducting and magnetic layers leads to an enhanced magnetoconductance of 0.4% under a magnetic field of 70 kOe.
 Please wait while we load your content...
Please wait while we load your content...

