
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEpitaxial growth of light-responsive azobenzene molecular crystal actuators on oriented polyethylene films†
Shaji
Varghese
 *a,
Sebastian
Fredrich
a,
Ghislaine
Vantomme
b,
Sugosh R.
Prabhu
c,
Joan
Teyssandier
c,
Steven
De Feyter
c,
John
Severn
ad,
Cees W. M.
Bastiaansen
ae and
Albertus P. H. J.
Schenning
*ab
*a,
Sebastian
Fredrich
a,
Ghislaine
Vantomme
b,
Sugosh R.
Prabhu
c,
Joan
Teyssandier
c,
Steven
De Feyter
c,
John
Severn
ad,
Cees W. M.
Bastiaansen
ae and
Albertus P. H. J.
Schenning
*ab
aStimuli-responsive Functional Materials and Devices, Department of Chemical Engineering and Chemistry, Eindhoven University of Technology, P.O. Box 513, 5600 MB Eindhoven, The Netherlands. E-mail: shaji_varghese@fas.harvard.edu; A.P.H.J.Schenning@tue.nl
bInstitute for Complex Molecular Systems, Eindhoven University of Technology, P.O. Box 513, 5600 MB Eindhoven, The Netherlands
cDivision of Molecular Imaging and Photonics, Department of Chemistry, KU Leuven, Celestijnenlaan 200F, B 3001, Leuven, Belgium
dDSM Materials Science Center, NL-6160 MD Geleen, The Netherlands
eSchool of Engineering and Materials Science, Queen Mary, University of London, Mile End Road London, E1 4NS, London, UK
First published on 9th December 2019
Abstract
We report on the epitaxial growth of photoresponsive alkyl-substituted azobenzene fibres on top of uniaxially oriented polyethylene (PE) films. In these fibres, the alkyl chains are oriented parallel with regard to the drawing direction of PE, whereas the azobenzene moieties pack into a roughly 60° angle. The bilayer films act as a light responsive actuator generating an actuation stress of about 3 MPa.
Introduction
Controlling the organisation of matter at the molecular scale such as the alignment of molecules on a polymeric substrate has many advantages. Aligned self-assembled molecule arrays on polymer films might result in functional materials with anisotropic properties.1–6 Common techniques to achieve a uniform directionality of the deposited molecules on films include the use of electric7 or magnetic fields,8 and photoalignment9,10 as well as rubbing of the surface,11 or shear forces.12Highly anisotropic epitaxial growth of organic crystals on substrates13 is a fast and easy way of aligning molecules to produce composite materials. In comparison with other alignment techniques, epitaxial growth does not need to be externally activated and remains physically stable. However, the substrate needs to be (partly) crystalline in order to induce the growth of crystals on its surface. Anisotropic epitaxial growth of organic crystals has applications for example in organic semiconductors14 and biomimetic surfaces15 with unidirectional wetting16 and switchable adhesion properties.17,18 So far, such materials have not been applied as soft actuators.
The conversion of light energy into mechanical work in a cheap and simple fashion is of importance in the field of soft actuator research. Regarding untethered actuation upon application of light, the integration of light responsive molecular crystal actuators onto flexible high modulus crystalline substrates is appealing for the development of photo-responsive functional materials. A common way to modify the properties of soft materials by a light stimulus is to incorporate photochromic moieties, such as azobenzenes in these polymers.19 Azobenzene molecules are known to barely switch in the crystalline state due to the large geometrical changes upon isomerisation accompanied by different crystal lattices or unit cells of both isomers. Some examples of azobenzene (co-)crystal actuators are known, which undergo a shape change or deforming crack-formation upon irradiation.20,21 In addition photo-induced melting of alkyl substituted azobenzene crystals can be observed upon irradiation.22,23
It was previously shown that n-alkanes (paraffin waxes) can grow epitaxially on oriented PE24,25 if their symmetry matches,26,27 and that small molecules like pentacene can grow on photoaligned polyimide.28,29 We now report on the epitaxial crystal growth of photo-responsive azobenzene derivatives with long aliphatic chains on highly oriented polyethylene (o-PE) films. The molecular crystals have a rod-like shape with a preferred orientation on the flexible PE films. The PE films act as light responsive actuators able to generate a stress of about 3 MPa.
Results and discussion
Considering the van der Waals interaction of n-alkanes with o-PE, we designed an alkyl chain of 20 carbon atoms on each side of the azobenzene core (1). Azobenzene was chosen as the rigid core in order to create a light-responsive functionality. Additionally, an o-hydroxy azobenzene unit with C20-chains on either side (2) was chosen to study the effect of molecular symmetry and also to compare the effect of the thermal cis–trans back isomerisation lifetime on the epitaxial morphology (Scheme 1a). The fast thermal relaxation of o-hydroxy azobenzene is caused by a partial single bond character of the diazo bond due to a tautomeric structure with the hydroxy-proton shifting to the opposite nitrogen atom simultaneously creating a ketone as the driving force.30 Therefore, no long-lasting change of the morphology is expected and the energy of the absorbed light is mainly transformed into heat, rendering azobenzene 2 a photo-thermal agent.19 In general, azobenzene derivatives 1 and 2 were designed to be crystallographically analogous to PE.31 They were synthesised via etherification of di- or trihydroxy azobenzene with 1-bromoeicosane (see the Experimental section for synthesis details) and were characterised by NMR spectroscopy and MALDI-TOF mass spectrometry (see Fig. S1–S3 in the ESI†).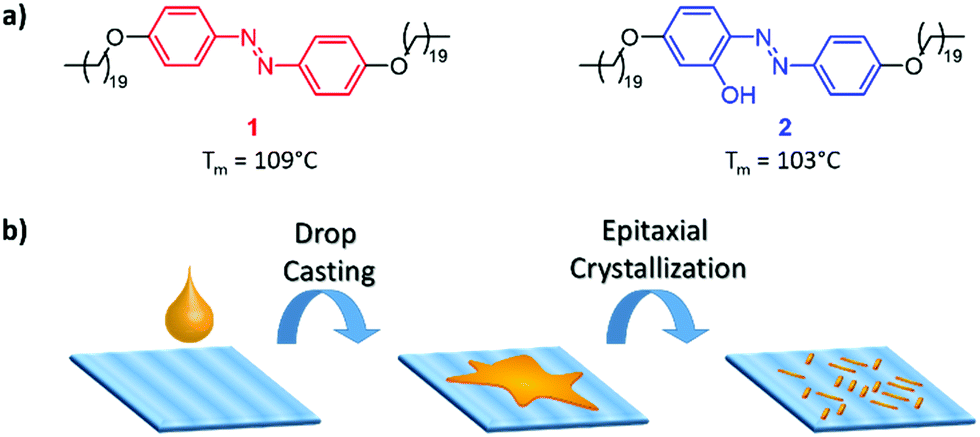 | ||
| Scheme 1 (a) Chemical structures of 1 and 2 employed in this study. (b) Schematic illustration of the epitaxial crystallisation of 1 and 2 on uniaxially drawn PE. | ||
Highly oriented UHMW-PE thin films were chosen as the substrate for the epitaxial crystal growth of alkane substituted azobenzenes 1 and 2 (see Scheme 1b, for results obtained with HDPE as the substrate, see the ESI†). These PE films can be aligned in one direction by uniaxial solid state drawing at 120 °C (see the Experimental section for the detailed procedure).32,33 The scanning electron microscopy (SEM) images of drawn UHMW-PE show the unidirectionally aligned fibrillar structures (see Fig. S12, ESI†).
When a hot toluene solution of 1 or 2 was deposited on the PE substrate at 80 °C, followed by slow cooling, formation of a herringbone-like structure was observed under an optical microscope (Fig. 1). Short rod-like crystallites cover the polymer at angles of either about 60° and 120° with regard to the alignment direction of the PE-layer. These short rods can be assigned to azobenzene domains. It is important to note that the oriented azobenzene crystallites homogeneously cover the entire samples. The feasibility to generate large area well-ordered layers of azobenzenes is clearly confirmed by these observations.
 | ||
| Fig. 1 Optical micrographs of the epitaxial layers of (a) 1 and (b) 2 on the surface of oriented UHMW-PE. The drawing direction of PE is indicated by the arrows. | ||
The surface morphology of the bilayer films was characterised using SEM (Fig. 2). The highly oriented PE chains in the background can be assigned to a flat drawn film surface, which is covered with uniformly distributed rod shaped or needle-like crystallites of specific orientation with an average diameter of 0.3 μm and a length between 5 to 8 μm in the case of 1 (Fig. 2a). Their orientation of 60° compared to the PE direction becomes even more obvious here. Interestingly, the formed crystallites are much larger for 2 with lengths of around 15 μm (Fig. 2b). The formation of the cross-hatch arrangement of the needles is a result of epitaxial growth. Atomic force microscopy (AFM) of the composite materials allows the determination of the vertical thickness of the crystallites, which was determined to be around 200 nm in the case of 1 and 150 nm in the case of 2 (Fig. 2c and d). Therefore, the size difference between both samples is mainly limited to the lateral dimensions and might be attributed to the stronger hydrogen bonds between molecules of 2 compared to mainly van der Waals forces between molecules of 1. The aligned surface of the PE induces epitaxial crystallisation. The fact that the needles are only about twice as wide as high indicates a weak preference of the azobenzene crystals to grow on azobenzene and PE compared to just azobenzene. The use of more or higher concentrated toluene solution for the dip coating results in the formation of multi-layered crystals and less order.
 | ||
| Fig. 2 SEM images of epitaxially grown crystals (a) of 1 on UHMW-PE; (b) of 2 on UHMW-PE. The large scale AFM topography images of epitaxially grown crystals of (c) 1 on UHMW-PE and height profile over the indicated blue line and (d) 2 on UHMW-PE and the height profile over the indicated blue line. | ||
The uniaxial alignment of the azobenzenes on the oriented PE substrate was confirmed by the polarised UV-Vis absorption spectra (see Fig. S5, ESI†), where the oriented azobenzenes absorb light predominantly along the drawing direction of PE. This indicates that the azobenzene molecules are aligned along the drawing direction. Additionally, this finding was supported by the polarised IR spectra, which showed a difference in the CH2 stretching of the PE chains and the aromatic regions parallel and perpendicular to the drawing direction (see Fig. S6, ESI†). The intensities of the CH2 stretching vibration of the PE chains at 2918 and 2848 cm−1 are higher in the perpendicular direction than those parallel to the drawing direction.
X-ray scattering measurements (Fig. S21, ESI†) revealed that the epitaxial crystallisation of the azobenzenes on PE is based on a similar orthorhombic unit cell of the alkyl chains of 1 (a) 0.668 and (b) 0.453 nm and drawn PE (a) 0.670 and (b) 0.444 nm. Therefore, the 60° angle of the crystallites originates from a common packing of aligned molecules with the alkyl chains following the direction of PE as depicted in Fig. 3a. Alkyl-substituted azobenzenes are known to form crystals with a packing angle with regard to the molecular orientation of the azobenzene core.22
 | ||
| Fig. 3 (a) Schematic depiction of the potential origin of the 60° angle of the crystals with regard to the drawing direction of UHMW-PE (large grey arrow). (b) AFM phase image of one epitaxially grown crystal of 1 (horizontally oriented) on UHMW-PE. (c) Fourier transformation of b revealing the periodicity along the crystal width. The corresponding spots are highlighted by red arrows. | ||
AFM analysis of the internal structure of the crystallites of 1 reveals indeed lamellar features parallel to their main axis with a periodicity of 6.2 ± 0.8 nm (Fig. 3b and c), which is in good agreement with the theoretical length of one azobenzene derivative molecule (6.25 nm, see also Fig. S18–S20, ESI†). For these observations, phase images were used for their better contrast and to exclude a contribution from the potential height variations of the crystal surfaces. The perfect alignment of the lamellae and the absence of the observed domain boundaries inside the crystallites show their single-crystalline nature.
In order to investigate if the composites can be used as light responsive actuators, the films with an epitaxial layer of 1 were exposed to UV light at 365 nm for 30 min. The light exposure led mainly to the disappearance of the crystallites (Fig. 4d) accompanied by a colour change from yellow to orange which is characteristic of the trans–cis isomerisation of azobenzenes and a proof for the photo-melting of the crystals. Furthermore, no significant photo-bending of the crystals was observed.
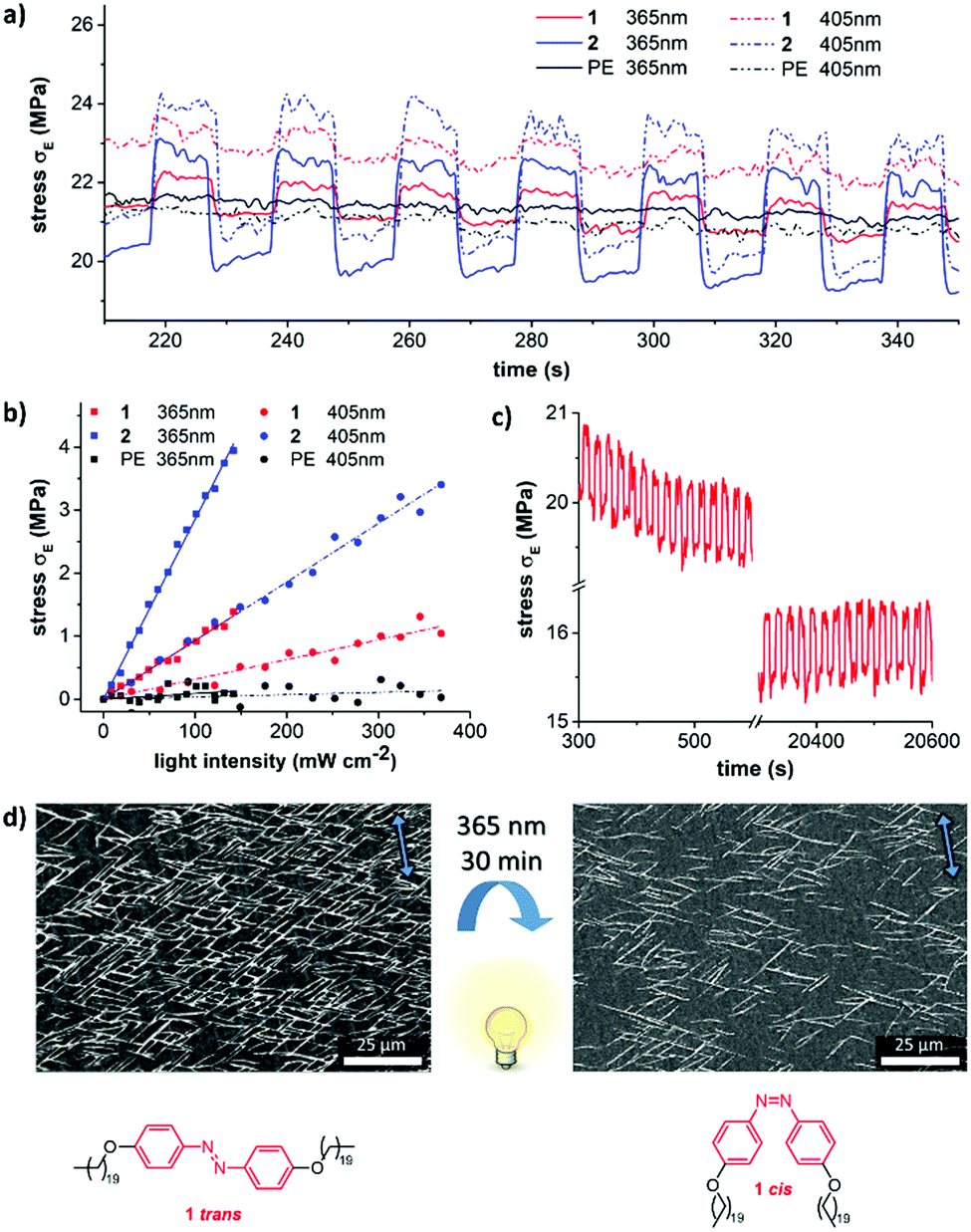 | ||
| Fig. 4 (a) Periodic photo-induced stress of drawn PE-films with epitaxially grown 1 (red), 2 (blue), or of pristine drawn UHMW-PE (black) upon irradiation at 365 nm (solid) or 405 nm (dashed). A pre-strain of 1% and a pre-load of 0.3 N was applied. (b) Dependence of the light-induced actuation stress on the intensity of the irradiated light for 1 (red), 2 (blue), and pristine PE (black) when exposed to 365 nm (squares) and 405 nm (circles) light; (c) periodic photo-induced stress of 1 over 2000 cycles (using 365 nm light); (d) photo-induced changes of the molecular self-assembly of 1 on a drawn PE-film upon irradiation at 365 nm. The arrows indicate the molecular chain direction of PE. | ||
Photo-induced mechanical changes of the aligned epitaxial films were studied using a dynamic mechanical analyzer (DMA) by subjecting the bilayer films to a constant strain thereby inducing stress relaxation. Comparison between the stress–strain curves of both epitaxial films and plain oriented UHMW-PE films can provide information about the light-induced stress. It was recorded as a function of time in order to check the stability of the crystal layers on UHMW-PE films (as shown in Fig. 4a). After clamping the ends of the film, a pre-strain of 1% and a pre-load of 0.3 N were applied. The actuation stress reached 3 MPa in the case of films with crystallites of 2 upon irradiation with light at 405 nm. The observed photo-induced stress is 10 times higher than the actuation stress of natural muscles (∼0.35 MPa).6,34 However, the actuation stress was found to be only 1 MPa in the case of photochromic compound 1 (365 nm). As expected in the control experiment, no photo-induced stress (neither using 365 nor 405 nm) was observed for naked UHMW-PE-films (Fig. 4a, black curve).
The photo-induced stress proved to be linear depending on the intensity of the irradiated light over a broad intensity range (Fig. 4b). Independent of the individual absorption spectra of both azobenzene molecules, the actuation was stronger at 365 nm compared to the same intensity at 405 nm. The heat transfer along the PE drawing direction towards the metal clamps is very fast and therefore impedes the accurate measurement of the film temperature during the actuation experiment. This is also the reason for the immediate regain of the initial stress level in the dark state upon switching off the light and the corresponding rectangular shape of the switching cycles in Fig. 4a and c. These cycles can be repeated more than 2000 times without relevant fatigue for both composites (Fig. 4c and Fig. S15, ESI†). Even continuous irradiation over 1 h with light at 365 nm does not lead to degradation (Fig. S16, ESI†). It should be noted that the actuation stress of the presented actuators is quite high, but the strain of the films is modest. While human muscles or liquid crystal actuators have strains around 20%, our actuators rather resemble conducting polymers, ionic polymer metal composites,35 or metal/metal oxide actuators with strains around one percent.36
In the case of the photo-thermal azobenzene 2 on oriented PE, a temperature up to 60 °C was recorded upon illumination with 405 nm light at 300 mW cm−2 accompanied by the disappearance of the azobenzene crystals. It is worthwhile to note that the epitaxial crystallites reappeared spontaneously in the dark. The same melting/recrystallisation was observed upon heating the films of 2-PE in the absence of light to 108 °C and then slowly cooling down (Fig. 5 and Fig. S17, ESI† for similar behaviour of 1). The fact that the crystallites really disappeared can be supported by the different arrangement of the needles on PE before and after the heating (though still strictly following the 60° angle with regard to the drawing direction). The discrepancy between the required temperatures to melt the crystallites of 2 on PE in the presence and absence of light suggests a considerable contribution of photoisomerisation and therefore facilitation of the melting upon irradiation.
 | ||
| Fig. 5 Optical microscopy images of 2 on UHMW-PE at (a) 25 °C (b) 100 °C (c) 108 °C (d) cooling back to 25 °C. Between taking the images b and c, the sample was heated to 112 °C to guarantee the melting of all crystals. | ||
Conclusions
We have described the anisotropic epitaxial growth of azobenzenes with long aliphatic chains on highly oriented PE films. The well-ordered crystal structure of the azobenzene derivatives with their long axis preferentially parallel to the chain direction of the PE surface has been studied by optical microscopy and SEM accompanied by X-ray diffraction. The observed epitaxial growth morphologies of the azobenzene derivatives are found to uniformly cover the whole PE films. This simple technique allows us to have a greater control of the two-dimensional oriented structures and symmetries of the photo-responsive crystals. We have also demonstrated that the films act as light responsive soft actuators. The photo-induced stress generated by the epitaxial films was found to be considerably higher than that of natural muscles. The high elastic modulus of the supportive UHMW-PE layer makes the system potentially useful for a mechanically flexible and very strong crystal photo-actuator that can apply pressure to an object without deforming itself. The simplicity of the proposed epitaxial bilayer system makes it a promising candidate for future photo-responsive fabrication of dynamic surfaces, remote control of surface hydrophobicity for self-cleaning coatings, artificial muscle actuators and soft-robotics.Experimental
UHMW-PE with a molecular weight average of approximately Mw = 3.5 × 106 g mol−1 was obtained from DSM (Geleen, The Netherlands). HDPE was obtained from Borealis (Burghausen, Germany) VS4580 with a number and molecular weight average of approximately 3.7 × 104 and 1.34 × 105 g mol−1, respectively. All other solvents and chemicals were purchased from commercial suppliers and used as received. UV-Vis measurements were performed using a PerkinElmer Lambda 750 UV-Vis-NIR spectrophotometer. The nuclear magnetic resonance (NMR) spectra were recorded on a Varian Mercury 400 MHz at room temperature or an Oxford NMR AS500 at 50 °C with a working frequency of 500 MHz (1H NMR) and 125 MHz (13C NMR), respectively. Chemical shifts were reported in ppm and referenced to tetramethylsilane. MALDI-TOF mass spectrometry was carried out using an Autoflex speed-Bruker spectrometer with α-cyano-4-hydroxycinnamic acid or trans-2-[3-(4-tert-butylphenyl)-2-methyl-2-propenylidene]malononitrile as a matrix in reflector mode. The optical microscopy images were obtained using a Leica CTR 6000 microscope, equipped with a DFC420C camera and a Linkam THMS600 hot-stage to control temperature. Wide angle X-ray scattering (WAXS) measurements were performed using a Ganesha lab instrument equipped with a Genix-Cu ultra-low divergence source producing X-ray photons with a wavelength of 1.54 Å and a flux of 1 × 108 photons per second. Diffraction patterns were collected using a Pilatus 300 K silicon pixel detector with 487 × 619 pixels of 172 μm2 placed at a sample-to-detector distance of 91 mm. The detector consists of three plates with a spacing of 17 pixels in between, resulting in two dark bands on the image. Atomic force microscopy (AFM) measurements were performed in air using a Multimode AFM with a Nanoscope VIII controller (Veeco/Digital Instruments) in intermittent contact mode. Olympus silicon cantilevers (AC160TS-R3), with a resonance frequency of around 300 kHz and a spring constant of around 26 N m−1, were used. AFM data analysis was performed using WSxM 5.0.37 Scanning tunneling microscopy (STM) experiments were carried out at room temperature (21–23 °C) at the 1-phenyloctane (Sigma 98%)/graphite interface using a Multimode Nanoscope III-d STM (Veeco) instrument operating in constant-current mode. Prior to imaging, a drop of solution of 1 was placed onto a freshly cleaved surface of highly oriented pyrolytic graphite (HOPG, grade ZYB, Advanced Ceramics Inc., Cleveland, USA). STM tips were prepared by mechanical cutting from Pt/Ir wire (80%/20%, diameter 0.2 mm). The imaging parameters are indicated in figure captions: sample bias (Vbias) and tunneling current (Iset). For analysis purposes, recording of a monolayer image was followed by imaging the graphite substrate underneath it under the same experimental conditions, except for increasing the current and lowering the bias. The images were corrected for drift via Scanning Probe Image Processor (SPIP) software (Image Metrology ApS), using the recorded graphite images for calibration purposes, allowing a more accurate unit cell determination. The molecular model provided in Fig. S16 (ESI†) was built using Hyperchem Professional 7.5., and SEM-images were obtained using a Jeol JSM 6010 LA, at 5 or 10 kV. The stress–relaxation curves were obtained at room temperature using a Discovery DMA 850. Differential Scanning Calorimetry (DSC) was carried out under a nitrogen flow using a DSC Q1000 instrument. Three cycles of heating and cooling between the temperature range from 0 to 150 °C were applied. The polymer films and the azobenzene samples 1 and 2 were heated and cooled at a constant rate of 5 °C min−1. Thermogravimetric analysis (TGA) measurements were performed using a TA Q500 instrument at a constant heating rate of 10 °C min−1 and at a constant air flow rate of 50 mL min−1.Uniaxially drawn UHMW-PE films32,38
2 g of UHMW-PE was added as a powder to xylene (200 mL) and the mixture was degassed for about 1 hour at room temperature. To form a gel, the resultant solution was heated at 140 °C. Subsequently, the solutions were cast and quenched to room temperature. A dry UHMW-PE sheet was obtained after the evaporation of solvents at room temperature. The sheets of the solution-cast UHMW-PE films were drawn uniaxially using a thermostatically controlled hot plate at 120 °C. A draw ratio (λ) of 30 (final length/initial length of the film) was determined by measuring the displacement of ink-marks.Synthesis of azobenzene derivatives 1 and 2
2,4,4′-Trihydroxyazobenzene was synthesised according to the reported procedure.391H-NMR (500 MHz, CDCl3, 50 °C) δ[ppm] = 7.88 (d, 4H, J(H,H) = 8.6 Hz), 6.98 (m, 4H), 4.03 (t, 4H, J(H,H) = 6.6 Hz), 1.81 (quin, 4H, J(H,H) = 6.8 Hz), 1.47 (m, 4H), 1.40–1.20 (m, 64H), 0.92–0.85 (t, 6H, J(H,H) = 6.8 Hz).
MS (MALDI-TOF MS): m/z = 775.72 (calc. 775.708 [C52H91N2O2]+).
1H-NMR (500 MHz, CDCl3, 50 °C) δ[ppm] = 13.54 (s, 1H), 7.80–7.68 (m, 3H), 7.03–6.91 (m, 2H), 6.62–6.52 (m, 1H), 6.48–6.41 (m, 1H), 4.02 (q, J(H,H) = 6.6 Hz, 4H), 1.80 (h, J(H,H) = 6.5 Hz, 4H), 1.47 (d, J(H,H) = 6.1 Hz, 13H), 1.27 (s, 70H), 0.97–0.77 (m, 6H).
13C-NMR (126 MHz, CDCl3, 50 °C) δ[ppm] = 162.9, 161.2, 155.8, 144.6, 134.0, 133.9, 132.9, 123.3, 123.3, 115.1, 108.2, 108.2, 102.1, 102.0, 68.5, 31.9, 29.7, 29.7, 29.6, 29.4, 29.3, 29.2, 26.0, 22.7, 14.0.
MS (MALDI-TOF MS): m/z = 792.30 (calc. 791.702 [C52H91N2O3]+).
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported partly by DSM. S. R. P. acknowledges financial support through a Marie Skłodowska-Curie Individual Fellowship (EU project 797156). We would like to thank M. M. R. M. Hendrix for the WAXS/GIWAXS measurements. The authors would also like to thank L. Shen, A. A. F. Froyen, S. J. A. Houben, and R. C. P. Verpaalen for helpful discussion.Notes and references
- M. Dai, O. T. Picot, J. M. N. Verjans, L. T. de Haan, A. P. H. J. Schenning, T. Peijs and C. W. M. Bastiaansen, ACS Appl. Mater. Interfaces, 2013, 5, 4945–4950 CrossRef CAS PubMed
.
- R. C. P. Verpaalen, M. G. Debije, C. W. M. Bastiaansen, H. Halilović, T. A. P. Engels and A. P. H. J. Schenning, J. Mater. Chem. A, 2018, 6, 17724–17729 RSC
- F. Cheng, R. Yin, Y. Zhang, C.-C. Yen and Y. Yu, Soft Matter, 2010, 6, 3447–3449 RSC
- A. Agrawal, T. Yun, S. L. Pesek, W. G. Chapman and R. Verduzco, Soft Matter, 2014, 10, 1411–1415 RSC
- M. Yamada, M. Kondo, J. Mamiya, Y. Yu, M. Kinoshita, C. J. Barrett and T. Ikeda, Angew. Chem., 2008, 120, 5064–5066 CrossRef
-
S. Varghese, J. R. Severn and A. P. H. J. Schenning, Photoresponsive Polyolefins, in Photoactive Functional Soft Materials: Preparation, Properties, and Applications, ed. Q. Li, John Wiley & Sons, 2019, pp. 319–340 Search PubMed
- A. Ural, Y. Li and H. Dai, Appl. Phys. Lett., 2002, 81, 3464–3466 CrossRef CAS
- J. S. Moore and S. I. Stupp, Macromolecules, 1987, 20, 282–293 CrossRef CAS
- K. Ichimura, Chem. Rev., 2000, 100, 1847–1874 CrossRef CAS PubMed
- T. Seki, Polym. J., 2014, 46, 751 CrossRef CAS
- K.-W. Lee, S.-H. Paek, A. Lien, C. Durning and H. Fukuro, Macromolecules, 1996, 29, 8894–8899 CrossRef CAS
- J. H. Laurer, B. S. Pinheiro, D. L. Polis and K. I. Winey, Macromolecules, 1999, 32, 4999–5003 CrossRef CAS
- A. K. Geim and I. V. Grigorieva, Nature, 2013, 499, 419 CrossRef CAS PubMed
- J. Yang and D. Yan, Chem. Soc. Rev., 2009, 38, 2634–2645 RSC
- S. Shin, J. Seo, H. Han, S. Kang, H. Kim and T. Lee, Mater, 2016, 9 Search PubMed
- N. A. Malvadkar, M. J. Hancock, K. Sekeroglu, W. J. Dressick and M. C. Demirel, Nat. Mater., 2010, 9, 1023 CrossRef CAS PubMed
- S. Reddy, E. Arzt and A. del
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) Campo, Adv. Mater., 2007, 19, 3833–3837 CrossRef CAS
Campo, Adv. Mater., 2007, 19, 3833–3837 CrossRef CAS - X. He, M. Aizenberg, O. Kuksenok, L. D. Zarzar, A. Shastri, A. C. Balazs and J. Aizenberg, Nature, 2012, 487, 214 CrossRef CAS PubMed
- M. Pilz da Cunha, E. A. J. van Thoor, M. G. Debije, D. J. Broer and A. P. H. J. Schenning, J. Mater. Chem. C, 2019, 7, 13502–13509 RSC
- N. K. Nath, M. K. Panda, S. C. Sahoo and P. Naumov, CrystEngComm, 2014, 16, 1850–1858 RSC
- N. K. Nath, L. Pejov, S. M. Nichols, C. Hu, N. Saleh, B. Kahr and P. Naumov, J. Am. Chem. Soc., 2014, 136, 2757–2766 CrossRef CAS PubMed
- Y. Norikane, E. Uchida, S. Tanaka, K. Fujiwara, E. Koyama, R. Azumi, H. Akiyama, H. Kihara and M. Yoshida, Org. Lett., 2014, 16, 5012–5015 CrossRef CAS PubMed
- E. Uchida, R. Azumi and Y. Norikane, Nat. Commun., 2015, 6, 7310 CrossRef CAS PubMed
- H. Li and S. Yan, Macromolecules, 2011, 44, 417–428 CrossRef CAS
- M. Campione, A. Sassella, M. Moret, A. Papagni, S. Trabattoni, R. Resel, O. Lengyel, V. Marcon and G. Raos, J. Am. Chem. Soc., 2006, 128, 13378–13387 CrossRef CAS PubMed
- J. Willems and I. Willems, Nature, 1956, 178, 429–430 CrossRef CAS
- M. Masnadi and S. G. Urquhart, Langmuir, 2013, 29, 6302–6307 CrossRef CAS PubMed
- D. Guo, K. Sakamoto, K. Miki, S. Ikeda and K. Saiki, Appl. Phys. Lett., 2007, 90, 102117 CrossRef
- D. Guo, K. Sakamoto, K. Miki, S. Ikeda and K. Saiki, Phys. Rev. Lett., 2008, 101, 236103 CrossRef PubMed
- W. R. Brode, J. H. Gould and G. M. Wyman, J. Am. Chem. Soc., 1952, 74, 4641–4646 CrossRef CAS
- M. M. Unterlass, E. Espinosa, F. Boisson, F. D’Agosto, C. Boisson, K. Ariga, I. Khalakhan, R. Charvet and J. P. Hill, Chem. Commun., 2011, 47, 7057–7059 RSC
- C. Bastiaansen, H.-W. Schmidt, T. Nishino and P. Smith, Polymer, 1993, 34, 3951–3954 CrossRef CAS
- L. Shen, S. S. D. Lafleur, S. J. A. Houben, J. N. Murphy, J. R. Severn and C. W. M. Bastiaansen, Langmuir, 2017, 33, 14592–14598 CrossRef CAS PubMed
- J. D. Madden, R. A. Cush, T. S. Kanigan and I. W. Hunter, Synth. Met., 2000, 113, 185–192 CrossRef CAS
- J. D. W. Madden, N. A. Vandesteeg, P. A. Anquetil, P. G. A. Madden, A. Takshi, R. Z. Pytel, S. R. Lafontaine, P. A. Wieringa and I. W. Hunter, IEEE J. Ocean. Eng., 2004, 29, 706–728 CrossRef
- K. W. Kwan and A. H. W. Ngan, Adv. Mater. Technol., 2019, 1900746 CrossRef
- I. Horcas, R. Fernández, J. M. Gómez-Rodríguez, J. Colchero, J. Gómez-Herrero and A. M. Baro, Rev. Sci. Instrum., 2007, 78, 13705 CrossRef CAS PubMed
- P. Smith, P. J. Lemstra, J. P. L. Pijpers and A. M. Kiel, Colloid Polym. Sci., 1981, 259, 1070–1080 CrossRef CAS
- A. H. Gelebart, D. J. Mulder, M. Varga, A. Konya, G. Vantomme, E. W. Meijer, R. L. B. Selinger and D. J. Broer, Nature, 2017, 546, 632 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9tc05407c |
| This journal is © The Royal Society of Chemistry 2020 |