
N-Heterocyclic carbene-ended polymers as surface ligands of plasmonic metal nanoparticles†
Srinivas
Thanneeru‡
a,
Kaitlynn M.
Ayers‡
a,
Murali
Anuganti
a,
Lei
Zhang
a,
Challa V.
Kumar
 a,
Gaël
Ung
*a and
Jie
He
*ab
a,
Gaël
Ung
*a and
Jie
He
*ab
aDepartment of Chemistry, University of Connecticut, Storrs, CT 06269, USA. E-mail: Gael.ung@uconn.edu; Jie.he@uconn.edu
bPolymer Program, Institute of Materials Science, University of Connecticut, Storrs, CT 06269, USA
First published on 7th January 2020
Abstract
A facile methodology to prepare N-heterocyclic carbene (NHC)-terminated polymers as surface ligands to functionalize gold nanoparticles (AuNPs) is reported. Our method highlights a mild, aerobic synthesis of NHC-functionalized polymers and a simple ligand exchange approach towards surface modification of AuNPs prepared in aqueous solution. Two methods, including end-group functionalization of halogen-ended polymers from a conventional atom transfer radical polymerization (ATRP) and post-polymerization functionalization of imidazole-containing polymers using imidazole-containing ATRP initiator, have been investigated to prepare imidazolium-ended polymers. Using a one-step, oxygen and moisture tolerant procedure, the polymer–NHC–Cu(I) species can be synthesized from imidazolium-ended polymers and readily bind to citrate-capped AuNPs likely through transmetalation, yielding robust polymer-stabilized AuNPs. Our synthetic method significantly simplifies the preparation and use of polymer–NHC ligands for surface functionalization of metal NPs. Our methodology is general and potentially applicable to any polymers prepared by ATRP to functionalize metal NPs via NHC–metal coordination; therefore, it will likely broaden the applications of polymer–NHC ligands for metal nanoparticles in the fields of catalysis and nanomedicine.
1. Introduction
Surface ligands play a key role in stabilizing nanoparticles (NPs) in solution to prevent coalescence. A variety of either organic or inorganic molecules/ions (e.g., surfactants, polymers, proteins, nucleic acids and inorganic ions) bound to surface atoms of NPs via metal–ligand coordination can be used as surface ligands for NPs. Synthetic polymers as ligands of NPs can enrich surface functionality, biocompatibility and responsiveness,1–8 which are key features for applications in nanomedicine,9,10 catalysis11,12 and photonic materials.13,14 Addition of polymer ligands to NPs is often achieved through ligand exchange of small-molecule-capped NPs by either end-functionalized polymers (“grafting to” method),15–22 or by molecular initiators used to grow polymers later (“grafting from” method).23–25 Thiol-ended polymers are the most popular polymer ligands for metal NPs, in particular when using the “grafting to” approach, due to the binding strength of the metal–thiolate interaction and the immediate availability of thiol-ended polymers prepared from reversible addition–fragmentation chain-transfer polymerization. However, early experiments in self-assembly monolayers suggest that thiol ligands are unstable and they are readily oxidized by oxygen or peroxides.26,27N-Heterocyclic carbenes (NHCs) have gained continuous attention as surface ligands of metal NPs.28–39 Due to the strong σ-donation of the carbene, NHCs bind strongly to metals via metal–carbon interactions.40–42 Notably, NHC-supported NPs are more stable than thiol-capped NPs under harsh oxidative conditions43 and have been shown to modulate the surface electron density of metal NPs as well as their catalytic activity.11,12,44–47 Although ligand exchange using free NHCs to modify metal NPs (such as Au,28,48 Pd,44,49 and Pt38) have been reported, profound synthetic challenges remain in the “grafting to” approach, especially for polymer–NHC ligands. Surface modification of metal NPs utilizing free NHCs typically requires anaerobic and anhydrous conditions since free NHCs are air and water sensitive. This largely limits the use of free NHCs particularly for metal NPs synthesized and dispersed in aqueous solution. On the other hand, one can prepare polymer–NHC modified gold NPs (AuNPs) using chemical reduction of polymeric NHC–Au precursors,34 although pre-synthesized AuNPs with well-defined size cannot be used directly. Moreover, our group recently demonstrated the “grafting to” approach for polymer NHC ligands using imidazolium-terminated polymers,50 similar to small molecular NHC ligands.30,51
As an alternative, we herein present another robust and facile methodology to prepare NHC-functionalized polymers as surface ligands to functionalize AuNPs. Our method utilizes mild, aerobic synthetic conditions to prepare NHC-functionalized polymers and perform surface modification of metal NPs using NHC-ended polymers in aqueous solution. This potentially removes the synthetic obstacles of polymer–NHC ligand chemistry and makes polymer–NHC ligands accessible without complex syntheses. The synthetic strategy involves the preparation of polymer–NHC–Cu(I) complexes by post-polymerization functionalization of polymers prepared via atom transfer radical polymerization (ATRP) with an imidazolium end group, followed by treatment with Cu(I). Four polymer–NHC–Cu(I) complexes were prepared, including, polystyrene (PS), poly(methyl methacrylate) (PMMA), poly(n-butyl acrylate) (PnBA), and poly(2-(2-methoxyethoxy)ethyl methacrylate) (PMEO2MA). These polymer–NHC–Cu(I) complexes were likely transmetalated to AuNPs at the water/oil interface, yielding robust polymer-stabilized AuNPs. The merit of our polymer–NHC ligand synthesis and exchange protocol is its versatility, since it can be extended to any polymers prepared by ATRP to functionalize metal NPs via NHC–metal coordination.
2. Experimental
2.1 Materials
All chemicals were purchased from Aldrich unless otherwise noted. Methyl methacrylate (MMA, 99%) and styrene (St, 99%) were passed through a basic aluminum oxide column prior to use. N,N,N′,N′′,N′′-Pentamethyldiethylenetriamine (PMDETA, >99%), tert-butyl α-bromoisobutyrate (BBiB, ≥98%), 2-bromo-2-methylpropionyl bromide (BMPB, 98%), copper(I) bromide (CuBr, 99.999% trace metal basis), anisole (anhydrous, 99.7%), copper(I) chloride (CuCl, ≥90% ACS reagent), gold(III) chloride hydrate (HAuCl4, ≥99.0%), sodium citrate tribasic dihydrate (≥99.0%), dodecanethiol (DDT), potassium carbonate (K2CO3), phosphoric acid (H3PO4, >85%), glyoxal (40% in water), 13C-paraformaldehyde (99%), and 1,4-dioxane (99.8%) were used as received. Methylamine (35.4% in water) was purchased from Alfa Aesar and ammonium chloride (NH4Cl, crystals) were purchased from Macron Chemical. Plain gold SPR Reichert slides were purchased from Reichert technology. Deionized water (Millipore Milli-Q grade) with a resistivity of 18.0 MΩ was used in all the experiments.2.2 Synthesis of PS and end-group functionalization
2.3 Synthesis of AuNPs and surface modification of AuNPs
2.4 Characterizations
SEC measurements were performed on a Waters SEC-1 (1515 HPLC pump and Waters 717Plus auto injector) equipped with a Varian 380-LC evaporative light scattering detector and three Jordi Gel fluorinated DVB columns (1–100 K, 2–10 K, and 1–500 Å). THF was used as an eluent at a flow rate of 1.25 mL min−1, and PS standards were used for molecular weight and molecular weight distribution calibration. The data sets were processed using Empower SEC software (Waters, Inc.). 1H NMR spectroscopy was recorded on a Bruker AVANCE 400 MHz spectrometer. 13C NMR spectroscopy was recorded on a Bruker AVANCE 500 MHz equipped with a 5 mm BBFO SmartProbe. The heteronuclear multiple bond correlation (HMBC) experiments were conducted on Varian INOVA 600 MHz spectrometer equipped with a triple-resonance 1H, 13C, 15N cryogenic probe. An HMBC coupling constant (J) between 1H and 13C was set to 12 Hz. Transmission electron microscopy (TEM) was carried out on a JEOL 2010 TEM with an accelerating voltage of 200 kV. UV-vis spectra were recorded on a Cary 60 UV-vis spectrophotometer. For grafting density, thermogravimetric analysis (TGA) was performed using TA Instruments Q-500. Each sample was first dried at 100 °C for 60 min to remove any residual solvent. The analysis was performed by heating the samples from 100 to 700 °C at a heating rate of 10 °C per min under N2.3. Results and discussion
We first envisioned synthesizing imidazolium-ended polymers using end group functionalization of halogen-ended polymers prepared via ATRP. A bromine-ended PS65–Br (Mn = 11![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 900 g mol−1 and Đ = 1.18, Fig. S1, ESI†) was synthesized using tert-butyl α-bromoisobutyrate as an ATRP initiator and the end bromide was substituted with N-methylimidazole (Fig. 1a). The conversion of the Br end group to imidazolium (PS65–Br to PS65–Im) was confirmed using 1H NMR spectroscopy (Fig. 1b). New peaks (d at 10.9 ppm and b, c at 7.8 ppm) are assigned to the N–CH–N, and vinyl protons of the imidazolium, respectively. The conversion of the Br end groups was estimated to be ∼25% by integrating peak d from the imidazolium against peak a, the protons on the aromatic ring of PS. PS65–Im was then treated with CuCl in presence of K2CO3 using a procedure similar to the one developed by Cazin and co-workers.52 The disappearance of peak d of PS65–Im (Fig. 1b) and shift of protons b and c indicates the formation of PS65–NHC–Cu(I), as similar shifts are observed with a small molecule analogue (see Fig. S2, ESI†). We used Cu instead of Ag since Cu is more abundant and cheaper than Ag, while still being able to perform transmetalation in molecular complexes. In addition, Ag can form bis-carbene complexes with NHC. We ruled out the possibility of a bis-carbene [(PS65–NHC)2Cu(I)]+ complex by studying the coordination chemistry of a small molecule analogue (Fig. S2 and Section S1 in ESI†).
900 g mol−1 and Đ = 1.18, Fig. S1, ESI†) was synthesized using tert-butyl α-bromoisobutyrate as an ATRP initiator and the end bromide was substituted with N-methylimidazole (Fig. 1a). The conversion of the Br end group to imidazolium (PS65–Br to PS65–Im) was confirmed using 1H NMR spectroscopy (Fig. 1b). New peaks (d at 10.9 ppm and b, c at 7.8 ppm) are assigned to the N–CH–N, and vinyl protons of the imidazolium, respectively. The conversion of the Br end groups was estimated to be ∼25% by integrating peak d from the imidazolium against peak a, the protons on the aromatic ring of PS. PS65–Im was then treated with CuCl in presence of K2CO3 using a procedure similar to the one developed by Cazin and co-workers.52 The disappearance of peak d of PS65–Im (Fig. 1b) and shift of protons b and c indicates the formation of PS65–NHC–Cu(I), as similar shifts are observed with a small molecule analogue (see Fig. S2, ESI†). We used Cu instead of Ag since Cu is more abundant and cheaper than Ag, while still being able to perform transmetalation in molecular complexes. In addition, Ag can form bis-carbene complexes with NHC. We ruled out the possibility of a bis-carbene [(PS65–NHC)2Cu(I)]+ complex by studying the coordination chemistry of a small molecule analogue (Fig. S2 and Section S1 in ESI†).
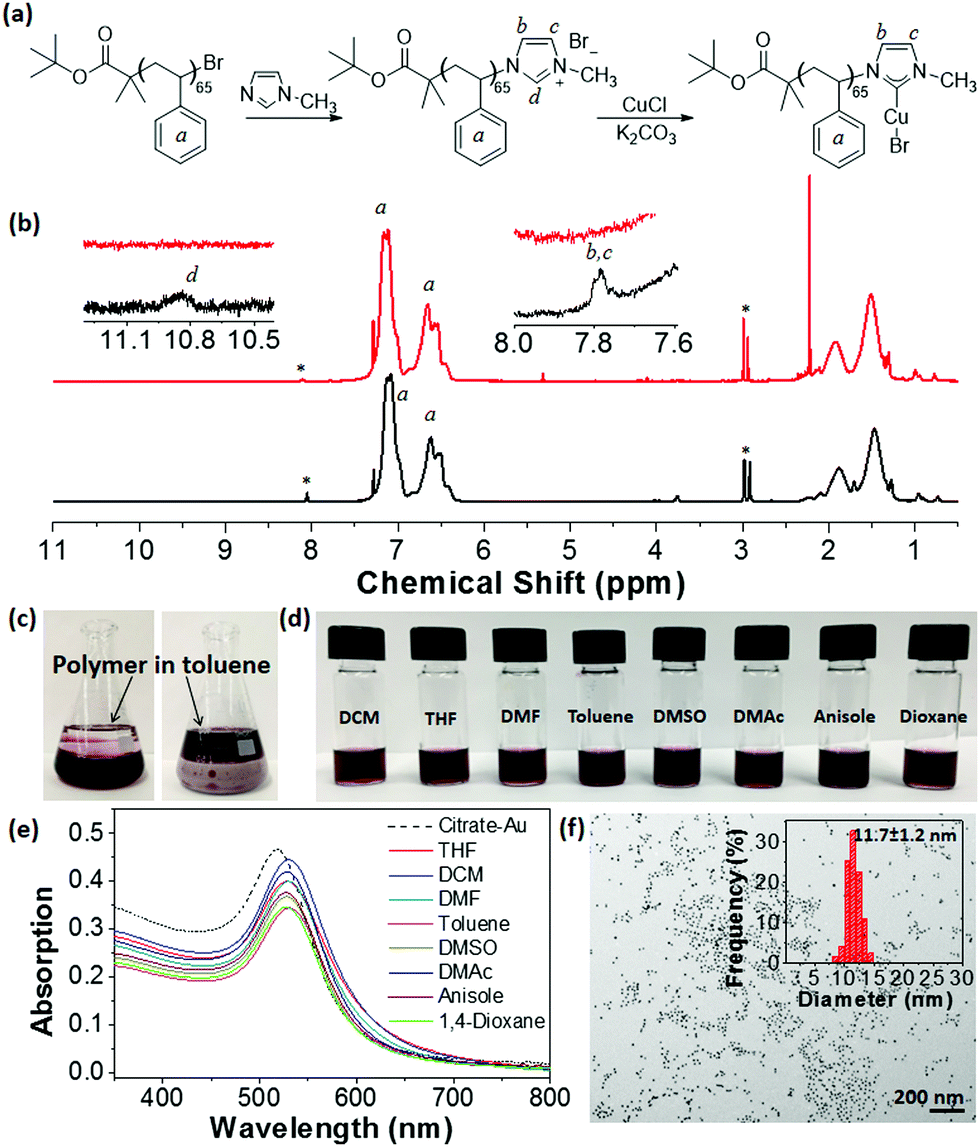 | ||
| Fig. 1 (a) Synthesis of PS65–NHC–Cu(I) using end group functionalization to convert Br to NHC–Cu(I). (b) 1H NMR spectra of PS65–Im (black, bottom) and PS65–NHC–Cu(I) (red, top) measured in d-CHCl3. *: residual DMF. (c) Images showing the transfer of citrate-capped AuNPs in water (bottom layer) to toluene (top layer) using PS65–NHC–Cu(I). (d) Images showing the dispersion of PS65–NHC modified AuNPs in various solvents. (e) UV-vis spectra showing the SPR absorption of AuNPs in various solvents. (f) TEM image of PS65–NHC–Au in THF. The inset shows the average size of AuNPs. | ||
The “grafting to” surface modification of citrate-capped AuNPs with PS65–NHC–Cu(I) was performed using a biphasic ligand exchange method in a water/toluene mixture.53,54 PS65–NHC–Cu(I) was dissolved in toluene at a concentration of 1 mg mL−1 and added to an aqueous solution of citrate-capped AuNPs (0.093 mg mL−1, ∼12 nm in diameter, synthesized using a reported method21). After sonication, the mixture was stirred overnight. The phase transfer of AuNPs from water to toluene is evident from the deep red color in the toluene layer (Fig. 1c). After removal of the aqueous layer, AuNPs were collected by centrifugation and purified by five more centrifugations in chloroform. As a control, similar experiments were run using PS65–Br and PS65–Im in toluene. Phase transfer of AuNPs was not observed in these cases, indicating that the NHC–Cu(I) moiety is essential for surface modification (Fig. S3, ESI†).
PS-modified AuNPs were dispersed in several organic solvents to confirm the surface modification of AuNPs with PS65–NHC. As shown in Fig. 1d, PS65–NHC–Au is readily re-dispersed in any good solvent for PS, such as DCM, THF, DMF, toluene, dimethyl sulfoxide (DMSO), dimethylacetamide (DMAc), anisole and 1,4-dioxane. The corresponding UV-vis absorption spectra of PS65–NHC–Au in various solvents clearly show the surface plasmon resonance (SPR) peak at 525–530 nm (Fig. 1e), indicative of excellent dispersity of AuNPs after ligand exchange. A slight redshift of the SPR peak compared to the original citrate-capped AuNPs (518 nm) is attributed to the presence of polymers on the surface and differences in the refractive indexes of the solvents compared to water. The TEM image in Fig. 1f shows that PS65–NHC–Au casted from THF is well-dispersed. The size of AuNPs measured from the TEM image is 11.7 ± 1.2 nm, comparable with that of citrate-capped AuNPs. Those results confirm the successful replacement of citrate ligands on the AuNPs by PS65–NHC.
The change in surface hydrophobicity of PS65–NHC–Au was investigated using water titration. When adding water to a THF solution of PS65–NHC–Au, the solvent quality changes from good to poor for PS, resulting in the collapse of PS and the aggregation of AuNPs. The UV-vis spectra in Fig. 2a shows a clear red-shift in the SPR absorption peak from 528 nm to 565 nm when the concentration of water reached 38 vol%. The critical water concentration (CWC ∼20%) was estimated by the onset position of a sudden increase in the SPR peak when plotting the SPR peak absorption as a function of water concentration (Fig. 2b). This CWC value is close to that observed in AuNPs capped with thiol-ended PS, further confirming the presence of PS as surface ligands.13 The red shift in SPR absorption reveals the formation of larger aggregates of AuNPs as confirmed by TEM (Fig. 2c). The stability of PS65–NHC–Au towards the competing binding with 1-dodecanethiol was also investigated. It is stable to 10 mM of 1-dodecanethiol for 24 h, comparable to other NHC–AuNPs (Fig. S4, ESI†).48,55
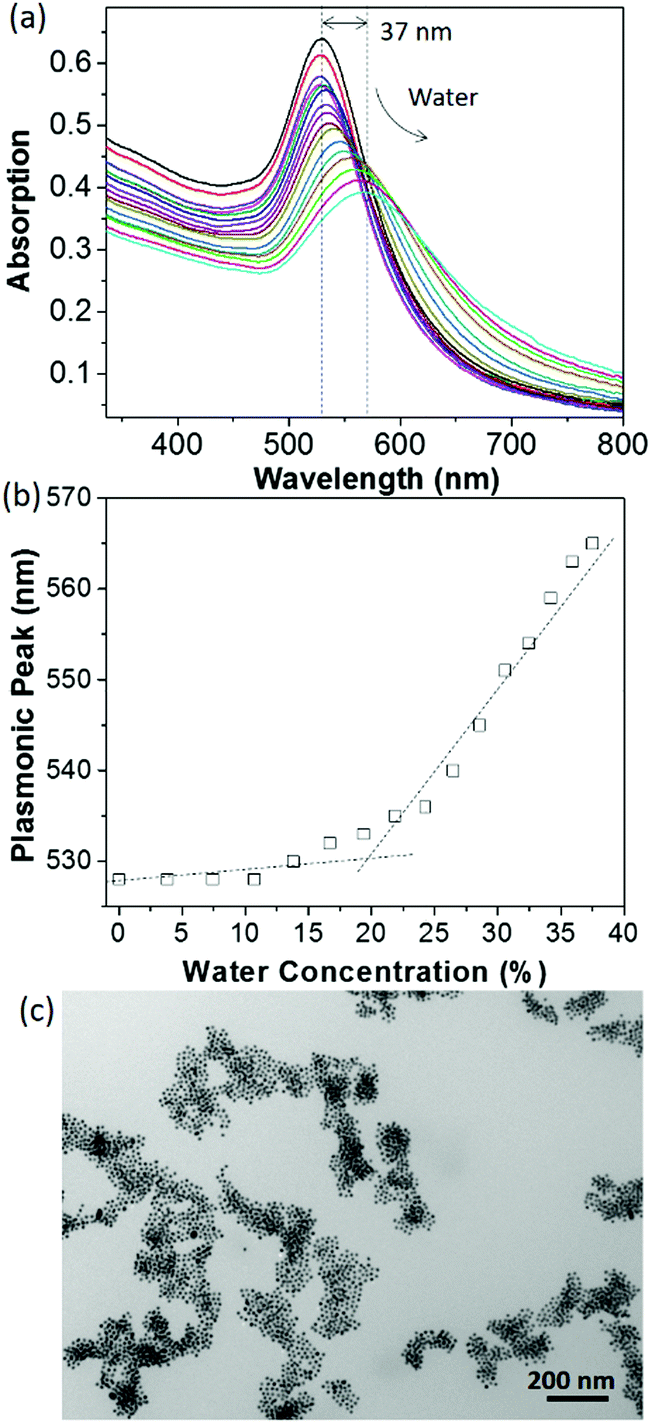 | ||
| Fig. 2 (a) UV-vis spectra of PS65–NHC modified AuNPs in THF when titrating with water. The spectra were collected by adding 80 μL of water each time into 2 mL of the THF solution of AuNPs. (b) Plotting the plasmonic peak of PS65–NHC modified AuNPs as a function of water concentration to determine the critical water concentration. (c) TEM image of PS65–NHC modified AuNPs in THF/water (1:1 vol) mixture. | ||
A 13C-labeled analogue, PS65–13CNHC–Cu(I), was used to examine the surface binding of polymer to AuNPs. The isotopically enriched imidazole N–13CH–N was used to prepare PS65–13CIm (Fig. 3). The synthetic details and characterization of 13C-labeled 1-methylimidazole are given in the ESI† (Section S3). The labeling and identity of PS65–13CIm was confirmed through both the chemical shift of N–13CH–N in PS65–13CIm at 135 ppm in the 13C NMR spectrum (Fig. 3c), and the proton peak of N–13CH–N in PS65–13CIm appearing as a doublet at ca. 9.9–10.4 ppm in the 1H NMR spectrum (Fig. S5, ESI†). Synthesis of the labeled carbene–Cu(I) complex (PS65–13CNHC–Cu(I)) was also confirmed by NMR spectroscopy. In the 13C NMR spectrum, the N–13C–N resonance shifted to 177 ppm (Fig. 3c). In the 1H NMR spectrum, the diagnostic doublet for the proton peak of N–13CH–N was no longer seen. The shifts and coupling in both 1H and 13C NMR spectra are consistent with changes observed in the small molecules and non-labeled polymers.
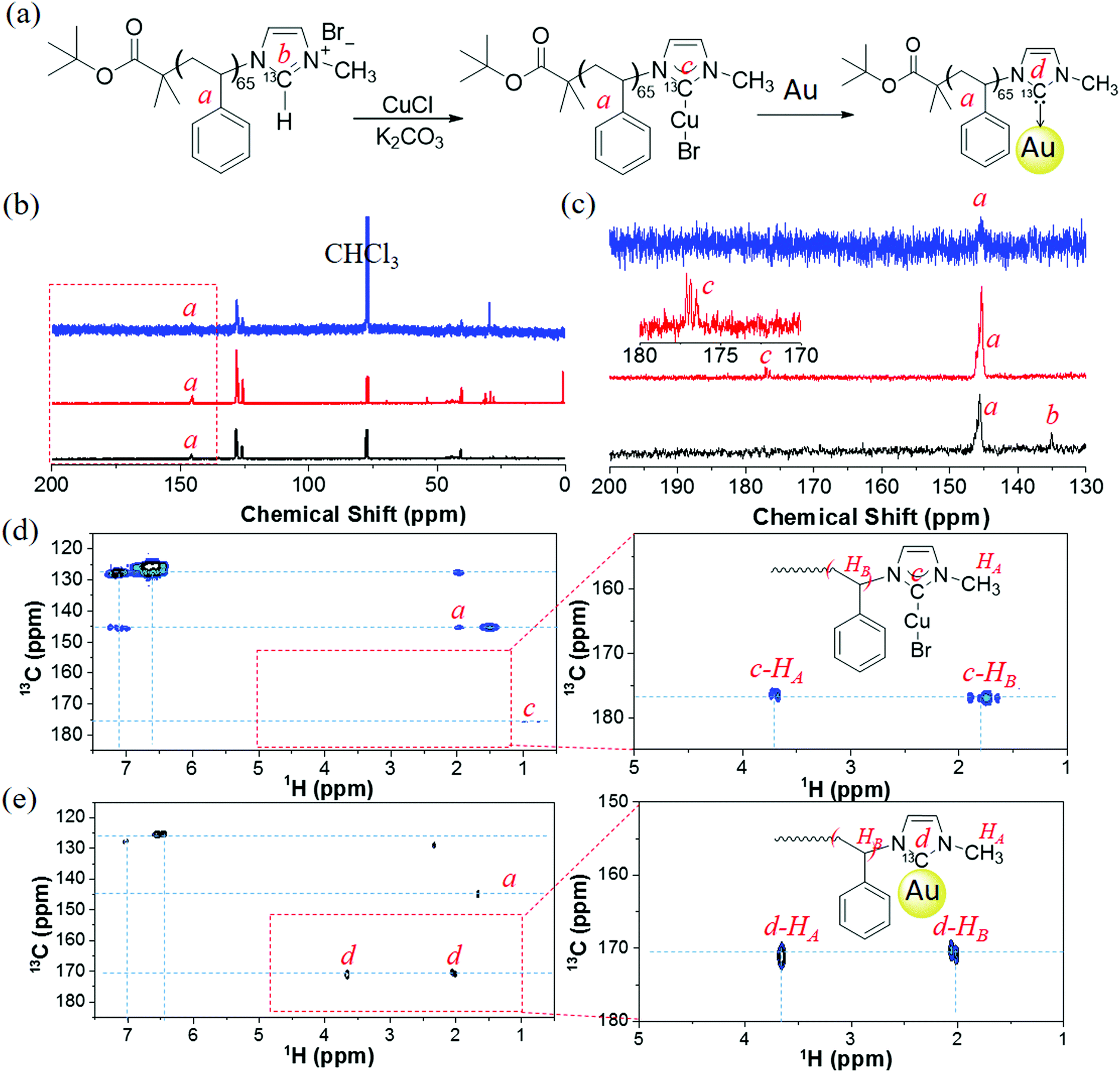 | ||
| Fig. 3 (a) Synthesis of 13C-labeled PS65–13C–NHC–Au using 13C–Im. (b and c) 13C NMR spectra of PS65–13C–Im (black, bottom), PS65–13C–NHC–Cu(I) (red, middle) and PS65–13C–NHC–Au (blue, top) in CDCl3. 13C NMR spectra in (b) in the range of 130–200 ppm is zoomed in (c). (d) 2D HMBC spectrum of non-labeled PS65–NHC–Cu(I). (e) 2D HMBC spectrum of 13C-labeled PS65–13CNHC–Au. | ||
After modification of AuNPs with PS65–13CNHC–Cu(I), the 13C NMR spectrum of PS65–13CNHC–Au did not show a clear peak for the N–13CAu–N resonance, due to the relatively low grafting amount of polymers on the surface of the NPs and the immobilization that causes inefficient relaxation of the carbene carbon. A higher concentration was also difficult to obtain because of the scarcity of labeled materials, arising from the high cost of the labeled precursor. Therefore, a two dimensional 1H–13C HMBC experiment was performed to detect the N–13CAu–N through its coupling to 1H. Several protons could potentially correlate with N–13CAu–N, including N–CH3 and N–CH–CH2 substituents of the imidazole. The experiment was calibrated for a J coupling of 12 Hz, in the range of typical imidazole 3JH–C (8–12 Hz).56,57 Two strong cross peaks were observed as displayed in Fig. 3e. The first one is between a proton at 3.67 ppm (HA) and a carbon at 171 ppm, which is assigned to the coupling of N–CH3 with N–13CAu–N. The chemical shift of N–CH3 is closed to that shown in our model compound (Fig. S2, ESI†). The second cross peak is observed between a proton signal at 2.01 ppm (HB) and the same carbon at 171 ppm, attributed to the coupling of benzyl protons of PS with N–13CAu–N. The chemical shift suggests that, there is likely virtual coupling along the main chain of PS, since the chemical shift of the N–CH–CH2 should appear at 5–6 ppm (Fig. S2, ESI†).
The new carbon resonance at 171 ppm is different than the signal of the N–13CCu–N precursor (177 ppm, Fig. 3b). To validate the observation of the 13C signal through its coupling to HA, we also ran the HMBC experiment with PS65–NHC–Cu(I). Cross peaks between the N–13CCu–N carbon signal at 177 ppm and protons at 3.65 ppm (HA, N–CH3), 1.6–1.95 ppm and 0.9 ppm (main chain of PS) were observed (Fig. 3d). The resonance peak was identical with that from PS65–13CNHC–Cu(I) in Fig. 3c (the peak c in red line). We note that, the coupling of N–CH–CH2 with N–13CAu–N is weaker compared to that with free polymers PS65–NHC–Cu(I) (refers to polymers before bound to AuNPs). This is presumably due to virtual coupling in free PS65–NHC–Cu(I) (referred to non-immobilized polymers). Moreover, the chemical shift of N–13CAu–N of PS65–13CNHC–Au is in the range of monometallic NHC–Au complexes,58,59 NHC–AuNPs,60,61 and NHC–Au clusters.60,62 The upfield shift has also been reported previously when transferring NHC–Cu(I) to NHC–Au(I).52,63 Therefore, the transmetalation from NHC–Cu(I) to Au(I) on the surface of AuNPs is likely the mechanism of our “grafting to” approach. However, on the other hand, the formation of metal–metal (Cu–Au) could be possible as well from a recent study using scanning tunneling microscope.64 We do not outline this mechanism in the scheme of Fig. 3 to distinguish the surface modification on AuNPs from free PS65–NHC–Cu(I). Regardless of NHC–Cu–Au or NHC–Au, PS65–NHC–Cu(I) can readily modify AuNPs. We also note that the chemical shift of N–13CAu–N in our results is much lower than those reported values from NHC–Au(0).51,65,66 It is possible that polymers only form adsorbed molecular species on AuNPs, other than strongly bounded species.
The grafting density of PS65–NHC ligands was evaluated by TGA (Fig. S6, ESI†). The density of PS65–NHC ligands was estimated to be 0.33 chains nm−2, corresponding to ∼144 PS chains per AuNP. Compared to our results with thiol polymers that have similar molecular weight (1–4 polymer chains per nm2),16 the grafting density of PS65–NHC ligands is low, likely due to the steric hindrance of the carbene. Nevertheless, another polymer possessing a longer block length, PS186–NHC–Cu(I), was successfully utilized for the surface modification of AuNPs using the same methodology (Fig. S7, ESI†). These results demonstrate that our method can be generalized to transform halogen end groups to NHC–Cu(I) for polymers prepared via ATRP and use the NHC–Cu(I) end groups to functionalize metal NPs through a facile “grafting to” approach.
Although successful, our initial methodology is restricted by the low conversion of the bromine substitution by N-methylimidazole. We sought an alternative synthesis that incorporates the imidazole moiety as an end group. Instead of end-group post-polymerization, the imidazole end group can be included from an imidazole-containing ATRP initiator, 2-(1H-imidazol-1-yl)ethyl-2-bromo-2-methylpropanoate. Using this ATRP initiator, three imidazole-ended (meth)acrylate polymers were synthesized (see ESI† and Fig. S8 for details), including PMMA85–Im (Mn = 7700 g mol−1 and Đ = 1.16), PnBA104–Im (Mn = 19900 g mol−1 and Đ = 1.09) and PMEO2MA125–Im (Mn = 20000 g mol−1 and Đ = 1.12) (see Fig. S9–S12, ESI†). Methylation using iodomethane followed by the metalation procedure presented above yielded all three corresponding NHC–Cu(I)-ended polymers. The incorporation of NHC–Cu(I) is much more efficient (close to 100%), compared to the substitution of end Br groups described above. Using PMMA85–Im as an example, Fig. 4b shows the 1H NMR spectra of PMMA85–Im and metalated PMMA85–NHC–Cu(I). The disappearance of peak f at 10.4 ppm, along with the shift of signals d and e confirms the coordination to Cu(I).
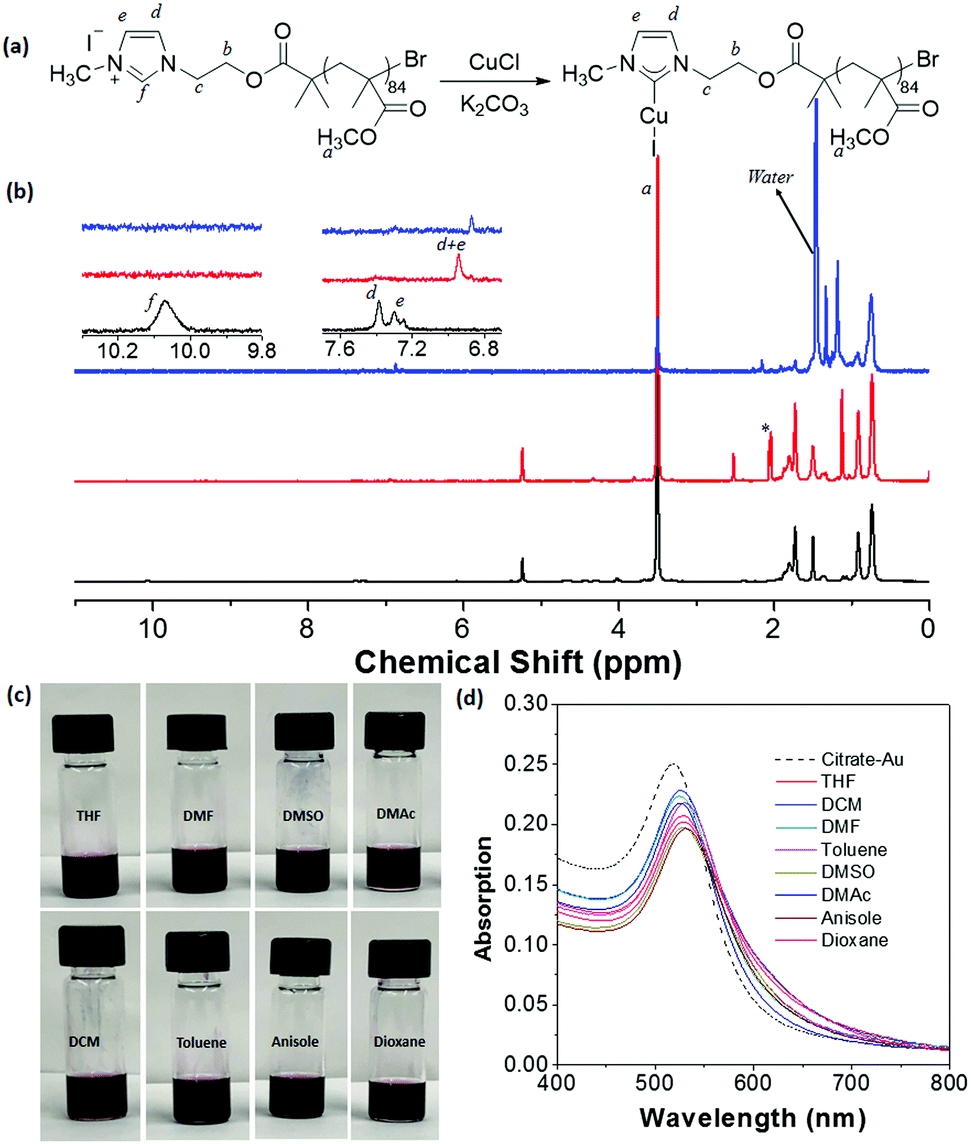 | ||
| Fig. 4 (a) Synthesis of PMMA85–NHC–Cu(I) using an imidazole-containing ATRP initiator. (b) 1H NMR spectra of PMMA85–Im (black, bottom), PMMA85–NHC–Cu(I) (red, middle) and PMMA85–NHC–Au (blue, top) in CD2Cl2. (c) Images showing PMMA85–NHC–Au dispersed in various organic solvents. (d) UV-vis spectra showing the SPR absorption of PMMA85–NHC–Au in various solvents. | ||
Surface modification of AuNPs with PMMA85–NHC–Cu(I) was performed using a similar ligand exchange method at the interface of DCM and water. Because of the absence of aromatic groups in the polymer, this surface modification can be investigated by 1H NMR spectroscopy. The two vinyl protons N–CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH–N from the imidazole ring of PMMA85–NHC–Cu(I) have a chemical shift of 6.95 ppm (Fig. 4b).28,67,68 After bound with AuNPs, the resonance of the two protons downshift to 6.87 ppm. The shift of the two vinyl protons towards values close to those reported for AuNPs28,68 also confirms the surface binding with AuNPs. PMMA85–NHC modified AuNPs show good dispersity in good solvents for PMMA such as THF, DMF, DMSO, DMAc, DCM, toluene, anisole and 1,4-dioxane (Fig. 4c). The UV-vis spectra of PMMA85–NHC–Au dispersed in various solvents shows that the SPR absorption of our modified AuNPs is similar to that of citrate-capped AuNPs in water, indicating that there is no aggregation after dispersion in organic solvents (Fig. 4d). Likewise, stable and dispersed PnBA104–NHC–Au was obtained utilizing the same methodology (Fig. S12, ESI†). It is worth noting that no successful examples have been reported using thiol-ended (meth)acrylate polymers solely to modify metal NPs through the “grafting to” method.
CH–N from the imidazole ring of PMMA85–NHC–Cu(I) have a chemical shift of 6.95 ppm (Fig. 4b).28,67,68 After bound with AuNPs, the resonance of the two protons downshift to 6.87 ppm. The shift of the two vinyl protons towards values close to those reported for AuNPs28,68 also confirms the surface binding with AuNPs. PMMA85–NHC modified AuNPs show good dispersity in good solvents for PMMA such as THF, DMF, DMSO, DMAc, DCM, toluene, anisole and 1,4-dioxane (Fig. 4c). The UV-vis spectra of PMMA85–NHC–Au dispersed in various solvents shows that the SPR absorption of our modified AuNPs is similar to that of citrate-capped AuNPs in water, indicating that there is no aggregation after dispersion in organic solvents (Fig. 4d). Likewise, stable and dispersed PnBA104–NHC–Au was obtained utilizing the same methodology (Fig. S12, ESI†). It is worth noting that no successful examples have been reported using thiol-ended (meth)acrylate polymers solely to modify metal NPs through the “grafting to” method.
To demonstrate the versatility of our method, hydrophilic PMEO2MA125–NHC–Cu(I) was also investigated to modify AuNPs (Fig. 5 and Fig. S13, ESI†). The PMEO2MA125–NHC–Au can be dispersed in a wide variety of organic solvents and water (Fig. 5b), suggesting that polymer ligands control the surface hydrophilicity of AuNPs. PMEO2MA125 modified AuNPs are stable in water and their aqueous solution can be stored for months without aggregation. The aqueous stability of PMEO2MA125–NHC–Au was examined against electrolyte-induced aggregation. While citrate-capped AuNPs aggregate when the NaCl concentration reached ∼45 mM, PMEO2MA125–NHC–Au showed no sign of aggregation even at 265 mM NaCl (Fig. 5d and e), further confirming the stabilizing power of long-hydrophilic polymer ligands.
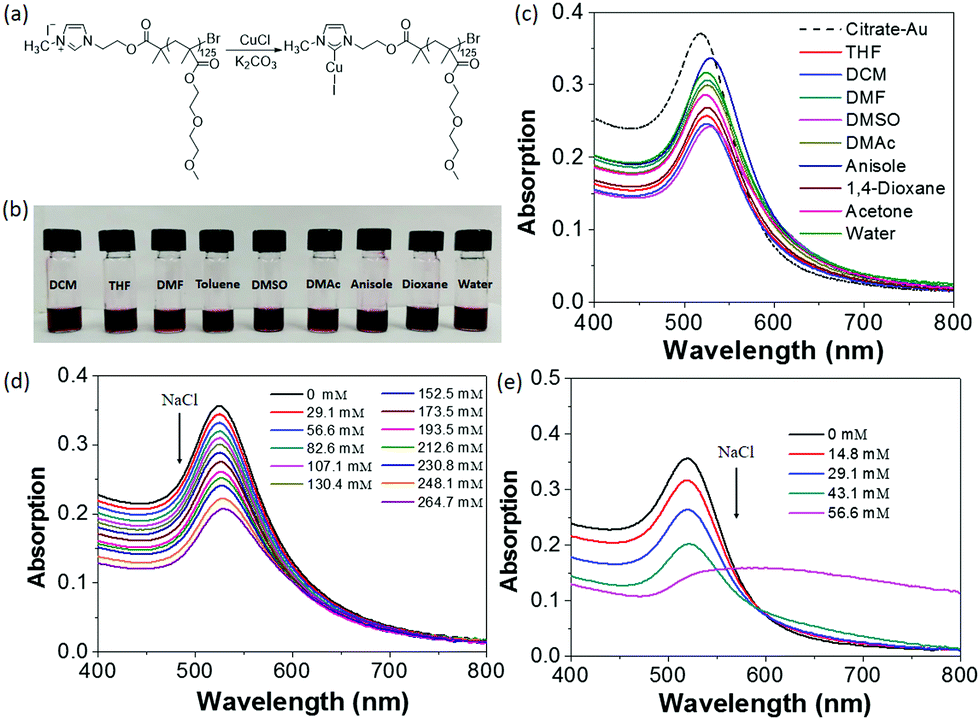 | ||
| Fig. 5 (a) Synthesis of PMEO2MA125–NHC–Cu(I) using an imidazole-containing ATRP initiator. (b) Image showing solutions of PMEO2MA125–NHC–Au in various organic solvents and water. (c) UV-vis spectra showing the SPR peaks of AuNPs dispersed in various solvents. (d and e) Titration experiments showing the stability of PMEO2MA125–NHC–Au (d) and citrate-capped AuNPs (e) upon addition of 1 M NaCl. | ||
The binding strength of PMEO2MA125–NHC with the Au surface was estimated using SPR spectroscopy. PMEO2MA125–NHC–Cu(I) aqueous solutions at different concentrations were flowed over a plain Au surface sensor chip while monitoring the change of the SPR signal to follow the adsorption. Note that, it is a flat Au surface instead of the curved surface of AuNPs. After the adsorption reached equilibrium, the surface of the Au chip was washed with DI water and the dissociation of ligands was recorded (see details in Section S4 in ESI†). The SPR response of the adsorption of the PMEO2MA174–SH is approximately two times the response of PMEO2MA125–NHC–Cu(I) at the same concentration (Fig. 6), indicating a higher grafting density for the thiolated polymer to Au. The apparent binding constants are (Ka) is 7.7 × 107 M−1 and 7.1 × 108 M−1 for PMEO2MA–NHC–Cu(I) and PMEO2MA174–SH, respectively (Table S2, ESI†). Although both values represent strong binding, the high grafting density of thiol groups on the surface of the Au chip will reduce the polymer chain dynamics, which in turn leads to a lower off-rate constant.
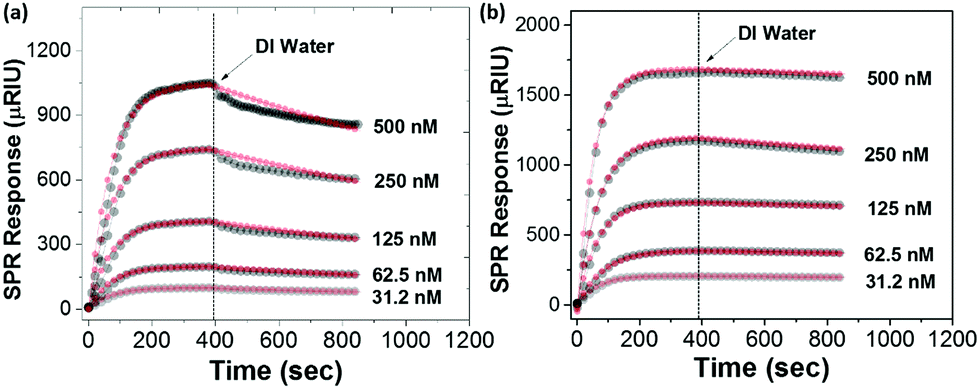 | ||
| Fig. 6 SPR traces showing the binding kinetics of (a) PMEO2MA125–NHC–Cu(I) and (b) PMEO2MA174–SH to Au. After the flow of polymer solution, the DI water was flowed over the Au sensor to trace the dissociation. The thin red lines are the fit curves of the kinetic data. | ||
4. Conclusion
To summarize, we demonstrated a simple yet powerful methodology to prepare polymer–NHCs as surface ligands to functionalize AuNPs through a “grafting to” approach. Our method highlights the use of NHC–Cu(I) terminated polymers to modify AuNPs prepared in aqueous solution. Two approaches were outlined to directly convert polymers prepared from ATRP to imidazolium-ended polymers. In the first approach, the halogen end group on conventional polymers prepared through ATRP could be functionalized using N-methylimidazole to generate the imidazolium end group; while for the second approach, the imidazole end groups of polymers were converted to imidazolium through quaternization with CH3I. Four polymers with NHC–Cu(I) end groups were obtained and readily bound to AuNPs through a biphasic ligand exchange method. The bonding from polymer–NHC–Cu(I) to polymer–NHC–Au was spectroscopically confirmed using HMBC with 13C labeling on polymer–NHCs, likely through transmetalation. Syntheses of all materials from the initial end-group functionalization to the polymer–NHC supported AuNPs were carried out on the bench without any precautions taken to exclude air or moisture. Our methods can therefore be applied to virtually any polymers prepared from ATRP, which will greatly extend the scope of polymer–NHC ligand chemistry and enrich the library of polymer ligands to metal NPs.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is supported by the startup fund of the University of Connecticut and ACS Petroleum Research Fund. The authors thank NMR facility coordinator Dr Vitaliy Gorbatyuk for his kind help on 2D HMBC measurements. JH is grateful for the support from National Science Foundation (CBET-1705566). The SEM/TEM studies were performed using the facilities in the UConn/FEI Center for Advanced Microscopy and Materials Analysis (CAMMA).Notes and references
- R. Shenhar, T. B. Norsten and V. M. Rotello, Adv. Mater., 2005, 17, 657–669 CrossRef CAS.
- J. Shan and H. Tenhu, Chem. Commun., 2007, 4580–4598 RSC.
- A. Sánchez-Iglesias, M. Grzelczak, T. Altantzis, B. Goris, J. Perez-Juste, S. Bals, G. Van Tendeloo, S. H. Donaldson Jr, B. F. Chmelka and J. N. Israelachvili, ACS Nano, 2012, 6, 11059–11065 CrossRef.
- J. Hu, T. Wu, G. Zhang and S. Liu, J. Am. Chem. Soc., 2012, 134, 7624–7627 CrossRef CAS PubMed.
- R. M. Choueiri, E. Galati, H. Thérien-Aubin, A. Klinkova, E. M. Larin, A. Querejeta-Fernández, L. Han, H. L. Xin, O. Gang and E. B. Zhulina, Nature, 2016, 538, 79–83 CrossRef CAS PubMed.
- B. Liu, S. Thanneeru, A. Lopes, L. Jin, M. McCabe and J. He, Small, 2017, 13, 1700091 CrossRef PubMed.
- A. Sánchez-Iglesias, N. Claes, D. M. Solís, J. M. Taboada, S. Bals, L. M. Liz-Marzán and M. Grzelczak, Angew. Chem., 2018, 130, 3237–3240 CrossRef.
- C. Yi, Y. Yang, B. Liu, J. He and Z. Nie, Chem. Soc. Rev., 2020 10.1039/C9CS00725C.
- J. Song, P. Huang, H. Duan and X. Chen, Acc. Chem. Res., 2015, 48, 2506–2515 CrossRef CAS.
- Y. Liu, J. He, K. Yang, C. Yi, Y. Liu, L. Nie, N. M. Khashab, X. Chen and Z. Nie, Angew. Chem., 2015, 127, 16035–16038 CrossRef.
- Z. Cao, D. Kim, D. Hong, Y. Yu, J. Xu, S. Lin, X. Wen, E. M. Nichols, K. Jeong, J. A. Reimer, P. Yang and C. J. Chang, J. Am. Chem. Soc., 2016, 138, 8120–8125 CrossRef CAS.
- Z. Cao, J. Derrick, J. Xu, R. Gao, E. Nichols, P. Smith, X. Liu, X. Wen, C. Coperet and C. Chang, Angew. Chem., Int. Ed., 2018, 57, 1–6 CrossRef.
- J. He, X. Huang, Y.-C. Li, Y. Liu, T. Babu, M. A. Aronova, S. Wang, Z. Lu, X. Chen and Z. Nie, J. Am. Chem. Soc., 2013, 135, 7974–7984 CrossRef CAS PubMed.
- G. Chen, Y. Wang, M. Yang, J. Xu, S. J. Goh, M. Pan and H. Chen, J. Am. Chem. Soc., 2010, 132, 3644–3645 CrossRef CAS PubMed.
- A. B. Lowe, B. S. Sumerlin, M. S. Donovan and C. L. McCormick, J. Am. Chem. Soc., 2002, 124, 11562–11563 CrossRef CAS.
- M. K. Corbierre, N. S. Cameron and R. B. Lennox, Langmuir, 2004, 20, 2867–2873 CrossRef CAS PubMed.
- E. R. Zubarev, J. Xu, A. Sayyad and J. D. Gibson, J. Am. Chem. Soc., 2006, 128, 15098–15099 CrossRef CAS PubMed.
- Z. Nie, D. Fava, E. Kumacheva, S. Zou, G. C. Walker and M. Rubinstein, Nat. Mater., 2007, 6, 609 CrossRef CAS PubMed.
- Z. Nie, D. Fava, M. Rubinstein and E. Kumacheva, J. Am. Chem. Soc., 2008, 130, 3683–3689 CrossRef CAS PubMed.
- B. Wang, B. Li, B. Zhao and C. Y. Li, J. Am. Chem. Soc., 2008, 130, 11594–11595 CrossRef CAS PubMed.
- J. He, Y. Liu, T. Babu, Z. Wei and Z. Nie, J. Am. Chem. Soc., 2012, 134, 11342–11345 CrossRef CAS PubMed.
- R. Liang, J. Xu, R. Deng, K. Wang, S. Liu, J. Li and J. Zhu, ACS Macro Lett., 2014, 3, 486–490 CrossRef CAS.
- T. K. Mandal, M. S. Fleming and D. R. Walt, Nano Lett., 2002, 2, 3–7 CrossRef.
- J. Raula, J. Shan, M. Nuopponen, A. Niskanen, H. Jiang, E. I. Kauppinen and H. Tenhu, Langmuir, 2003, 19, 3499–3504 CrossRef CAS.
- J. Song, L. Cheng, A. Liu, J. Yin, M. Kuang and H. Duan, J. Am. Chem. Soc., 2011, 133, 10760–10763 CrossRef CAS PubMed.
- M.-T. Lee, C.-C. Hsueh, M. S. Freund and G. S. Ferguson, Langmuir, 1998, 14, 6419–6423 CrossRef CAS.
- G. E. Poirier, T. M. Herne, C. C. Miller and M. J. Tarlov, J. Am. Chem. Soc., 1999, 121, 9703–9711 CrossRef CAS.
- E. C. Hurst, K. Wilson, I. J. Fairlamb and V. Chechik, New J. Chem., 2009, 33, 1837–1840 RSC.
- J. Vignolle and T. D. Tilley, Chem. Commun., 2009, 7230–7232 RSC.
- K. V. Ranganath, J. Kloesges, A. H. Schäfer and F. Glorius, Angew. Chem., Int. Ed., 2010, 49, 7786–7789 CrossRef CAS PubMed.
- P. Lara, O. Rivada-Wheelaghan, S. Conejero, R. Poteau, K. Philippot and B. Chaudret, Angew. Chem., 2011, 123, 12286–12290 CrossRef.
- C. Richter, K. Schaepe, F. Glorius and B. J. Ravoo, Chem. Commun., 2014, 50, 3204–3207 RSC.
- S. G. Song, C. Satheeshkumar, J. Park, J. Ahn, T. Premkumar, Y. Lee and C. Song, Macromolecules, 2014, 47, 6566–6571 CrossRef CAS.
- M. J. MacLeod and J. A. Johnson, J. Am. Chem. Soc., 2015, 137, 7974–7977 CrossRef CAS PubMed.
- A. V. Zhukhovitskiy, M. J. MacLeod and J. A. Johnson, Chem. Rev., 2015, 115, 11503–11532 CrossRef CAS PubMed.
- M. D. de los Bernardos, S. Perez-Rodriguez, A. Gual, C. Claver and C. Godard, Chem. Commun., 2017, 53, 7894–7897 RSC.
- S. Engel, E.-C. Fritz and B. J. Ravoo, Chem. Soc. Rev., 2017, 46, 2057–2075 RSC.
- K. Klauke, I. Gruber, T.-O. Knedel, L. Schmolke, J. Barthel, H. Breitzke, G. Buntkowsky and C. Janiak, Organometallics, 2018, 37, 298–308 CrossRef CAS.
- C. A. Smith, M. R. Narouz, P. A. Lummis, I. Singh, A. Nazemi, C.-H. Li and C. M. Crudden, Chem. Rev., 2019, 119, 4986–5056 CrossRef CAS PubMed.
- D. Bourissou, O. Guerret, F. P. Gabbaï and G. Bertrand, Chem. Rev., 2000, 100, 39–92 CrossRef CAS PubMed.
- T. Rovis and S. P. Nolan, Synlett, 2013, 1188–1189 CrossRef CAS.
- S. P. Nolan, N-Heterocyclic Carbenes: Effective Tools for Organometallic Synthesis, John Wiley & Sons, 2014 Search PubMed.
- C. M. Crudden, J. H. Horton, I. I. Ebralidze, O. V. Zenkina, A. B. McLean, B. Drevniok, Z. She, H.-B. Kraatz, N. J. Mosey and T. Seki, Nat. Chem., 2014, 6, 409 CrossRef CAS PubMed.
- A. Ferry, K. Schaepe, P. Tegeder, C. Richter, K. M. Chepiga, B. J. Ravoo and F. Glorius, ACS Catal., 2015, 5, 5414–5420 CrossRef CAS.
- J. B. Ernst, C. Schwermann, G.-i. Yokota, M. Tada, S. Muratsugu, N. L. Doltsinis and F. Glorius, J. Am. Chem. Soc., 2017, 139, 9144–9147 CrossRef CAS PubMed.
- H. Rothfuss, N. D. Knofel, P. W. Roesky and C. Barner-Kowollik, J. Am. Chem. Soc., 2018, 140, 5875–5881 CrossRef CAS PubMed.
- R. Ye, A. V. Zhukhovitskiy, R. V. Kazantsev, S. C. Fakra, B. B. Wickemeyer, F. D. Toste and G. A. Somorjai, J. Am. Chem. Soc., 2018, 140, 4144–4149 CrossRef CAS PubMed.
- R. W. Y. Man, C.-H. Li, M. W. A. MacLean, O. V. Zenkina, M. T. Zamora, L. N. Saunders, A. Rousina-Webb, M. Nambo and C. M. Crudden, J. Am. Chem. Soc., 2018, 140, 1576–1579 CrossRef CAS PubMed.
- A. Rühling, K. Schaepe, L. Rakers, B. Vonhören, P. Tegeder, B. J. Ravoo and F. Glorius, Angew. Chem., Int. Ed., 2016, 55, 5856–5860 CrossRef PubMed.
- L. Zhang, Z. Wei, S. Thanneeru, M. Meng, M. Kruzyk, G. Ung, B. Liu and J. He, Angew. Chem., Int. Ed., 2019, 58, 15834–15840 CrossRef CAS PubMed.
- M. R. Narouz, K. M. Osten, P. J. Unsworth, R. W. Man, K. Salorinne, S. Takano, R. Tomihara, S. Kaappa, S. Malola and C.-T. Dinh, Nat. Chem., 2019, 11, 419 CrossRef CAS PubMed.
- O. Santoro, A. Collado, A. M. Z. Slawin, S. P. Nolan and C. S. J. Cazin, Chem. Commun., 2013, 49, 10483–10485 RSC.
- M. R. Furst and C. S. Cazin, Chem. Commun., 2010, 46, 6924–6925 RSC.
- Y. D. Bidal, O. Santoro, M. Melaimi, D. B. Cordes, A. M. Z. Slawin, G. Bertrand and C. S. J. Cazin, Chem. – Eur. J., 2016, 22, 9404–9409 CrossRef CAS PubMed.
- K. Salorinne, R. W. Y. Man, C. H. Li, M. Taki, M. Nambo and C. M. Crudden, Angew. Chem., Int. Ed., 2017, 56, 6198–6202 CrossRef CAS PubMed.
- R. E. Wasylishen and G. Tomlinson, Biochem. J., 1975, 147, 605–607 CrossRef CAS PubMed.
- K. Pachler, R. Pachter and P. Wessels, Org. Magn. Reson., 1981, 17, 278–284 CrossRef CAS.
- M. E. Garner, W. Niu, X. Chen, I. Ghiviriga, K. A. Abboud, W. Tan and A. S. Veige, Dalton Trans., 2015, 44, 1914–1923 RSC.
- V. Lewe, M. Preuss, E. A. Woźnica, D. Spitzer, R. Otter and P. Besenius, Chem. Commun., 2018, 54, 9498–9501 RSC.
- M. Rodríguez-Castillo, D. Laurencin, F. Tielens, A. van der Lee, S. Clément, Y. Guari and S. Richeter, Dalton Trans., 2014, 43, 5978–5982 RSC.
- Z. Cao, D. Kim, D. Hong, Y. Yu, J. Xu, S. Lin, X. Wen, E. M. Nichols, K. Jeong and J. A. Reimer, J. Am. Chem. Soc., 2016, 138, 8120–8125 CrossRef CAS PubMed.
- M. Rodríguez-Castillo, G. Lugo-Preciado, D. Laurencin, F. Tielens, A. van der Lee, S. Clément, Y. Guari, J. M. López-de-Luzuriaga, M. Monge and F. Remacle, Chem. – Eur. J., 2016, 22, 10446–10458 CrossRef PubMed.
- M. R. L. Furst and C. S. J. Cazin, Chem. Commun., 2010, 46, 6924–6925 RSC.
- E. A. Doud, M. S. Inkpen, G. Lovat, E. Montes, D. W. Paley, M. L. Steigerwald, H. c. Vázquez, L. Venkataraman and X. Roy, J. Am. Chem. Soc., 2018, 140, 8944–8949 CrossRef CAS PubMed.
- D. S. Weinberger, M. Melaimi, C. E. Moore, A. L. Rheingold, G. Frenking, P. Jerabek and G. Bertrand, Angew. Chem., Int. Ed., 2013, 52, 8964–8967 CrossRef CAS PubMed.
- L. Jin, D. S. Weinberger, M. Melaimi, C. E. Moore, A. L. Rheingold and G. Bertrand, Angew. Chem., Int. Ed., 2014, 53, 9059–9063 CrossRef CAS PubMed.
- S. Díez-González, E. C. Escudero-Adán, J. Benet-Buchholz, E. D. Stevens, A. M. Slawin and S. P. Nolan, Dalton Trans., 2010, 39, 7595–7606 RSC.
- J. Crespo, Y. Guari, A. Ibarra, J. Larionova, T. Lasanta, D. Laurencin, J. M. López-de-Luzuriaga, M. Monge, M. E. Olmos and S. Richeter, Dalton Trans., 2014, 43, 15713–15718 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: Synthetic details of polymers, polymer–NHC–Cu(I) and surface ligand exchange methods as well as the additional characterization of polymer–NHC capped AuNPs. See DOI: 10.1039/c9tc04776j |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2020 |