
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Tuneable peptide cross-linked nanogels for enzyme-triggered protein delivery†
Lucia
Massi‡
a,
Adrian
Najer‡
 a,
Robert
Chapman
b,
Christopher D.
Spicer§
a,
Valeria
Nele
a,
Junyi
Che
a,
Marsilea A.
Booth
a,
James J.
Doutch
c and
Molly M.
Stevens
*a
a,
Robert
Chapman
b,
Christopher D.
Spicer§
a,
Valeria
Nele
a,
Junyi
Che
a,
Marsilea A.
Booth
a,
James J.
Doutch
c and
Molly M.
Stevens
*a
aDepartment of Materials, Department of Bioengineering, and Institute of Biomedical Engineering, Imperial College London, London SW7 2AZ, UK. E-mail: m.stevens@imperial.ac.uk
bCentre for Advanced Macromolecular Design (CAMD) and the Australian Centre for Nanomedicine (ACN), School of Chemistry, UNSW Sydney, Australia
cISIS Neutron and Muon Source, STFC, Rutherford Appleton Laboratory, Didcot OX11 ODE, UK
First published on 3rd September 2020
Abstract
Many diseases are associated with the dysregulated activity of enzymes, such as matrix metalloproteinases (MMPs). This dysregulation can be leveraged in drug delivery to achieve disease- or site-specific cargo release. Self-assembled polymeric nanoparticles are versatile drug carrier materials due to the accessible diversity of polymer chemistry. However, efficient loading of sensitive cargo, such as proteins, and introducing functional enzyme-responsive behaviour remain challenging. Herein, peptide-crosslinked, temperature-sensitive nanogels for protein delivery were designed to respond to MMP-7, which is overexpressed in many pathologies including cancer and inflammatory diseases. The incorporation of N-cyclopropylacrylamide (NCPAM) into N-isopropylacrylamide (NIPAM)-based copolymers enabled us to tune the polymer lower critical solution temperature from 33 to 44 °C, allowing the encapsulation of protein cargo and nanogel-crosslinking at slightly elevated temperatures. This approach resulted in nanogels that were held together by MMP-sensitive peptides for enzyme-specific protein delivery. We employed a combination of cryogenic transmission electron microscopy (cryo-TEM), dynamic light scattering (DLS), small angle neutron scattering (SANS), and fluorescence correlation spectroscopy (FCS) to precisely decipher the morphology, self-assembly mechanism, enzyme-responsiveness, and model protein loading/release properties of our nanogel platform. Simple variation of the peptide linker sequence and combining multiple different crosslinkers will enable us to adjust our platform to target specific diseases in the future.
Introduction
The development of smart and controlled nanocarriers for targeted and triggered drug delivery provides therapeutic opportunities for the treatment of major diseases such as infections, inflammation, and cancer.1 Synthetic amphiphilic copolymers, able to undergo self-assembly into nanostructures, are ideal carriers for drug delivery due to the flexibility and diversity of polymer chemistry. Various carrier morphologies can be engineered with tuneable stabilities and surface chemistries, which include facile functionalisation with targeting ligands, in addition to triggerable drug release via stimuli-responsive copolymers.2–5 Typical morphologies of polymer-based structures used for drug delivery include micelles (spherical and worm-like), vesicles (polymersomes), capsules, polyion complex (PIC) micelles and vesicles, nanoparticles, and nanogels. The key advantages offered by nanogels are their ease of preparation that yields uniform and tuneable sizes, relatively high encapsulation efficiency, stability in the presence of serum, and the possibility of simple introduction of stimuli responsiveness.6 The properties and formation method of nanogels make them suitable candidates for protein delivery.7,8To yield high level of control of drug loading and triggered release, temperature and/or enzyme-responsive motifs can be incorporated in polymer-based drug delivery.9 With respect to temperature-triggered systems, poly(N-isopropylacrylamide) (PNIPAM) remains one of the most studied polymers incorporated in the design of temperature-responsive materials for biomedical applications due to its fast temperature-induced phase transition at a lower critical solution temperature (LCST) of ∼32 °C.10,11 Copolymers of PNIPAM with a temperature-independent hydrophilic block allow rapid assembly of nanostructures upon heating either after or during the polymerisation because of the thermally induced collapse of the PNIPAM block at temperatures above the LCST.12 However, PNIPAM-based nanoassemblies are limited to transition around 32 °C; the LCST cannot be changed easily. Copolymerisation of NIPAM with other co-monomers is one option to modulate the LCST.13,14 However, temperature is not an ideal trigger for cargo release given our fixed body temperature. Hence, modulation of the self-assembly temperatures (LCST) using a range of different copolymers, combined with a suitable stimuli-responsive cross-linking strategy, represents an interesting alternative platform that could be employed both above and below the LCST.
Our strategy uses temperature changes as a simple and fast organic solvent-free nanogel formation method to encapsulate biomolecules. Poly(N-cyclopropylacrylamide) (PNCPAM), closely related to PNIPAM, consists of pendant cyclopropylamine groups instead of the isopropylamine groups in PNIPAM and possesses a LCST of ∼49 °C.10 We propose herein that the copolymerisation of NCPAM and NIPAM to form block copolymers together with a hydrophilic block—consisting of a short block with functional reactive handles and polyethylene glycol (PEG)—provides an efficient method to precisely control the LCST, and hence the self-assembly temperature, from 32 up to 49 °C. Fine-tuning of the transition temperature using a library of copolymers, with precise control of the LCST around body temperature, enables application-dependent selection of a certain copolymer from the library to create customised vehicles for loading and release of sensitive therapeutic protein cargoes.
A cross-linking strategy was further included to (i) stabilise the nanocarrier, (ii) avoiding premature release of cargo, and (iii) incorporating stimuli-responsiveness. This additional cross-linking strategy serves as an additional trigger besides temperature for cargo release, through degradation of the responsive cross-links. These are important considerations when taking into account the immensely complex environment faced by nanocarriers upon in vivo administration. We employed a copper-catalysed azide–alkyne cycloaddition (CuAAC) cross-linking strategy, which remains one of the most popular “click” reactions for cross-linking and functionalisation in the biomaterial and biomedical fields due to high efficiency, specificity, and simplicity, to incorporate the necessary functional handles.15–17 Our covalent cross-linking strategy provided the fundamental advantage of keeping our nanogels intact above and below the LCST, which in turn allows fast and simple loading of cargo and subsequent purification.
With respect to cross-linker backbone chemistry, peptides are particularly attractive due to their simple solid phase synthesis, incorporation of non-canonical functional groups for the introduction of “clickable” handles,18,19 and the possibility of using enzyme-specific cleavable peptide sequences.19,20 Due to their intrinsic involvement in biological and metabolic processes, enzymes have emerged as suitable triggers for drug delivery.21 Different classes of enzymes (e.g. proteases, phospholipases, oxidoreductases) have been leveraged for the purpose of triggered drug release from nanocarriers.22 A specific class of proteolytic enzymes, matrix metalloproteinases (MMPs), are associated with several disease conditions, including infections, inflammation, cardiovascular diseases and cancer.23 Overexpression of MMPs at disease sites can be exploited to release drugs at the target site by selective rupture of engineered MMP-responsive delivery platforms.22,24
Herein, we successfully synthesised libraries of PNIPAM-, PNCPAM- and P(NIPAM-co-NCPAM)-based triblock copolymers with tuneable compositions via reversible addition–fragmentation chain-transfer (RAFT) polymerisation. The structure of the final block copolymers was carefully designed to include these temperature-responsive blocks, a functional block for nanogel cross-linking and a hydrophilic outer block. The thermo-responsive block is hydrophilic below its LCST and turns hydrophobic above the LCST, thus switching the block copolymer from hydrophilic to amphiphilic, which promotes the formation of self-assembled nanostructures. The choice over amphiphilicity or hydrophilicity of degradation products at body temperature, through selecting a copolymer with a transition temperature below or above 37 °C, offers means of potentially modulating elimination pathways upon in vivo administration in the future. Choosing a suitable NIPAM–NCPAM ratio in the thermo-responsive block will further allow one to adjust the LCST to only slightly below body temperature, which could be a means of speeding up cargo release at the disease site, when close to the surface, through locally applied cold patches. To increase nanoparticle stability and incorporate responsiveness to a model MMP enzyme (MMP-7), azido-bi-functionalised peptides were synthesised and used to cross-link the nanoparticles via the alkyne groups present in the functional middle block using CuAAC.
A considerable challenge when deciphering the exact morphology of copolymer assemblies is the structural ambiguity that is provided by certain routine characterisation techniques such as conventional dynamic light scattering (DLS) and negative stain transmission electron microscopy (TEM). However, accurate information on morphology is of great importance since it directly affects choice of suitable disease targets, drug cargo, delivery route, stability and release mechanism. Thus, the use of additional complementary characterisation techniques is needed to characterise the morphology of soft self-assembled nanostructures. TEM at cryogenic temperatures (cryo-TEM)25,26 and small-angle neutron scattering (SANS)27 are two complementary methods to study these types of nanostructures in their native hydrated state.28–30 A third technique, fluorescence correlation spectroscopy (FCS) is a diffusion-based single-molecule analysis technique.31,32 FCS allows accurate characterisation of nanoparticle assembly/disassembly—including in the presence of enzymatic triggers—surface functionalisation, cargo loading, and release.33–35 Our thoroughly characterised nanogel system is shown to serve as a versatile protein delivery platform with a simple and gentle temperature induced formation and loading protocol, and triggered release through stimuli-responsiveness provided by enzymatically cleavable cross-links. This modular platform can be adjusted for disease-specific applications by choosing the appropriate copolymer, peptide cross-linker sequence, and therapeutic protein cargo.
Results and discussion
Synthesis of peptide cross-linkable block copolymers with fine-tuned temperature-responsiveness
We designed triblock copolymers to confer the final self-assembled nanogels with favourable properties for enzyme-triggered drug delivery applications (Scheme 1). Conceptually, an inert hydrophilic block (Scheme 1, dark blue), which we expected to form the corona of the assembled nanogels, was connected to a thermo-sensitive block (Scheme 1, light blue/green) which would drive self-assembly at temperatures above its LCST. We chose to use PEG as the hydrophilic block and either PNIPAM, PNCPAM or copolymers of the two P(NIPAM-co-NCPAM) as the thermo-responsive block. Between these two blocks we incorporated a functional block (Scheme 1, red) composed of a mixture of methacrylic acid (MAA) and trimethylsilylpropargylmethacrylate (TMSPMA) to provide a latent handle for cross-linking with azide-bi-functionalised peptide after self-assembly and desilylation.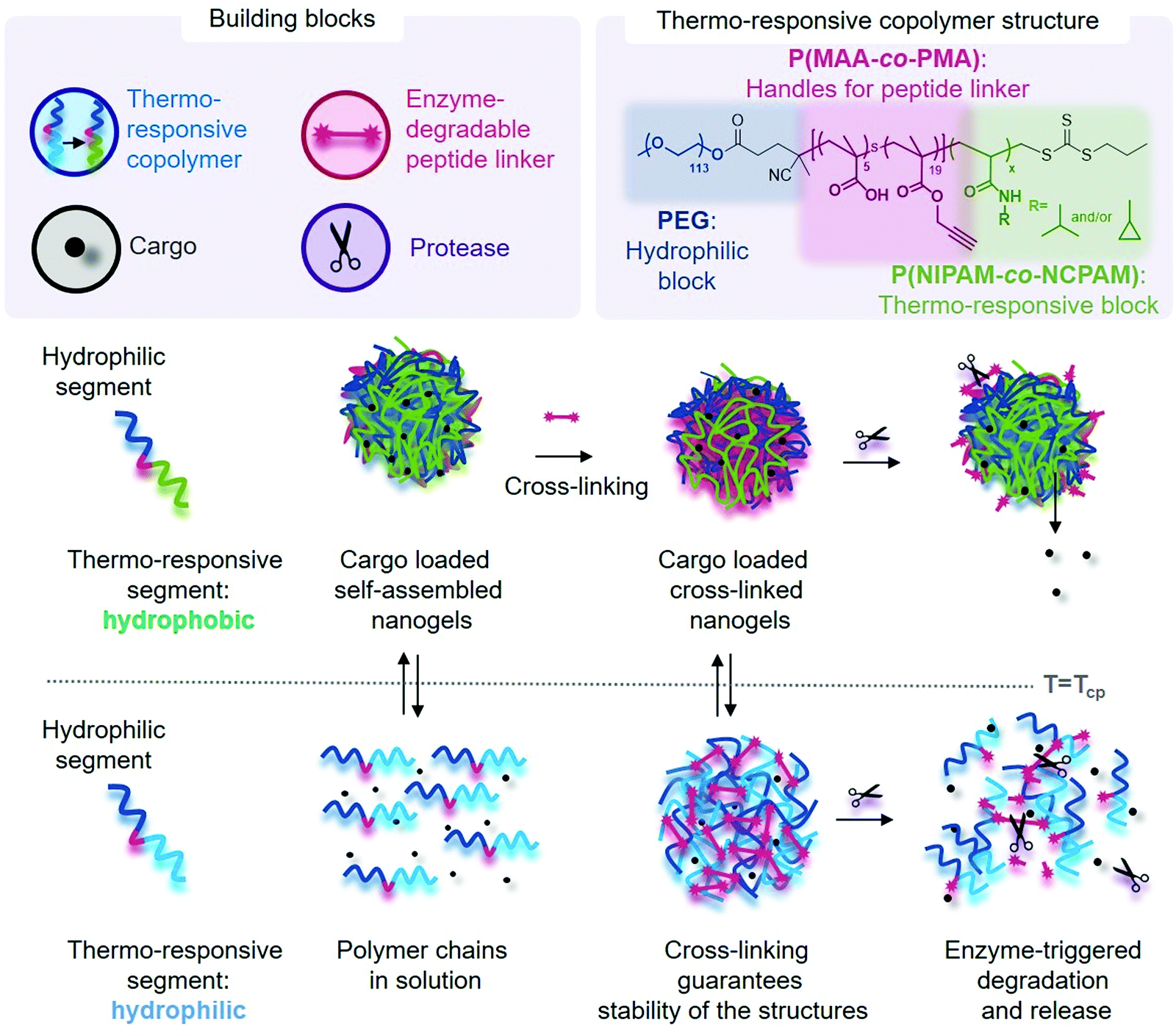 | ||
| Scheme 1 Illustration of copolymer design, and biomolecule loading and release mechanism of the dual-responsive nanogel platform. Thermo-responsive triblock copolymers self-assemble above the LCST (Tcp: cloud point temperature as measured by DLS), which was used to encapsulate biomolecule cargo. Subsequent crosslinking via enzyme-responsive peptides increases the nanogel stability below and above the LCST and provides enzyme-triggered cargo release. | ||
We used RAFT polymerisation for the synthesis of these polymers, due to the ease of preparing well-defined multi-block copolymers and PNIPAM via this route.11,36–40 The detailed synthetic procedure is shown in Scheme 2.
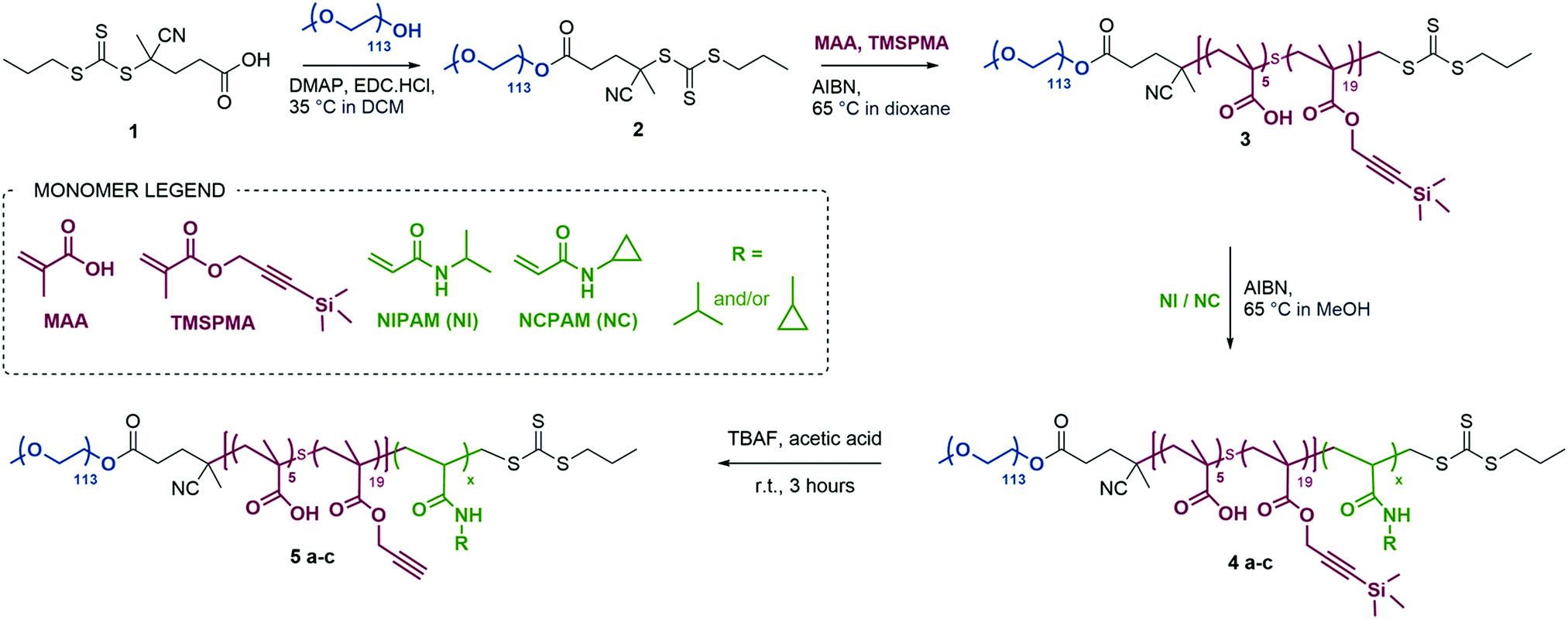 | ||
| Scheme 2 Synthetic route for P(NIPAM)-, P(NCPAM)- and P(NIPAM-co-NCPAM)-based triblock copolymers. Copolymer names correspond to the specific thermo-responsive block repeating units: (a) for NIPAM, (b) for NCPAM and (c) for NIPAM/NCPAM copolymers. | ||
The first step consisted of the synthesis of the RAFT agent 1, following a procedure previously reported in the literature,41 which was then conjugated to a PEG block (113 units) via esterification to form a macroRAFT agent. Chain extension of this macroRAFT agent with a random copolymer of MAA and TMSPMA enabled the introduction of carboxylic groups and terminal alkyne functionalities (in their protected form) as functional handles. Good control over the polymerisation of PEG-b-P(MAA-co-TMSPMA) 3 was evidenced by low dispersity Đ = 1.07, and a clean shift in the gel permeation chromatography traces (GPC, Fig. S1a, ESI†). The same polymer (with 113 PEG units, 5 MAA units and 19 TMSPMA units) was used throughout the remainder of the study and chain extended to produce three different libraries of triblock copolymers: (i) a library of PNIPAM-based block copolymers (5a1–5a5) with increasing number of NIPAM units; (ii) a library of PNCPAM-based block copolymers (5b1–5b5) with increasing number of NCPAM units; (iii) a library of P(NIPAM-co-NCPAM)-based block copolymers (5c1–5c3) with different NIPAM/NCPAM (NI/NC) ratios. We hypothesised that the incorporation of cyclopropyl moieties (NCPAM) would lead to an increase in the LCST providing a Tcp (cloud point temperature as measured by DLS) for nanogel self-assembly/disassembly at temperatures just above body temperature.
Successful synthesis of the three block copolymer libraries was confirmed by 1H NMR and GPC yielding degree of polymerisation (DP), number average molecular weight (Mn), and dispersity (Đ) (Table 1). Full composition and characterisation details of selected 5a4/5c1–3 block copolymers are reported in Table 1, whilst data for the remaining block copolymers can be found in Table S6 of the ESI.†
| # | NI/NC ratioa | DP (NMR) | M n (Da) (NMR) | M n (Da) (GPC) | Đ (Mw/Mn) (GPC) | D H (nm) (DLS) | T cp (°C) |
|---|---|---|---|---|---|---|---|
| a NI/NC ratio is NIPAM to NCPAM ratio; DP is degree of polymerisation calculated by 1H NMR; Mn is the number average molecular weight calculated by GPC and NMR; Mw/Mn represents the dispersity obtained by GPC; DH in nm represent the hydrodynamic diameters of the resulting self-assembled nanostructures (T above Tcp: 5a4 = 34 °C, 5c1 = 60 °C, 5c2 = 55 °C, 5c3 = 55 °C) reported by DLS (number/%) and Tcp is the cloud point temperature as measured by DLS. | |||||||
| 5a4 | NI1.0NC0.0 | 421 | 55![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 800 800 |
66600 |
1.41 | 74 ± 7 | 33 |
| 5c1 | NI0.8NC0.2 | 421 | 55600 |
61900 |
1.31 | 80 ± 2 | 37 |
| 5c2 | NI0.5NC0.5 | 395 | 52500 |
77800 |
1.34 | 179 ± 20 | 44 |
| 5c3 | NI0.4NC0.6 | 357 | 48100 |
58700 |
1.29 | 295 ± 74 | — |
For the P(NIPAM-co-NCPAM)-based copolymers (5c1–3), full chain extension was observed for all the copolymers with a minimal amount of macro-RAFT agent left behind and minimal shouldering in the GPC traces (Fig. S1a, ESI†). Due to the structural similarity and similar degree of reactivity between NIPAM and NCPAM, the formation of a random block stemming from their simultaneous polymerisation rather than two distinct blocks was assumed. In general, the dispersity was Đ < 1.5. The length of the thermo-responsive block was successfully modulated in the separate NIPAM and NCPAM libraries (Table S6, ESI†), whilst the copolymers shown in Table 1 were of similar length, as shown by NMR and GPC. The evolution of Mn and Đ versus DP is shown in Fig. S1b (ESI†). We also determined the block copolymer cloud point temperatures Tcpvia temperature sweeps and monitoring by dynamic light scattering (DLS). The corresponding sizes of the self-assembled nanostructures resulting from heating the block copolymer solutions above the Tcp are included in Table 1 and Table S6 (ESI†). The small difference in the number mean size between 5a4 (69.0 ± 2.7 nm, n = 3, N = 3) and 5c1 (98.1 ± 9.6 nm, n = 3, N = 3) above the LCST is significant (*p = 0.043, Student's t-test, n = 3, N = 3) when comparing three independent samples.
Modulation of self-assembly temperature via P(NIPAM-co-NCPAM)-based copolymers
Temperature-responsive assembly/disassembly of nanostructures around body temperature is a valuable means of controlling encapsulation/release of therapeutic cargo. Modulation of the transition temperature allows for application-specific optimisation of cargo loading and delivery, which are critical considerations in the drug delivery field. The synthesised P(NIPAM-co-NCPAM)-based triblock copolymers were designed to exhibit increasing transition temperatures. DLS was used to confirm successful modulation of the self-assembly temperature for the P(NIPAM-co-NCPAM)-based triblock copolymers containing increasing amounts of the cyclopropyl-based repeating unit (NCAPM from 0 to 60%) (5a4/5c1–3) (Fig. 1). By dissolving these copolymers in PBS at 25 °C and increasing the temperature to 60 °C (above the LCST), a clear increase in the turbidity of the solution could be observed by eye and by light scattering (derived count rate, kcps) recorded using DLS, which suggests a coil-to-globule transition (Fig. 1b). Since larger structures scatter more light than dissolved copolymers, the self-assembly of the copolymers could be followed by monitoring the scattering intensity. The inflection points of each curve represent the Tcp of the corresponding copolymers. DLS measurements at temperatures below/above the LCST indicate formation of stable and well-defined nanostructures when heated above the LCST (Fig. 1c), whilst bigger structures were formed with increasing amounts of NCPAM (mean hydrodynamic diameters between 74 and 295 nm). Due to the formation of large structures/aggregates that led to some precipitation for 5c3 when heated/cooled slowly (1 °C temperature ramp with 5 min equilibration time), the intensity curves are not smooth (Fig. 1b) and no accurate Tcp can be given for this copolymer. However, it appears to be between 45 to 50 °C when estimating based on both curves (up/down, heating and cooling). The dependence of particle size on the heating rate during the self-assembly process was also confirmed for two copolymers that form smaller nanoparticles in general (5a4 and 5c1, Fig. 1d). The final assembled particles tended to be larger when subjected to slower heating. Comparing the transition temperature of 5a4 (33 °C) with previously reported PEG-b-PNIPAM (33.7 °C),42 it can be concluded that the short functional middle block in our copolymers did not affect the LCST.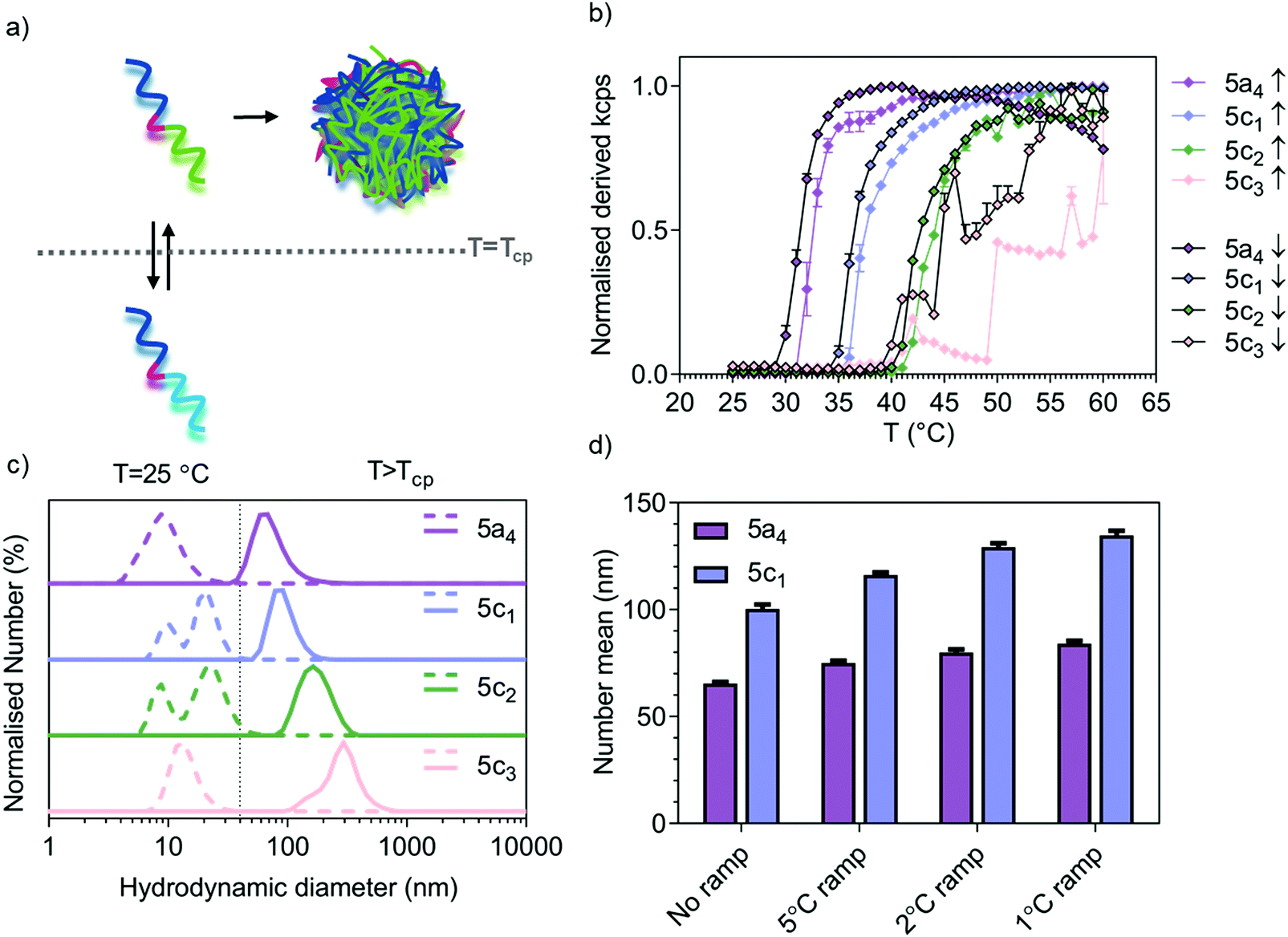 | ||
| Fig. 1 Thermo-responsive behaviour of P(NIPAM-co-NCPAM)-based triblock copolymers. (a) Schematic representation of temperature-triggered self-assembly of acrylamide-based block copolymers. (b) Temperature trend from 25 to 60 °C of P(NIPAM-co-NCPAM)-based copolymers measured by DLS demonstrating the evolution of normalised derived count rate versus increasing temperature (the inflection point of each curve indicates Tcp). (c) Normalised size distribution based on dissolved copolymers in PBS below (dashed line) and above (continuous line) the LCST (Tcp) as measured by DLS (number %). (d) Effect of heating rate on self-assembled nanogel hydrodynamic diameter for 5a4 and 5c1 block copolymers as shown by measuring the hydrodynamic diameter (number mean ± SD in nm, n = 3) by DLS. | ||
The cyclopropyl-dependent size increase might be caused by the higher rigidity and lower rotational freedom of the cyclopropyl group when compared to the isopropyl group, which might affect the packing parameters to yield larger structures. The self-assembly temperature of the resulting triblock copolymers was successfully tuned from 33 °C, corresponding to the Tcp of the 5a4 copolymer with 0% NCPAM content, to a Tcp of about 47 °C for the 5c3 copolymer with 60% NCPAM content. It can be concluded that by increasing the number of cyclopropyl units in the copolymers, the self-assembly temperature increases proportionally. However, when the NCPAM content exceeds 50%, the stability of the resulting self-assembled structures is compromised. Formation of large particles/aggregates, which led to sedimentation was observed (Fig. 1b). The self-assembly process of PNIPAM- and P(NIPAM-co-NCPAM)-based copolymers can be reversed by cooling the solution below the transition temperature (down to 25 °C) causing complete nanogel dissolution yielding free polymer chains in solution (Fig. 1b and Fig. S1c, ESI†). The slight hysteretic behaviours in Fig. 1b could be explained by a slightly slower globule-to-coil transition compared to the coil-to-globule transition. According to this initial DLS analysis, the selected block copolymers (5a4/5c1–3) revealed an LCST-type, reversible thermo-responsive behaviour that yielded nanogels at temperatures above their LCST. We also tested all the other copolymers from the separate PNIPAM (5a1 to 5a5) and PNCPAM (5b1 to 5b5) series for the formation of nanogels at temperatures above the LCST (Table S6, ESI†). All copolymers except 5b1 assembled into nanogels, however, no clear trends regarding polymer lengths and corresponding nanostructure sizes could be found.
Characterisation of morphology and self-assembly process by cryo-TEM and SANS
Accurate shape, size, and morphology characterisation of drug delivery systems is crucial for understanding their behaviour, especially for dynamic self-assembled systems. This will help to define their potential as drug delivery vehicles. As an example, morphology and shape can affect cargo loading and cellular uptake.43 Conventional DLS and TEM can give an indication of nanostructure size, polydispersity, and shape, but complementary techniques are needed to decipher the internal morphology of self-assembled particles. SANS allows more detailed structural information to be acquired in an artefact-free manner to obtain bulk properties such as morphology and polydispersity. Cryo-TEM is a useful tool for imaging self-assembled nanostructures to obtain a snapshot in their native hydrated state. Often, the drying step in the sample preparation procedure needed for negative stain TEM introduces drying artefacts, which may prevent accurate morphology determination.44 A combination of SANS and cryo-TEM is often used when characterising block copolymer assemblies in solution, e.g. when studying vesicle shape transformations at the nanoscale.28–30 We used these two techniques to further analyse the self-assembly process by first studying the copolymers before (‘coil’ state) and after self-assembly (‘globule’ state) (Fig. 2). We chose the copolymers 5a4 and 5c1 for this cryo-TEM and SANS analysis because these two copolymers could be cross-linked successfully using our peptide cross-linker (see next section).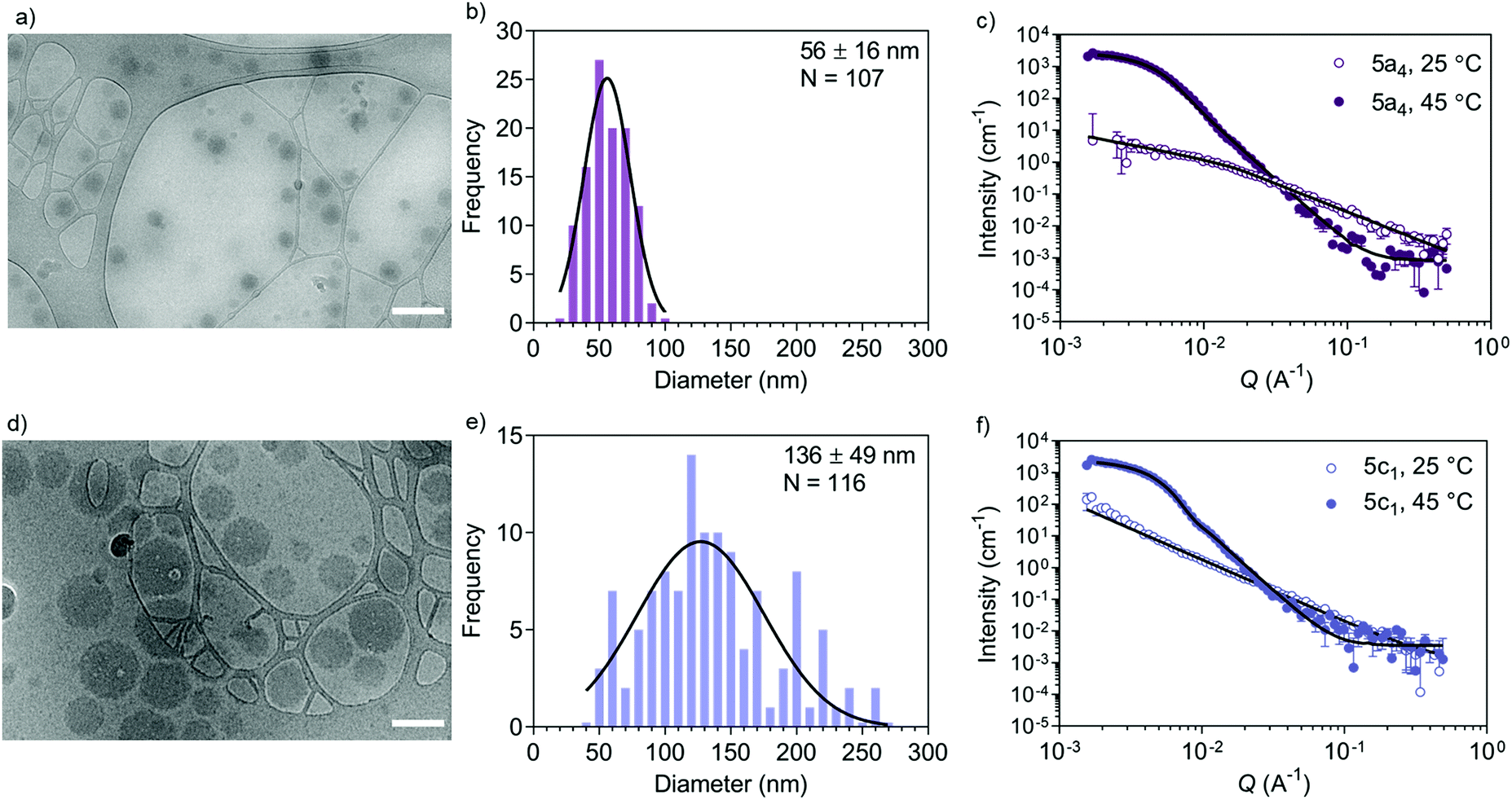 | ||
| Fig. 2 Self-assembled nanogel morphology characterisation. (a) Representative cryo-TEM image of self-assembled 5a4 copolymer (scale bar: 200 nm), (b) size distribution histogram from representative images, (c) SANS analysis below and above the LCST. (d) Cryo-TEM image of self-assembled 5c1 copolymer (scale bar: 200 nm), (e) size distribution histogram from representative images, (f) SANS analysis below and above the LCST. | ||
Cryo-TEM of self-assembled nanostructures made from copolymers 5a4 and 5c1 revealed a spherical morphology with homogenous contrast across whole particles (Fig. 2a and d). This indicates formation of dense, polymer-filled, non-vesicular spherical nanogels. The sizes of the nanogels (Fig. 2b and e) made from both copolymers were measured via image analysis and were in good agreement with the hydrodynamic diameters obtained by DLS. The increase in size of the self-assembled structures when NCPAM is present, as measured by DLS (Fig. 1c and d), was confirmed by cryo-TEM. We additionally studied the zeta potential of self-assembled nanostructures above the LCST (45 °C), which yielded slightly negative values (5a4 = −5.5 ± 0.2 mV; 5c1 = −16.0 ± 0.5 mV).
SANS enabled us to capture the morphology change from coil-to-globule (Fig. 2c and f), which is induced by the temperature-triggered transition in agreement with literature on other PEG-b-PNIPAM-based block copolymer systems.45–47 Neutron scattering data of the two selected block copolymers below and above the LCST (25 and 45 °C, respectively) are shown (Fig. 2c and f). Both copolymers, either without cyclopropyl units (5a4) or including cyclopropyl units (20%, 5c1) in the thermo-responsive block, revealed a drastic change in the scattering profiles at the two different temperatures. Below the LCST (at 25 °C), SANS confirmed that the block copolymers are present as chains in solution. Two shape-independent models, Guinier–Porod (5a4; Porod exponent of 1.77 ± 0.04; radius of gyration Rg of 4.6 ± 0.7 nm; dimension variable s is 0.86 ± 0.07) and power law (5c1; power law exponent of 1.96 ± 0.01) were used to fit the scattering profiles at 25 °C. Scattering data of copolymer 5a4 revealed a clear Guinier region, therefore we could fit the data with the Guinier–Porod model to obtain accurate Rg values. The data obtained for 5a4 suggests that this block copolymer has fully swollen chains in good solvent conditions at 25 °C (below the LCST) in agreement with literature on similar copolymers.46 A power law exponent of 1.96 ± 0.01 for block copolymer 5c1 is explained by the presence of polymer chains in theta conditions with equally favoured polymer–polymer and polymer–solvent interactions. This might also explain the bimodal distribution in the DLS data below the LCST (Fig. 1c), which was not observed in case of 5a4 (monomodal DLS) and is supported by the SANS data (see discussion above).
When heated above the LCST (45 °C), the copolymers are expected to transition from hydrated and flexible coils to collapsed globules.48–50 SANS data for copolymers 5a4 and 5c1 above the LCST were successfully fitted with a Sphere model. This indicates formation of dense spherical polymer nanogels rather than vesicles (polymersomes), which confirms the conclusions from the cryo-TEM images (Fig. 2a and d). The insertion of 20% cyclopropyl repeating units (5c1) did not affect the final morphology and the scattering data could again be fitted with the simple Sphere model. The best-fit diameters were 47.6 ± 1.0 nm (5a4) and 78.0 ± 0.5 nm (5c1), respectively. Data for size obtained by SANS, cryo-TEM and DLS are similar and the formation of larger structures for the NCPAM-containing copolymer 5c1 compared to 5a4 was consistent across all the measurements. SANS typically shows the smallest diameter of all the three methods, because it does not account for the hydration shell in the measurement.
None of our synthesised block copolymers self-assembled into vesicular morphologies, which would have been observed as membranous structures in the cryo-TEM micrographs and distinct SANS curves that could be fitted with a Hollow sphere model as shown previously.28 This is in disagreement with previous studies on similar PEG-b-PAA-b-PNIPAM block copolymer assemblies,40,51 which highlights the challenge of using theoretical rules to predict the final structure and encourages the use of various complementary techniques to precisely evaluate morphology. A combination of techniques such as DLS, cryo-TEM and scattering techniques like SANS, small-angle X-ray scattering (SAXS) and combining dynamic and static light scattering (DLS/SLS) is necessary to obtain a clear picture of the size and morphology of self-assembled nanostructures, which allows identification of the type of nanostructure formed.
Nanogel cross-linking via peptides to increase stability and introduce enzyme-responsiveness
Several cross-linking strategies have been adopted in the drug delivery field to increase the stability of self-assembled nanostructures and introducing stimuli-responsive properties to the delivery system.19,40,52 The dynamic nature of self-assembled nanostructures can compromise their stability in biological environments due to the presence of other entities (e.g. proteins) and dilution upon injection in the body.16,53 This can lead to premature payload leakage. Stabilisation of amphiphilic assemblies by covalent bonds is a versatile method to address the above-mentioned challenges, whilst stimuli-responsive motifs can be incorporated simultaneously. A stimuli-responsive cross-linker domain that is cleaved in the presence of the desired stimuli allows triggered structure disassembly and controlled payload delivery. Herein, we leveraged natural enzymes as triggers for cargo release due to their inherent involvement in disease progression. The functional alkyne handles, introduced into the copolymers as described above, were used as cross-linking points using azido-bi-functionalised peptides bearing a protease sensitive sequence. A model matrix metalloproteinase was selected as our initial target due to their involvement in many diseases.23 Matrix metalloproteinase 7 (MMP-7) was chosen due to its small size with a Mw of only 19 kDa in its active form,54 which minimises steric hinderance to allow the enzyme to access the cross-links in the nanogels.Three of the block copolymers (5a4/5c1/5c2) were used for the development of peptide cross-linked nanogels because these copolymers formed stable structures upon heating above their LCST (Fig. 1b and c). The peptide cross-linker was prepared by solid phase peptide synthesis inserting an azido-lysine residue at both the C- and N-termini according to literature procedures.18 An MMP-7-cleavable sequence (K(N3)PLELRAK(N3), see Fig. S2a for the structure, ESI†) was installed into the peptide backbone enabling enzyme-triggered nanogel degradation. LC-MS spectra of the functionalised MMP-7-cleavable peptide after purification is shown in Fig. S2b (ESI†). In order to prepare the cross-linked nanogels, we developed a two-step protocol. First, the formation of self-assembled nanogels was induced by heating the block copolymer solution to 45 °C (above the LCST). A solution of the cross-linker was then added, together with small amounts of CuSO4·5H2O and (+)-sodium-L-ascorbate, leading to covalent crosslinking; the mixture was subsequently purified by size exclusion chromatography. Between the different samples we expect similar cross-linking due to the availability of the same number of alkynes per chain for all the copolymers and use of same equivalents of all the reagents for cross-linking. Upon cooling below the LCST, our nanogels therefore retained their structure, despite the solubilisation of the polymer chains. Details of the cross-linking strategy are schematically shown in Fig. 3a.
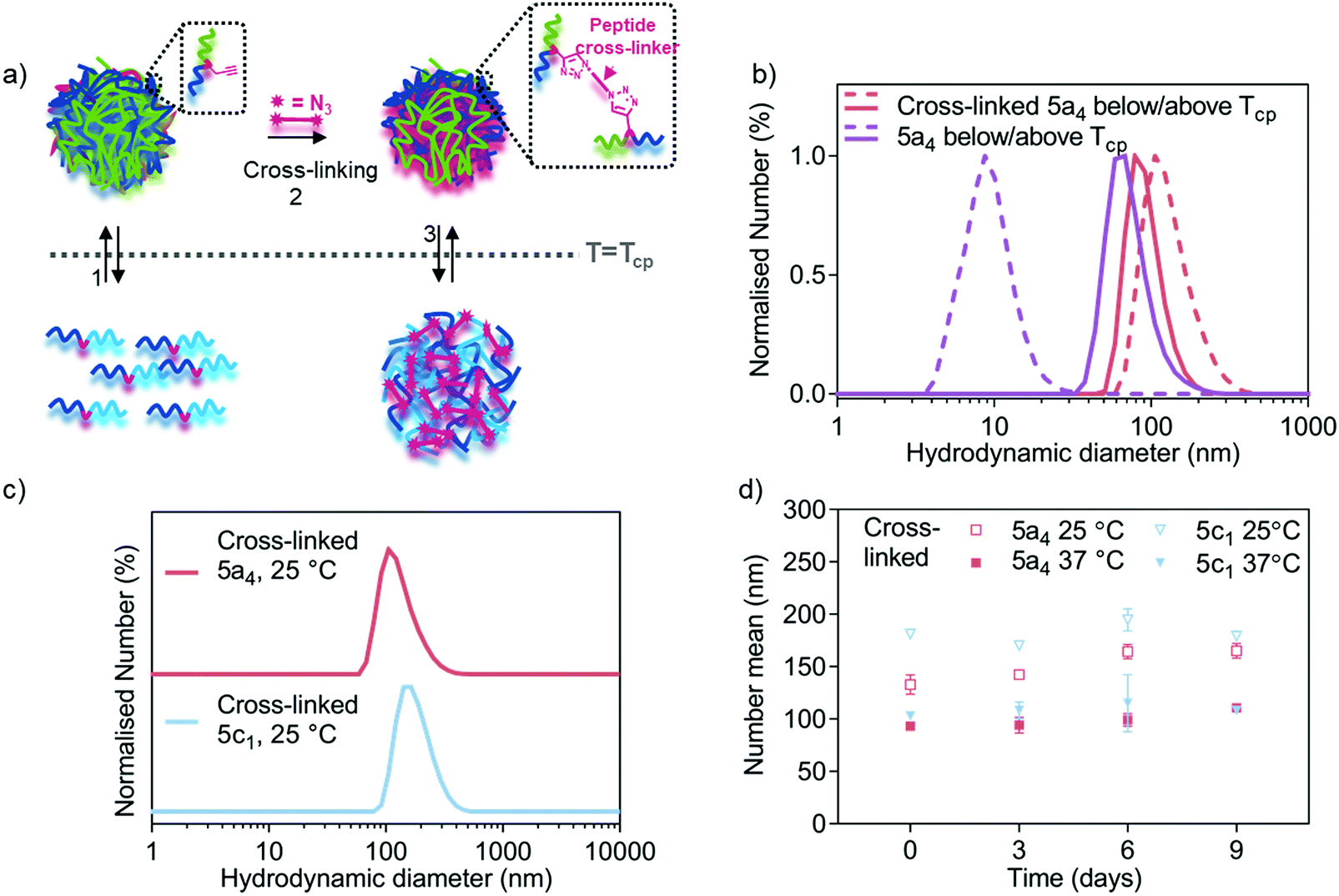 | ||
| Fig. 3 Nanogel cross-linking via enzyme-responsive peptides using azide-bi-functionalised peptides via CuAAC. (a) Schematic representation of the cross-linking strategy showing how cross-linking enhances nanogel stability below the LCST. (b) DLS traces comparing size distribution of 5a4-based self-assembled (before cross-linking from Fig. 1c) and cross-linked nanogels below (dashed line) and above (solid line) the LCST (33 °C for 5a4). (c) DLS traces comparing size distribution of 5a4- and 5c1-based cross-linked nanogels at 25 °C (below the LCST). (d) Study of long-term stability of cross-linked 5a4- and 5c1-based nanogels in PBS incubated at 37 °C and measured by DLS at 25 °C (open symbols) and 37 °C (closed symbols) at each timepoint (N = 1, n = 3). | ||
After the cross-linking reaction, the resulting polymer nanogels were analysed by DLS, both above and below the LCST, which is shown in Fig. 3b and c. A small difference in the hydrodynamic diameter was observed between the cross-linked (93 ± 2 nm) and self-assembled 5a4 nanogels (74 ± 7 nm, before cross-linking) above the LCST. In contrast, DLS measurements at 25 °C (below the LCST) revealed successful cross-linking had taken place. The non-cross-linked polymer nanogels were disassembled when the temperature was lowered below the LCST, as described above (Fig. 1b and c), but nanogels remained stable when cross-linked. When comparing the DLS traces of the cross-linked 5a4 nanogels at 37 °C and 25 °C, a size increase can be observed (93 ± 2 nm compared to 133 ± 9 nm). The slight size increase with decreasing temperature can be attributed to the thermo-responsive phase transition of the PNIPAM block, which switches from hydrophobic nature above the LCST to hydrophilic below the LCST. This could cause relaxation and water-induced swelling of the polymer chains, as previously observed in the literature.40,52 These networks are expected to swell and relax up to a limit determined by constraints imposed by the crosslinking, which will be dependent on the length and chemical structure of the peptide, and the cross-linking density.
The developed cross-linking protocol was then applied to the preparation of cross-linked polymer nanogels composed of the other P(NIPAM-co-NCPAM)-based block copolymers. Fig. 3c shows DLS characterisation of 5c1 (NI0.8NC0.2)-based nanogels after cross-linking. 5c1-based nanogels could be cross-linked successfully, yielding larger sizes compared to 5a4-based nanogels when measured below the LCST (181 ± 4 nm vs. 133 ± 9 nm). Cross-linking of 5c2-based nanogels could not be achieved due to severe aggregation and precipitation. Hence, detailed morphological analysis was restricted to the two copolymers that could also be cross-linked successfully (5a4 and 5c1). Long-term stability of cross-linked nanogels made from these two block copolymers when incubated in PBS at 37 °C was demonstrated by DLS (Fig. 3d). The hydrodynamic diameters did not decrease when observed over 9 days, both below and above the LCST, which demonstrates integrity of the structure and the absence of nanogel disassembly.
Besides obtaining morphological information at fixed temperatures above and below the LCST, as also demonstrated previously with similar PNIPAM-based block copolymers,46,47 SANS was applied herein to dynamically monitor the transition using one selected copolymer. During SANS measurements, 5c1 block copolymers and the corresponding cross-linked nanogels were subjected to a temperature ramp from 25 to 45 °C and from 32 to 42 °C, respectively (Fig. 4 and Tables S7, S8, ESI†).
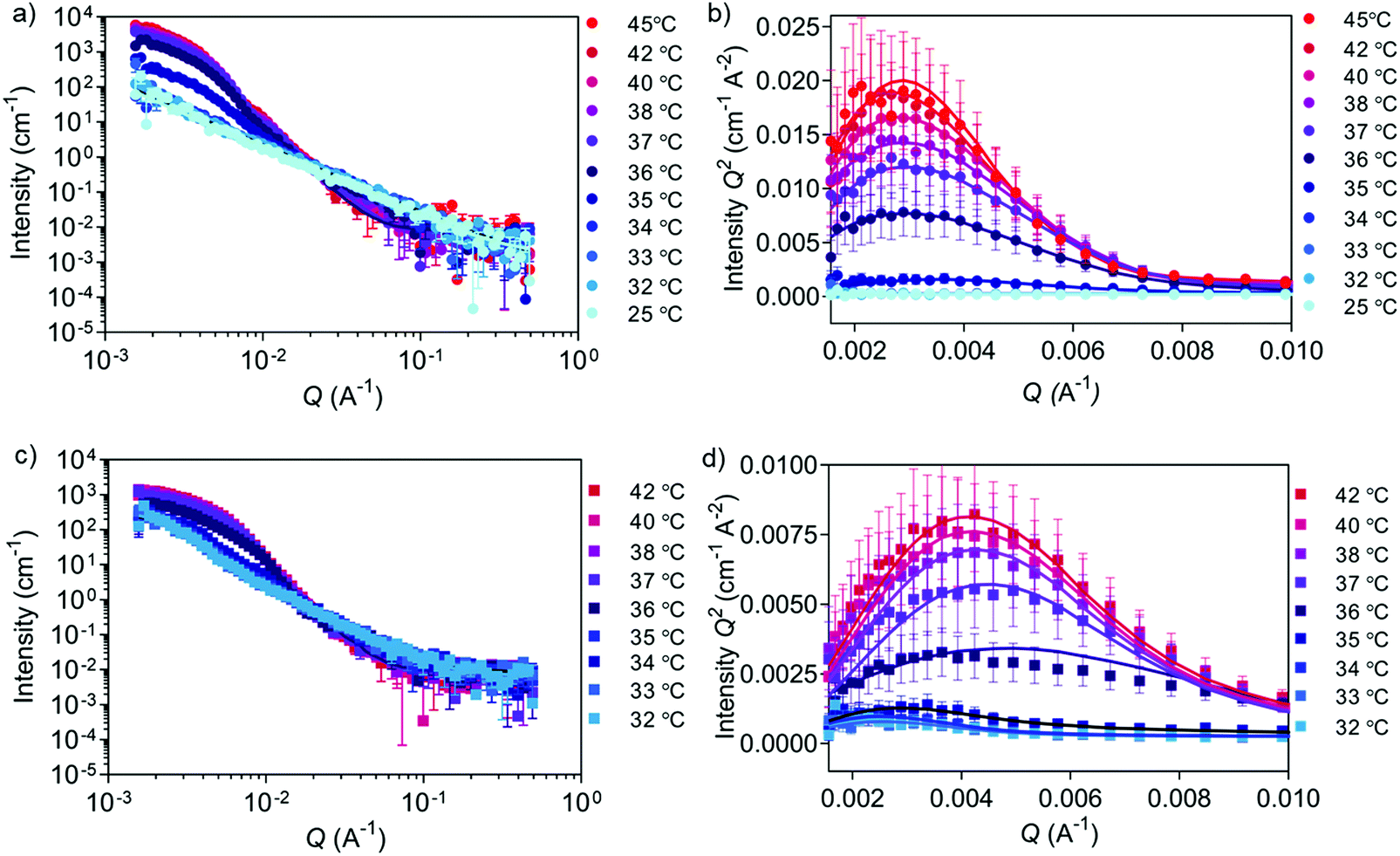 | ||
| Fig. 4 Temperature-dependent SANS analysis of self-assembled versus cross-linked 5c1 block copolymers. (a) Evolution of neutron scattering data collected for 5c1 block copolymers at temperatures from 25 to 45 °C (lines are fitted curves; data points are symbols; errors are ± SD). (b) Kratky plot for (a) showing an increase of globularity as the temperature rises from 25 to 45 °C. (c) Evolution of neutron scattering data collected for 5c1-based cross-linked nanogels at temperatures from 32 to 42 °C. (d) Kratky plot for (c) showing an increase of globularity as the temperature rises from 32 to 42 °C. | ||
SANS data of 5c1 block copolymers at temperatures between 25 and 34 °C (Fig. 4a and b) were fitted with the power law model (power law exponent of ∼1.9 obtained for all), which suggests the presence of Gaussian polymer chains in theta solvent conditions and the absence of self-assembly. Upon heating to 35 °C, the scattering data was better fitted using the Unified power Rg model. The obtained power law exponent of 2.40 ± 0.03 indicates a transition from Gaussian chains to a more entangled polymer network (Rg of 52.0 ± 0.6 nm) indicating the start of the coil-to-globule transition. At higher temperatures (36 and 45 °C) data were fitted using a triaxial ellipsoid model, which suggests the formation of non-perfect spherical nanogels, with an observable trend of ellipse transitioning to more sphere shape with increasing temperature (Table S7, ESI†). From these experiments a Tcp of 35–36 °C was obtained, which was similar to the value found by DLS (37 °C). Multiplying the scattering intensity by Q2 allowed us to obtain the Kratky plot with linear x- and y-axes (Fig. 4b and d). This plot is useful for studying nanogel degree of globularity, flexibility and compactness.29,55 The peak appearing in the Kratky plot at higher temperatures suggests the formation of globular particles above 35 °C. As the temperature further increases, the peak becomes more defined suggesting an increase in globularity.
The effect of cross-linking can be seen in the temperature-dependent SANS data of peptide cross-linked 5c1 nanogels (Fig. 4c, d and Table S8, ESI†) when compared to the purely self-assembled nanogels (Fig. 2f, 4a, b and Table S7, ESI†). We measured a narrower temperature range around the LCST, because we already knew the transition temperature from the non-cross-linked nanogels (Fig. 4a and b). The power law exponent value below 2 for the non-cross-linked copolymer compared to a value of ∼2.4 for the cross-linked nanogels, reveals the difference of the two populations at lower temperatures. As expected, by cross-linking the polymer chains a stable polymer network was formed, and the chains remained interconnected between each other also at temperatures below the LCST. When comparing the two Kratky plots (Fig. 4b and d) it can be concluded that the self-assembled and cross-linked nanogels both transition from coil to globule at 36 °C, whilst the cross-linked particles were slightly smaller after the transition as seen by the shift of the peak to the higher Q region. This trend is also confirmed by cryo-TEM (Fig. 2d and Fig. S3, ESI†). An explanation for this smaller size could be that the peptide cross-linkers restrict chain flexibility, keeping the polymer chains tightly connected.56
Enzyme-triggered nanogel degradation
MMP-triggered nanogel disassembly was analysed using MMP-7 as a model MMP. We first confirmed that the free peptide in solution can be cleaved by MMP-7 as shown using LC-MS (Fig. S2b, ESI†). We next established the necessary concentrations of MMP-7 to successfully degrade the cross-links of the nanogels, which revealed that physiologically relevant nanomolar concentrations are sufficient for degradation (Fig. S4, ESI†). At the lowest concentrations (9 pM, 90 pM), no degradation was observed, whilst the higher concentrations (9 nM, 90 nM, 900 nM) completely disassembled the nanogels. The intermediate concentration of 0.9 nM marks the transition point where a size increase was measured. This can be explained by the partial cleavage of the cross-links, which caused the nanogels to relax into a larger overall structure (still partially cross-linked) because the measurement was conducted below the LCST. The kinetics of MMP-7-responsive nanogel disassembly at a fixed enzyme concentration (90 nM) were subsequently studied by DLS (normalised derived count rate (in kcps) vs. incubation time). The kinetic study (Fig. 5a) reveals a drop in the scattering intensity over time only in the presence of the enzyme, indicating a decrease in nanogel concentration and/or size. The kcps value remained unvaried in the control nanogel sample without enzyme (red symbols). A further control, incubating the cross-linked nanogels at 37 °C in 5% (v/v) fetal bovine serum (FBS) revealed very high stability of the nanogels in the presence of proteins over at least two days (Fig. S5, ESI†). This confirms the ability of the nanogels to undergo enzyme-triggered degradation.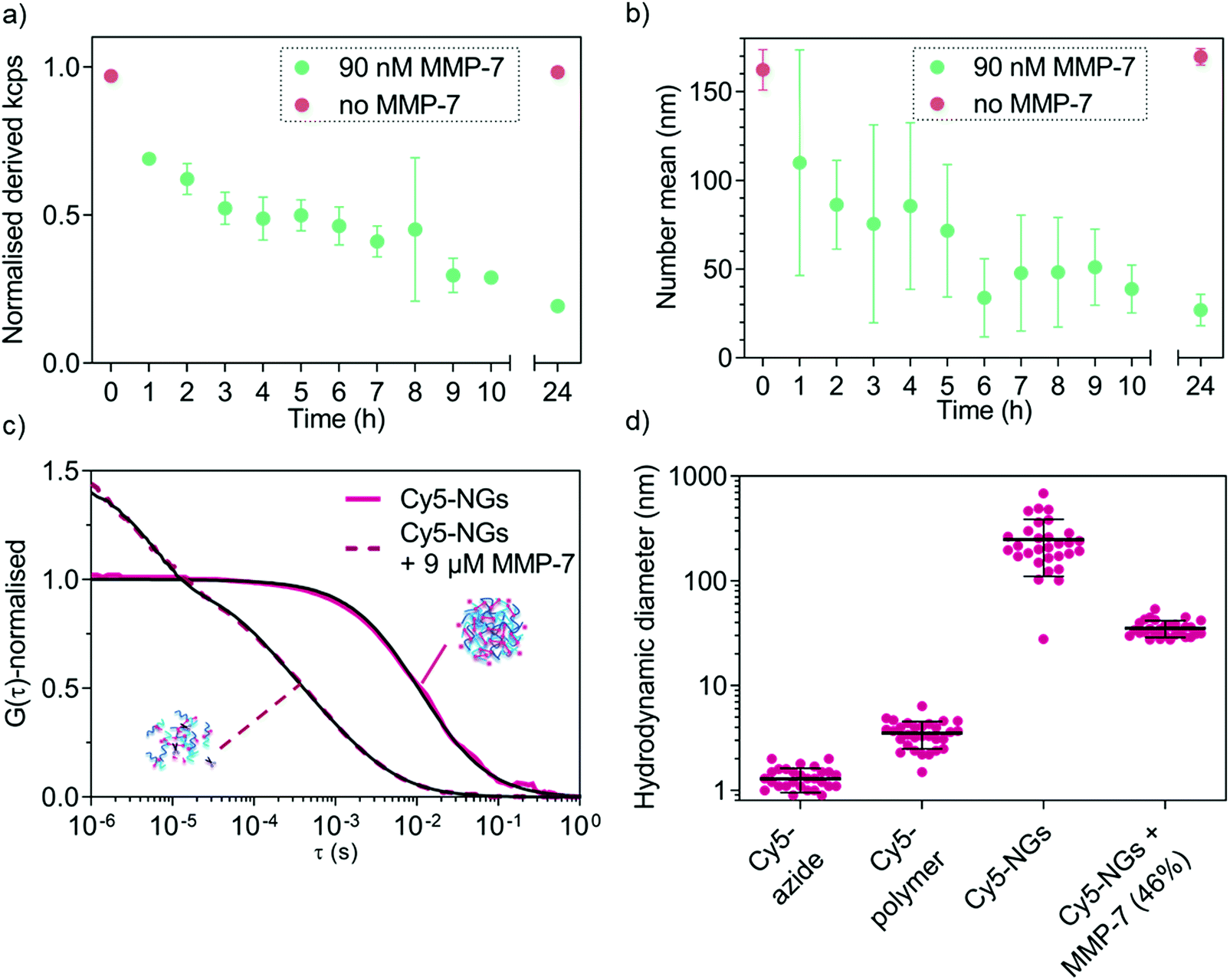 | ||
| Fig. 5 DLS and FCS analysis of MMP-7 triggered degradation of cross-linked 5a4-based nanogels. (a) Evolution of normalised derived count rate upon incubation with 90 nM MMP-7 over 24 hours indicating degradation kinetics as monitored by DLS. (b) Evolution of cross-linked nanogel average size (number mean) over time upon incubation with 90 nM MMP-7. (c) Normalised autocorrelation curves (coloured lines) and corresponding fits (black lines) from FCS data of Cy5-labelled nanogels and MMP-7-degraded (9 μM) Cy5-labelled nanogels in enzyme buffer. (d) Hydrodynamic diameters of Cy5-labelled nanogels and degraded Cy5-labelled nanogels with controls (Cy5-azide and Cy5-polymer) as calculated from FCS data. In the case of MMP-7 degraded nanogels, no more nanogels of the original size were detected; a two-component fit revealed only free Cy5-polymer diffusion (fixed diffusion time, 54% of diffusing species) and possibly entangled debris of slightly intermediate sizes between intact nanogels and free chains (46%, shown in graph). Values are mean of 30 curves ±SD. | ||
From the evolution of scattering intensity in Fig. 5a it is apparent that degradation has already occurred to some extent after 1 h incubation with MMP-7. The number mean size of the nanogels has already decreased (measured below the LCST), which suggests the presence of differently sized fragments and partial degradation. Within the next few hours of incubation with enzyme, a mixture of differently sized particles/fragments/polymers (large errors) was detected. After 24 h incubation, a clear size difference between the control and enzyme incubated sample became apparent (Fig. 5b) and the small error indicates that an endpoint has been reached. Indeed after 24 h incubation, the fragment size (27 ± 9 nm) is much closer to that of the corresponding free block copolymer chains (10 ± 2 nm).
Following DLS analysis, we also monitored nanogel degradation by a single-molecule detection method called fluorescence correlation spectroscopy (FCS). We previously used FCS for a detailed study of MMP-responsive degradation using another peptide-based nanostructure for diagnostic applications.35 Herein, FCS was employed to first observe nanogel degradation and subsequently to quantitatively analyse model biomolecule loading and release. In order to study MMP-7 triggered degradation of nanogels by FCS, cross-linked nanogels were labelled with Cy5-azide and incubated with excess MMP-7 (9 μM). Successful labelling of nanogels with Cy5-azide can be seen when calculating the hydrodynamic diameters from the FCS autocorrelation analysis (Fig. 5c) that yielded nanogel size rather than free dye (Fig. 5d). Further, non-crosslinked Cy5-azide labelled copolymers in non-assembled form revealed an intermediate diffusion time between free dye and nanogels (Fig. S6, ESI†). FCS curves of control experiments to check for non-specific interactions of dyes with nanogels/copolymers yielded diffusion times similar to free dye (Fig. S6, ESI†), which confirms successful specific labelling in the case of Cy5-azide (Fig. 5c and d).
After MMP-7 incubation, the corresponding autocorrelation curves shifted to faster diffusion times indicating cleavage of the peptide bonds (Fig. 5c). In the presence of MMP-7, a two-component fit of the autocorrelation curves revealed complete disappearance of any structures with original nanogel sizes. We only detected the presence of free Cy5-polymer chains (54% of total diffusing species) and some bigger, possibly entangled degradation products (DH value of 35 ± 6 nm, 46%, shown in Fig. 5d), demonstrating MMP-7-dependent peptide cleavage and complete nanogel disassembly below the LCST. By comparing the nanogel signal intensity (counts per particle, CPP in kHz) to the dye intensity, the number of dye molecules per single nanogel could be determined. Upon enzyme-mediated nanogel degradation, the calculated number of dyes per particle changed from about 54 dyes per particle before degradation, to about 2 dyes per particle for the degradation products. This is a further indication of nanogel disassembly. Together with the DLS analysis (Fig. 5a and b), enzyme-responsiveness of our nanogel system was successfully demonstrated.
Biomolecule loading and release from enzyme-responsive nanogels
After confirming the possibility of enzyme-triggered degradation of the herein developed peptide-cross-linked nanogels using a disease-relevant enzyme of the MMP class, loading and release properties of a model protein cargo (fluorescent Oregon Green-labelled bovine serum albumin, OG-BSA) was studied using one selected copolymer (5a4). UV-vis spectroscopy revealed a loading efficiency of 18 ± 8% (average ± SD of three independent batches) after purification by size exclusion chromatography. This represents better loading efficiencies for nanogels compared to polymersomes made via standard film rehydration, that often have low loading efficiencies of a few percent for hydrophilic cargo such as proteins.57 Further, the film rehydration method for polymersome formation can take several days, whilst solvent injection methods might harm sensitive protein cargo during self-assembly. Our rapid procedure to yield protein-loaded, well-defined nanogels at high polymer concentrations without the use of any organic solvent represents a suitable strategy to load sensitive therapeutic proteins for biomedical applications in the future. Loading and release of our model protein OG-BSA was further studied in detail by FCS (Fig. 6).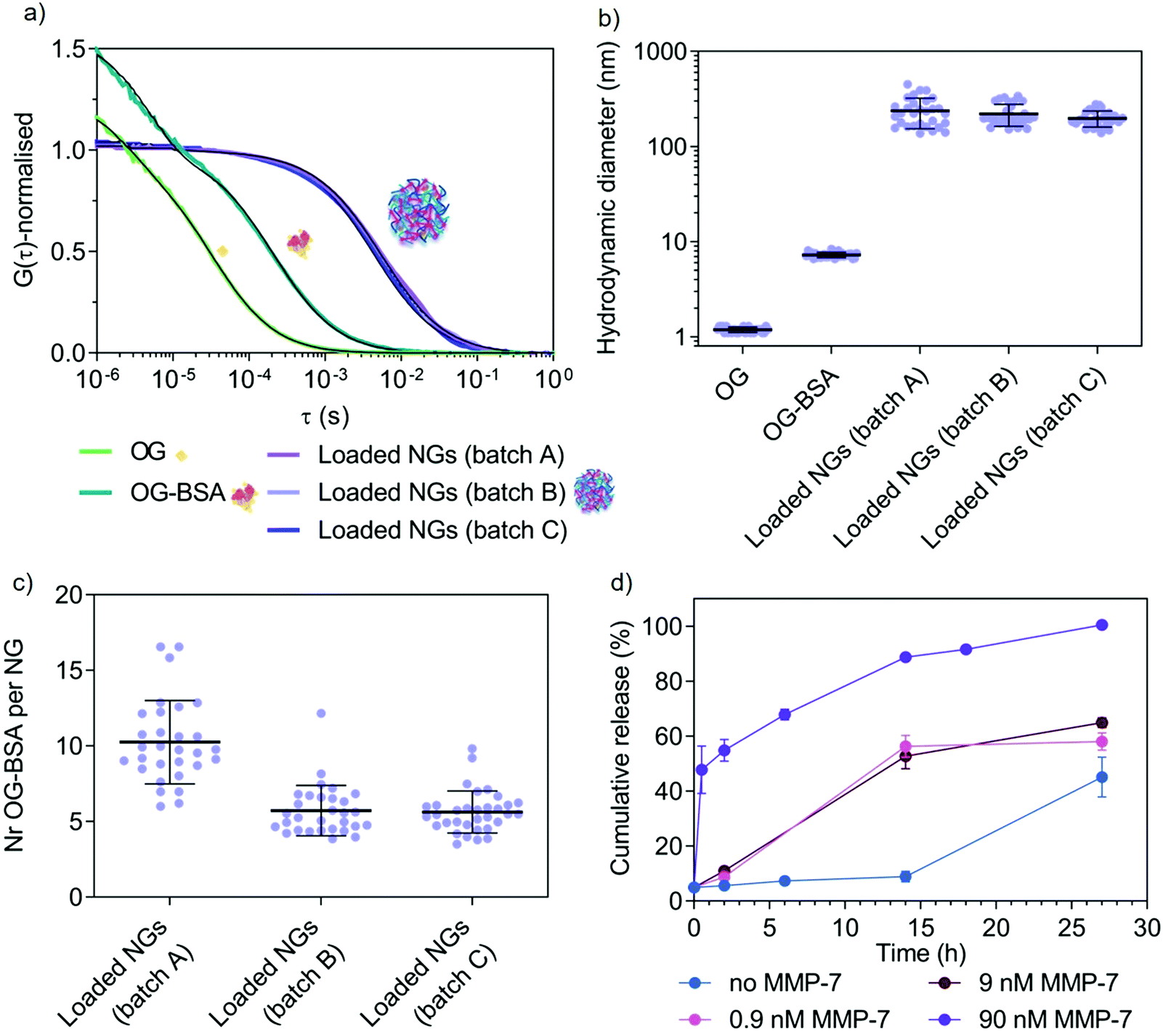 | ||
| Fig. 6 Study of model protein (OG-BSA) loading and MMP-7-triggered release by FCS. (a) Normalised autocorrelation curves (coloured lines) and corresponding fits (black lines) from FCS data of three nanogel batches (A–C). (b) Hydrodynamic diameter of the three different batches of OG-BSA loaded nanogels with controls (free OG and free OG-BSA) as calculated from FCS data. (c) Number of OG-BSA per nanogel for the three nanogel batches as calculated from FCS data. (d) Cumulative release (%) of OG-BSA over time. This graph shows the release from loaded nanogels A, B and C upon incubation with increasing concentration of MMP-7 (0.9 nM, 9 nM and 90 nM) as compared with diffusion-mediated release (no MMP-7). These results are calculated from the FCS autocorrelation curves displayed in Fig. S7 (ESI†). Error bars are SEM (N = 3, n = 30). | ||
Successful loading of OG-BSA into peptide cross-linked 5a4 nanogels was observed by FCS. Free dye, OG-BSA and OG-BSA incorporated in nanogels can already be distinguished clearly in the fitted autocorrelation curves (Fig. 6a). When calculating the corresponding diffusion times (τD) and hydrodynamic diameters (DH) the size difference between the diffusing species was obtained (Fig. 6b). In the case of the nanogel measurements, the diffusing species correspond to the nanogel hydrodynamic diameter obtained in other measurements (Fig. 3c and 5c, d), which indicates successful incorporation of OG-BSA. Comparing the signal intensity per nanogel (counts per particle, CPP in kHz) with that of free OG-BSA, the number of loaded OG-BSA per single nanogel was determined and shows that multiple protein cargos were loaded per nanogel (Fig. 6c). Release studies were performed in triplicate by incubating OG-BSA loaded nanogels (batches A–C) with various MMP-7 concentrations in a physiologically relevant nanomolar range.58,59 FCS curves recorded at different time points in the presence or absence of MMP-7 revealed the release kinetics (Fig. 6d and Fig. S7, ESI†). This allowed us to compare diffusion-driven release (non-specific release from the nanogels over time) to MMP-7 triggered release.
FCS curves at the different time points were fitted using a two-component fit to account for both diffusing species (loaded nanogels and released OG-BSA). Further, a correction was included to account for the disproportional effect of bright slow diffusing species (loaded nanogels) compared to the less bright free OG-BSA.60 We propose herein (see Materials and methods in ESI†) to simply combine the fraction of free OG-BSA (directly from the two-component fit) with the decrease in the number of cargo for the remaining nanogels (via CPP), which is a second indicator for OG-BSA release (number of proteins per nanogel is decreasing). This correction is necessary because nanogels with several loaded proteins overlap with the free protein signal, as demonstrated in the literature for mixtures of differently sized nanoparticles of varying brightness.60 This finally yielded the percentage of free OG-BSA and allowed us to further calculate the cumulative release profile over time (Fig. 6d). A clear difference between 90 nM MMP-7-triggered release and diffusion-based profiles was detected, whilst the release profile in the presence of 9 and 0.9 nM MMP-7 gave intermediate results as expected for an enzyme-triggered release. Diffusion-based release (no MMP-7) reached 87 ± 4% after 39 h (N = 3, n = 30). This successful enzyme-triggered release profile justifies the further study of these copolymer libraries using therapeutic cargo and studying release behaviour at various temperatures, accompanied by corresponding delivery data at the cell and tissue level but this is beyond the scope of this paper. It is also clear that MMP-7 triggered release profiles will vary significantly in vivo compared to the in vitro data, due to a much higher level of environmental complexity that will cause differences in cross-linker degradation and hence release. Reasons for these differences include the presence of other substrates and specific cofactors, as well as continuous turn-over of enzymes. Release studies in more complex environments, including in the presence of cells and tissue, are needed in the future to choose an optimal copolymer system for the delivery of a specific drug for a chosen disease that involves upregulation of MMP enzymes at the disease site. Cytocompatibility of new materials is one critical aspect for moving towards biomedical applications. We have tested cytotoxicity of cross-linked nanogels (5a4) in a macrophage cell line (RAW 264.7 cells) (Fig. S8, ESI†), where we found no cytotoxic effects.
Due to the involvement of MMPs, such as MMP-7, in several disease conditions, including infections, inflammatory diseases, cardiovascular disease and cancer, our MMP-7-responsive platform could be used for local MMP-7 dependent drug delivery. Additionally, the tuneable temperature-range for nanogel disassembly would allow the formulation of nanogels with purely hydrophilic nature at body temperature (LCST above 37 °C), potentially enabling rapid clearance from the body after disassembly. Alternatively, nanogels with LCST just slightly below body temperature could be used to achieve MMP7-responsive release that could be accelerated by applying e.g. cold patches locally. Simply changing the peptide cross-linker sequence will further allow application of our nanogel platform using other enzymes as triggers, as relevant in specific diseases.
Conclusion
In this work, we took advantage of the versatility of polymer chemistry for developing a tuneable, temperature-responsive, peptide-crosslinked polymeric nanogel platform for enzyme-triggered biomolecule delivery. Via copolymerisation of NIPAM with NCPAM we could tune the thermal behaviour of the resulting copolymers resulting in a range of Tcp values around body temperature (from 33 to 44 °C). A peptide-based cross-linking approach was developed to covalently constrain the nanogel-forming copolymer chains to increase stability and equip the nanocarrier platform with enzyme-responsiveness. We obtained high-quality morphological information on the self-assembled structures, successfully studied dynamic behaviours upon temperature changes and cross-linking, as well as model protein loading and enzyme-triggered release by employing a combination of cryo-TEM, SANS and FCS. This detailed analysis yielded a morphology of filled spheres (imaged with cryo-TEM) that relaxed to an interconnected polymer network upon cross-linking and lowering the temperature below the LCST as found by SANS measurements. Even further, the coil-to-globule transition of one selected copolymer was successfully monitored in real-time by SANS. Model enzyme-triggered (MMP-7) particle disassembly and accompanying increase in cargo release kinetics was determined by employing FCS. Our modular platform features the following key characteristics: reproducible, simple and fast temperature-induced nanogel assembly, modulation of the self-assembly temperature via copolymerisation of NIPAM and NCPAM in the thermo-sensitive block, containing two distinct handles (carboxylic groups and alkynes) for cross-linking and functionalisation, as well as enzyme-triggered release mechanism through specific peptide cross-links, which can be adapted to a specific disease when choosing appropriate peptide sequences and drug cargo.Conflicts of interest
The authors declare no competing interests.Acknowledgements
M. M. S. and L. M. acknowledge funding from the European Union's Horizon 2020 research and innovation programme under Marie Skłodowska-Curie grant agreement no. 642414. A. N. acknowledges support from his previous Swiss National Science Foundation Early Postdoc Mobility Fellowship (P2BSP2_168751) and current Sir Henry Wellcome Postdoctoral Fellowship (209121_Z_17_Z) from the Wellcome Trust. R. C. acknowledges support from the Australian Research Council DECRA (DE170100315). C. D. S. and M. M. S. acknowledge the British Heart Foundation Cardiovascular Regenerative Medicine Centre (RM/13/1/30157). V. N. acknowledges support from the Ermenegildo Zegna Founder's Scholarship program. V. N. and M. M. S. acknowledge support from the Rosetrees Trust. J. C. acknowledges support from the China Scholarship Council. M. A. B. acknowledges the Freemasons Foundation of New Zealand through the Royal Society of New Zealand-Rutherford Foundation (MAB, RFT-ICT 1501-FF). M. A. B. and M. M. S. acknowledge support from the grant from the UK Regenerative Medicine Platform “Acellular Approaches for Therapeutic Delivery” (MR/K026682/1) and the Engineering and Physical Science Research Council (EPSRC) grant “Bio-functionalised nanomaterials for ultrasensitive biosensing” (EP/K020641/1). M. M. S. acknowledges support from the Wellcome Trust Senior Investigator Award (098411/Z/12/Z).Experiments at the ISIS Neutron and Muon Source were supported by beamtime allocations from the Science and Technology Facilities Council (RB1810203). This work benefited from the use of the SasView application, originally developed under NSF award DMR-0520547. SasView contains code developed with funding from the European Union's Horizon 2020 research and innovation programme under the SINE2020 project, grant agreement 654000. The authors acknowledge the use of microscopy facilities within the Harvey Flower Electron Microscopy Suite at Imperial College London. The Light Microscopy Facilities at the Francis Crick Institute London is acknowledged for providing access to the FCS machine. The authors wish to acknowledge Dr Ulrike Kauscher Pinto and Dr Margaret N. Holme for assistance with cryo-TEM, and Dr Michael R. Thomas and Dr Hanna M. G. Barriga for assistance with SANS experiments.
Raw data are available upon reasonable request from E-mail: rdm-enquiries@imperial.ac.uk.
References
- T. M. Allen and P. R. Cullis, Science, 2004, 303, 1818–1822 CrossRef CAS.
- Y. Mai and A. Eisenberg, Chem. Soc. Rev., 2012, 41, 5969–5985 RSC.
- D. J. Keddie, Chem. Soc. Rev., 2014, 43, 496–505 RSC.
- V. Delplace and J. Nicolas, Nat. Chem., 2015, 7, 771 CrossRef CAS.
- C. D. Spicer, C. Jumeaux, B. Gupta and M. M. Stevens, Chem. Soc. Rev., 2018, 47, 3574–3620 RSC.
- I. Neamtu, A. G. Rusu, A. Diaconu, L. E. Nita and A. P. Chiriac, Drug Delivery, 2017, 24, 539–557 CrossRef CAS.
- L. Arnfast, C. G. Madsen, L. Jorgensen and S. Baldursdottir, Ther. Delivery, 2014, 5, 691–708 CrossRef CAS.
- M. Molina, M. Asadian-Birjand, J. Balach, J. Bergueiro, E. Miceli and M. Calderón, Chem. Soc. Rev., 2015, 44, 6161–6186 RSC.
- S. Mura, J. Nicolas and P. Couvreur, Nat. Mater., 2013, 12, 991–1003 CrossRef CAS.
- D. Roy, W. L. A. Brooks and B. S. Sumerlin, Chem. Soc. Rev., 2013, 42, 7214–7243 RSC.
- P. Kujawa, F. Segui, S. Shaban, C. Diab, Y. Okada, F. Tanaka and F. M. Winnik, Macromolecules, 2005, 39, 341–348 CrossRef.
- C. A. Figg, A. Simula, K. A. Gebre, B. S. Tucker, D. M. Haddleton and B. S. Sumerlin, Chem. Sci., 2015, 6, 1230–1236 RSC.
- H. Feil, Y. H. Bae, J. Feijen and S. W. Kim, Macromolecules, 1993, 26, 2496–2500 CrossRef CAS.
- Y. K. Kim, E. J. Kim, J. H. Lim, H. K. Cho, W. J. Hong, H. H. Jeon and B. G. Chung, Nanoscale Res. Lett., 2019, 14, 77 CrossRef.
- C. D. Spicer and B. G. Davis, Nat. Commun., 2014, 5, 4740 CrossRef CAS.
- Y. Zhao, Langmuir, 2016, 32, 5703–5713 CrossRef CAS.
- S. Hiki and K. Kataoka, Bioconjugate Chem., 2010, 21, 248–254 CrossRef CAS.
- C. Byrne, P. A. McEwan, J. Emsley, P. M. Fischer and W. C. Chan, Chem. Commun., 2011, 47, 2589–2591 RSC.
- Y. Wang, S.-L. Qiao and H. Wang, ACS Appl. Nano Mater., 2018, 1, 785–792 CrossRef CAS.
- B. A. Badeau, M. P. Comerford, C. K. Arakawa, J. A. Shadish and C. A. DeForest, Nat. Chem., 2018, 10, 251 CrossRef CAS.
- J. Hu, G. Zhang and S. Liu, Chem. Soc. Rev., 2012, 41, 5933 RSC.
- Q. Hu, P. S. Katti and Z. Gu, Nanoscale, 2014, 6, 12273–12286 RSC.
- C. J. Malemud, Front. Biosci., 2006, 11, 1696–1701 CrossRef CAS.
- D. G. Vartak and R. A. Gemeinhart, J. Drug Targeting, 2007, 15, 1–20 CrossRef CAS.
- K. Murata and M. Wolf, Biochim. Biophys. Acta, Gen. Subj., 2018, 1862, 324–334 CrossRef CAS.
- C. J. Newcomb, T. J. Moyer, S. S. Lee and S. I. Stupp, Curr. Opin. Colloid Interface Sci., 2012, 17, 350–359 CrossRef CAS.
- J. P. Patterson, M. P. Robin, C. Chassenieux, O. Colombani and R. K. O’Reilly, Chem. Soc. Rev., 2014, 43, 2412–2425 RSC.
- R. Salva, J.-F. Le Meins, O. Sandre, A. Brûlet, M. Schmutz, P. Guenoun and S. Lecommandoux, ACS Nano, 2013, 7, 9298–9311 CrossRef CAS.
- M. Sedlak, P. Falus, M. Steinhart, J. Gummel, P. Stepanek and S. K. Filippov, Macromol. Chem. Phys., 2013, 214, 2841–2847 CrossRef CAS.
- K. Mortensen and Y. Talmon, Macromolecules, 2002, 28, 8829–8834 CrossRef.
- D. Magde, E. Elson and W. W. Webb, Phys. Rev. Lett., 1972, 29, 705–708 CrossRef CAS.
- R. Rigler, Ü. Mets, J. Widengren and P. Kask, Eur. Biophys. J., 1993, 22, 169–175 CrossRef CAS.
- P. Rigler and W. Meier, J. Am. Chem. Soc., 2006, 128, 367–373 CrossRef CAS.
- A. Najer, D. Wu, M. G. Nussbaumer, G. Schwertz, A. Schwab, M. C. Witschel, A. Schäfer, F. Diederich, M. Rottmann, C. G. Palivan, H.-P. Beck and W. Meier, Nanoscale, 2016, 8, 14858–14869 RSC.
- C. N. Loynachan, A. P. Soleimany, J. S. Dudani, Y. Lin, A. Najer, A. Bekdemir, Q. Chen, S. N. Bhatia and M. M. Stevens, Nat. Nanotechnol., 2019, 14, 883–890 CrossRef CAS.
- A. J. Convertine, B. S. Lokitz, Y. Vasileva, L. J. Myrick, C. W. Scales, A. B. Lowe and C. L. McCormick, Macromolecules, 2006, 39, 1724–1730 CrossRef CAS.
- R. Hoogenboom and H. Schlaad, Polym. Chem., 2017, 8, 24–40 RSC.
- M. Glassner, K. Lava, V. R. de la Rosa and R. Hoogenboom, J. Polym. Sci., Part A: Polym. Chem., 2014, 52, 3118–3122 CrossRef CAS.
- G. Vancoillie, D. Frank and R. Hoogenboom, Prog. Polym. Sci., 2014, 39, 1074–1095 CrossRef CAS.
- H. Xu, F. Meng and Z. Zhong, J. Mater. Chem., 2009, 19, 4183–4190 RSC.
- R. Chapman, A. J. Gormley, M. H. Stenzel and M. M. Stevens, Angew. Chem., 2016, 128, 4576–4579 CrossRef.
- W. Zhang, L. Shi, K. Wu and Y. An, Macromolecules, 2005, 38, 5743–5747 CrossRef CAS.
- S. Venkataraman, J. L. Hedrick, Z. Y. Ong, C. Yang, P. L. R. Ee, P. T. Hammond and Y. Y. Yang, Adv. Drug Delivery Rev., 2011, 63, 1228–1246 CrossRef CAS.
- P. Renz, M. Kokkinopoulou, K. Landfester and I. Lieberwirth, Macromol. Chem. Phys., 2016, 217, 1879–1885 CrossRef CAS.
- A. Papagiannopoulos, J. Zhao, G. Zhang, S. Pispas and A. Radulescu, Polymer, 2013, 54, 6373–6380 CrossRef CAS.
- S. K. Filippov, A. Bogomolova, L. Kaberov, N. Velychkivska, L. Starovoytova, Z. Cernochova, S. E. Rogers, W. Man Lau, V. V. Khutoryanskiy and M. T. Cook, Langmuir, 2016, 32, 5314–5323 CrossRef CAS.
- J. Clara-Rahola, A. Fernandez-Nieves, B. Sierra-Martin, A. B. South, L. A. Lyon, J. Kohlbrecher and A. Fernandez Barbero, J. Chem. Phys., 2012, 136, 214903 CrossRef CAS.
- Y. Maeda, T. Nakamura and I. Ikeda, Macromolecules, 2001, 34, 8246–8251 CrossRef CAS.
- K. Jain, R. Vedarajan, M. Watanabe, M. Ishikiriyama and N. Matsumi, Polym. Chem., 2015, 6, 6819–6825 RSC.
- X. Wang, X. Qiu and C. Wu, Macromolecules, 1998, 31, 2972–2976 CrossRef CAS.
- R. Cheng, F. Meng, S. Ma, H. Xu, H. Liu, X. Jing and Z. Zhong, J. Mater. Chem., 2011, 21, 19013 RSC.
- B. Aktan, L. Chambre, R. Sanyal and A. Sanyal, Biomacromolecules, 2017, 18, 490–497 CrossRef CAS.
- C. F. van Nostrum, Soft Matter, 2011, 7, 3246–3259 RSC.
- H.-J. Ra, S. Harju-Baker, F. Zhang, R. J. Linhardt, C. L. Wilson and W. C. Parks, J. Biol. Chem., 2009, 284, 27924–27932 CrossRef CAS.
- D. R. Ratnaweera, U. M. Shrestha, N. Osti, C.-M. Kuo, S. Clarson, K. Littrell and D. Perahia, Soft Matter, 2012, 8, 2176–2184 RSC.
- N. Gundlach and R. Hentschke, Polymers, 2018, 10, 446 CrossRef.
- C. G. Palivan, O. Fischer-Onaca, M. Delcea, F. Itel and W. Meier, Chem. Soc. Rev., 2012, 41, 2800–2823 RSC.
- K. J. Isaacson, M. Martin Jensen, N. B. Subrahmanyam and H. Ghandehari, J. Controlled Release, 2017, 259, 62–75 CrossRef CAS.
- G. Nagel, A. Sousa-Herves, S. Wedepohl and M. Calderón, Theranostics, 2020, 10, 91–108 CrossRef CAS.
- A. Tcherniak, C. Reznik, S. Link and C. F. Landes, Anal. Chem., 2009, 81, 746–754 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0tb01546f |
| ‡ Contributed equally. |
| § Current address: Department of Chemistry and York Biomedical Research Institute, University of York, Heslington, YO10 5DD, UK. |
| This journal is © The Royal Society of Chemistry 2020 |