
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSingle-molecule DNA origami aptasensors for real-time biomarker detection†
Keitel
Cervantes-Salguero
a,
Mark
Freeley
a,
Jorge L.
Chávez
*b and
Matteo
Palma
 *a
*a
aSchool of Biological and Chemical Sciences and Materials Research Institute, Queen Mary University of London, London, UK. E-mail: m.palma@qmul.ac.uk
bAir Force Research Laboratory, 711th Human Performance Wing, Wright Patterson Air Force Base, Dayton, Ohio, USA. E-mail: jorge.chavez_benavides.2@us.af.mil
First published on 17th July 2020
Abstract
Here we report the use of DNA nanostructures as platforms to monitor the inherent conformational changes of aptamers upon analyte binding, with single-molecule resolution and real-time capability. An aptasensor designed to sense cortisol was found to suffer from instability in solution, but this was reconciled via a rational design of a single-molecule sensing platform. In this regard, DNA origami was employed to immobilise individual aptasensors on a glass surface and to ensure adequate interaction with their environment, for single-molecule analysis. The strategy presented here can be applied to any aptamer obtained by the destabilisation of a duplex in a SELEX process, and hence employed in the rational design of single-molecule biosensors.
The development of highly sensitive and selective sensors to recognise analytes has been the focus of intense research for environmental control, medical diagnostics, and the monitoring of important physiological (dys)functions.1–9 Advances in nanoscale assembly strategies have opened up possibilities in the development of novel platforms for the fundamental understanding of bio-molecular recognition processes and for the detection of biological agents down to single-molecule sensitivity.10–23 The ability to investigate and sense target-analyte interactions with single-molecule control could permit the fabrication of biochips offering unmatched sensitivity, smaller test sample volumes, and high throughput analysis. Additionally, single-molecule investigations enable monitoring of biochemical processes in real time, characterisation of transient intermediates, and measurement of the distributions of molecular properties rather than their ensemble averages:24–26 fundamental aspects for the rational design of biosensing devices.
Aptamers are well-placed to be implemented as sensors, both in the bulk and at the single-molecule level, due to their high affinity and specificity for analytes as well as their facile synthesis.1,3,4,7,8,27,28 Moreover, there are numerous examples of single-molecule detection in electrical,10–14 fluorescent,15–18 and mechanical systems,19 as well as in atomic force microscopy (AFM)-based analysis.20–23 However, previously reported aptamer-based single-molecule sensors are typically based on aptamers with well-documented conformations or have not been observed in real-time, so the scope of applications has been limited.
To develop optical sensors, two types of aptasensor configurations have been mainly employed: split aptasensors,29–31 and duplexed aptasensors.32–34 Split aptasensors are based on aptamers which have been split into two or more fragments that are unstable in the absence of the target; typically, the sensing is monitored by tagging each fragment with fluorescent moieties that interact after stabilisation from the target. While these aptasensors lend themselves to high specificity, it is not always possible to implement aptamers in this way since their binding pockets or secondary structures may not be easily split into segments. Conversely, duplexed aptasensors can be more generally applicable. Typically, they contain a short sequence which partially binds to the aptamer recognition sequence; upon binding of the target the short sequence is displaced and a signal is generated.32 Another benefit of duplexed aptasensors is their tunability, as the short complimentary sequence can be varied to optimise the binding of the target. This is advantageous in tailoring the aptamer to detect the analyte at a specific concentration range and to account for the physiological environment.
Solution and surface-based duplexed aptasensors often suffer from leakage that limits their sensing capabilities, requiring statistical formulations to remove false positives.35 In order to employ these systems in surface-bound biochip sensing devices, we sought to transfer a known duplexed aptasensor from a solution-based sensor to a single-molecule sensor immobilised on a surface. In this regard, DNA origami offers a well-established approach for the organisation of molecules on surfaces,36–41 and there are examples of combining it with DNA aptamers for sensing applications.18,20–23,42–44
Herein, we explore the use of DNA nanostructures as platforms to monitor aptamers inherent conformational changes upon analyte binding, with single-molecule resolution and real-time capability. In particular, we designed a DNA origami with a single cortisol-sensing duplexed aptasensor on one face,27 which could be immobilised to surfaces via molecular anchors on the opposite face. Using this approach, we could be certain that a single aptamer would be present within the range of the origami, while protruding from the surface for adequate interaction with the environment. Moreover, the density of DNA origami placement could be tuned with concentration, allowing us to optically resolve single-molecule events without the need of super-resolution techniques. In our system, a fluorescent signal is generated upon binding of cortisol; single-molecule recognition events typically occurred within seconds, in contrast to longer incubation times for the aptasensor in solution.27 Most notably, the aptasensor was found to suffer from instability in solution, but this was reconciled via the rational design of a single-molecule sensing platform. The strategy we present is of general applicability for the use of duplexed aptasensors in biosensing systems with single-molecule control and nanoscale spatial resolution.
As a model aptamer, we selected a recently reported cortisol aptamer developed by the Stojanovic group.27 This aptamer was originally obtained through systematic evolution of ligands by exponential enrichment (SELEX) where prospective aptamers were released from capturing columns into solution upon target-binding. For our fluorescent in-solution aptasensor, we chose the native cortisol aptamer sequence and its complementary capture strand, i.e. a duplexed DNA aptasensor shown in Fig. 1A. The aptasensor comprises two oligonucleotides: the “aptamer”, which is labelled with a dye, and a partially complementary sequence called the “quencher”, which is labelled with a quencher. The aptamer is divided into three subsections called the “dye”, the “lock”, and the “trigger”, where the dye subsection is fully complementary to the quencher.
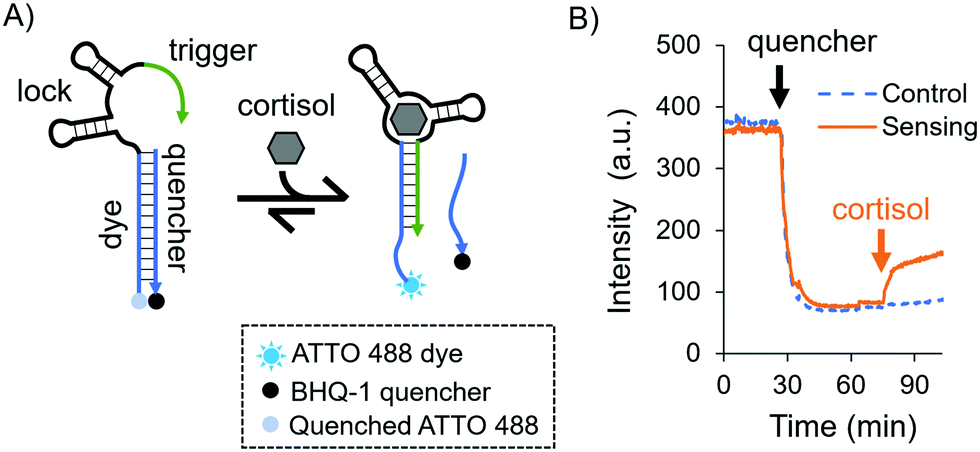 | ||
| Fig. 1 (A) Schematic of the solution-based duplexed DNA aptasensor. (B) Sensing experiment in solution using time-dependent fluorescence spectroscopy. The orange and blue curves represent the sensing and control experiments, respectively. For both curves: the ATTO 488-labelled aptamer in 1× DPBS was introduced and allowed to equilibrate. Then, the quencher was added, mixed, and let equilibrate again. Finally, for the orange curve a cortisol solution was added to a final concentration of 10 μM and mixed by pipetting, while for the blue dashed curve the same solution without cortisol was added and mixed by pipetting. | ||
Initially we investigated the sensing capabilities of the aptasensor via in-solution time-dependent fluorescence spectroscopy. The aptamer sequence was labelled with an ATTO 488 dye, and the quencher sequence was labelled with a BHQ-1 black quencher. Fig. 1B shows biomarker detection in 1× DPBS (Dulbecco's Phosphate-Buffered Saline) in the physiological range for pH and ionic strength [see the ESI,† and Fig. S1 for additional buffer conditions]. The dye-labelled aptamer sequence was allowed to equilibrate before the quencher was added and mixed thoroughly (black arrow). As expected, a sharp drop in fluorescence intensity was observed and this was again allowed to equilibrate. Finally, cortisol was added to the solution (orange arrow) eliciting a fluorescent response due to the dehybridisation of the dye/quencher duplex (90% of the total response is achieved after 7.5 minutes). The fluorescence does not recover fully to the initial intensity, possibly due to the presence of the quencher in solution even after its release from the aptasensor.
While an increase in fluorescence was expected when cortisol was added, a slow increase in intensity over time was also observed in the control experiments, where no cortisol was added: a leak reaction, which can be attributed to the dehybridisation of the dye/quencher duplex. Since the trigger was partially complimentary to the dye subsection, we investigated its influence on the stability of the dye/quencher duplex via in-solution time-dependent fluorescence spectroscopy (Fig. 2A).
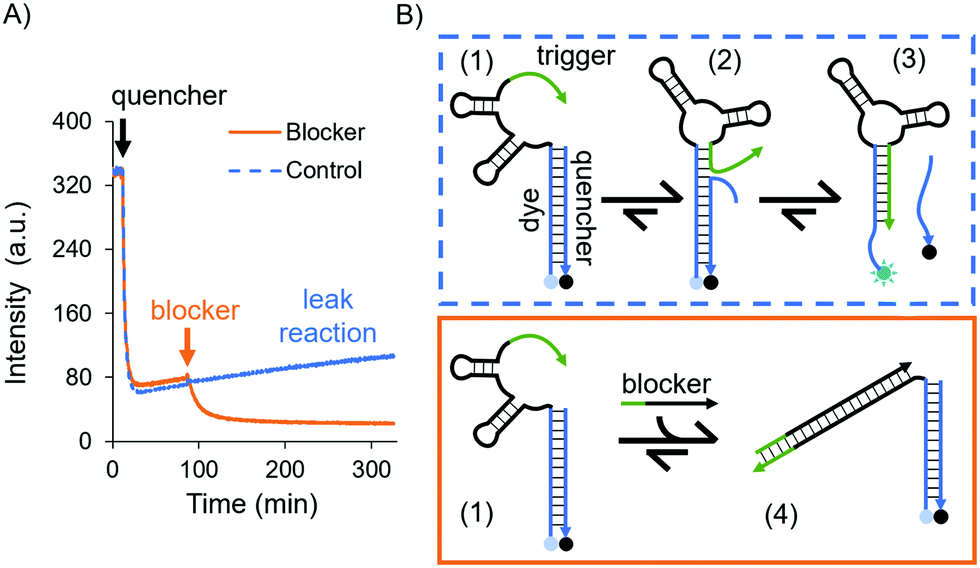 | ||
| Fig. 2 Testing the trigger's effect on the in-solution aptasensor. (A) Experiment in solution using time-dependent fluorescence spectroscopy. The blue and orange curves represent the leak reaction (control) and sequestering experiment, respectively. For both curves the ATTO 488-labeled aptamer in 1× DPBS was introduced and allowed to equilibrate. Then, the quencher was added after 10 min, mixed and let equilibrate again. Finally, for the orange curve the blocker was added (orange arrow) and mixed by pipetting, which disabled the trigger thus stabilising the dye/quencher duplex. For the blue curve the same buffer without the blocker was added and mixed by pipetting, and the system kinetics remained unresponsive, i.e. the trigger continued to destabilise the dye/quencher duplex. (B) Schematic of the trigger displacing the quencher (blue box) and of the blocker sequestering the trigger (orange box). | ||
A possible kinetic route is the dehybridisation of the quencher due to dynamic reconfigurations of the aptamer, i.e. the conformational change and branch migration (state 2 in Fig. 2B) allows the trigger to displace the quencher from the aptamer (state 3, Fig. 2B). This was evidenced by the increase in fluorescence intensity over time (see the blue curve in Fig. 2A).
To confirm that this process was due to the trigger, we designed a DNA sequence as a “blocker” which sequesters and deactivates the trigger permanently (state 4 in Fig. 2B). After the addition of the blocker the fluorescence intensity decreased, settling on a lower value (the orange curve in Fig. 2A); hence, the dye/quencher duplex remains hybridised. These results demonstrate the intramolecular effect of the trigger. To truly sense through induced fit, i.e. via a target-destabilised duplex, a key requirement is to have a stable dye/quencher duplex, either by tuning its sequence composition or by having an excess of the quencher in solution in order to create a kinetically stable duplex.
In order to monitor individual aptasensors, we designed a single-molecule surface-based system where we could address the destabilising effects of the trigger at the single-molecule level and ensure that the dye/quencher pair is stable. We devised a version of the aptasensor that was investigated in solution and integrated it into a triangular DNA origami nanostructure,37 which was then immobilised on a glass surface (Fig. 3A). The in-solution experiments gave us invaluable insight into the dynamic behaviour of the free aptasensor, which we could consequently address in our surface-based approach. At the single-molecule level, any destabilising effects from the trigger without the presence of the biomarker, would result in a false positive. To achieve duplex stability, the aptamer must be stably fixed to a surface and free to change conformation to allow target binding, while the duplex must remain hybridised. Furthermore, we designed our single-molecule biosensor to transduce a detection event into a highly fluorescent state.
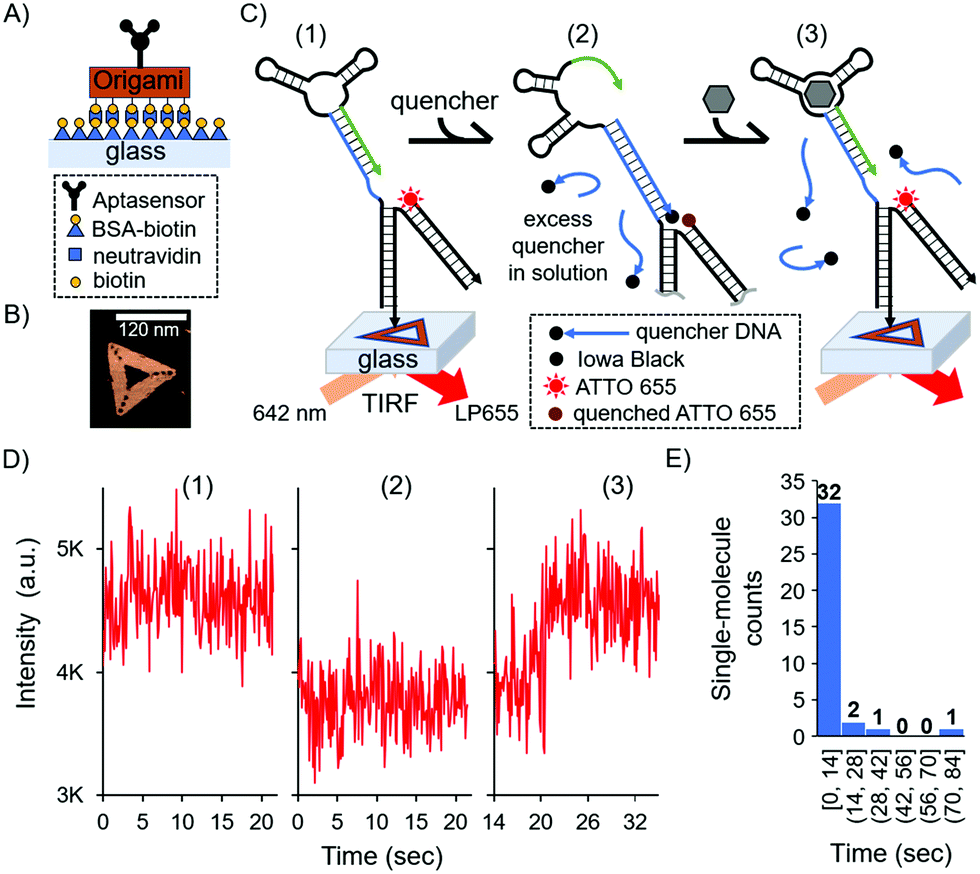 | ||
| Fig. 3 Single-molecule surface-based aptasensor via a DNA origami strategy. (A) Schematic of origami immobilisation on glass coverslip. (B) Atomic Force Microscopy image in liquid of the triangular DNA origami. (C) Schematic of the single-molecule detection on a glass surface. Structures were excited with 642 nm light in TIRF mode, and emission was filtered by a low pass filter with a threshold of 655 nm (LP655). (D) Representative single-molecule detection event for 10 μM cortisol by TIRF microscopy at three different states: (1) before adding the quencher, (2) after adding excess amount of quencher in solution, and (3) after addition of 10 μM cortisol. The states 1, 2 and 3 correspond to the states in (C). Detection was achieved 20.3 s after cortisol addition. Camera acquisition time was set at 100 ms. (E) Histogram of the single-molecule detection events, where the “single-molecule counts” correspond to the number of detection events, and “time” corresponds to the binning time in which the event was recorded. | ||
In this surface aptasensor, both the aptamer and a dye (ATTO 655)-labelled DNA are separately hybridised to the origami. This DNA origami strategy allows for the control of an individual aptamer sequence within an area of ca 6200 nm2 (area of the DNA triangle 120 nm by side); hence, avoiding potentially detrimental steric hindrance effects. Moreover, the density of DNA origami on the surface could be easily tuned with concentration, allowing us to optically resolve single-molecule events without the need of super-resolution techniques.
The glass surface of a custom-made chamber was passivated with bovine serum albumin (BSA)-biotin and functionalised with neutravidin (see the ESI,† and Fig. 3A). The DNA origami was synthesised with biotin groups on one face for the immobilisation of the origami on the surface, while the aptamer was present by design on the opposite face of the DNA nanostructure to ensure it was free to interact with the solution and the biomarker. Fig. 3B shows a representative atomic force microscopy (AFM) image of the DNA nanostructure (see also the ESI,† and Fig. S2). As with the bulk experiments, we investigated the duplexed aptasensor's response to cortisol in physiologically relevant buffer conditions.
Single-molecule events were imaged by Total Internal Reflection Fluorescence (TIRF) Microscopy in 1× DPBS. TIRF time series were acquired and divided in three stages which correspond to three fluorescent states (Fig. 3C and Fig. S3, ESI†): (1) the location of DNA origami via the single-molecule fluorescence of ATTO 655; (2) the addition of the quencher and consequent fluorescence loss; (3) the addition of the cortisol biomarker and its detection via fluorescence recovery (see also the ESI†). An excess amount of quencher in solution relative to the surface aptasensor was necessary to have a kinetically hybridised duplex, offsetting the trigger's destabilising effect. We observed the aforementioned three states for 36 single-molecule detection events, each representing a single DNA origami and aptasensor (see the ESI†).
For each single-molecule detection there is a series of intensity changes related to each stage (see representative single-molecule trace in Fig. 3D). Initially we monitored the fluorescence of a single ATTO 655 (state 1) which decreases after adding the quencher (state 2). Following the addition of the cortisol biomarker, the fluorescence initially remained unchanged relative to state 2 for the first ∼20 s, after which there was a recovery of fluorescence (state 3) to the original intensity (state 1); this final permanent increase in fluorescence indicated dehybridisation of the quencher due to biomarker detection. Additional single-molecule intensity traces can be seen in Fig. S4 (ESI†). The observed single-molecule detection events have a distribution over time (histogram in Fig. 3E), where most of the events were detected within 14 seconds (see the Fig. S4D, ESI† for a representative trace) compared to several minutes in the solution-based measurements (cf.Fig. 1B).
These results highlight the differences and advantages of the single-molecule system over the bulk aptasensor. The observed leakage in solution was efficiently addressed at the single-molecule level by adding excess quencher to the buffer, resulting in a higher stability between the quencher and the aptamer. In all aptasensors analysed at the single-molecule level no quencher was observed dehybridising prior to sensing the cortisol, indicating that sensing is purely due to the triggered reconfiguration of the aptasensor; this observation therefore serves as an intrinsic control.
This work elucidated the importance of a complementary solution-based characterisation for realising reliable surface-based aptamer biosensors with single-molecule sensitivity. Our aptasensor detected cortisol within seconds using DNA nanostructures as scaffolds, allowing for real-time single-molecule measurement via fluorescence microscopy. Moreover, an unwanted leak reaction was easily addressed at the single-molecule level via a large excess of quencher. To the best of our knowledge our work is the first demonstration of a fast, signal-on single-molecule aptasensor on a surface with real time detection.
The single-molecule approach presented here can be employed in similar aptamers where the target biomarker destabilises a duplex as a result of binding.27,32,34,45 Moreover, our strategy could be directly applied to any other new aptamer obtained by destabilising a duplex during SELEX, i.e. without the necessity of tuning the aptamer's sequence; this capability could be of further interest for the development of rapid tests for new pathogens.46,47 Such knowledge-based platforms can allow the rational engineering and optimisation of nucleic acid architectures in biosensing devices, also potentially in combination with molecular imaging technologies for cell-based studies.48
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors gratefully acknowledge financial support from the Air Force Office of Scientific Research under award FA9550-17-1-0179. This document was cleared for public release on 13 May 2020, Case Number: 88ABW-2020-1754.References
- J. Liu, Z. Cao and Y. Lu, Chem. Rev., 2009, 109, 1948–1998 CrossRef CAS PubMed.
- H. Pei, X. Zuo, D. Zhu, Q. Huang and C. Fan, Acc. Chem. Res., 2014, 47, 550–559 CrossRef CAS PubMed.
- A. D. Keefe, S. Pai and A. Ellington, Nat. Rev. Drug Discovery, 2010, 9, 537–550 CrossRef CAS PubMed.
- E. J. Cho, J.-W. Lee and A. D. Ellington, Annu. Rev. Anal. Chem., 2009, 2, 241–264 CrossRef CAS PubMed.
- M. Bauer, M. Strom, D. S. Hammond and S. Shigdar, Molecules, 2019, 24, 4377 CrossRef CAS PubMed.
- T. G. Drummond, M. G. Hill and J. K. Barton, Nat. Biotechnol., 2003, 21, 1192–1199 CrossRef CAS PubMed.
- Y. Y. Broza, X. Zhou, M. Yuan, D. Qu, Y. Zheng, R. Vishinkin, M. Khatib, W. Wu and H. Haick, Chem. Rev., 2019, 119, 11761–11817 CrossRef CAS PubMed.
- M. Xiao, W. Lai, T. Man, B. Chang, L. Li, A. R. Chandrasekaran and H. Pei, Chem. Rev., 2019, 119, 11631–11717 CrossRef CAS PubMed.
- Z. Zhou, Y. S. Sohn, R. Nechushtai and I. Willner, ACS Nano, 2020 DOI:10.1021/acsnano.0c04031.
- X. Guo, Adv. Mater., 2013, 25, 3397–3408 CrossRef CAS PubMed.
- S. Liu, X. Zhang, W. Luo, Z. Wang, X. Guo, M. L. Steigerwald and X. Fang, Angew. Chem., Int. Ed., 2011, 50, 2496–2502 CrossRef CAS PubMed.
- X. Chen, C. Zhou and X. Guo, Chin. J. Chem., 2019, 37, 897–902 CrossRef CAS.
- J. Y. Y. Sze, A. P. Ivanov, A. E. G. Cass and J. B. Edel, Nat. Commun., 2017, 8, 1–10 CrossRef CAS PubMed.
- X. Lin, A. P. Ivanov and J. B. Edel, Chem. Sci., 2017, 8, 3905–3912 RSC.
- F. D. Morris, E. M. Peterson, J. M. Heemstra and J. M. Harris, Anal. Chem., 2018, 90, 12964–12970 CrossRef CAS PubMed.
- D. Wu, E. Katilius, E. Olivas, M. Dumont Milutinovic and D. R. Walt, Anal. Chem., 2016, 88, 8385–8389 CrossRef CAS PubMed.
- A. Haller, R. B. Altman, M. F. Soulière, S. C. Blanchard and R. Micura, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 4188–4193 CrossRef CAS PubMed.
- D. Daems, I. Rutten, J. Bath, D. Decrop, H. Van Gorp, E. P. Ruiz, S. De Feyter, A. J. Turberfield and J. Lammertyn, ACS Sens., 2019, 4, 2327–2335 CrossRef CAS PubMed.
- P. M. Yangyuoru, S. Dhakal, Z. Yu, D. Koirala, S. M. Mwongela and H. Mao, Anal. Chem., 2012, 84, 5298–5303 CrossRef CAS PubMed.
- M. Godonoga, T. Y. Lin, A. Oshima, K. Sumitomo, M. S. L. Tang, Y. W. Cheung, A. B. Kinghorn, R. M. Dirkzwager, C. Zhou, A. Kuzuya, J. A. Tanner and J. G. Heddle, Sci. Rep., 2016, 6, 21266 CrossRef CAS PubMed.
- Z. Lu, Y. Wang, D. Xu and L. Pang, Chem. Commun., 2017, 53, 941–944 RSC.
- M. S. L. Tang, S. C. C. Shiu, M. Godonoga, Y. W. Cheung, S. Liang, R. M. Dirkzwager, A. B. Kinghorn, L. A. Fraser, J. G. Heddle and J. A. Tanner, Nanomedicine, 2018, 14, 1161–1168 CrossRef CAS PubMed.
- H.-K. Walter, J. Bauer, J. Steinmeyer, A. Kuzuya, C. M. Niemeyer and H.-A. Wagenknecht, Nano Lett., 2017, 17, 2467–2472 CrossRef CAS PubMed.
- M. Palma, J. J. Abramson, A. A. Gorodetsky, E. Penzo, R. L. Gonzalez, M. P. Sheetz, C. Nuckolls, J. Hone and S. J. Wind, J. Am. Chem. Soc., 2011, 133, 7656–7659 CrossRef CAS PubMed.
- F. Ritort, J. Phys.: Condens. Matter, 2006, 18, R531–R583 CrossRef CAS PubMed.
- R. D. Smiley and G. G. Hammes, Chem. Rev., 2006, 106, 3080–3094 CrossRef CAS PubMed.
- K. A. Yang, H. Chun, Y. Zhang, S. Pecic, N. Nakatsuka, A. M. Andrews, T. S. Worgall and M. N. Stojanovic, ACS Chem. Biol., 2017, 12, 3103–3112 CrossRef CAS PubMed.
- I. Willner and M. Zayats, Angew. Chem., Int. Ed., 2007, 46, 6408–6418 CrossRef CAS PubMed.
- H. Yu, J. Canoura, B. Guntupalli, X. Lou and Y. Xiao, Chem. Sci., 2017, 8, 131–141 RSC.
- A. Chen, M. Yan and S. Yang, TrAC, Trends Anal. Chem., 2016, 80, 581–593 CrossRef CAS.
- H. Yu, J. Canoura, B. Guntupalli, O. Alkhamis and Y. Xiao, Anal. Chem., 2018, 90, 1748–1758 CrossRef CAS PubMed.
- J. D. Munzar, A. Ng and D. Juncker, Chem. Soc. Rev., 2019, 48, 1390–1419 RSC.
- J. D. Munzar, A. Ng and D. Juncker, Nat. Commun., 2018, 9, 1–15 CrossRef CAS PubMed.
- J. D. Munzar, A. Ng, M. Corrado and D. Juncker, Chem. Sci., 2017, 8, 2251–2256 RSC.
- R. Weng, S. Lou, L. Li, Y. Zhang, J. Qiu, X. Su, Y. Qian and N. G. Walter, Anal. Chem., 2019, 91, 1424–1431 CrossRef CAS PubMed.
- D. Huang, K. Patel, S. Perez-Garrido, J. F. Marshall and M. Palma, ACS Nano, 2019, 13, 728–736 CrossRef CAS PubMed.
- D. Huang, M. Freeley and M. Palma, Sci. Rep., 2017, 7, 45591 CrossRef CAS PubMed.
- P. Zhan, T. Wen, Z. Wang, Y. He, J. Shi, T. Wang, X. Liu, G. Lu and B. Ding, Angew. Chem., Int. Ed., 2018, 57, 2846–2850 CrossRef CAS PubMed.
- S. Mishra, Y. Feng, M. Endo and H. Sugiyama, ChemBioChem, 2020, 21, 33–44 CrossRef CAS PubMed.
- A. M. Hung, C. M. Micheel, L. D. Bozano, L. W. Osterbur, G. M. Wallraff and J. N. Cha, Nat. Nanotechnol., 2010, 5, 121–126 CrossRef CAS PubMed.
- R. Wang, C. Nuckolls and S. J. Wind, Angew. Chem., Int. Ed., 2012, 51, 11325–11327 CrossRef CAS PubMed.
- N. Mohammad Danesh, M. Ramezani, A. Sarreshtehdar Emrani, K. Abnous and S. M. Taghdisi, Biosens. Bioelectron., 2016, 75, 123–128 CrossRef CAS PubMed.
- S. Ranallo, A. Porchetta and F. Ricci, Anal. Chem., 2019, 91, 44–59 CrossRef CAS PubMed.
- I. Mela, P. P. Vallejo-Ramirez, S. Makarchuk, G. Christie, D. Bailey, R. Henderson, H. Sugiyama, M. Endo and C. Kaminski, Angew. Chem., Int. Ed., 2020, 59, 12698–12702 CrossRef CAS PubMed.
- K. A. Yang, R. Pei, D. Stefanovic and M. N. Stojanovic, J. Am. Chem. Soc., 2012, 134, 1642–1647 CrossRef CAS PubMed.
- Y. Song, J. Song, X. Wei, M. Huang, M. Sun, L. Zhu, B. Lin, H. Shen, Z. Zhu and C. Yang, Anal. Chem., 2020, 92(14), 9895–9900 CrossRef CAS PubMed.
- N. Bhalla, Y. Pan, Z. Yang and A. F. Payam, ACS Nano, 2020 DOI:10.1021/acsnano.0c04421.
- F. Li, C. Li, X. Liu, Y. Chen, T. Bai, L. Wang, Z. Shi and S. Feng, Chem. – Eur. J., 2012, 18, 11641–11646 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Methods, materials, and additional data. See DOI: 10.1039/d0tb01291b |
| This journal is © The Royal Society of Chemistry 2020 |