
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIntracellular delivery of therapeutic antisense oligonucleotides targeting mRNA coding mitochondrial proteins by cell-penetrating peptides†
Carmine Pasquale
Cerrato‡
 *a,
Tove
Kivijärvi
b,
Roberta
Tozzi
a,
Tõnis
Lehto
a,
Maxime
Gestin
a and
Ülo
Langel
ac
*a,
Tove
Kivijärvi
b,
Roberta
Tozzi
a,
Tõnis
Lehto
a,
Maxime
Gestin
a and
Ülo
Langel
ac
aDepartment of Biochemistry and Biophysics, The Arrhenius Laboratories for Natural Sciences, Stockholm University, Svante Arrhenius väg 16B, SE-10691 Stockholm, Sweden. E-mail: carmine.pasquale.cerrato@dbb.su.se; carmine.cerrato@ki.se
bDepartment of Fibre and Polymer Technology, KTH Royal Institute of Technology, Teknikringen 56-58, SE-10044 Stockholm, Sweden
cLaboratory of Molecular Biotechnology, Institute of Technology, University of Tartu, Nooruse 1, 50411 Tartu, Estonia
First published on 6th November 2020
Abstract
Cell-penetrating peptides are a promising therapeutic strategy for a wide variety of degenerative diseases, ageing, and cancer. Among the multitude of cell-penetrating peptides, PepFect14 has been preferentially used in our laboratory for oligonucleotide delivery into cells and in vivo mouse models. However, this activity has mainly been reported towards cytoplasm and nuclei, while the mentioned disorders have been linked to mitochondrial defects. Here, we report a library generated from a combinatorial covalent fusion of a mitochondrial-penetrating peptide, mtCPP1, and PepFect14 in order to deliver therapeutic biomolecules to influence mitochondrial protein expression. The non-covalent complexation of these peptides with oligonucleotides resulted in nano-complexes affecting biological functions in the cytoplasm and on mitochondria. This delivery system proved to efficiently target mitochondrial genes, providing a framework for the development of mitochondrial peptide-based oligonucleotide technologies with the potential to be used as a treatment for patients with mitochondrial disorders.
Introduction
Antisense oligonucleotides (ASOs) are one of the many available RNA-based therapeutics, such as small-interfering (siRNAs), microRNAs (miRNAs), aptamers, and synthetic mRNAs. This technology has the potential to modulate or silence the expression of virtually any cellular gene. The first use of ASOs dates back to 1978 when Paul Zamecnik and colleagues used a 13-mer ASO containing a phosphodiester backbone to inhibit Rous sarcoma virus replication and cell transformation.1 From there, several chemical modifications have been developed, such as phosphorothioate (PS), modification of the 2′-hydroxyl (OH) to 2′-O-methyl (O-Me), 2′-fluoro (F), 2′-methoxyethyl (MOE) or bicyclic structures containing a 2′,4′-O-methylene bridge, termed as locked nucleic acid (LNA), and peptide nucleic acid (PNA).2–5 These modifications have significantly increased the stability, deliverability, potency, and pharmacologic properties of ASOs and its binding avidity to the target RNA. However, ASOs still have low bioavailability to traverse the phospholipid bilayer and enter cells. Despite the ability of some ASOs to enter certain specific cell types in naked form, a process named gymnosis,6 they lack reasonably efficient cell targeting and entry to the desired cell types, organelle or tissue. To address these problems, delivery of oligonucleotides into cells has been investigated using both viral and non-viral vectors, as well as liposome-mediated gene transfer.7 However, drawbacks such as viral toxicity and inefficient transfection rates, immune response to viral vectors, and difficulty in creating gene vector, have limited their usefulness in gene therapy.8 Furthermore, localizing a gene product within the cell has been a long-standing challenge within the field.Among the non-viral methods available for delivery of oligonucleotides, our group has specialized on cell-penetrating peptides (CPPs).9–16 CPPs appear to offer a method for the efficient and rapid transport of highly charged, polar compounds across virtually all cell membranes and tissues in a concentration-dependent manner. CPPs are well tolerated with only minimal cytotoxic effects seen at high concentrations in cell culture. The first CPP was discovered in 1988 when Frankel A. D. and Pabo C. O., at the same time as Green M. and Loewenstein P. M. demonstrated the ability of Tat protein derived from HIV to efficiently enter cells and promote the viral gene expression.17,18 Since then, a thousand new peptide delivery vectors have been discovered, synthesized, or chemically engineered to increase delivery and ameliorate pharmacological properties. The mechanisms for cellular entry and intracellular trafficking are still not fully elucidated but are reviewed thoroughly elsewhere.13,19,20 The main accepted uptake mechanism is endocytosis where the main limiting step is considered to be the entrapment in endosomes.
Another limiting step for organelle targeting other than cell entry is to cross one more lipid bilayer, and targeting mitochondria requires penetration through the mitochondrial double membrane. In 2004, Kesheng Zhao et al. reported the development of peptide antioxidants with the ability to cross the mitochondrial membrane and accumulate in these oragenelle.21 The Kelley laboratory has also been developing mitochondrial-penetrating peptides (MPPs).22 They have studied the sequence requirements for mitochondrial entry and modifications are responsible for impeding access to the organelle.23 They have also proved that MPPs can be used for the mitochondrial delivery of different drugs like chlorambucil, platinum-based anticancer agent, doxorubicin, desferrioxamine.24–27 In nature, the mitochondrial preproteins are synthesized in the cytosol with specific targeting signals. These signals are N-terminal presequences, which are proteolytically removed from the proteins by a protease in the matrix.28 The preproteins are recognized by receptors on the surface of mitochondria and transferred across the outer membrane by the translocase of the outer membrane (TOM) complex. Translocation of preproteins into or across the inner membrane is mediated by the translocase of the inner membrane (TIM) 23 complex.29 The TIM22 translocase inserts carrier proteins and subunits of the TIM complexes into the inner membrane.30 Some inner membrane proteins are initially translocated into the matrix and inserted into the inner membrane from the matrix side. The intracellular uptake and mitochondrial targeting of synthesized peptides change from peptide to peptide. Y. C. Kang et al. reported that some cell-penetrating artificial mitochondria-targeting peptide-conjugated to recombinant proteins were found in mitochondria as early as 3 h after incubation, whereas other constructs took up to 48 h of incubation.31 The protein import system can be involved for the import of small siRNAs as potential route for internalization of therapeutic molecules into mitochondria, if delivery systems are not employed. However, ASOs internalization by cells in vitro without the use of any transfection reagents have been predominately observed in the cytoplasm and not into mitochondria.2,32 Chinnery et al. showed the import of a fused cytochrome c oxidase subunit VIII presequence to a PNA into the inner membrane or the matrix of isolated mitochondria. Unfortunately, the methodology did not provide conclusive proof that the attachment of the presequence to PNA reached the mitochondrial matrix, essential for its functionality.33 Weissig and Torchilin reported DQAsome, vesicles formed from dequalinium (DQA), as a strategy to form complexes with DNA and for the release of DNA to the mitochondria.34,35 Lyrawati et al. also demonstrated the ability to use DQAsomes for the delivery of an artificial mini-mitochondrial genome to the mitochondria.36 Yamada et al. first reported the use of the MITO-Porter system to target the mitochondrial genome by delivering encapsulated DNase I to digest mtDNA,37,38 and successively reported an R8/GALA-modified MITO-Porter for the delivery of antisense RNA to mitochondria.39 The mitochondrial delivery of ASO and consequent knockdown of mitochondrial RNA would be expected to advance the development of mitochondrial therapeutic approaches.
In this work, we developed a peptide-based delivery system to target mitochondrial genes in different cellular compartments. We investigated the intracellular localization, splice correcting and antisense silencing ability of SCO, ASO targeting the uncoupling protein 2 (ASO [UCP2]), D-arm modified ASO targeting UCP2 (D-arm ASO [UCP2]), and D-arm modified ASO targeting cytochrome c oxidase subunit II (D-arm ASO [COXII]). D-arm is a D stem-loop import signal of tRNATyr(GUA) and has been shown to be efficiently imported into the mitochondrion of Leishmania, a kinetoplastid protozoan, as well as into mitochondrial matrix in isolated mitochondria.40–42 We modified the ASOs with this stem-loop import signal to facilitate the delivery and import inside the cells.
The silencing of UCP2 and COXII, a mitochondrial anion carrier able to modulate the reactive oxygen species (ROS) and a mitochondrial protein that make up complex IV of the respiratory chain related to maintaining the mitochondrial membrane potential respectively, yielded significant changes of the mitochondrial membrane potential and levels of ROS. We investigated the effects of ASO against UCP2 mRNA and COXII mRNA on mitochondria functionality as indicator of successful delivery of the complexes to the cytosol or to the mitochondria, an absolute requirement for mitochondrial antisense-mediated gene silencing. A schematic representation of the study is presented in Fig. 1. Given that many human diseases involve mitochondrial dysfunction, and there are currently no satisfactory methods to correct these defects, there is a need to develop mitochondrial therapies. These findings may have a significant implication for the design of therapeutic applications for mitochondrial diseases.
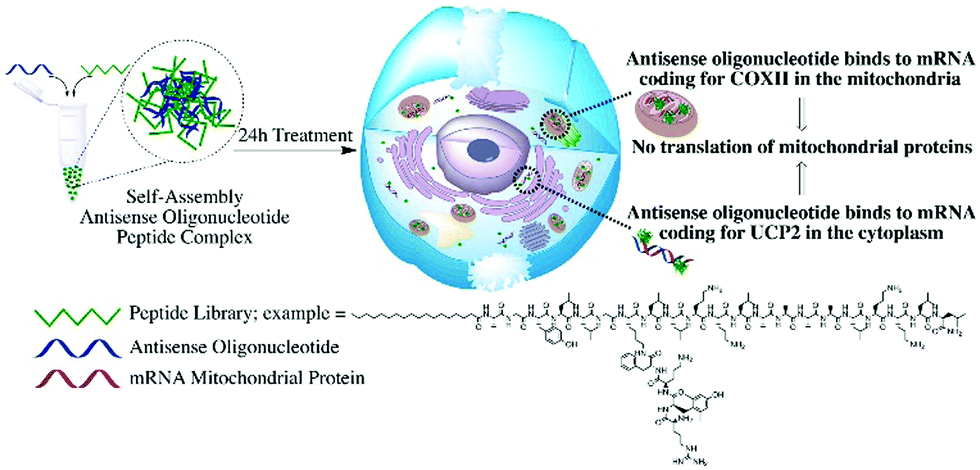 | ||
| Fig. 1 Schematic representation of the study. Development of a cell-penetrating peptide library to deliver antisense oligonucleotides affecting mitochondria functionalities by targeting genes coding for mitochondrial proteins in different cellular compartments. | ||
Results and discussion
Generation of a peptide library to target mRNA coding mitochondrial proteins
The rational design of the peptide library presented in this study is based on our most successful peptide for the delivery of oligonucleotides to the cytoplasm or nuclei, PepFect14 (PF14),43 and our cell-penetrating peptide targeting mitochondria, mtCPP1.44 By a combinatorial approach fusing the two peptides, we envisioned a purely peptide-based machinery to deliver oligonucleotides into the mitochondria or to target mRNA coding mitochondrial proteins. The library was derived from fusing mtCPP1 at the N-terminus, C-terminus, or on the side chain of PF14 (see Table 1 for peptide sequences). For side-chain conjugation, lysine 7 of PF14 was modified with mtCPP1. We also took into consideration different positions of the fatty acid of PF14, stearic acid. The absence or presence of stearic acid at the N-terminus, on lysine 7 directly or on the N-terminus of the branched mtCPP1 flanked from lysine 7 of PF14, was investigated.| Peptide | Sequence | Charge | MW (g mol−1) |
|---|---|---|---|
| Stearyl – stearic acid; r – D-arginine; Ya – 2,6-L-dimethyltyrosine; O – L-ornithine; NH2 – amidated C-terminus. | |||
| PF14 | Stearyl-AGYLLGKLLOOLAAAALOOLL-NH2 | +5 | 2404.40 |
| mtCPP1 | rYaOF-NH2 | +3 | 625.40 |
| mitFect1 | AGYLLGK(Stearyl-εN)LLOOLAAAALOOLL-NH2 | +5 | 2404.40 |
| mitFect2 | rYaOFAGYLLGK(Stearyl-εN)LLOOLAAAALOOLL-NH2 | +7 | 3014.97 |
| mitFect3 | Stearyl-AGYLLGK(rYaOF-εN)LLOOLAAAALOOLL-NH2 | +7 | 3014.97 |
| mitFect4 | Stearyl-rYaOFAGYLLGKLLOOLAAAALOOLL-NH2 | +7 | 3014.97 |
| mitFect5 | rYaOFAGYLLGKLLOOLAAAALOOLL-NH2 | +8 | 2745.74 |
| mitFect6 | AGYLLGK(rYaOF-εN)LLOOLAAAALOOLL-NH2 | +8 | 2745.74 |
| mitFect7 | AGYLLGK(Stearyl-rYaOF-εN)LLOOLAAAALOOLL-NH2 | +7 | 3014.97 |
| mitFect8 | Stearyl-AGYLLGKLLOOLAAAALOOLLrYaOF-NH2 | +7 | 3014.97 |
| mitFect9 | Stearyl-rYaOF-NH2 | +2 | 891.10 |
mitFect1/SCO and the mixture of mtCPP1 and PF14 efficiently induces SCO-mediated splice-correction in complete media in HeLa pLuc705 cell model
The first test was to prove the transfection ability and efficacy of the peptides in complex with SCO. To screen for the most optimal peptide/SCO ratio for splice-correction activity, peptide/SCO complexes were formed at different molar ratios (MRs). MR 3, 7 and 10 were tested in complete media with 100 nM SCO doses. LF2000 was used as positive control in all experiments according to manufacturer's protocol and it was used the same dose of SCO (100 nM). The assay for this test was based on the use of HeLa pLuc705 cell line, a stably transfected cell line with a recombinant plasmid carrying the luciferase-encoding gene interrupted by a mutated human beta-globin intron 2. This mutation causes aberrant splicing of luciferase pre-mRNA resulting in the synthesis of non-functional luciferase.45 Masking the aberrant splice site with PS-2′-OMe SCO (see Table 2 for details) redirects splicing towards the correct mRNA and consequently restores luciferase activity. We found that splice-correction was induced in a MRs-dependent manner, as expected. MR 5 previously resulted in the MR with the highest splice-correction activity in both serum free and media containing serum.43 mitFect1/SCO at MR 5 and MR 10 has a transfection efficacy of 20- and 30-fold increase compared to PF14 at the respective MRs. Remarkably, the mixture with mtCPP1 and PF14 at MR 5 exceeded PF14 by 20-fold increase in luminescence. The mitochondrial targeting peptide alone, mtCPP1, did not show the ability to deliver the SCO into the cells. Interestingly, mitFect1/SCO transfection efficacy at MR 10 was also 35-fold higher than LF2000. mitFect3 at MR 5 and MR 10 proved to be as efficient as PF14 to deliver the SCO (Fig. 2).| Oligonucleotide | Nucleotide sequence |
|---|---|
| SCO for HeLa pLuc705 cells | 5′-CCUCUUACCUCAGUUACA-3′ |
| AlexaFluor568-SCO for HeLa pLuc705 cells | 5′-AlexaFluor568-CCUCUUACCUCAGUUACA-3′ |
| SCO for mdx mouse myotubes | 5′-GGCCAAACCUCGGCUUACCU |
| ASO [UCP2] | 5′-CTTTGAAACCAACCATGATTC-3′ |
| D-arm ASO [UPC2] | 5′-GGGACUGUAGCUCAAUUGGUAGAGCAUCTTTGAAACCAACCATGATTC-3′ |
| D-arm ASO [COXII] | 5′-GGGACUGUAGCUCAAUUGGUAGAGCAUCUUGCGCUGCAUGUGCCAU-3′ |
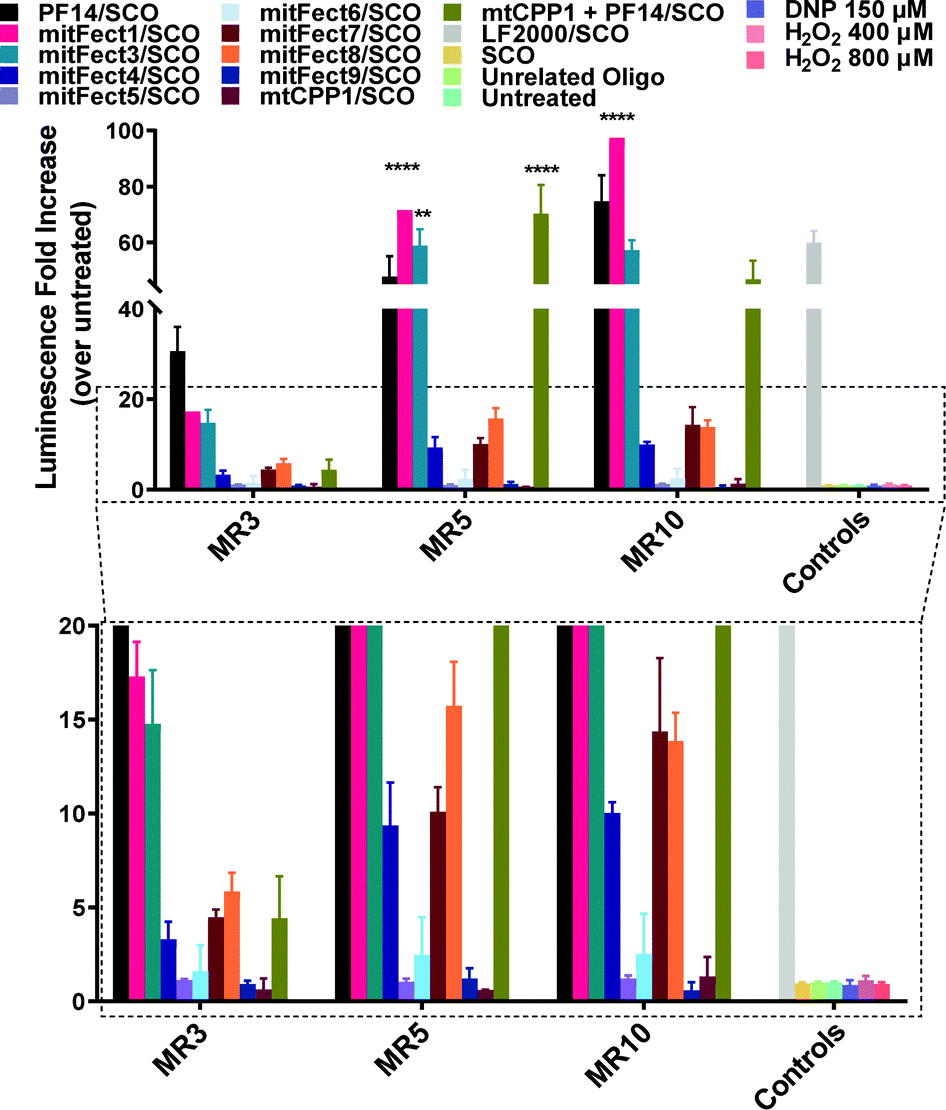 | ||
Fig. 2 Splice-correction activity of peptides/SCO nanocomplexes in HeLa pLuc705 cell line. HeLa pLuc705 cells (7 × 103) were seeded 24 h prior to experiments into 96-well plates. Cells were treated with peptides/SCO nanocomplexes at three different MRs (3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, 5:1, and 10:1) in complete media and incubated for 24 h. The controls used for this experiment were SCO alone (100 nM), unrelated ON (100 nM). LF2000 was used according to the manufacturer's protocol using 100 nM SCO concentration. Cells were lysed in 0.1% Triton X-100 and luciferase activity was measured. Splice-correction assay results are presented as fold-increase over untreated cells (control). The values represent the mean of at least three experiments performed in triplicate (mean ± SD, n = 3). **p ≤ 0.01, and ****p ≤ 0.0001. Ordinary two-way analysis of variance (ANOVA) with Dunnett's multiple comparison test vs. PF14/SCO luminescence fold increase at each MR. Complete statistical analysis data are presented in ESI† (Table S1). :1, 5:1, and 10:1) in complete media and incubated for 24 h. The controls used for this experiment were SCO alone (100 nM), unrelated ON (100 nM). LF2000 was used according to the manufacturer's protocol using 100 nM SCO concentration. Cells were lysed in 0.1% Triton X-100 and luciferase activity was measured. Splice-correction assay results are presented as fold-increase over untreated cells (control). The values represent the mean of at least three experiments performed in triplicate (mean ± SD, n = 3). **p ≤ 0.01, and ****p ≤ 0.0001. Ordinary two-way analysis of variance (ANOVA) with Dunnett's multiple comparison test vs. PF14/SCO luminescence fold increase at each MR. Complete statistical analysis data are presented in ESI† (Table S1). | ||
Many peptide complexes did not show a transfection efficacy higher than 5-fold (magnified panel Fig. 2). Uptake of peptides/Alexa Fluor 568 SCO (AF568SCO) was also quantified by spectrofluorometric analysis (Fig. S3, ESI†). Confocal microscopy was used to visualize the localization of the complexes in HeLa pLuc705 cells (Fig. S4, ESI†).
Evaluation of mitochondrial mRNA knockdown
We evaluated the knockdown of mRNA coding for mitochondrial proteins, following the ASO delivery by the library of peptides. In this experiment, we used three different oligonucleotides: antisense DNA targeting UCP2 (ASO [UCP2]), D-arm-modified 2′-OMe chimeric antisense targeting UCP2 (D-arm ASO [UCP2]), and D-arm modified antisense 2′-OMe RNA which targeted COXII (D-arm ASO [COXII]) (see Table 2 for sequences details). Mitochondrial uncoupling proteins (UCP) are members of the larger family of mitochondrial anion carrier proteins (MACPs). The mitochondrial proton leak is due to the UCPs by separating oxidative phosphorylation from ATP synthesis dissipating energy as heat. UCPs facilitate the transfer of anions from the inner to the outer mitochondrial membrane and the return transfer of protons from the outer to the inner mitochondrial membrane.46–48 They also reduce the mitochondrial membrane potential in mammalian cells. UCPs exhibit tissue specificity and the exact methods of how UCPs transfer H+/OH− are not known. UCPs contain three homologous protein domains of MACPs. This gene is expressed in many tissues, with the greatest expression in skeletal muscle.49 It is thought to play a role in nonshivering thermogenesis, obesity and diabetes.50 Skulachev V. reported that an increase in the proton leak of the mitochondrial membranes would lead to a decrease in force and kinetic constraints on respiratory chain and a stimulation of O2 consumption, which in turn decreases the formation of ROS.51 The role of the UCPs in keeping ROS production low by causing mild uncoupling has also been reported, showing that the inhibition of UCPs in mitochondria increases membrane potential and mitochondrial ROS production.52–55 UCP2 is a mitochondrial protein but the gene is located on chromosome 11.To test our hypothesis, the possibility to knockdown mRNA coding for mitochondrial proteins, we used the antisense oligonucleotides strategy. If our hypothesis was true, antisense oligonucleotides directed against UCP2 – cytosolic mRNA – or COXII – mitochondrial mRNA – must induce a change in membrane potential associated with changes of level of oxidative species.
Peptides efficiently deliver ASO [UCP2] with effects on mitochondrial membrane potential and ROS production
The assay we used is based on the evaluation of the mitochondrial membrane potential and ROS production. We prepared the complexes mixing the peptides with ASO [UCP2]. Tetramethylrhodamine methyl ester (TMRE) was used as fluorescent probe to measure the mitochondrial membrane potential to investigate the effects of the antisense oligonucleotide on HeLa pLuc705 cells.A 6 h treatment with UCP2 antisense oligonucleotide at 100 nM dose induced an increase in TMRE uptake at all the tested MRs (Fig. 3A); the 24 h treatment with ASO [UCP2] elicited an even higher increase uptake of TMRE in HeLa pLuc705 (Fig. 3B).
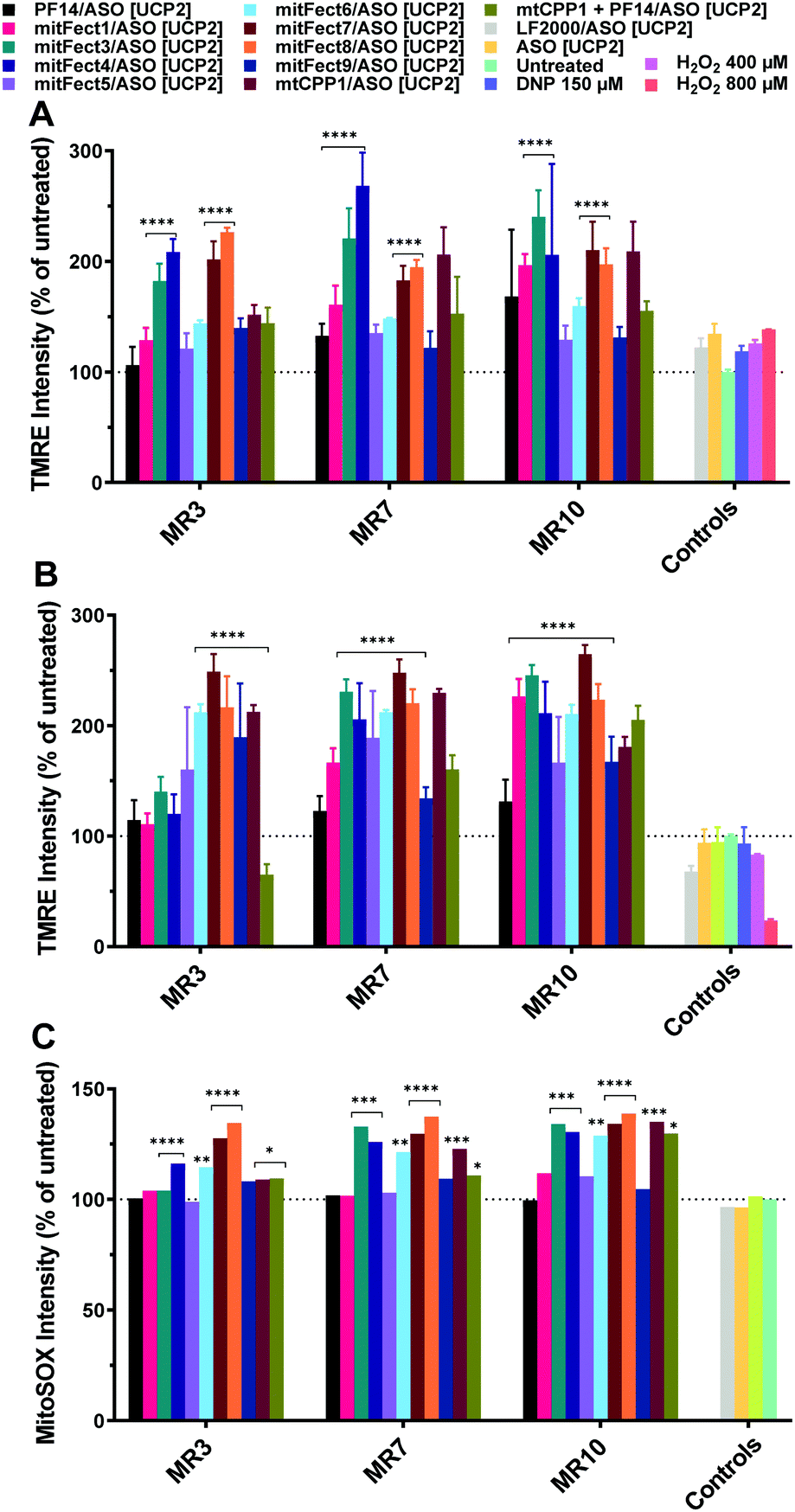 | ||
| Fig. 3 Effect of ASO [UCP2] on mitochondrial membrane potential and intracellular ROS production in HeLa pLuc705 cells. The oligonucleotide treatment was performed as described in the materials and methods section. HeLa pLuc705 were treated for 6 h (A) and 24 h (B), at MR 3, 7, and 10 with 100 nM ASO [UCP2] antisense, or ASO [UCP2] alone, or unrelated oligonucleotide, as indicated in the Materials and methods. The mitochondrial membrane potential was evaluated by TMRE uptake and intracellular ROS production was monitored by the fluorescent probe MitoSOX Red (C). Results are normalized and expressed as percentage of results obtained from untreated cells. The values represent the mean of one or more experiments performed in triplicate (mean ± SD, A and B: n = 3; C: n = 1). ****p ≤ 0.0001. Ordinary two-way analysis of variance (ANOVA) with Dunnett's multiple comparison test vs. untreated cells. Complete statistical analysis data are presented in ESI† (Table S2a and b). | ||
While a small effect was shown when cells were transfected for 6 h with LF2000 or the ASO [UCP2] alone, cells transfected for 24 h with LF2000 showed a small depolarization of the mitochondrial membrane potential and the ASO [UCP2] alone had no effect with TMRE uptake levels similar to untreated cells. As shown in Fig. 3, not all the peptides have the same efficacy to form the complexes and/or to deliver the ASO to the mitochondria. This could explain the increment of the mitochondrial membrane potential and the different detection of ROS levels after treatment with different peptides/oligonucleotide.
The changes in mitochondrial membrane potential and ROS levels in cells treated with ASO [UCP2] are in agreement with the previously reported data showing that UCP2 decreases ROS production by lowering the membrane potential and therefore reducing reverse electron transfer into complex I.56 Furthermore, these findings are also in agreement with the changes observed in mitochondrial function in ucp2 knockout animals.57 To confirm the efficient delivery of ASO [UCP2] we investigated the effect of the treatment on mitochondria ROS production measuring intracellular superoxide anion levels. To monitor intracellular ROS levels, we chose MitoSOX™ Red mitochondrial superoxide indicator, a fluorescent probe rapidly oxidized by superoxide resulting in red fluorescence. As expected, the intracellular production of ROS was enhanced by the 24 h treatment with ASO [UCP2] at all MRs and in a similar manner to the changes observed for the mitochondrial membrane potential; whereas no significant effects were observed when cells were treated with the unrelated ON or PF14/ASO [UCP2] (Fig. 3C). The complexes were also tested for cytotoxicity using the WST-1 reagent assay. None of the complexes showed to be toxic at any of the tested MRs with a cell viability over the 80% (Fig. S1B, ESI†). Taken together, these data demonstrate that treatment with ASO [UCP2] induce specific changes in mitochondrial function and ROS production into HeLa pLuc705 cells.
Peptides deliver D-arm ASO [UCP2] and cause stronger effects on mitochondrial membrane potential compared to ASO [UCP2]
The treatment with D-arm [UCP2] antisense oligonucleotide at 100 nM dose induced a stronger effect on the mitochondrial functionality. D-arm is a D stem-loop import signal of tRNATyr(GUA) and has been shown to be efficiently imported into the mitochondrion of Leishmania, a kinetoplastid protozoan, as well as into mitochondrial matrix in isolated mitochondria.17–19 We report an increase in TMRE uptake at all the tested MRs and time points (Fig. 4); the 6 h treatment with D-arm ASO [UCP2] at MR 3 elicited a 1-fold increase in TMRE uptake by HeLa pLuc705 (Fig. 4A) compared to the treatment with ASO [UCP2] at same conditions. At MR 7 and MR 10 was elicited a 4.5-fold increase in TMRE uptake over the untreated cells. The mitochondrial membrane potential is massively perturbed after 24 h treatment with D-arm ASO [UCP 2] delivered by some peptides (see Fig. 4B), with a maximum of 20-fold increase of TMRE uptake over untreated cells. A stronger effect was shown also when cells were transfected with LF2000 or the D-arm ASO [UCP2] alone, proving that the D-arm modification of the ASO [UCP2] is improving the cellular uptake. Cells treated with 2,4-dinitrophenol (DNP) at 150 μM shown a hyperpolarization of the mitochondrial membrane potential (positive control), while the unrelated oligonucleotide had no effect with TMRE uptake similar to untreated cells. As shown in Fig. 4, not all the peptides have the same effect on the mitochondrial membrane potential as result of different efficacy to form the complexes and or to deliver the D-arm ASO [UCP2] into the cells.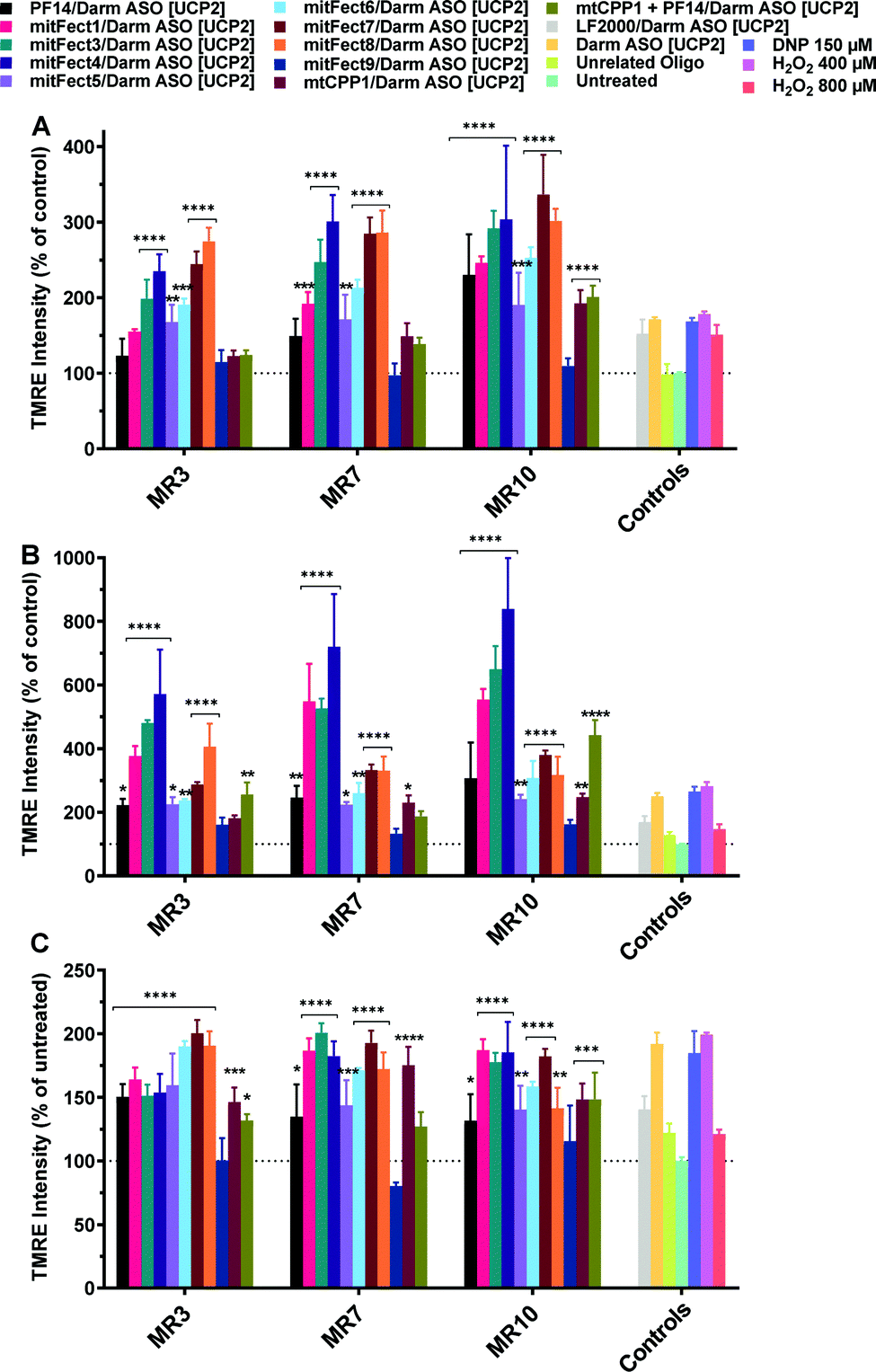 | ||
| Fig. 4 Effect of D-arm ASO [UCP2] on mitochondrial membrane potential in HeLa pLuc705 cells. The oligonucleotide treatment was performed as described in the Materials and methods section. HeLa pLuc705 were treated for 6 h (A), 24 h (B), and 48 h (C) at MR 3, 7, and 10 with 100 nM D-arm ASO [UCP2], or D-arm ASO [UCP2] alone, or unrelated oligonucleotide, or DNP as indicated in the materials and methods. Results are normalized and expressed as percentage of TMRE uptake in untreated cells. The values represent the mean of at least two experiments performed in triplicate (mean ± SD, n = 2). ns (p > 0.05), *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001, and ****p ≤ 0.0001. Ordinary two-way analysis of variance (ANOVA) with Dunnett's multiple comparison with untreated cells. All the bars under the draw line with **** have the indicated significance if not differently reported by another draw line with a specific significance. Complete statistical analysis data are presented in ESI† (Table S3a–c). | ||
Peptides efficiently deliver D-arm ASO [COXII] to mitochondria with effects on mitochondrial membrane potential
We investigated the effects of mitochondrial ASO delivery on mitochondrial functions using the same library of peptides (Table 1) by evaluating the mitochondrial membrane potential and the ROS production. If the delivery of D-arm ASO [COXII] was successful, the expression levels of the target mitochondrial protein would be decreased, followed by a decrease in mitochondrial functions such as maintaining mitochondrial membrane potential, and a slight increase of ROS production would consequently happen. The 6 h treatment with D-arm ASO [COXII] showed a strong decrease of TMRE uptake in HeLa pLuc705 cells, suggesting a depolarization of the mitochondrial membrane potential (Fig. 5A). In this experiment, not the LF2000, the antisense oligonucleotide alone, nor the unrelated oligonucleotide induced an effect on the mitochondrial membrane potential. All the peptides efficiently deliver the D-arm ASO [COXII] to the mitochondria, except for PF14. This was not confirmed when the cells where transfected for 6 h at MR 3 and MR 7 (Fig. S2, ESI†). After 24 h transfection of complexes, the TMRE uptake in HeLa pLuc705 cells was reduced (Fig. 5B) compared to the 6 h treatment, suggesting that the mitochondrial membrane potential is decreased but the cells have probably recovered over time. In case of the transfection with LF2000, D-arm ASO [COXII] or unrelated ON, the mitochondria membrane potential was not affected, and the results of TMRE uptake were similar to the results for untreated cells. It was also confirmed that the use of the D-arm ASO [COXII] increases the ROS production (Fig. 5C). These results were further confirmed by analyzing gene expression using quantitative reverse transcription polymerase chain reaction (RT-qPCR). The RT-qPCR results showed a strong effect of the treatment with mitFect3 in complex with the antisense oligonucleotide against COXII. The RNA level of COX-II got almost knocked out by the delivery of the antisense oligonucleotide whereas the level of UCP2 did not significantly change (Fig. 6). | ||
| Fig. 5 Effect of D-arm ASO [COXII] on mitochondrial membrane potential and intracellular ROS production in HeLa pLuc705 cells. The oligonucleotide treatment was performed as described in the Materials and methods section. HeLa pLuc705 were treated for 6 h (A) and 24 h (B) at MR10 with 100 nM D-arm ASO [COXII], or LF2000, D-arm ASO [COXII] alone, or unrelated oligonucleotide, as indicated in the materials and methods. The mitochondrial membrane potential was evaluated by TMRE uptake and intracellular ROS production was monitored by the fluorescent probe MitoSOX Red (C). Results are normalized and expressed as percentage of results obtained from untreated cells. The values represent the mean of at least one experiment performed in triplicate (mean ± SD, n = 2). ns (p > 0.05), *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001, and ****p ≤ 0.0001. Ordinary one-way analysis of variance (ANOVA) with Holm–Sidak's (A) or Dunnett's multiple comparison (B, and C), with a single pooled variance were compared with untreated cells. All the bars under the draw line with **** have the indicated significance. Complete statistical analysis data are presented in ESI† (Table S4a and b). | ||
 | ||
| Fig. 6 Fold change in the gene expression of UCP2 and COXII in HeLa pLuc705 cells (A). The difference in fold changes is between untreated HeLa pLuc 705 cells and treated with mitFect3/ASO [COXII]. Fold changes are expressed in a logarithmic base 2 scale. qPCR cycle thresholds have been normalized to GAPDH and UBC expressions before the fold change was calculated. In (B) is shown the western blot for UCP2, COX-II, and β-actin for both untreated (left column) and treated (right column) cells. The three blots come from the same gel and the bands are cropped from their original picture (the complete membrane pictures are shown in Fig. S5, ESI†). In (C) is shown the quantification of the protein level after two normalizations, first to the β-actin level, and then to untreated cells. | ||
This result indicates a good efficacy of mitochondrial gene therapy. The constant RNA level of UCP2 shows that the mitochondria's RNA producing machinery is not severed by the treatment and that the decrease in COXII RNA is indeed an effect of the delivery of the antisense oligonucleotide. The western blot analysis showed an increase of 30% in the level of the protein COXII after the treatment with mitFect3 in complex with the antisense oligonucleotide D- arm ASO [COXII].
Instead, the protein levels of UCP2 did not change (Fig. 6B and C). These results suggest that the down regulation of the COXII after transfection of D-arm ASO [COXII] results in an imbalance among the subunit in complex IV with effects on mitochondrial functions such as maintaining the mitochondrial membrane potential and the production of ROS.
Dynamic light scattering analysis and zeta potential measurements of peptides/ASO [UCP2], peptides/D-arm ASO [UCP2] and peptides/D-arm ASO [COXII] complexes
To investigate the formation of nanocomplexes in the formulation, dynamic light scattering (DLS) studies and ζ potential measurements were performed. These assessments were conducted in water to find a correlation between particles physical properties and the transfection efficacy. The diameter, Đ (dispersity, an indicator of particle-size distribution) and the ζ potential of the peptides/ASO [UCP2] nanoparticles are listed in Table 3. Average diameter of the particles formed is different at different MRs, with size decreasing at increasing MR. The ζ potential is also different for the different formulations. While it was negative at MR 3 with an average value of ∼−20 mV, the ζ potential changed to positive values at MR 7 and increased to a value of ∼20 mV at MR 10 for all the formulations, except for mitFect4/ASO [UCP2], mitFect8/ASO [UCP2], mtCPP1/ASO [UCP2], and mtCPP1 + PF14/ASO [UCP2] at MR 7 or mitFect9/ASO [UCP2], mtCPP1/ASO [UCP2], and mtCPP1 + PF14/ASO [UCP2] at MR 10. Based on these results, positively charged nanoparticles with more homogenous structures were prepared at the MR 7 and MR 10, which are more efficiently internalized, in accordance also with the biological effects tested on the HeLa pLuc705 cells.| Complex | MR3 | MR7 | MR10 | ||||||
|---|---|---|---|---|---|---|---|---|---|
| d | Đ | ζ | d | Đ | ζ | d | Đ | ζ | |
| d – diameter (nm); Đ – dispersity; ζ – ζ potential (mV). The complexes were formed at peptide/D-arm ASO [COXII] MR 3, 7, and 10 in MQ water at 1 μM of oligonucleotide. After 1 h of incubation the complexes were diluted 10× in MQ and number mean size, Đ, and ζ potential were measured from the same sample (mean, n = 1). | |||||||||
| PF14/ASO [UCP2] | 422.6 | 0.47 | −19.4 | 266.0 | 0.57 | 20.7 | 81.5 | 0.45 | 27.6 |
| mitFect1/ASO [UCP2] | 307 | 0.54 | −27.1 | 209.6 | 0.98 | 15.9 | 123.9 | 0.53 | 18.6 |
| mitFect3/ASO [UCP2] | 216.5 | 0.36 | −18.6 | 167.9 | 0.35 | 26.5 | 91.4 | 0.34 | 22.7 |
| mitFect4/ASO [UCP2] | 287.4 | 0.61 | −27.0 | 210.6 | 0.54 | −19.0 | 113.4 | 0.56 | 21.2 |
| mitFect5/ASO [UCP2] | 301.7 | 0.58 | −19.7 | 102.8 | 0.33 | 7.84 | 133.4 | 0.38 | 7.42 |
| mitFect6/ASO [UCP2] | 254.6 | 0.52 | −14.5 | 96.3 | 0.36 | 13.0 | 54.38 | 0.32 | 8.08 |
| mitFect7/ASO [UCP2] | 323.9 | 0.64 | −19.6 | 223.5 | 0.49 | 19.4 | 115.8 | 0.35 | 20.8 |
| mitFect8/ASO [UCP2] | 300.3 | 0.53 | −17.6 | 166.6 | 0.41 | −24.9 | 132.6 | 0.34 | 18.7 |
| mitFect9/ASO [UCP2] | 198 | 0.44 | −28.9 | 197.7 | 0.46 | 31.0 | 95.9 | 0.27 | −21.5 |
| mtCPP1/ASO [UCP2] | — | — | −4.38 | — | — | −35.5 | — | — | −12.1 |
| mtCPP1 + PF14/ASO [UCP2] | 241.2 | 0.51 | −27.0 | 216.6 | 0.64 | −24.0 | 417.6 | 0.83 | −18.2 |
It has been reported that increased positive charge lead to improved cellular uptake against various cell lines via electrostatic interaction with negatively charged surface.9 The preparation of positively charged nanoparticles is a requirement to increase the cellular uptake. The mitochondrial uptake of peptides or nanoparticles is strongly dependent on the highly negative potential of the inner mitochondrial membrane. The nanoparticle physicochemical properties, like size, shape, surface properties as well as colloidal stability of nanoparticles in aqueous solution and biological media, influence the cellular and/or organelle uptake.58
The same measurements have been performed also for the complexes formed by peptides/D-arm ASO [UCP2] and peptides/D-arm ASO [COXII] reported in Tables 4 and 5, respectively. Similar size changes of the complexes were seen at increased MRs. The complexes of peptides/D-arm ASO [UCP2] and peptides/D-arm ASO [COXII] differ in ζ potential from the complexes of peptides/ASO [UCP2]. The majority of ζ potentials are negative even at increasing MRs with only few complexes switching from negative to positive values at MR 10.
| Complex | MR 3 | MR 7 | MR 10 | ||||||
|---|---|---|---|---|---|---|---|---|---|
| d | Đ | ζ | d | Đ | ζ | d | Đ | ζ | |
| d – diameter (nm); Đ – dispersity; ζ – ζ potential (mV). The complexes were formed at peptide/D-arm ASO [UCP2] MR 3, 7, and 10 in MQ water at 1 μM of oligonucleotide. After 1 h of incubation the complexes were diluted 10× in MQ and number mean size, Đ, and ζ potential were measured from the same sample (mean, n = 1). | |||||||||
| PF14/D-arm ASO [UCP2] | 117.7 | 0.76 | −15.8 | 31.9 | 0.34 | −12.5 | 50.9 | 0.39 | −17.5 |
| mitFect1/D-arm ASO [UCP2] | 51.8 | 0.38 | −28.4 | 45.1 | 0.42 | −20.2 | 57.8 | 0.56 | −24.7 |
| mitFect3/D-arm ASO [UCP2] | 123.5 | 0.57 | −19.9 | 48.5 | 0.38 | −17.1 | 99.8 | 0.85 | −17.2 |
| mitFect4/D-arm ASO [UCP2] | 25.4 | 0.57 | −22.9 | 54.8 | 0.34 | −22.5 | 54.3 | 0.54 | −21.2 |
| mitFect5/D-arm ASO [UCP2] | 81.1 | 0.62 | −23.0 | 48.5 | 0.54 | −21.6 | — | — | 2.6 |
| mitFect6/D-arm ASO [UCP2] | 124.9 | 0.54 | −17.2 | 41.3 | 0.45 | −21.1 | 37.9 | 0.34 | 8.5 |
| mitFect7/D-arm ASO [UCP2] | 39.9 | 0.42 | −21.5 | 47.3 | 0.45 | −22.3 | 40.9 | 0.35 | −19.1 |
| mitFect8/D-arm ASO [UCP2] | 56.7 | 0.62 | −21.5 | 34.6 | 0.43 | −16.7 | 26.1 | 0.56 | −23.2 |
| mitFect9/D-arm ASO [UCP2] | 80.4 | 0.51 | −19.2 | 79.3 | 0.06 | −16.0 | 36.9 | 0.8 | −5.2 |
| mtCPP1/D-arm ASO [UCP2] | — | — | — | — | — | — | — | — | — |
| mtCPP1 + PF14/D-arm ASO [UCP2] | 42.1 | 0.42 | −22.0 | 38.8 | 0.23 | −17.2 | 36.9 | 0.57 | −20.2 |
| Complex | MR 3 | MR 7 | MR 10 | ||||||
|---|---|---|---|---|---|---|---|---|---|
| d | Đ | ζ | d | Đ | ζ | d | Đ | ζ | |
| d – diameter (nm); Đ – dispersity; ζ – ζ potential (mV). The complexes were formed at peptide/D-Arm [COXII] MR 3, 7, and 10 in MQ water at 1 μM of oligonucleotide. After 1 h of incubation the complexes were diluted 10× in MQ water and the number mean size Đ, and ζ potential were measured from the same sample (mean, n = 1). | |||||||||
| PF14/D-arm ASO [COXII] | 66.2 | 0.36 | −19.6 | 43.1 | 0.42 | −20.5 | 29.4 | 0.47 | −20.1 |
| mitFect1/D-arm ASO [COXII] | 55.6 | 0.32 | −24.2 | 45.2 | 0.49 | −24.2 | 69.6 | 0.41 | −15.6 |
| mitFect3/D-arm ASO [COXII] | 67.9 | 0.47 | −18.9 | 71.9 | 0.32 | 27.8 | 135.6 | 0.23 | −1.8 |
| mitFect4/D-arm ASO [COXII] | 69.6 | 0.42 | −19.4 | 35.9 | 0.32 | −18.7 | 23.7 | 0.39 | −17.0 |
| mitFect5/D-arm ASO [COXII] | 86.7 | 0.94 | −22.9 | 31.5 | 0.56 | −22.6 | 96.1 | 0.88 | −18.5 |
| mitFect6/D-arm ASO [COXII] | 82.3 | 0.63 | −20.1 | 30.1 | 0.57 | −18.7 | 137.9 | 0.61 | 6.49 |
| mitFect7/D-arm ASO [COXII] | 79.3 | 0.68 | −23.3 | 35.3 | 0.40 | −18.5 | 84.8 | 0.81 | −20.3 |
| mitFect8/D-arm ASO [COXII] | 54.5 | 0.55 | −18.1 | 48.3 | 0.41 | −19.8 | 101.8 | 0.47 | 12.9 |
| mitFect9/D-arm ASO [COXII] | 76.4 | 0.32 | −19.1 | 34.2 | 0.34 | −21.1 | 52.9 | 0.41 | −35.6 |
| mtCPP1/D-arm ASO [COXII] | — | — | −36.7 | 6.8 | 0.76 | −0.5 | 0.6 | 1 | 0.1 |
| mtCPP1 + PF14/D-arm ASO [COXII] | 53.2 | 0.47 | −20.7 | 49.1 | 0.44 | −19.9 | 20.3 | 0.40 | −21.4 |
Conclusions
We report a truly peptide-based mitochondrial gene targeting using antisense oligonucleotides to affect mitochondrial functions. We have shown that the chemically modified CPPs are able to efficiently deliver SCO, ASO [UCP2], D-arm ASO [UCP2], and D-arm ASO [COXII] to HeLa pLuc705 cells. We did not study the intracellular trafficking in this work but has been previously studied for the backbone peptide (PF14).43 It is still not very clear or possible to generalize which endocytic pathway is used for internalization but very often it has been observed that more than one pathway operates in parallel; by using inhibitors for one of the internalization route, it is still possible to observe the internalization of nano-complexes. It has been suggested that the inhibition of some receptors/internalization route would cause the uptake by other routes as a compensatory cellular mechanism.59–63 We have successfully formulated peptides/oligonucleotides nano-complexes as a proof-of-principle that could be used as a pharmaceutical form for peptide-based therapeutics. If we can build upon these findings, we could make a significant difference to the lives of patients with mitochondrial diseases.Experimental
Design, synthesis, purification, and analysis of peptides
The library of peptides was designed based on our previous published peptides, mtCPP1 and PF14. Peptides were synthesized on a 0.1 mmol scale following fluorenylmethyloxycarbonyl (Fmoc) solid phase peptide synthesis (SPPS) chemistry. The synthesis was performed manually or using Biotage Alstra plus microwave assisted peptide synthesizer (Biotage, Sweden) with ChemMatrix Rink Amide resin (0.50 mmol g−1, Biotage, Sweden) as a solid support to obtain C-terminally amidated peptides. The resin was swollen in N,N-dimethylformamide (DMF, VWR, Radnor, Pennsylvania, USA) for 5 min at 70 °C with oscillating mixer on. At each coupling step Fmoc-protected D- or L-amino acids were dissolved in DMF, and ethyl 2-cyano-2-(hydroxyimino)acetate (OxymaPure Novabiochem, Merck Millipore, Burlington, Massachusetts, USA) and carbodiiamide (DIC, Iris Biotech GmbH, Germany), used as coupling reagents, for 5 min at 75 °C. The Fmoc group was removed by treatment with piperidine (20% v/v, Iris Biotech GmbH, Germany) in DMF (programmed in two consecutive cycles; the first reaction at 45 °C for 2 min and the second at room temperature for 12 min). The final cleavage was performed using a standard cleavage protocol (95% trifluoroacetic acid (TFA, Iris Biotech GmbH, Germany)), 2.5% triisopropylsilane (TIS, Merck KGaA, Darmstadt, Germany) and 2.5% H2O for 4 h at room temperature. Peptides were precipitated in cold diethyl ether (VWR, Radnor, Pennsylvania, USA) and purified by reversed-phase high performance liquid chromatography (RP-HPLC) using a BioBasic C8 column (Thermo Scientific, Sweden) or Zorbax SB-300 C3 column (Agilent Technologies, Santa Clara, CA, USA). The crude peptides were eluted using gradients of acetonitrile in water (0.1% TFA). The purified peptides were lyophilized, and the molecular mass was determined by UHPLC-MS (Agilent 1260 Infinity, Agilent Technologies, Santa Clara, California, USA).Cell cultures
HeLa pLuc705 (human cervical carcinoma cell line) cells, in which a mutated beta-globin intron, carrying an aberrant splice site at nucleotide 705, interrupts the coding region of the luciferase reporter gene inserted in HeLa pLuc705 cells, were maintained in complete medium which is Dulbecco's modified Eagle's medium (DMEM) with glutamax supplemented with 0.1 mM non-essential amino acids, 100 units per mL penicillin, 100 μg mL−1 streptomycin, and 10% fetal bovine serum (FBS). Cells were cultured at 37 °C in a 5% CO2 atmosphere. HeLa pLuc705 cells were seeded on plates or dishes for each experiment. Immediately before transfection, the media was replaced with fresh media. Media and chemicals were purchased from Thermo Fisher Scientific, Waltham, MA, USA.Cell proliferation
Cell proliferation was assessed by the Roche Wst-1 proliferation assay (Roche Diagnostics Scandinavia AB, Sweden) according to the manufacturer's instructions. Briefly, 7 × 103 HeLa pLuc705 cells per well were seeded 1 day prior to the experiment on a 96-well plate. Cells were treated with peptide/oligonucleotide nanocomplexes at different MRs for 24 h in complete media, incubated at 37 °C and 5% CO2. LF2000 was used according to the manufacturer's protocol. Cells incubated with dimethylsulfoxide (DMSO) 20% in complete media (v/v) and hydrogen peroxide (H2O2) at 400 μM were used as positive controls. Wst-1 was added according to the manufacturer's protocol, to each well at final dilution 1:10. Wst-1 measures the activity of the mitochondrial dehydrogenases to convert tetrazolium salts to formazan and cell proliferation is directly correlated to the amount of formazan dye that is formed. After 4 h incubation at 37 °C and 5% CO2, absorbance was measured at 450 nm on Sunrise microplate absorbance reader (Tecan, Switzerland). The percentage of viable cells was determined by normalizing the values obtained for treated cells versus untreated cells.
Peptide/oligonucleotide complexes formation
PS-2′-OMe SCO, ASO [UCP2], D-arm ASO [UCP2], or D-arm ASO [COXII] were mixed with each peptide at different MRs in MilliQ-water in 10% of the final treatment volume. Complexes were formed for 1 h at room temperature and meanwhile the cell medium was replaced in the wells with fresh complete media. Thereafter, complexes were added to each well. When using Lipofectamine 2000 (Invitrogen, Sweden), the complexes were prepared according to the manufacturer's protocol.Splice-correction assay
HeLa pLuc705 cells (7 × 103) were seeded 24 h prior to experiments into 96-well plates. Cells were treated with peptide/SCO nanocomplexes at different MRs (3:1, 5:1, 10:1) and incubated for 24 h in complete media. Thereafter, the cells were lysed using 20 μL 0.1% Triton X-100 in HEPES-buffered Krebs Ringer (HKR) buffer for 30 min at room temperature. Luciferase activity was measured using Promega's luciferase assay system on GLOMAX™ 96 microplate luminometer (Promega, Sweden).
Determination of mitochondrial membrane potential
Mitochondrial membrane potential (Δψm) was evaluated using the fluorescent probe TMRE (mitochondrial potential membrane assay kit, Abcam, United Kingdom). The cell positively charged permeant dye, TMRE, readily accumulates in active mitochondria due to their relative negative charge. The dye fails to be sequestered into depolarized and/or non-functional mitochondria due to a decreased mitochondrial membrane potential. Briefly, HeLa pLuc705 cells were seeded into 96-well plates 24 h before the treatments. Cells were treated with nanocomplexes at three MRs (3:1, 7:1, and 10:1) and 100 nM ASO dose for 24 h. H2O2 400 μM, 800 μM, and DNP were used as positive controls. TMRE 400 nM was added to the complete medium and cells were incubated for 20 min at 37 °C, 5% CO2 protected from light. The media was removed, and cells were washed once with fresh media to remove background fluorescence from the cell culture media. 100 μL per well of fresh media was added. The plate was read on a fluorescence reader (Flex Station II, Molecular Device; peak excitation = 549 nm, peak emission = 575 nm). Δψm of treated cells was expressed as percentage of Δψm of untreated cells.
Determination of reactive oxygen species in cells
The ROS production was determined by using the fluorescent probe MitoSOX™ Red mitochondrial superoxide indicator (Invitrogen Detection technologies). MitoSOX™ Red reagent is rapidly and selectively targeted to the mitochondria. Once in the mitochondria, MitoSOX™ Red reagent is oxidized by superoxide and exhibits red fluorescence (Lifetechnologies, Molecular Probes®). According to the manufacturer's protocol, a vial of MitoSOX™ reagent was dissolved in 13 μL of dimethylsulfoxide (DMSO) to make a 5 mM MitoSOX™ reagent stock solution. The 5 mM stock solution was diluted in HBSS/Ca/Mg to obtain a 5 μM MitoSOX™ Red reagent working solution. Briefly, HeLa pLuc705 cells were seeded in 96-well plates at a concentration of 7 × 103 cells per well containing 100 μL of complete media and allowed to recover for 24 h. Cells were treated with nanocomplexes at three MRs (3:1, 7:1, and 10:1) and 100 nM dose ASO for 24 h. MitoSOX™ Red reagent 5 μM was added to the cells and they were incubated for 10 min at 37 °C and 5% CO2, protected from light. After the staining with MitoSOX™ Red, the media containing the fluorescent probe was removed and the cells were washed once with fresh media to remove background fluorescence, and finally 100 μL per well of fresh media was added. The plate was read with a fluorescence reader (Flex Station II, Molecular Device) with settings suitable for MitoSOX™ Red (peak excitation = 510 nm, peak emission = 580 nm).
Fluorescence microscopy
The digital images were obtained with LSM 5 Pascal laser scanning microscope (Zeiss, Germany) at 2048 × 2048 pixels. Images were taken using 10×, 40× dry objective lens and 63× oil immersion objective lens and optical section was <1 μm. Draq5™ fluorescent probe was excited by the 646 nm line of an argon laser, and the emission at 697 was recorded for the nuclear staining. To localize mitochondria, cells were loaded with 200 nM MitoTracker Green, which distributes into the negatively charged cellular compartment, for 20 min at 37 °C. MitoTracker Green fluorescence was excited by laser at 490 nm and recorded at 516 nm. AlexaFluor568 labelled oligonucleotides were excited at 568 nm and emission was read at 603 nm. A blue pseudo color was applied to visualize the nuclear stain. Green and red pseudo colors were applied to visualize mitochondria and the complexes into the cells, respectively. All the records were performed at controlled temperature (35 °C to 37 °C). Images were analyzed with the software program ImageJ.RNA extraction and cDNA synthesis
In a 10 cm Petri dish, 2 × 106 HeLa pLuc705 cells were seeded in 9 mL media. The cells were treated with the complex mitFect3/ASO in the same way as previously described. A control was prepared with untreated cells. After 24 h the media was removed, the cells harvested with the use of trypsin and resuspended in PBS. A pellet was formed by centrifugation and total RNA was extracted using Qiagen RNeasy kit. The two samples were then cleaned and purified with Qiagen clean and concentrator kit. RNA purity was verified by an absorbance measurement at 230 nm, 260 nm and 280 nm using an Implen P330 nanophotometer. Immediately after, the RNA samples were turned into cDNA using RevertAid Minus First Strand cDNA Synthesis kit and the purity of the cDNAs was verified by absorbance measurements.Real time qPCR
The real time qPCR measurements have been achieved with primers for uncoupling protein 2 (UCP 2) and cytochrome c oxidase subunit 2 (COX-II). The sequences of the primers used for the RT-qPCR assay are reported in Table S5 (ESI†). Every experiment has been performed in duplicates. The two cDNA samples concentration were normalized to 50 ng μL−1. 100 ng of each cDNA sample were mixed in a PCR plate containing the lyophilized primers with 10 μL SYBR green probe and MilliQ water to 20 μL. The plate was placed in a BioRad iQ5 reader and the amplification cycles were run. Two primers for GAPDH and UBC were used as home keeping gene reference.Protein extraction and western blot
In a 10 cm Petri dish, 2 × 106 HeLa pLuc705 cells were seeded in 9 mL media. The cells were treated with the complex mitFect3/ASO in the same way as previously described. A control was prepared with untreated cells. After 24 h the media was removed, and the cells were washed with PBS. PBS was then removed, and 1 mL of ice-cold lysis buffer was added to the dishes. The adherent cells were scraped with a cold plastic cell scraper and the suspensions were transferred to a pre-cooled centrifuge tube. An agitation was maintained for 30 min on ice prior to a centrifugation at 12000 rpm for 20 min. The supernatant was kept, and the protein concentration was measured with a Bradford assay using BSA as a standard. The proteins were denatured with the use of a 4× Laemmli buffer at 95 °C for 5 min and 10 μg of protein from each sample were loaded on a BioRad 4–15% Mini-PROTEAN® TGX™ Precast Protein Gel. The gel was run for 45 minutes in SDS-PAGE buffer and then transferred to a membrane. The membrane was blocked with a solution containing 1% of non-fat skimmed milk. The membrane was then incubated with a rabbit polyclonal anti-UCP2 antibody (ab97931, Abcam) overnight at 4 °C. The membrane was then washed 3 times for 5 min each with Tris-buffered saline containing 0.05% Tween 20 (TBST) before addition of a goat anti-rabbit IgG H&L conjugated with horse radish Peroxidase (HRP) (ab205718, Abcam) for 1 h. The membrane was washed 3 times with TBST and then incubated with the substrate for HRP for 5 min before imaging. The antibodies were then stripped from the membrane by the use of a medium stripping buffer containing SDS, Tween 20 and glycine. The same procedure was used for the blot of COX-II (ab91317, Abcam) and β-actin. The results have been quantified for each lane using the software ImageJ and then normalized first to β-actin level and then to the untreated sample.
Characterization of nanoparticles
DLS was used to determine the hydrodynamic mean diameter and the zeta-potential (ζ potential) of peptide/ASO. The complexes were formed in MilliQ water. Measurements were carried out using a Zetasizer Nano ZS apparatus (Malvern Instruments, United Kingdom). Samples were assessed in disposable low-volume cuvettes. All data were plotted as size distribution by intensity.Statistical analysis
All statistical results are expressed as mean ± SD. Ordinary one-way analysis of variance (ANOVA) tests were used for multiple comparisons of the mean in each group with that of the standard (untreated cells). Dunnett's multiple comparisons test with a single pooled variance and corrections with 95% confidence intervals and significance were used. Two-way ANOVA tests were used for multiple comparisons of the mean in each group at each MR with that of the standard (untreated cells). Dunnett's or Holm–Sidak's multiple comparisons test were used for multiple comparisons. All statistical tests were performed with GraphPad Prism software (Version 8.1.1(224)), and statistical significance was defined as follows: ns (p > 0.05), *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001, and ****p ≤ 0.0001.Author contributions
C. P. C. and Ü. L. formulated the idea and designed the study; C. P. C., T. K., R. T., T. L., and M. G. performed the experimental work; C. P. C. analyzed the data and wrote the manuscript. T. K. proof-read, edited, and commented on the manuscript. All authors contributed to the discussion and proof-reading of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Swedish Research Council for Natural Sciences (621-2011-5902), the Swedish Research Council for Medical Research (K2012-66X-21148-04-5), and the Swedish Cancer Foundation (CAN 2014/259).References
- P. C. Zamecnik and M. L. Stephenson, Proc. Natl. Acad. Sci. U. S. A., 1978, 75, 280–284 CrossRef CAS.
- N. Dias and C. A. Stein, Mol. Cancer Ther., 2002, 1, 347–355 CAS.
- E. Urban and C. R. Noe, Farmaco, 2003, 58, 243–258 CrossRef CAS.
- V. K. Sharma, R. K. Sharma and S. K. Singh, MedChemComm, 2014, 5, 1454–1471 RSC.
- X. Shen and D. R. Corey, Nucleic Acids Res., 2018, 46, 1584–1600 CrossRef CAS.
- C. A. Stein, J. B. Hansen, J. Lai, S. Wu, A. Voskresenskiy, A. Høg, J. Worm, M. Hedtjärn, N. Souleimanian, P. Miller, H. S. Soifer, D. Castanotto, L. Benimetskaya, H. Ørum and T. Koch, Nucleic Acids Res., 2010, 38, e3 CrossRef CAS.
- R. L. Juliano, Nucleic Acids Res., 2016, 44, 6518–6548 CrossRef.
- C. E. Thomas, A. Ehrhardt and M. A. Kay, Nat. Rev. Genet., 2003, 4, 346–358 CrossRef CAS.
- D. M. Copolovici, K. Langel, E. Eriste and Ü. Langel, ACS Nano, 2014, 8, 1972–1994 CrossRef CAS.
- C. P. Cerrato, K. Künnapuu and Ü. Langel, Expert Opin. Drug Delivery, 2016, 2, 245–255 Search PubMed.
- C. P. Cerrato, K.-L. Veiman and Ü. Langel, Future Sci., 2015, 160–171 Search PubMed.
- T. Lehto, K. Kurrikoff and Ü. Langel, Expert Opin. Drug Delivery, 2012, 9, 823–836 CrossRef CAS.
- C. P. Cerrato, T. Lehto and Ü. Langel, Biomol. Concepts, 2014, 5, 479–488 CAS.
- C. P. Cerrato and U. Langel, Cell-penetrating peptides targeting mitochondria, 2018 Search PubMed.
- M. Dowaidar, M. Gestin, C. P. Cerrato, M. H. Jafferali, H. Margus, P. A. Kivistik, K. Ezzat, E. Hallberg, M. Pooga, M. Hällbrink and U. Langel, Sci. Rep., 2017, 7(1), 12635 CrossRef.
- C. P. Cerrato and Ü. Langel, Mol. Ther.–Methods Clin. Dev., 2017, 5, 221–231 CrossRef CAS.
- A. D. Frankel and C. O. Pabo, Cell, 1988, 55, 1189–1193 CrossRef CAS.
- M. Green and P. M. Loewenstein, Cell, 1988, 55, 1179–1188 CrossRef CAS.
- F. Madani, S. Lindberg, Ü. Langel, S. Futaki and A. Gräslund, J. Biophys., 2011, 2011, 1–10 CrossRef.
- T. Lehto, K. Ezzat, M. J. A. Wood and S. E. L. Andaloussi, Adv. Drug Delivery Rev., 2016, 106, 172–182 CrossRef CAS.
- K. Zhao, G.-M. Zhao, D. Wu, Y. Soong, A. V. Birk, P. W. Schiller and H. H. Szeto, J. Biol. Chem., 2004, 279, 34682–34690 CrossRef CAS.
- K. L. Horton, K. M. Stewart, S. B. Fonseca, Q. Guo and S. O. Kelley, Chem. Biol., 2008, 15, 375–382 CrossRef CAS.
- L. F. Yousif, K. M. Stewart, K. L. Horton and S. O. Kelley, ChemBioChem, 2009, 10, 2081–2088 CrossRef CAS.
- S. B. Fonseca, M. P. Pereira, R. Mourtada, M. Gronda, K. L. Horton, R. Hurren, M. D. Minden, A. D. Schimmer and S. O. Kelley, Chem. Biol., 2011, 18, 445–453 CrossRef CAS.
- S. P. Wisnovsky, J. J. Wilson, R. J. Radford, M. P. Pereira, M. R. Chan, R. R. Laposa, S. J. Lippard and S. O. Kelley, Chem. Biol., 2013, 20, 1323–1328 CrossRef CAS.
- G. R. Chamberlain, D. V. Tulumello and S. O. Kelley, ACS Chem. Biol., 2013, 8, 1389–1395 CrossRef CAS.
- R. Y. P. Alta, H. A. Vitorino, D. Goswami, C. W. Liria, S. P. Wisnovsky, S. O. Kelley, M. T. Machini and B. P. Espósito, PLoS One, 2017, 12, 1–15 CrossRef.
- W. Neupert and J. M. Herrmann, Annu. Rev. Biochem., 2007, 76, 723–749 CrossRef CAS.
- M. Donzeau, K. Káldi, A. Adam, S. Paschen, G. Wanner, B. Guiard, M. F. Bauer, W. Neupert and M. Brunner, Cell, 2000, 101, 401–412 CrossRef CAS.
- P. Kovermann, K. N. Truscott, B. Guiard, P. Rehling, N. B. Sepuri, H. Müller, R. E. Jensen, R. Wagner and N. Pfanner, Mol. Cell, 2002, 9, 363–373 CrossRef CAS.
- Y. C. Kang, M. Son, S. Kang, S. Im, Y. Piao, K. S. Lim, M. Y. Song, K. S. Park, Y. H. Kim and Y. K. Pak, Exp. Mol. Med., 2018, 50, 105 CrossRef.
- J. K. Watts and D. R. Corey, J. Pathol., 2012, 226, 365–379 CrossRef CAS.
- P. F. Chinnery, R. W. Taylor, K. Diekert, R. Lill, D. M. Turnbull and R. N. Lightowlers, Gene Ther., 1999, 7, 813 CrossRef.
- G. G. M. D’Souza, R. Rammohan, S. M. Cheng, V. P. Torchilin and V. Weissig, J. Controlled Release, 2003, 92, 189–197 CrossRef.
- V. Weissig and V. P. Torchilin, J. Drug Targeting, 2001, 9, 1–13 CrossRef CAS.
- D. Lyrawati, A. Trounson and D. Cram, Pharm. Res., 2011, 28, 2848–2862 CrossRef CAS.
- Y. Yamada, R. Furukawa, Y. Yasuzaki and H. Harashima, Mol. Ther., 2011, 19, 1449–1456 CrossRef CAS.
- Y. Yamada, E. Kawamura and H. Harashima, J. Nanopart. Res., 2012, 14 DOI:10.1007/s11051-012-1013-3.
- R. Furukawa, Y. Yamada, E. Kawamura and H. Harashima, Biomaterials, 2015, 57, 107–115 CrossRef CAS.
- S. Mahapatra, S. Ghosh, S. K. Bera, T. Ghosh, A. Das and S. Adhya, Nucleic Acids Res., 1998, 26, 2037–2041 CrossRef CAS.
- S. Nath Bhattacharyya, S. Chatterjee and S. Adhya, Mol. Cell. Biol., 2002, 22, 4372–4382 CrossRef.
- S. N. Bhattacharyya, S. Mukherjee and S. Adhya, Mol. Cell. Biol., 2000, 20, 7410–7417 CrossRef CAS.
- K. Ezzat, S. E. L. Andaloussi, E. M. Zaghloul, T. Lehto, S. Lindberg, P. M. D. Moreno, J. R. Viola, T. Magdy, R. Abdo, P. Guterstam, R. Sillard, S. M. Hammond, M. J. A. Wood, A. A. Arzumanov, M. J. Gait, C. I. E. Smith, M. Hällbrink and Ü. Langel, Nucleic Acids Res., 2011, 39, 5284–5298 CrossRef CAS.
- C. P. Cerrato, M. Pirisinu, E. N. Vlachos and Ü. Langel, FASEB J., 2015, 29, 4589–4599 CrossRef CAS.
- S. Kang, M. Cho, R. Kole, N. Carolina, C. Hill and N. Carolina, Biochemistry, 1998, 37, 6235–6239 CrossRef CAS.
- M. Klingenberg, Trends Biochem. Sci., 1990, 15, 108–112 CrossRef CAS.
- D. G. Nicholls and R. M. Locke, Physiol. Rev., 1984, 64, 1–64 CrossRef CAS.
- R. Ricquier, L. Casteilla and F. Bouillaud, FASEB J., 1991, 5, 2237–2242 CrossRef.
- D. Langin, D. Larrouy, P. Barbe, L. Millet, N. Viguerie-Bascands, F. Andreelli, M. Laville and H. Vidal, Int. J. Obes., 1999, 23, S64–S67 CrossRef CAS.
- J. Liu, J. Li, W.-J. Li and C.-M. Wang, J. Diabetes Res., 2013, 2013, 1–7 Search PubMed.
- V. P. Skulachev, Q. Rev. Biophys., 1996, 29, 169–202 CrossRef CAS.
- M. D. Brand and T. C. Esteves, Cell Metab., 2005, 2, 85–93 CrossRef CAS.
- A. J. Kowaltowski, A. D. T. Costa and A. E. Vercesi, FEBS Lett., 1998, 425, 213–216 CrossRef CAS.
- A. Negre-Salvayre, C. Hirtz, G. Carrera, R. Cazenave, M. Troly, R. Salvayre, L. Penicaud and L. Casteilla, FASEB J., 1997, 11, 809–815 CrossRef CAS.
- D. A. Talbot, A. J. Lambert and M. D. Brand, FEBS Lett., 2004, 556, 111–115 CrossRef CAS.
- C. Y. Zhang, G. Baffy, P. Perret, S. Krauss, O. Peroni, D. Grujic, T. Hagen, A. J. Vidal-Puig, O. Boss, Y. B. Kim, X. X. Zheng, M. B. Wheeler, G. I. Shulman, C. B. Chan and B. B. Lowell, Cell, 2001, 105, 745–755 CrossRef CAS.
- D. Arsenijevic, H. Onuma, C. Pecqueur, S. Raimbault, B. S. Manning, B. Miroux, E. Couplan, M. C. Alves-Guerra, M. Goubern, R. Surwit, F. Bouillaud, D. Richard, S. Collins and D. Ricquier, Nat. Genet., 2000, 26, 435–439 CrossRef CAS.
- J. Zhao and M. H. Stenzel, Polym. Chem., 2018, 9, 259–272 RSC.
- A. Eguchi, B. R. Meade, Y.-C. Chang, C. T. Fredrickson, K. Willert, N. Puri and S. F. Dowdy, Nat. Biotechnol., 2009, 27, 567–571 CrossRef CAS.
- K. Ezzat, H. Helmfors, O. Tudoran, C. Juks, S. Lindberg, K. Padari, S. E. L. Andaloussi, M. Pooga and Ü. Langel, FASEB J., 2012, 26, 1172–1180 CrossRef CAS.
- P. Arukuusk, L. Pärnaste, H. Margus, N. K. J. Eriksson, L. Vasconcelos, K. Padari, M. Pooga and U. Langel, Bioconjugate Chem., 2013, 24, 1721–1732 CrossRef CAS.
- H. Margus, K. Padari and M. Pooga, Adv. Drug Delivery Rev., 2013, 65, 1031–1038 CrossRef CAS.
- C. Juks, A. Lorents, P. Arukuusk, Ü. Langel and M. Pooga, FASEB J., 2017, 31(3), 975–988 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0tb01106a |
| ‡ Current affiliation: Division of Molecular Toxicology, Institute of Environmental Medicine, Karolinska Institutet, Nobel väg 13, Stockholm, SE-17177, Sweden. |
| This journal is © The Royal Society of Chemistry 2020 |