
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePhotonic hyperthermal and sonodynamic nanotherapy targeting oral squamous cell carcinoma†
Jiaxin
Zuo
a,
Minfeng
Huo
*b,
Liying
Wang
b,
Jia
Li
a,
Yu
Chen
 *c and
Ping
Xiong
*a
*c and
Ping
Xiong
*a
aDepartment of Ultrasound, Shanghai Ninth People's Hospital, Shanghai Jiaotong University School of Medicine, Shanghai, 200011, P. R. China. E-mail: xiongp1281@sh9hospital.org.cn
bState Key Laboratory of High Performance Ceramics and Superfine Microstructure, Shanghai Institute of Ceramics, Chinese Academy of Sciences, Shanghai, 200050, P. R. China. E-mail: mfhuo@mail.sic.ac.cn
cSchool of Life Sciences, Shanghai University, Shanghai, 200444, P. R. China. E-mail: chenyuedu@shu.edu.cn
First published on 21st August 2020
Abstract
Nanomedicine that enables multiple synergetic treatments provides effective non-invasive treatment modalities for cancer therapy. Yet treatments for oral squamous cell carcinoma (OSCC) are rarely reported. Here, we designed OSCC-targeting multi-functional nanomedicines to overcome the therapeutic obstacles during OSCC treatments, including ineffective chemotherapy, and the traumatic surgery and radiotherapy. The urokinase plasminogen activator receptor (uPAR)-targeting ligand AE105 decorated dendritic mesoporous silica nanoparticles (DMSN) encapsulating photonic active ultrasmall Cu2−xS NPs and sonosensitizer Rose Bengal (RB) have been rationally designed and constructed (designated as Cu2−xS-RB@DMSN-AE105, abbreviated as CRDA). These CRDAs initially target the uPAR, which is overexpressed in the OSCC cell membrane, to increase the localized accumulation of CRDAs at tumor sites. Under the irradiation of both near-infrared laser and ultrasound, the in situ photonic-hyperthermal and sonodynamic effects are respectively enabled to induce the cell death of OSCC. Upon both in vitro/in vivo challenges, tumor cells/xenografts have been efficiently eradicated, achieving the targeting and synergetic treatment modality against the OSCC with satisfactory biocompatibility.
Introduction
The application of “nanomedicine” against cancer is one of the medical research favorites at present. Various nanoplatforms have been constructed using unique theranostic functionalities to meet sophisticated biomedical circumstances.1–4 In particular, multifunctional nanoparticles (NPs) combined with various therapeutic modalities, including photonic hyperthermia, photodynamic,1 chemotherapy,5 magnetotherapy6 and sonodynamic therapy,7,8 can generate multiple theranostic modalities in practical use to achieve a synergistic therapeutic outcome. Among diverse nanoplatforms, the biomedical applications of mesoporous silica nanoparticles (MSNs) have received considerable attention during the past decades.9–12 MSNs feature a well-defined mesoporous structure that can not only encapsulate therapeutic drugs but also functional nanoparticles to support the construction of multifunctional nanoplatforms. This progress provides an important basis for the clinical transformation of nanomedicine with appealing therapeutic potentials.Oral squamous cell carcinoma (OSCC) is the leading cause of death in head and neck squamous cell carcinoma (HNSCC) with high malignancy, easy metastasis, and high recurrence.13,14 One of the reasons for the poor prognosis of OSCC-patients is the inadequate therapeutic efficacy for complete OSCC destruction.13,15 The most frequently adopted treatments are surgery supplemented with radiotherapy and/or chemotherapy. Surgical dissection is associated with considerable trauma and serious side-impacts, which substantially affect the post-treatment life quality of the patients.15–17 Therefore, applications of nanomedicine with great medical significance for the treatment of OSCC can effectively ameliorate the prognosis. Photonic hyperthermal therapy (PTT) typically refers to the use of near-infrared light to enable photonic hyperthermia in the presence of photonic active agents. These agents such as two-dimensional nanosheets,18,19 organic semiconductors,20 and other inorganic metal NPs (e.g., gold NPs,21–23 CuS24,25 and Fe3O426) can convert incident light energy into heat for tumor destruction. Among these nanoparticulate photonic active agents, ultrasmall Cu2−xS NPs (∼5 nm) feature high therapeutic prospects.27 Although it has been demonstrated that PTT could generate a prominent therapeutic effect against tumors, yet the uneven distribution of heat and poor tissue penetration depth enabled by the laser during treatment may not possibly lead to complete tumor eradiation, ultimately resulting in tumor reoccurrence.28,29 In order to guarantee a therapeutic outcome and satisfactory prognosis, enhanced tumor-site accumulations of therapeutic nanoagents are urgently demanded, specifically by targeting engineering, which can simultaneously reduce the side impacts while improving the therapeutic outcome.
The present research in the field of nanomedicine for OSCC treatment is mostly focused on PTT with chemotherapy.30,31 Statistical data have shown that, over the past 40 years, the application of cytotoxic chemotherapeutic drugs has not significantly ameliorated the survival or life quality of patients with oral cancer.32,33 Therefore, exploring new synergistic therapeutic modalities to achieve safe and effective treatment for OSCC, as well as to ameliorate the prognosis, is highly urgent and necessary. Reactive oxygen species (ROS) play critical roles in cell apoptosis,7 which could be generated by sonodynamic therapy (SDT).29 Recently, ultrasound (US)-triggered SDT was found to be promising for practical use because of its noninvasive nature and high tissue-penetrating capability.29,34 For instance, McEwan et al. designed RB containing sonodynamic nanoplatforms PLGA MB-RB,35 Oxy MB + RB36 and realized satisfactory SDT performance with a minimal RB dose of as low as 1 μM. Therefore, the elaborate combination of laser-triggered photonic hyperthermia and US-activated sonodynamic treatment is expected to achieve highly efficient OSCC treatment outcomes.
It has been documented that epidermal growth factor receptors (EGFRs) are overexpressed in OSCC, and the inhibition of the EGFRs has been a desirable therapeutic target for OSCC treatment. Currently, cetuximab is the only EGFR-targeting drug approved by the US Food and Drug Administration. However, secondary drug resistance would inevitably occur during the medication.37 Recently, another OSCC-targeting site, urokinase plasminogen activator receptor (uPAR) has been discovered. The over-expression of uPAR in the OSCC cell membrane can reinforce the signal transmission of the uPAR-integrin pathway, a critical transmembrane pathway for OSCC metastasis and proliferation.38–40 At the same time, uPAR exhibits a higher OSCC specificity when compared to EGFR. Therefore, uPAR can be used as an alternative biocompatible target against OSCC with better performance.
In this work, we report uPAR-targeting PTT/SDT synergistic therapeutic Cu2−x-RB@DMSN-AE105 (abbreviated as CRDA) nanoplatforms for targeted PTT/SDT synergistic therapy of OSCC. As the drug delivery system, biocompatible dendritic mesoporous silica NPs (DMSN) can biodegrade slowly in the acidic microenvironment of the tumor,9 achieving the cargo release. DMSNs were employed to encapsulate photonic active Cu2−xS NPs27 and sonosensitive Rose Bengal (RB)41 to realize synergistic PTT/SDT nanotherapeutics. For OSCC targeting design, AE105 (H-Asp-Cha-Phe-(d)Ser-(d)Arg-Tyr-Leu-Trp-Ser-OH) peptide features prominent targeting against uPAR that originates from the competing binding towards uPAR.42–44 When modified on the surface of DMSNs, OSCC targeting of CRDAs can be additionally achieved, providing an intriguing treatment modality against OSCC with targeting and synergistic performance.
Materials and methods
Materials
Cetyltrimethylammonium chloride (CTAC, Sigma-Aldrich Co.), tetraethyl orthosilicate (TEOS, Sinopharm Chemical Reagent Co.), triethanolamine (TEA, Sinopharm Chemical Reagent Co.), sodium salicylate (NaSal, Sigma-Aldrich Co.), hydrochloric acid (Sinopharm Chemical Reagent Co.), sulfur powder (Aladdin Biochemical Technology Co., Ltd), oleylamine (Aladdin Biochemical Technology Co., Ltd), chloroform (Sinopharm Chemical Reagent Co.), silane–PEG–carboxyl (silane–PEG2000–COOH, Shanghai Ponsure Biotech, lnc.), AE105 peptide (D-Cha-FsrYLWS, Nanjing Peptide Biotechnology Co., Ltd), Red Bengal (RB, Aladdin Biochemical Technology Co., Ltd).Synthesis of DMSNs
In a typical procedure, TEA (0.068 g) was initially mixed with deionized water (5 mL). Then, 0.5 mL of the above solution was mixed with 24.5 mL of deionized water, and placed in a water bath with a magnetic stirrer (300 rpm at 80 °C for 30 min). CTAC (0.38 g), as the structure-directing agent, was added to the solution. After complete dissolution, NaSal (0.168 g) was added to the solution and the reaction was allowed to continue for 1 h, followed by the dropwise addition of TEOS (2 mL). After another 2 h, the product was collected by centrifugation. CTAC was removed using a mixture of ethanol and concentrated HCl (6%) by extracting for 24 h at 70 °C three times. Finally, the product was collected by centrifugation, washed with water several times, and dispersed in 20 mL of ethanol.Synthesis of ultrasmall Cu2−xS NPs
Cu2−xS NPs with diameter 2–5 nm were synthesized according to the reported literature.27 Initially, sulfur (64 mg) was immersed in oleylamine (12 mL) and placed in a water bath at 70 °C with a magnetic stirrer until a dark red-brown solution was formed. A chloroform solution (40 mL) containing copper acetylacetonate (523.5 mg) and oleylamine (10 mL) was added to the above solution in a dropwise manner. During the reaction of 0.5 h at 70 °C, the mixture turned from brown-yellow to dark-green. Ethanol was added to precipitate the reaction. Finally, the product was collected by centrifugation, washed with water several times, and re-dispersed in 20 mL of chloroform.Fabrication of CRDAs
First, 2 mL of Cu2−xS NPs dispersed in chloroform (10 mg mL−1) was added to 2 mL of DMSNs dispersed in chloroform (5 mg mL−1) with agitation. After 12 h of encapsulation, Cu2−xS@DMSN NPs were formed and collected by centrifugation. Then, Cu2−xS@DMSN NPs (20 mg) were added to 10 mL ethanol containing 10 mg mL−1 of silane–PEG–NH2 (molecular weight = 2000) at 60 °C with magnetic stirring. After 24 h of PEGylation, the PEGylated Cu2−xS@DMSN NPs were centrifuged and washed several times to remove residual PEG. Decoration of AE105 onto DMSNs was accomplished by the reaction between AE105 and PEGylated Cu2−xS@DMSN NPs in MES solution in the presence of EDC and NHS for 12 h. After the centrifugation and purification process, Cu2−xS@DMSN-AE105 NPs were collected. 2 mL of Cu2−xS@DMSN-AE105 NPs re-suspended in ethanol (1 mg mL−1) was mixed with RB (4 mg) for 12 h to encapsulate the RB sonosensitizer. Finally, the Cu2−xS-RB@DMSN-AE105 NPs (CRDAs) were obtained. To produce Cu2−xS-RB@DMSN NPs (CRDs), 2 mL of PEGylated Cu2−xS@DMSN NPs in ethanol (1 mg mL−1) was mixed with RB (4 mg). After an additional 12 h of encapsulation, the CRDs were collected by centrifugation.To explore the release profile of RB, 10 mL PBS solutions at pH 7.4 and 5.5, respectively, containing CRDAs (RB: 10 μg mL−1), were stirred on a magnetic stirrer. At pre-determined time points (0.5, 1, 2, 4, 8, 12, 24 h), aliquots of the solution were centrifuged and the supernatant was used for UV-vis measurements.
Characterization
Transmission electron microscopy (TEM) images, energy-dispersive X-ray spectroscopy (EDS) and corresponding EDS-mapping were collected on a JEM-2100F transmission electron microscope operated at 300 kV. DLS was conducted on a Nano ZS90 Zetasizer (Malvern Instruments, Malvern, UK). UV-vis-NIR absorption spectra were recorded on a UV-3600 Shimadzu spectrometer. Zeta potential was tested on a Zetasizer Nanoseries instrument (Nano ZS90). The nitrogen (N2) adsorption–desorption isothermal curve and the corresponding pore-size distribution of DMSNs and Cu2−xS@DMSN NPs were calculated using Micromeritics Tristar 3000 system. Inductively coupled plasma atomic emission spectrometry (ICP-AES) was conducted on a VISTA instrument (Varian Company, American). ESR characterization was performed on a Bruker EMX electron paramagnetic resonance (EPR) spectrometer.Sonodynamic and photonic hyperthermia performance of CRDs
In this study, DPBF was employed to evaluate the singlet oxygen (1O2) generation induced by RB and CRDs respectively upon US irradiation. Briefly, RB or CRDs (RB: 10 μg mL−1) was mixed with DPBF (50 μM in DMF), diluted with ethanol. Then, the above solution was exposed to US irradiation (1 MHz, 2.0 W cm−2, 50% duty cycle, 2 min) in the dark. The absorbance of the solution was then recorded on a UV-vis spectrometer.For ESR assays, triacetonediamine (TEMP, Aladdin Biochemical Technology Co., Ltd) was used to trap 1O2 generated by RB and CRDAs using an ultrasound trigger. Typically, dimethyl formamide (DMF) solution containing RB and CRDAs (RB: 8 μg mL−1) were exposed to US irradiation (1.0 MHz, 2.0 W cm−2, 50% duty cycle, 2 min) in the presence of TEMP (20 μM). Aliquots of the assay solution were then transferred into the quartz tube for ESR measurements.
For the assessment of the photonic hyperthermia performance of CRDs, the localized temperature elevation profile of CRDs (0, 25, 50, 100 and 200 μg mL−1) under NIR laser irradiation (Shanghai Connect Fiber Optics, 1064 nm, 1.5 W cm−2, 5 min) was recorded on an infrared thermal imaging camera (A325SC; FLIR Systems, Wilsonville, OR, USA). The photonic hyperthermia stability of CRDs was revealed by the negligible fluctuations of the maximum temperature during five sequential laser on/off cycles against CRDs (400 μg mL−1, 1064 nm, 1.5 W cm−2, 5 min).
In vitro synergetic performance of PTT/SDT of CRDs against OSC-19 cells
OSC-19 cells were grown in DMEM with 10% FBS and 1% penicillin/streptomycin in an incubator at 37 °C in an atmosphere of 5% CO2. Cells were initially seeded into 96-well plates with a density of 104 cells per well for 24 h. CRDs (0, 25, 50, 100, 200, 400 and 800 μg mL−1)-containing fresh DMEM were replaced for further co-incubations. For cytocompatibility investigations, medium was discarded 24 h or 48 h post-incubation and cells were rinsed with PBS. Cell viability was performed using cell-counting kit-8 (CCK-8) according to the protocols.For in vitro PTT/SDT therapeutics, cells co-cultured with CRDs (0, 25, 50, 100, 200 and 400 μg mL−1) for 4 h were divided into three groups according to the respective treatments: US group (US irradiation); laser group (laser irradiation); US + laser group (US irradiation, followed by laser irradiation). The parameters of US irradiation were 1.0 MHz, 2.0 W cm−2, 50% duty cycle, 2 min. The parameters of laser irradiation were 1064 nm, 1.5 W cm−2, 5 min. Then, the medium was discarded and cells were rinsed with PBS several times. Cell viability was determined using a cell-counting kit-8 (CCK-8) according to the protocols.
In vitro synergetic performance of PTT/SDT of CRDs against OSC-19 cells by confocal laser scanning microscopy (CLSM)
Cells were initially seeded into confocal disks for 12 h with a density of 105 cells per disk. Then, these disks were divided into seven groups for respective treatments: control group (untreated); US (US irradiation only); laser (laser irradiation only); NPs group (co-incubation with CRDs for 4 h); CRDs + laser group (laser irradiation followed by co-incubation of CRDs for 4 h); CRDs + US group (US irradiation followed by co-incubation of CRDs for 4 h); NP + US + laser group (co-incubation of CRDs for 4 h with subsequent US irradiation and laser irradiation). CRDs: 100 μg mL−1, laser: 1064 nm, 1.5 W cm−2, 5 min, US: 1.0 MHz, 2.0 W cm−2, 50% duty cycle, 2 min. Then, the cell culture medium was replaced with non-fluorescent DCFH-DA solution and incubated for 30 min. Finally, cells were washed gently with PBS and observed using CLSM on a FV1000 system (Olympus, Tokyo, Japan).For visualization of live/dead OSC-19 cells by CLSM, OSC-19 cells were stained by calcein-AM/PI (calcein-AM (40 μM) and PI (60 μM) in PBS). After 10 min of staining, cells were washed thrice with PBS prior to CLSM observation.
In vivo synergetic therapeutics of CRDAs against OSCC xenografts in mice
Procedures involving animal experiments were in accordance with the guidelines set by the Animal Care and Use Committee of Shanghai Ninth People's Hospital within the School of Medicine of Shanghai Jiao Tong University (Shanghai, China). Healthy male BALB/c mice (4 weeks) were purchased by Shanghai Xipuer-Beikai Experimental Animals (Shanghai, China). They were randomly divided into four groups (n = 4 in each group) followed by intravenous injection of CRDAs at elevated doses (0 mg kg−1 (control), 5 mg kg−1, 10 mg kg−1, and 20 mg kg−1) to assess in vivo biocompatibility. These mice were sacrificed after 15 days of feeding for whole blood panel tests, biochemical analyses and H&E-stained organ slices.For in vivo biodistribution analysis, 8 healthy male BALB/c nude mice were divided into 2 groups (n = 4) randomly. These mice were then injected with OSC-19 cells (106 cells per site, subcutaneous injection (s.c.)) in the right hind limb to establish OSCC-xenograft models. When the average volume of the tumors exceeded 150 mm3, mice were injected intravenously (i.v.) with CRDAs or CRDs (20 mg kg−1) in PBS for each group respectively. At predetermined time-points, mice were sacrificed at 4, 8, 12 and 24 h post-injection. Dissected organs and tumors were weighted and homogenized, followed by ICP-AES analysis to determine the concentration of Cu. The distribution of CRDAs/CRDs in organs and the tumor-targeting efficiency were calculated as the species concentration. For in vivo tumor-targeting analyses, in vivo photoacoustic imaging was performed on two male BALB/c nude mice bearing an OSCC xenograft on the right hind limb. When the volume of the tumors exceeded 50 mm3, mice were injected intravenously (i.v.) with CRDAs or CRDs (20 mg kg−1) in PBS for each group respectively. At predetermined time-points (0, 4, 8, 12 and 16 h post-injection), photoacoustic imaging of the nude mice was performed.
For in vivo PTT/SDT therapeutics, 42 OSCC-bearing mice were randomly divided into 7 groups (n = 6 in each group): control group: i.v. injection of saline; US group: i.v. injection of saline, followed by US irradiation; laser group: i.v. injection of saline, followed by laser irradiation; CRDAs group: i.v. injection of CRDAs; CRDAs + laser group: i.v. injection of CRDAs, followed by laser irradiation; CRDAs + US group: i.v. injection of CRDAs, followed by US irradiation; CRDAs + US + laser group: i.v. injection of CRDAs, followed by sequential US irradiation and laser irradiation. Treatment parameters: injection doses of CRDAs are 10 mg kg−1 (laser: 1064 nm, 1.5 W cm−2, 5 min; and US: 1.0 MHz, 2.0 W cm−2, 50% duty cycle, 2 min). Tumor tissues were collected from one mouse in each group at 24 h after treatment for ultrathin section preparation and histopathological staining using H&E, TUNEL, and antigen Ki-67 respectively. After respective treatment, mice were weighed and the tumor sizes of mice in different groups were recorded every other day during the 15 days evaluation period. The length (L) and width (W) of tumors were measured and the tumor volume (V) was calculated according to the following formula:
| V = L × W2/2 |
To evaluate the survival rate of the mice, the number of the living mice was recorded every other day. Following the guidelines of the animal experiments, when the tumor volume exceeded 1000 mm3, the mice were sacrificed. All the mice were sacrificed after the two-month evaluation period.
Statistical analyses
Data are the mean ± standard deviation. Significance of the data was obtained according to the Student's t-test. P < 0.05 was considered as significant.Results and discussion
Design, synthesis, and characterization of CRDAs
The design and synthesis of CRDAs are illustrated in Fig. 1a. DMSNs and Cu2−xS NPs were initially fabricated respectively. Featuring large mesopores of DMSNs, the Cu2−xS NPs and RB could be enabled to be sequentially encapsulated. Further decoration of the uPAR-targeting ligand AE105 leads to the fabrication of CRDAs, which guarantee the targeting biosafety and synergistic therapeutic performance of CRDAs for efficient tumor therapy both in vitro and in vivo. | ||
| Fig. 1 Schematic diagram of the synthesis of CRDAs in combating OSCC. (a) The synthetic route for Cu2−xS NPs and RB encapsulated, AE105 decorating CRDAs. (b) Therapeutic modalities of CRDAs for targeted photonic hyperthermal/sonodynamic therapy against OSCC. CRDAs can target and accumulate in the uPAR-overexpressed OSCC tumor sites to produce synergistic PTT/SDT therapeutics upon NIR laser and US irradiations. | ||
After intravenous injection, the CRDAs could accumulate into tumor tissue for nanotherapies (Fig. 1b). Typically, CRDAs could penetrate into tumor tissue via the enhanced permeability and retention (EPR) effect and the active targeting by uPAR-targeting ligand AE105. Then, the Cu2−xS NPs of CRDAs could generate the photonic hyperthermal effect for localized hyperthermia against OSCC under laser irradiation at 1064 nm, while the sonosensitive RB could induce the sonodynamic effect to produce singlet oxygen species under US irradiation. With these prominent effects, the OSCC cells/tissues are considered to be completely eradicated.
Transmission electron microscopy (TEM) and dynamic light scattering (DLS) were employed to characterize the morphology, size and composition of Cu2−xS NPs, DMSNs, and CRDAs respectively (Fig. 2). Typically, Cu2−xS NPs with a size of 2–5 nm were synthesized via the pyrolysis method.27 The synthetic Cu2−xS are hydrophobic NPs coated with an oleylamine layer (Fig. 2a and Fig. S1a, b, ESI†). DMSNs with a size of approximately 100 nm were characterized with dendritic mesoporous structures that were suitable for the encapsulation of Cu2−xS NPs and RB, as confirmed by the TEM (Fig. 2b and Fig. S1c, ESI†) and dark field images (Fig. S1d, ESI†). The morphology of CRDAs (Fig. 2c and Fig. S1e, f, ESI†) was similar to that of DMSNs according to the microscopy images, illustrating that the loading of Cu2−xS NPs did not alter the three-dimensional architecture of DMSNs. The loading mechanism of RB in CRDAs was based on the electrostatic attraction and pore encapsulations by DMSNs. Since RB is negatively charged, an electrostatic attraction between RB and host nanoparticles could facilitate the encapsulation of RB inside the pores of the Cu2−xS@DMSN-PEG-AE105 nanoparticles (Fig. S2, ESI†). The pore diameter of DMSNs was calculated to be 21.5 nm, determined via the N2 adsorption–desorption technique using Barrett–Joyner–Halenda methods (Fig. S3a, ESI†). Comparatively, the specific surface area (SSA) of DMSNs and Cu2−xS@DMSN NPs are determined using the N2 adsorption–desorption technique using Brunauer–Emmett–Teller methods (Fig. S3b, ESI†). DMSNs and Cu2−xS@DMSN NPs showed specific surface areas (SSAs) of as high as 574.3 m2 g−1 and 482.7 m2 g−1, respectively, illustrating that ultrasmall Cu2−xS NPs had been encapsulated inside the DMSNs nanoparticles. EDS (Fig. 2g) and corresponding EDS-mapping (Fig. 2h) exhibited uniformly distributed Si, O, Cu, and S elements, illustrating the dispersive loading of Cu2−xS NPs. The dynamic light scattering (DLS) data acquired in aqueous solutions demonstrate that the average hydrodynamic diameters (Dh) of DMSNs, and CRDAs were ca. 197 and 198 nm respectively (Fig. 2e). The XRD pattern of Cu2−xS NPs most imitates the standard diffraction pattern of Cu5S8 (Fig. S4, ESI†). To further quantify the loading amounts of Cu2−xS NPs and RB, inductively coupled plasma atomic emission spectrometry (ICP-AES) and UV-vis spectrometer were employed respectively. It has been determined that the encapsulation percentage of Cu2−xS NPs (within CRDAs) is approximately 38.8%, while that of RB sonosensitizer is ca. 8.2% (within CRDAs) (Fig. S5a, b and S6, ESI†). Although DMSNs can degrade faster than mesoporous silica of other configuration structures, it can maintain the configuration for several days in neutral and acidic environments.9 The release profile of RB from CRDAs under different pH conditions showed that RB can be released prior to the degradation of DMSNs (Fig. S7, ESI†). The successful encapsulation of both photonic active Cu2−xS NPs and sonodynamically active RB sonosensitizers into the DMSNs enables therapeutic purposes in the future.
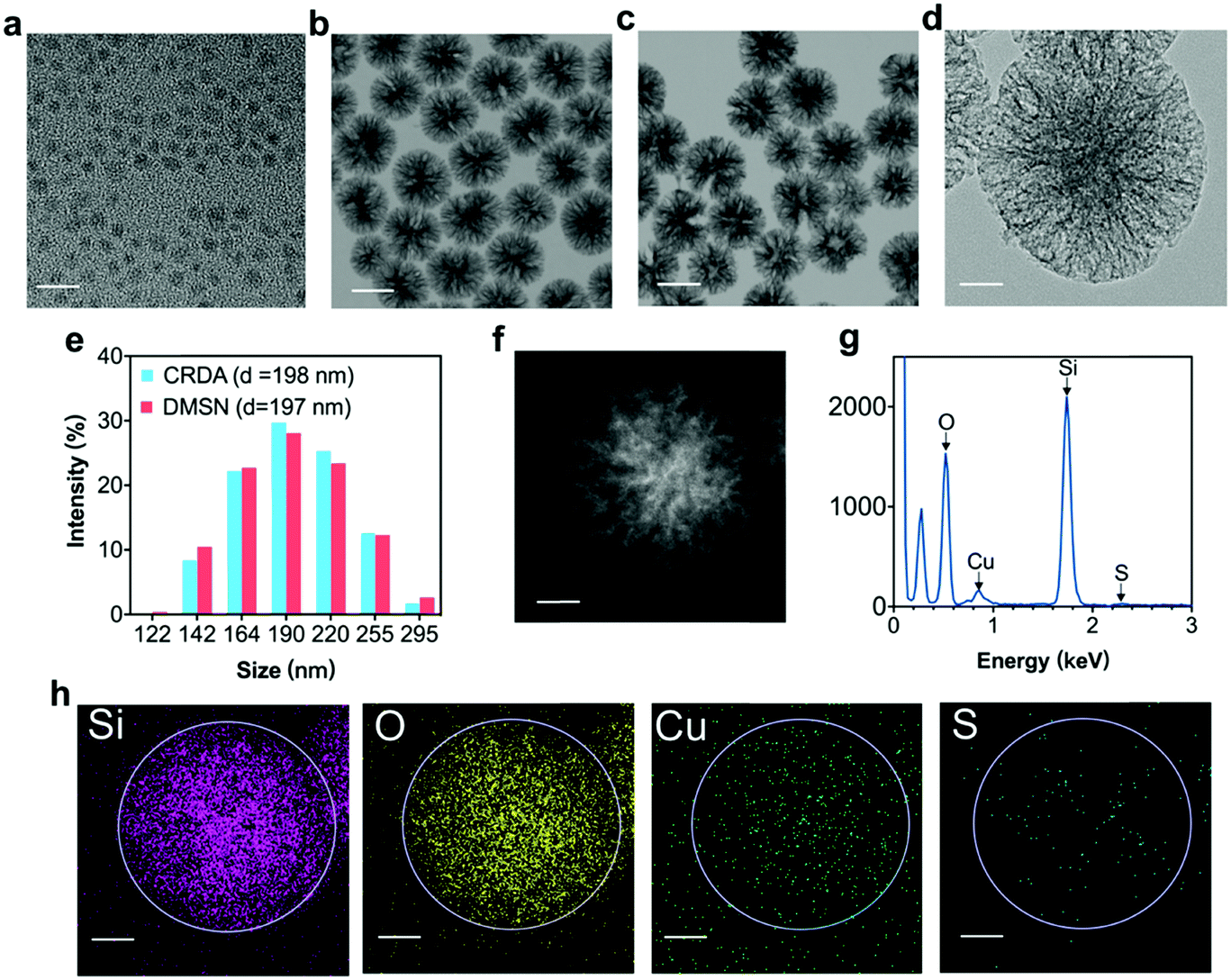 | ||
| Fig. 2 Structural and composition characterizations of Cu2−xS NPs, DMSNs, CRDs and CRDAs. (a) TEM image of Cu2−xS NPs, (b) DMSNs and (c) CRDAs. (d) TEM image of CRDAs. (e) Size distribution profile of CRDAs and DMSNs measured by DLS. (f) Dark-field image (DFI), (g) energy dispersive spectrometry (EDS) and (h) corresponding elemental mapping (EDS-mapping) of CRDAs. Scale bar: (a), (d), (f) and (h): 20 nm; (b) and (c): 100 nm. | ||
Sonodynamic and photonic hyperthermia performance of CRDs
As stimulated by the US and/or laser irradiations, Cu2−xS-RB@DMSN NPs (CRDs) can enable ROS generation (via SDT process) and localized temperature elevation respectively (Fig. 3a) since SDT is essentially a sonosensitizer-mediated radical-generating process for tumor destruction upon US irradiation. The singlet oxygen (1O2) generation by RB sonosensitizers45 is the dominant factor to evaluate the sonodynamic performance of CRDs. Combined with ESR results (Fig. S8, ESI†), it can be certified that CRDs as sonosensitizers can generate singlet oxygen (1O2) under US irradiation to induce the toxic effect and achieve the therapeutic function afterward. Therefore, 1,3-diphenylisobenzofuran (DPBF) was used exclusively to evaluate the generation level of 1O2. The decline of the characteristic absorptions of DPBF was recorded via UV-vis spectrometry at the wavelength of 410 nm after US irradiation when DPBF mixtured sequentially with RB (8 μg mL−1) (Fig. 3b) and CRDs ([RB] = 2 μg mL−1) (Fig. 3c) were subjected to the dark, indicating that CRDs with RB could efficiently induce ROS generation during the SDT process.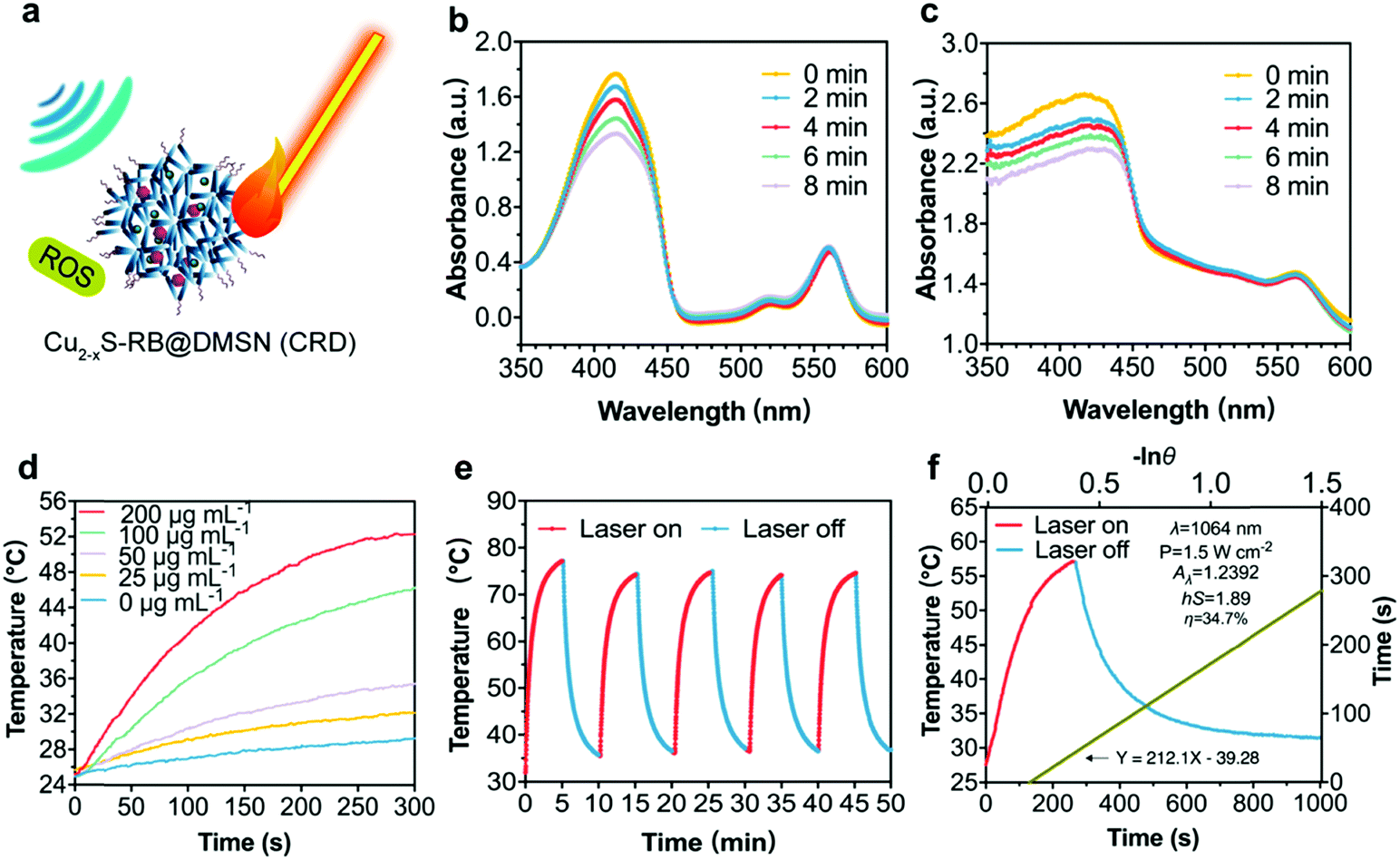 | ||
| Fig. 3 Investigation of the PTT/SDT performance of CRDs. (a) Schematic illustration of synergistic PTT/SDT of CRDs. UV-vis absorption spectra of DPBF mixed with (b) RB and (c) CRDs after US irradiation (1 MHz, 2.0 W cm−2, 50% duty cycle) for 0, 2, 4, 6, 8 min. (d) Localized temperature elevation curves of CRDs at a series of CRDs concentrations under the exposure to laser irradiation (1.5 W cm−2). (e) Photonic hyperthermia cycling profiles of CRDs during five sequential laser on and off cycles (1.5 W cm−2). (f) Specific period of heating and cooling curves of CRDs (250 μg mL−1) and the linear fitting of time constants derived from the cooling curve. | ||
Photonic active Cu2−xS NPs encapsulated inside DMSNs are capable of converting laser photon energy into heat. The hyperthermia performance of CRDs was assessed by the localized temperature elevation and photonic hyperthermia stability. Upon laser irradiation (1064 nm, 1.5 W cm−2, 5 min), the temperature of CRD aqueous suspension (at varied CRD concentrations: 25, 50, 100 and 200 μg mL−1) increased by 4 to 20 degrees in a dose-dependent manner, as compared with the control group (without CRDs) (Fig. 3d). In addition, the excellent photonic hyperthermia stability of CRDs was revealed by the negligible fluctuations of the maximum temperature between five sequential heating and cooling cycles (Fig. 3e). Finally, after detailed calculations, the photonic hyperthermal-conversion efficiency (η) of CRDs under 1064 nm laser was determined to be 34.7% according to the generally accepted calculation method18 (Fig. 3f).
In vitro synergetic performance of PTT/SDT enabled by CRDs against OSC-19 cells
According to the literature, the production of ROS and the generation of hyperthermia could kill cancer cells effectively (Fig. 4a). In order to evaluate the in vitro therapeutic performance against OSCC, we employed OSC-19 cells for CRDs co-incubation. The cytotoxicity of CRDs was initially assessed by Cell Counting Kit-8 (CCK-8). According to the results, no obvious cytotoxicity was observed even when the concentration of CRDs reached as high as 800 μg mL−1 after co-incubation with OSC-19 cells for 24 and 48 h, indicating their high biocompatibility (Fig. 4b). Next, the therapeutic effects of SDT and PTT against CRDs-treated OSC-19 cells were estimated, irradiated with US and/or laser respectively (Fig. 4c–e). The cell viability of OSC-19 cells decreased along with the elevated concentration of CRDs after US and/or laser irradiation. At a dosage of 400 μg mL−1 of CRDs, the viabilities were 77.1% (US group), 45.9% (laser group), and 29.0% (US + laser group) respectively. According to in vitro cellular experiment, the dose–viability fitting curves of the three groups of cell therapy can reflect the therapeutic effect (Fig. S9, ESI†). The decline in the curve of the US + laser group was significantly greater than that of the US group and the laser group. Therefore, combinative treatment of PTT/SDT against CRDs-treated OSC-19 cells induces the highest cytotoxicities, indicating that the synergetic SDT and PTT could improve the therapeutic efficacy over the respective monotherapy.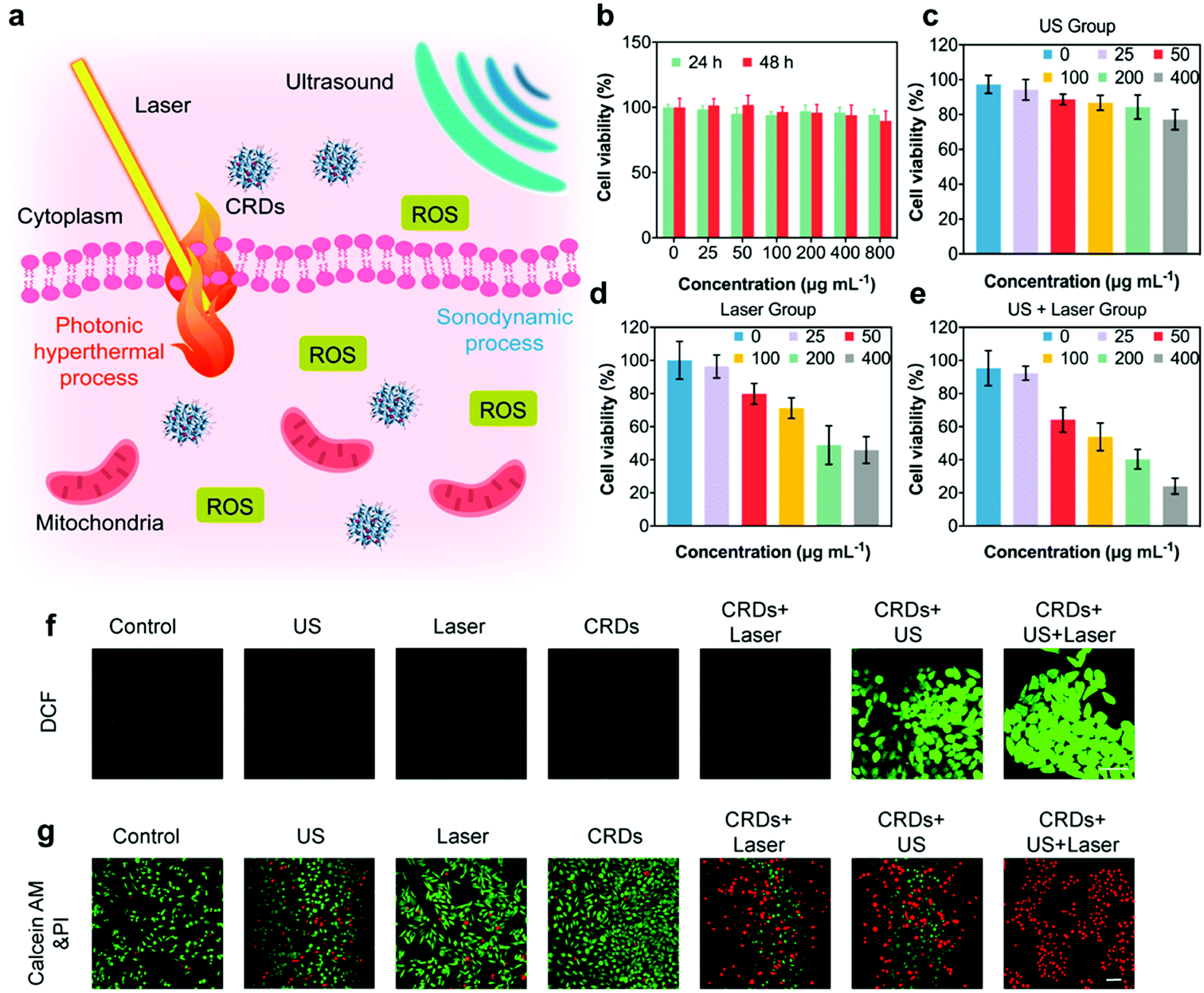 | ||
| Fig. 4 In vitro cellular experiment of CRDs against OSC-19 cells. (a) Schematic diagram of the in vitro CRDs-based therapeutics against OSC-19 cells. Confocal images of OSC-19 cells stained with (f) DCFH-DA and (g) calcium-AM&PI after respective treatments as indicated in the image. (b) Cell viabilities of OSC-19 cells after co-incubation with varied concentrations of CRDs for 24 and 48 h. (c–e) Cell viabilities of OSC-19 cells co-incubated with varied concentrations of CRDs under US-only (US group) (c), laser-only (laser group) (d) and combinative US and laser (US + laser group) (e) treatments. Scale bar of (f and g): 50 μm. | ||
To visualize the SDT performance and therapeutic outcome, a DCFH-DA probe was used to indicate the cellular generation of ROS upon sonodynamic treatment, which is attributed to the transformation of non-fluorescent DCFH-DA to green fluorescent 2,7-dichlorofluorescein (DCF) in the presence of ROS.18 After co-incubating the cells with CRDs, DCFH-DA was supplemented to stain the cellular ROS for confocal observations. It has been observed that the cells in CRDs + US, CRDs + US + laser groups displayed the strongest green fluorescence signals, revealing the prominent generation of ROS (Fig. 4f and Fig. S10, ESI†). Comparatively, the cancer cells in other treatment groups exhibited weaker green fluorescence signals, implicating the inefficient ROS-generating capacity. To directly visualize the anti-tumor effects, calcein-AM and PI cell stains were supplemented after respective treatments (Fig. 4g and Fig. S11, ESI†). As indicated, a major proportion of dead cells (stained in red fluorescence) was observed in the synergetic CRDs + US + laser treatment group, as compared to that of the other treatment groups. Collectively, these experiments solidly validated the prominent synergetic therapeutic performance enabled by the in vitro PTT and SDT as enabled by the constructed CRDs.
In vivo synergetic PTT/SDT cancer therapeutics enabled by CRDAs
An in vivo biocompatibility assay was conducted to guarantee further in vivo therapeutics of CRDAs. Sixteen healthy BALB/c mice were randomly divided into four groups (n = 4 in each group) followed by intravenous injection of CRDAs at elevated doses (0 mg kg−1 (as the control group), 5 mg kg−1, 10 mg kg−1, and 20 mg kg−1). These mice were sacrificed at the end of the 15 day-evaluation period for harvesting the whole blood and major organs (i.e., heart, liver, spleen, lungs, and kidneys). As compared to the control group, the bodyweight profiles of mice between each group show no obvious variations during the therapeutics (Fig. 5a), and no significant variations in complete blood panel tests and biochemical analyses could be indicated (Fig. 5b). In addition, no substantial tissue damage or inflammatory lesions were discovered from hematoxylin and eosin (H&E)-staining ultrathin sections of different major organs (Fig. 5c), suggesting that the injection of CRDAs (with a dose of as high as 20 mg kg−1) featured satisfactory in vivo biocompatibility. To identify the in vivo targeting effects of CRDAs, the biodistribution of CRDAs was evaluated by analyzing the concentration of Cu species (Fig. 5d and e) in the main organs and tumors at 4, 8, 12, 24 h post-injection. The in vivo biodistribution evaluation suggests that CRDAs could effectively accumulate into tumor sites with a distribution dosage percentage of 10.99% (Fig. 5d) 4 h post-injection, which was significantly higher than that of CRDs without AE105 targeting design (3.03%, Fig. 5e). Compared to nontargeting CRDs, the targeting CRDAs can accumulate higher and faster at the tumor site, while causing less pathological impact against other organs. In addition, the ICP-AES results showed that CRDAs and CRDs accumulated inside the tumor sites in 4 h. By imaging the in vivo tumor sites via photoacoustics,27 targeting accumulation of CRDAs could also be indicated (Fig. S12a–c, ESI†). The specifically enhanced accumulations of the nanoplatforms could further benefit the prominent therapeutics with mitigated cytotoxicity.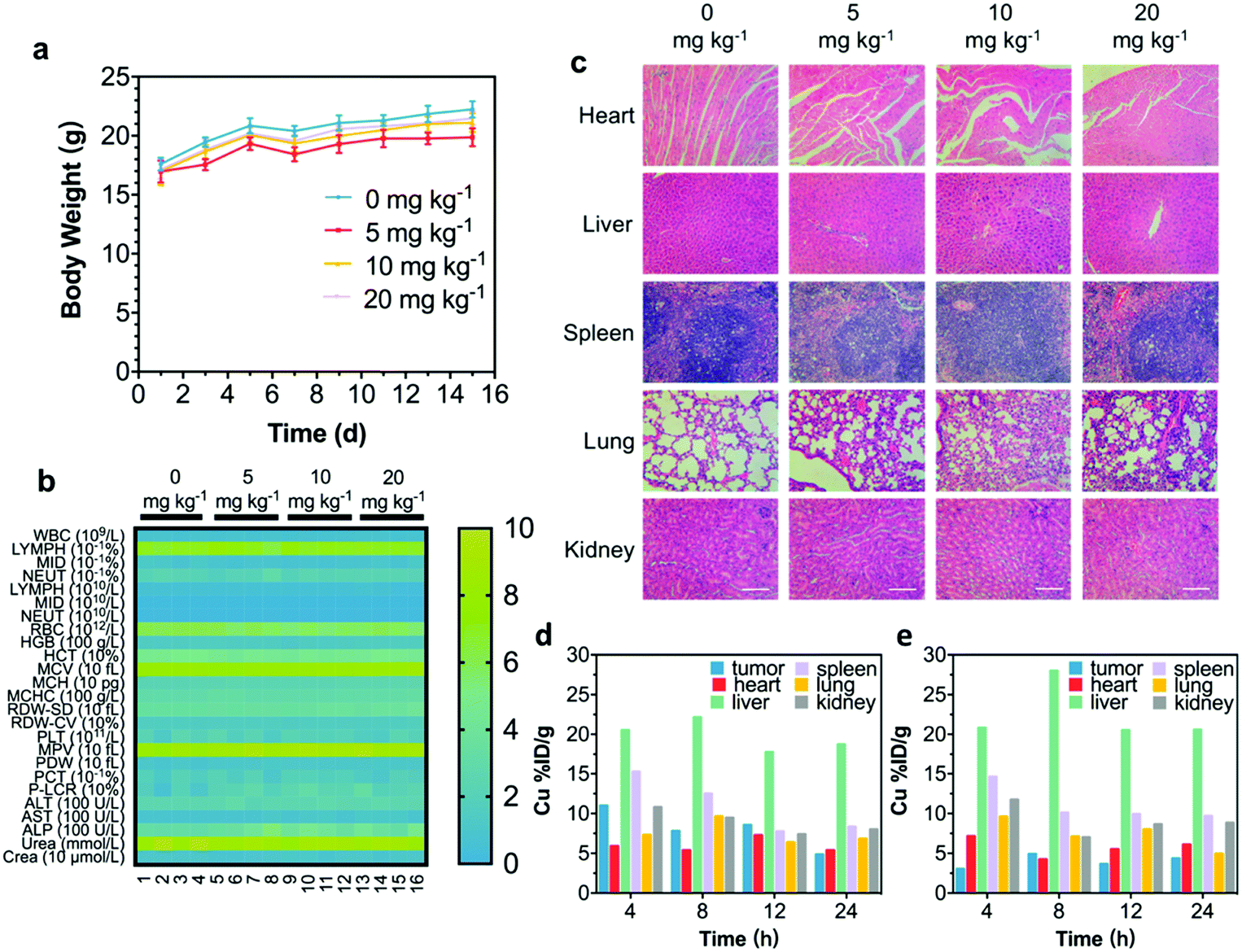 | ||
| Fig. 5 In vivo biocompatibility assay and biodistribution of CRDAs. (a) Body weight of mice during 15 days in vivo biocompatibility evaluation experiments. (b) Whole blood panel tests and biochemical analyses. (c) H&E-stained organ slices from mice after in vivo biocompatibility evaluation experiments. (d and e) In vivo biodistribution profiles of nontargeted CRDs (d) and targeted CRDAs (e) at different time-points post-injection. Scale bar of (c): 100 μm. | ||
The in vitro synergetic PTT and SDT by CRDs and their desirable in vivo biocompatibility encourage further systematic in vivo therapeutics of CRDAs against OSC-19 cells. During the investigations, forty-two BALB/c nude mice were randomly divided into seven groups (n = 6 in each group) prior to the tumor xenograft establishment. When the volume of the tumor xenografts reached 50 mm3, these nude mice were injected with saline (for mice in control, US, laser groups) and CRDAs (for mice in CRDAs, CRDAs + US, CRDAs + laser, CRDAs + US + laser groups) respectively. CRDA-treated mice were further exposed to external stimuli irradiation including US and/or laser correspondingly (Fig. 6a). During NIR-laser irradiation (for CRDAs + US + laser group), the localized temperature of tumor sites increased from 24 °C to 54 °C while for mice in the laser group, merely 5 degrees of temperature increase was recorded (Fig. 6b). During the 15 day-therapeutics, the body weight of mice and the dimension of the tumors were recorded every other day. The body weight profiles of mice between each group show no obvious variations during the therapeutics, implying the biocompatibility of the therapeutic modalities (Fig. 6c). The tumor xenografts of mice in control, US, laser and CRDAs groups grew extensively within 15 days, while mice receiving synergistic PTT/SDT treatments enabled by CRDAs in CRDAs + US + laser group exhibited intense therapeutic efficacy with an inhibition rate (IR) of 103.4% (meaning destruction rather than inhibition), significantly higher than that of the CRDAs + US group (49.79%) and CRDAs + laser group (61.89%) (Fig. 6d), verifying the effectiveness of the synergetic therapeutic consequence of PTT/SDT enabled by the targeting CRDAs.
 | ||
| Fig. 6 In vivo antitumor experiments of CRDAs against OSCC tumor xenografts. (a) Schematic schedule diagram of the in vivo synergetic PTT/SDT therapeutics. (b) Infrared thermal images of mice during PTT treatment along the irradiation time. (c) Body weight profile and (d) tumor volume profile of mice in each group during the in vivo therapeutic treatment period. (e) Histological microscopic images of tumor sections stained with H&E, TUNEL and antigen Ki-67 of mice. (f) Digital photographs of tumors of mice in each group collected at the end of the in vivo therapeutic experiment. Scale bar of (e): 100 μm. | ||
At 24 h post-treatments, one tumor-bearing mouse from each group was euthanized and the major organs and tumors were dissected for detailed pathological analyses by H&E, TdT-mediated dUTP Nick-End Labeling (TUNEL) and antigen Ki-67 immunostaining respectively (Fig. 6e). As compared to the control, US, laser and CRDAs groups, mice in the monotherapeutic CRDAs + US and CRDAs + laser groups exhibited reduction of the tumor cells, enlarged proportions of the apoptotic area (in H&E and TUNEL staining microscopy images), and mitigated expressions of the cell proliferation (in antigen Ki-67 staining microscopy images). Comparatively, the synergetic PTT/SDT therapy was conceived to deliver much prominent consequence. The xenografts of mice in the CRDAs + US + laser group were almost eradicated, as revealed by, H&E, TUNEL, antigen Ki-67 staining images and the digital photographs (Fig. 6f). The survival rate suggested that photonic hyperthermal and sonodynamic targeting therapy can extend the living time of mice (Fig. S13, ESI†). These results collectively demonstrate the prominent therapeutic effects of the synergetic PTT/SDT therapeutics enabled by CRDAs against OSCC with satisfactory biocompatibility.
Conclusions
In summary, we have constructed an intriguing uPAR-targeting ligand AE105-decorated DMSNs-based delivery system co-encapsulated with photonic active Cu2−xS nanoagents and sonosensitizer RB (abbreviated as CRDAs) for high-performance OSCC-targeting and synergetic PTT/SDT nanotherapeutics. Based on the prominent hyperthermia and sonodynamic performance of the integrated Cu2−xS NPs and RB respectively, the rationally engineered CRDAs could efficiently enable satisfactory tumor destruction against the OSCC both in vitro and in vivo. Featuring the targeting performance of AE105, the enhanced accumulation of CRDAs into tumor xenografts was achieved on an in vivo OSCC tumor xenograft. Together with the high biocompatibility of the designed CRDAs, a prominent tumor inhibition rate of 103.4% was realized. The present synergetic strategy has illustrated a satisfactory therapeutic modality to treat oral squamous cell carcinoma with both effectiveness and biocompatibility.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We greatly acknowledge the financial support from the National Natural Science Foundation of China (Grant No. 81971618, 51672303, and 51722211), National Key R&D Program of China (Grant No. 2016YFA0203700), Program of Shanghai Subject Chief Scientist (Grant No. 18XD1404300) and China Postdoctoral Science Foundation (Grant No. 2020M671243).Notes and references
- S. Monro, K. L. Colon, H. Yin, J. Roque, 3rd, P. Konda, S. Gujar, R. P. Thummel, L. Lilge, C. G. Cameron and S. A. McFarland, Chem. Rev., 2019, 119, 797–828 CrossRef CAS.
- H. Wu, L. Song, L. Chen, W. Zhang, Y. Chen, F. Zang, H. Chen, M. Ma, N. Gu and Y. Zhang, Acta Biomater., 2018, 74, 302–311 CrossRef CAS.
- W. Yue, L. Chen, L. Yu, B. Zhou, H. Yin, W. Ren, C. Liu, L. Guo, Y. Zhang, L. Sun, K. Zhang, H. Xu and Y. Chen, Nat. Commun., 2019, 10, 2025 CrossRef.
- M. Zhao, Y. Xu, M. Xie, L. Zou, Z. Wang, S. Liu and Q. Zhao, Adv. Healthcare Mater., 2018, 7, e1800606 CrossRef.
- Z. Zhao, K. Xu, C. Fu, H. Liu, M. Lei, J. Bao, A. Fu, Y. Yu and W. Zhang, Biomaterials, 2019, 219, 119379 CrossRef CAS.
- Z. Jin, K. Wu, J. Hou, K. Yu, Y. Shen and S. Guo, Biomaterials, 2018, 153, 49–58 CrossRef CAS.
- F. Gao, G. He, H. Yin, J. Chen, Y. Liu, C. Lan, S. Zhang and B. Yang, Nanoscale, 2019, 11, 2374–2384 RSC.
- J. Shi, Z. Chen, B. Wang, L. Wang, T. Lu and Z. Zhang, ACS Appl. Mater. Interfaces, 2015, 7, 28554–28565 CrossRef CAS.
- M. Huo, L. Wang, Y. Chen and J. Shi, Nat. Commun., 2017, 8, 357 CrossRef.
- T. Suteewong, H. Sai, R. Hovden, D. Muller, M. S. Bradbury, S. M. Gruner and U. Wiesner, Science, 2013, 340, 337–341 CrossRef CAS.
- F. Tang, L. Li and D. Chen, Adv. Mater., 2012, 24, 1504–1534 CrossRef CAS.
- Y. Wang, Q. Zhao, N. Han, L. Bai, J. Li, J. Liu, E. Che, L. Hu, Q. Zhang, T. Jiang and S. Wang, Nanomedicine, 2015, 11, 313–327 CrossRef CAS.
- C. M. Carnielli, C. C. S. Macedo, T. De Rossi, D. C. Granato, C. Rivera, R. R. Domingues, B. A. Pauletti, S. Yokoo, H. Heberle, A. F. Busso-Lopes, N. K. Cervigne, I. Sawazaki-Calone, G. V. Meirelles, F. A. Marchi, G. P. Telles, R. Minghim, A. C. P. Ribeiro, T. B. Brandao, G. de Castro, Jr., W. A. Gonzalez-Arriagada, A. Gomes, F. Penteado, A. R. Santos-Silva, M. A. Lopes, P. C. Rodrigues, E. Sundquist, T. Salo, S. D. da Silva, M. A. Alaoui-Jamali, E. Graner, J. W. Fox, R. D. Coletta and A. F. Paes Leme, Nat. Commun., 2018, 9, 3598 CrossRef.
- R. L. Siegel, K. D. Miller and A. Jemal, Ca-Cancer J. Clin., 2016, 66, 7–30 CrossRef.
- D. A. Mahvi, R. Liu, M. W. Grinstaff, Y. L. Colson and C. P. Raut, Ca-Cancer J. Clin., 2018, 68, 488–505 CrossRef.
- S. R. Gvetadze, P. Xiong, M. Lv, J. Li, J. Hu, K. D. Ilkaev, X. Yang and J. Sun, Dentomaxillofac. Radiol., 2017, 46, 20160345 CrossRef.
- J. A. Ochoa-Alvarez, H. Krishnan, J. G. Pastorino, E. Nevel, D. Kephart, J. J. Lee, E. P. Retzbach, Y. Shen, M. Fatahzadeh, S. Baredes, E. Kalyoussef, M. Honma, M. E. Adelson, M. K. Kaneko, Y. Kato, M. A. Young, L. Deluca-Rapone, A. J. Shienbaum, K. Yin, L. D. Jensen and G. S. Goldberg, Oncotarget, 2015, 6, 9045–9060 CrossRef.
- C. Dai, S. Zhang, Z. Liu, R. Wu and Y. Chen, ACS Nano, 2017, 11, 9467–9480 CrossRef CAS.
- H. Lin, W. Qiu, J. Liu, L. Yu, S. Gao, H. Yao, Y. Chen and J. Shi, Adv. Mater., 2019, 31, e1903013 CrossRef.
- X. Wang, Z. Geng, H. Cong, Y. Shen and B. Yu, ChemBioChem, 2019, 20, 1628–1636 CrossRef CAS.
- Y. Cao, S. Li, C. Chen, D. Wang, T. Wu, H. Dong and X. Zhang, Biomaterials, 2018, 158, 23–33 CrossRef CAS.
- R. He, Y. C. Wang, X. Wang, Z. Wang, G. Liu, W. Zhou, L. Wen, Q. Li, X. Wang, X. Chen, J. Zeng and J. G. Hou, Nat. Commun., 2014, 5, 4327 CrossRef CAS.
- W. Xu, J. Qian, G. Hou, A. Suo, Y. Wang, J. Wang, T. Sun, M. Yang, X. Wan and Y. Yao, ACS Appl. Mater. Interfaces, 2017, 9, 36533–36547 CrossRef CAS.
- W. Gao, Y. Sun, M. Cai, Y. Zhao, W. Cao, Z. Liu, G. Cui and B. Tang, Nat. Commun., 2018, 9, 231 CrossRef.
- Q. L. Li, Y. Sun, L. Ren, X. Wang, C. Wang, L. Li, Y. W. Yang, X. Yu and J. Yu, ACS Appl. Mater. Interfaces, 2018, 10, 29314–29324 CrossRef CAS.
- R. Ge, C. Liu, X. Zhang, W. Wang, B. Li, J. Liu, Y. Liu, H. Sun, D. Zhang, Y. Hou, H. Zhang and B. Yang, ACS Appl. Mater. Interfaces, 2018, 10, 20342–20355 CrossRef CAS.
- J. Mou, P. Li, C. Liu, H. Xu, L. Song, J. Wang, K. Zhang, Y. Chen, J. Shi and H. Chen, Small, 2015, 11, 2275–2283 CrossRef CAS.
- Z. Zhou, Y. Yan, K. Hu, Y. Zou, Y. Li, R. Ma, Q. Zhang and Y. Cheng, Biomaterials, 2017, 141, 116–124 CrossRef CAS.
- X. Qian, Y. Zheng and Y. Chen, Adv. Mater., 2016, 28, 8097–8129 CrossRef CAS.
- J. Nam, S. Son, L. J. Ochyl, R. Kuai, A. Schwendeman and J. J. Moon, Nat. Commun., 2018, 9, 1074 CrossRef.
- Y. Wang, W. Zhang, P. Sun, Y. Cai, W. Xu, Q. Fan, Q. Hu and W. Han, Theranostics, 2019, 9, 391–404 CrossRef.
- S. D. da Silva, M. Hier, A. Mlynarek, L. P. Kowalski and M. A. Alaoui-Jamali, Front. Pharmacol., 2012, 3, 149 Search PubMed.
- L. A. Garraway and P. A. Janne, Cancer Discovery, 2012, 2, 214–226 CrossRef CAS.
- B. Yang, Y. Chen and J. Shi, Chem. Rev., 2019, 119, 4881–4985 CrossRef CAS.
- C. McEwan, C. Fowley, N. Nomikou, B. McCaughan, A. P. McHale and J. F. Callan, Langmuir, 2014, 30, 14926–14930 CrossRef CAS.
- C. McEwan, J. Owen, E. Stride, C. Fowley, H. Nesbitt, D. Cochrane, C. C. Coussios, M. Borden, N. Nomikou, A. P. McHale and J. F. Callan, J. Controlled Release, 2015, 203, 51–56 CrossRef CAS.
- C. R. Leemans, B. J. Braakhuis and R. H. Brakenhoff, Nat. Rev. Cancer, 2011, 11, 9–22 CrossRef CAS.
- F. Blasi and P. Carmeliet, Nat. Rev. Mol. Cell Biol., 2002, 3, 932–943 CrossRef CAS.
- M. A. Pavon, I. Arroyo-Solera, M. V. Cespedes, I. Casanova, X. Leon and R. Mangues, Oncotarget, 2016, 7, 57351–57366 CrossRef.
- H. W. Smith and C. J. Marshall, Nat. Rev. Mol. Cell Biol., 2010, 11, 23–36 CrossRef CAS.
- Y. W. Chen, T. Y. Liu, P. H. Chang, P. H. Hsu, H. L. Liu, H. C. Lin and S. Y. Chen, Nanoscale, 2016, 8, 12648–12657 RSC.
- A. Christensen, K. Juhl, M. Persson, B. W. Charabi, J. Mortensen, K. Kiss, G. Lelkaitis, N. Rubek, C. von Buchwald and A. Kjaer, Oncotarget, 2017, 8, 15407–15419 CrossRef.
- M. C. Kriegbaum, M. Persson, L. Haldager, W. Alpizar-Alpizar, B. Jacobsen, H. Gardsvoll, A. Kjaer and M. Ploug, Curr. Drug Targets, 2011, 12, 1711–1728 CrossRef CAS.
- M. Persson, M. Hosseini, J. Madsen, T. J. Jorgensen, K. J. Jensen, A. Kjaer and M. Ploug, Theranostics, 2013, 3, 618–632 CrossRef.
- B. McCaughan, C. Rouanet, C. Fowley, N. Nomikou, A. P. McHale, P. A. McCarron and J. F. Callan, Bioorg. Med. Chem. Lett., 2011, 21, 5750–5752 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Supplementary figures contain STEM image and dark-field images of Cu2−xS NPs, STEM image and SEM image of DMSNs and CRDAs, XRD pattern of Cu2−xS NPs, UV-vis absorption spectra of CRDs, CRDAs and Cu2−xS@DMSN-AE105 NPs, UV-vis absorption spectra of varied RB doses and the corresponding dose–absorption standard curve. See DOI: 10.1039/d0tb01089h |
| This journal is © The Royal Society of Chemistry 2020 |