
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Self-recovering dual cross-linked hydrogels based on bioorthogonal click chemistry and ionic interactions†
Henan
Zhan
a,
Shanshan
Jiang
a,
Anika M.
Jonker
a,
Imke A. B.
Pijpers
b and
Dennis W. P. M.
Löwik
 *a
*a
aRadboud University Nijmegen, Institute for Molecules and Materials, Bio-organic Chemistry, Heyendaalseweg 135, 6525 AJ Nijmegen, The Netherlands. E-mail: dennis.lowik@ru.nl
bEindhoven University of Technology, Department of Biomedical Engineering, Bio-organic Chemistry, De Groene Loper 3, 5612 AE Eindhoven, The Netherlands
First published on 9th June 2020
Abstract
The biocompatible, injectable and high water-swollen nature of hydrogels makes them a popular candidate to imitate the extracellular matrix (ECM) for tissue engineering both in vitro and in vivo. However, commonly used covalently cross-linked hydrogels, despite their stability and tunability, are elastic and deteriorate as bulk material degrades which would impair proper cell function. To improve these deficiencies, here, we present a self-recovering cross-linked hydrogel formed instantaneously with functionalized poly(ethylene glycol) as a basis. We combine covalent cross-links introduced via a strain-promoted azide–alkyne cycloaddition (SPAAC) click reaction and non-covalent links between phosphonate groups and calcium ions. By adjusting the ratios of non-covalent and covalent cross-links, we synthesized these dual cross-linked (DC) hydrogels that displayed storage moduli below ∼2000 Pa and relaxation times from seconds to minutes. The gels recovered to 41–96% of their initial mechanical properties after two subsequent strain failures. Cryo-scanning electron microscopy revealed that DC hydrogels containing approximately equal amounts of covalent and non-covalent cross-links displayed phase separation. Finally, we functionalized the DC hydrogels by incorporating an integrin binding motif, RGDS, to provide a biocompatible environment for human mesenchymal stem cells (HMSCs) by facilitating adhesion inside the gel network. Inside these DC gels HSMCs displayed a viability up to 73% after five days of cell culture.
Introduction
In the last several decades, synthetic biomaterial research has for a large part been carried out to realize the unmet challenges in tissue engineering that call for highly tailorable three-dimensional (3D) extracellular microenvironments to direct cellular behavior and function.1–3 Due to their biocompatibility, injectability and biomimetic properties, hydrogels – highly water – swollen 3D polymeric networks – are considered to be attractive candidates for native tissue mimicry.4 Among the tissues in the human body, the extracellular matrix (ECM) plays an essential role to support and direct cell behavior by providing a dynamic environment of various fibrous proteins, proteoglycans, glycoproteins and soluble macromolecules (growth factors and cytokines) secreted by adjacent cells.3,5 The fact that permanently cross-linked synthetic hydrogels are stable and easily tuned in both biophysical and biochemical respect, they form a particularly suitable basis for mimicking the natural ECM.6 Dynamics are introduced via degradation mechanisms such as passive hydrolytic degradation7–9 or cell-mediated enzymatic degradation10,11 to allow cell activities like spreading, proliferation and migration. And even though synthetic hydrogels are now widely used for 3D cell culture, they are still unsatisfactory in certain respects. A considerable flaw is that the mechanical properties deteriorate as the bulk material degrades and these evolving mechanical properties have their effect on cell behavior.12,13 Furthermore, unlike the viscoelastic natural ECM, the covalent cross-links in many synthetic gels decrease the flexibility of the polymer chains by restricting their movements to result in elastic materials which prevent cells cultured inside from performing functions like undergoing morphology and fate transformations.14–18To overcome the structural failure caused by bulk material degradation and to recover the dynamic viscoelastic properties of the ECM, adaptable hydrogels also known as self-recovering hydrogels with reversible cross-links have been devised. These reversible linkages can be re-formed after a fracture to generate, in this way, a transient polymer network. Various mechanisms to introduce such adaptable bonds, including dynamic covalent cross-linkers and non-covalent ionic interactions, have been widely developed in applications of such self-recovering hydrogels. Despite of their stability towards a variety of conditions (e.g. temperature induced sol–gel transition,19 pH20 and UV light21), these reversible covalent connections still have many limitations such as suffering from incompatible reaction conditions, slow kinetics of bond cleavage and formation, and requirement of catalysis to achieve rapid equilibrium.6,12,15 In contrast, hydrogels based on ionic cross-links can self-recover via electrostatic interactions between oppositely charged ions under mild conditions,22 and have been frequently used for cell encapsulation. In this respect alginate hydrogels are archetypal as a well-known basis for ionically cross-linked hydrogels, that is able to form reversible linkages through the exchange with cations, typically calcium ions.23 Considering the rather poor stability of simple ionically cross-linked hydrogels, recent studies tend to lean more toward blending ionic cross-links in a covalently cross-linked network. For instance, Sun et al. designed an extremely stretchable and tough hydrogel by mixing alginate and poly-acrylamide (PAAm).24 They first loaded and unloaded a certain amount of stress on their hydrogels, then sealed the gels and stored them at temperatures ranging from 20–80 °C. They found their gels healed better from the internal damage (74%) before re-loading after one-day storage at 80 °C. Another study, reported by Ghanian et al.,25 introduced a self-recovering hydrogel constituted from calcium-binding alginate and four-arm poly(ethylene glycol)–maleimide used to take part in a Diels–Alder click reaction. By changing the degree of the substitution they were able to prepare hydrogel systems with a tunable sol–gel transition time ranging from 45 to 140 minutes. Nevertheless, to meet the requirements for cell encapsulation in designing hydrogels,12 some work still needs to be done to create cyto-compatible gels that possess fast gelation kinetics, rapid self-recovery under physiological conditions and form in situ. Although the bio-compatibility of these hydrogels prepared from natural components are better, when compared to synthetic hydrogels, the possibility to tune their (mechanical) properties is still dissatisfactory.
Among the materials used to build synthetic non-covalent hydrogel networks, bisphosphonates based components have been employed as a substitute for materials involving bone, tooth and calcium metabolism.26 Currently, they are increasingly employed in the clinic to treat a variety of conditions such as skeletal disorders, osteoporosis and Paget's disease by inhibiting bone resorption by chelating calcium ions at the bone hydroxyapatite surface.27,28 Due to this strong ability to bind calcium, they have recently been used to create hydrogels with flexible and transient cross-links.29,30 Lopez-Perez et al. grafted bisphosphonate moieties on 8-armed poly(ethylene glycol) (PEG) polymers binding calcium ions to afford hydrogels that can form instantaneously.29 They showed these gels could recover between 70 to 100% of their original storage and loss modulus after rupture. Their study, however, on bisphosphonate–calcium bonded networks focused on polymers of large molecular weight at relatively high concentrations. This may be less suitable when working towards tissue regeneration and for cell culture applications. To further develop this type of fast forming, biocompatible and self-recovering hydrogel system with well-defined mechanical properties, here in this study, we present a dual cross-linked hydrogel network containing non-covalent ionic bonds (bisphosphonates/Ca2+) as physical cross-links combined with chemical cross-links introduced via covalent click reactions – i.e. a strain-promoted azide–alkyne cycloaddition (SPAAC). The ionic bonds support the covalent cross-links by effective dissipation of energy under loading through unzipping,24 and return rapidly after the load is released. At the same time, the covalent chemical links provide sufficient mechanical property to resist high strain. We evaluated the mechanical properties and microscopic morphology of a variety of dual cross-linked hydrogel networks by changing the ratio between the chemically and physically cross-linked parts. Furthermore, to study their potential for mimicking the extracellular matrix, we functionalized the hydrogels with a cell adhesion motif in the form of an RGDS peptide sequence to stimulate cell adhesion and examined their ability to support stem cell growth.
Experimental
General information
4-Armed poly(ethylene glycol)-NH2·HCl (10 kDa) (star-PEG-NH2) and 4-armed poly(ethylene glycol) succinimidyl glutaramide (star-PEG-SGA) were obtained from Jenkem Thechnology (USA). Dibenzocyclooctyne-N-hydroxysuccinimidyl ester was purchased from Lumiprobe GmbH (Germany). Alendronic acid ((4-amino-1-hydroxybutane-1,1-diyl)di-phosphonic acid) was purchased from Fluorochem Ltd (UK). 2-Chloro-trityl resin was purchased from Bachem (Bubendorf, Switzerland). All amino acids were purchased from Nova-biochem (EMD Chemicals, Gibbstown, USA) or Bachem. 1H-NMR spectra were measured on a 400 MHz Varian Inova NMR and 31P-NMR spectra were measured on a 400 MHz Bruker Avance NMR. DCl3 was used as solvents and purchased from Sigma-Aldrich. Mass spectra were recorded on a Bruker Biflex MALDI-TOF (Bruker Daltronik, Bremen, Germany). α-Cyano-4-hydroxycinnamic acid was used as the matrix. LCMS was performed on a Thermo Finnigan LCQ-Fleet ESI-ion trap (Thermo Fischer, Breda, The Netherlands) equipped with a Phenomenex C18 column, 50 × 2.0 mm, particle size 3 μM (Phenomenex, Utrecht, The Netherlands). An acetonitrile/water gradient containing 0.1% formic acid was used for elution (5–100%, 50 min, flow 0.2 mL min−1). Preparative HPLC was performed on a Shimadzu LC-20A Prominence system equipped with a Gemini-NX C18 column, 110A, 150 × 21.2 mm, particle size 10 μm (Phenomenex). Elution of the peptides was achieved with water/acetonitrile gradient containing 0.1% trifluoroacetic acid (5–100% in 5–55 min, flow 6 mL min−1). Samples were dissolved in acetic acid.Synthesis of star-PEG-alendronic acid
Alendronic acid (8 eq.) was dissolved in 20 mL PBS solution (2.67 mM KCl, 127.93 mM NaCl, 15 mM KH2PO4 and 85 mM Na2HPO4 in MilliQ, pH = 7.4), after which the pH was brought back to 7.4 with 1M NaOH. Subsequently star-PEG-SGA (1 eq.) was added to the solution and stirred at r.t. for 8 h. The solution was transferred to a dialysis tube (MW cutoff: 3.5 kDa) and dialyzed against water for one day. The polymer was obtained after lyophilisation in a 76% yielding. MALDI-TOF: Fig. S10 (ESI†). 31P NMR (400 MHz, CDCl3): Fig. S11 (ESI†).Synthesis of star-PEG-DBCO
Star-PEG-NH2 (1 eq.), dibenzocyclooctyne-N-hydroxysuccinimidyl ester (6 eq.), and triethylamine (12 eq.) were dissolved in dry DCM (10 mL), and stirred o/n under nitrogen atmosphere. The reaction mixture was consecutively washed with KHSO4 and brine, and dried over MgSO4. Subsequently, volatiles were removed under reduced pressure, and the crude mixture was purified using column chromatography (2–30% MeOH in DCM). Lyophilization from 1,4-dioxane yielded the DBCO-functionalized polymer in a 56% yield. Rf = 0.26 (10% MeOH in DCM). MALDI-TOF (α-cyano-4-hydroxycinnamic acid): Fig. S12 (ESI†). 1H NMR (400 MHz, CDCl3): Fig. S13 (ESI†).Peptide synthesis
The azido-RGDS peptide was synthesized on 1.0 g 2-chloro-trityl resin using standard Fmoc solid-phase peptide synthesis (SPPS) protocols. In short the resin was swollen in dry dichloromethane (DCM) for 20 min prior to use. The resin was functionalized with the first amino acid in 1 h with 2.0 eq. of the required Fmoc-Gly-OH and 2.2 eq. of N,N-diisopropylethylamine (DiPEA). This procedure was repeated after which capping took place with 2.2 eq. DiPEA in 10 mL 10% (v/v) methanol in DCM. Deprotection of the Fmoc groups was carried out by treatment with piperidine in N,N-dimethylformamide (DMF) (20%, v/v, three times 6 min). Subsequent couplings were performed with 3.0 eq. of the required amino acid, 3.3 eq. of N,N′-diisopropylcarbodiimide (DIPCDI) and 3.6 eq. of N-hydroxybenzotriazole (HOBt) in DMF. After each coupling and deprotection step, a Kaiser test was performed to ensure completion of the reaction.31 In the final step, 3.0 eq. of azido acetic acid was coupled using DIPCDI (3.3 eq.) and HOBt (3.6 eq.). The peptide was cleaved from the resin with a mixture of DCM, trifluoroethanol (TFE) and acetic acid (HOAc) (3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:1) for 2 h. After filtration the solvent was removed under reduced pressure and the product was co-evaporated with chloroform. The peptide was obtained as a white solid after purification using preparative HPLC and lyophilization from dioxane. AzAc-Arg(Pbf)-Gly-Asp(OtBu)-Ser(tBu)-Gly-OH. LC-MS: 938.64 [M + H]+ (calculated [M + H]+ = 938.06), Rt = 23.70 min (Fig. S14, ESI†).
:1:1) for 2 h. After filtration the solvent was removed under reduced pressure and the product was co-evaporated with chloroform. The peptide was obtained as a white solid after purification using preparative HPLC and lyophilization from dioxane. AzAc-Arg(Pbf)-Gly-Asp(OtBu)-Ser(tBu)-Gly-OH. LC-MS: 938.64 [M + H]+ (calculated [M + H]+ = 938.06), Rt = 23.70 min (Fig. S14, ESI†).
Star-PEG-N3, star-PEG-CF-N3 and star-PEG-RGDS-N3
Star-PEG-NH2 (1 eq.), BOP (8 eq.), DiPEA (12 eq.) and azidoacetic acid (8 eq.) or peptide AzAc-Arg(Pbf)-Gly-Asp(OtBu)-Ser(tBu)-Gly-OH (6 eq.) were added were dissolved in DCM (20 mL), and stirred at r.t. overnight. The solution was washed with KHSO4 and brine, and dried over MgSO4. Volatiles were removed under reduced pressure, and the crude mixture was purified using column chromatography over aluminum oxide (10% MeOH in DCM). Lyophilization from 1,4-dioxane yielded 59% star-PEG-N3. Star-PEG-RGDS-N3 was deprotected by the treatment with TFA/TIS/H2O (95:2.5:2.5) for 4 h, followed by precipitation in ether or by removal of the solvents under reduced pressure. Lyophilization from 1,4-dioxane yielded 59%. For polymer star-PEG-CF-N3 synthesis the same procedure as star-PEG-N3, besides adding 0.08 eq. carboxyfluorescein when mixing chemicals at the beginning. Lyophilization from 1,4-dioxane yielded 31%. Star-PEG-N3: Rf = 0.43 (10% MeOH in DCM). Star-PEG-RGDS-N3: Rf = 0.38 (10% MeOH in DCM). Star-PEG-CF-N3: Rf = 0.40 (10% MeOH in DCM). MALDI-TOF (α-cyano-4-hydroxycinnamic acid): Fig. S15 (ESI†). 1H NMR (400 MHz, CDCl3): Fig. S16 (ESI†).
Hydrogel formation
Chemical cross-linked hydrogels were prepared by mixing equal molar amounts of star-PEG-DBCO and star-PEG-N3 solutions both 50 mg mL−1 in medium. Physical cross-linked hydrogels were prepared by mixing an equal volume of 100 mg mL−1 star-PEG-AA in PBS and 80 mM CaCl2 in medium. For dual cross-linked hydrogels, the chemical parts and physical parts were mixed at volume ratios shown in Table 1 to yield a total polymer concentration of 50 mg mL−1.| Physical cross-links (%) | Chemical cross-links (%) | |
|---|---|---|
| 100c | — | 100 |
| 20p/80c | 20 | 80 |
| 40p/60c | 40 | 60 |
| 50p/50c | 50 | 50 |
| 60p/40c | 60 | 40 |
| 80p/20c | 80 | 20 |
| 100p | 100 | — |
Rheological characterization
The storage (G′) and loss modulus (G′′) of the hydrogels were measured using an AR2000ex rheometer (from TA instruments). All measurements were performed using a flat steel plate geometry (20 mm, gap size 500 mm) at a temperature of 37 °C. In general, oscillatory time sweep measurements were performed at a constant strain of 1% and an angular frequency of 10 rad s−1. Stress–relaxation measurements were performed at a constant strain of 15%. Oscillatory strain sweep measurements were performed from 0.1 to 1000% at 10 rad s−1 constant frequency. All rheology measurements were performed in triplo.For self-recovering measurements, we ran a protocol as below. First we formed the hydrogel via the procedure mentioned above on the rheometer and performed an oscillatory time sweep for 900 s, then followed by a 1000 s stress–relaxation step. After 120 s soaking time, samples were measured by an oscillatory strain sweep and an oscillatory time sweep (600 s) in sequence. These last two sweeps were repeated for one more time.
Cryo-scanning electron microscopy (JEOL JSM-6330F)32,33
Hydrogel samples, which were prepared as described above, were frozen in super-cooled liquid nitrogen and fractured to expose inner structures. After sublimating and coating with gold–palladium inside a vacuum cryo chamber images were acquired at 3 kV and 12 μA. Mesh sizes were analyzed with the Nano Measurer Software (Nano Measurer 1.2, Fudan University, China), the distributions were based on 100 mesh sizes for each sample.Swelling test34
Dual cross-linked Hydrogels (100 μL) were prepared following the hydrogel formation protocol using the polymer concentration of 50 mg mL−1. Hydrogels were set for 15 min, then swell in medium at 37 °C. Hydrogels were dabbed with tissue paper to remove extra liquid and weighed (Ws) at certain time point (1 h, 3 h, 7 h and 24 h). The swelling ratio was determined by the weight of the fully hydrated hydrogel (Ws) versus the weight of the dried sample (Wd) using the following equation: swelling ratio = (Ws − Wd)/Wd. All measurements were performed in triplo.Cell culture
Human mesenchymal stem cells (HMSCs) were cultured in sterile conditions in Dulbecco's Modified Eagle's medium (DMEM) (PAA laboratories, Pasching, Austria) supplemented with 10% heat in the activated fetal bovine serum (FBS) (Integro, Zaandam, the Netherlands), 2 mM L-glutamine,100 U mL−1 penicillin and 100 μg mL−1 streptomycin (P/S). HMSCs were cultured up to passage 6 to be used inside the hydrogels. All cells were maintained at 37 °C in a humidified atmosphere of 7.5% CO2. The cultured medium was changed twice a week and the cells were trypsinized (typsin/EDTA) when they reached 90% confluence. Cells were suspended in media and the number of cells was counted using an inverted microscope and a cell counting chamber (Fuchs-Rosenthal).Confocal microscopy (Leica TCS SP2 AOBS)
Cell viability and morphology were assessed by confocal microscopy (SP8x, Leica-microsystems, CTR advanced) using a live/dead assay. Human mesenchymal stem cells were cultured in the hydrogels formed by the protocol of the hydrogel formation described previously, followed by an incubation period of 5 days. For live/dead staining, we diluted calcein-AM and ethidium homodimer-1 (EthD-1) in PBS to a concentration of 2 μM. After an incubation period of 30 min, the stain and solution were removed, and samples were analyzed using confocal laser scanning microscopy. Wavelengths of 488 and 532 nm were used to excite calcein and EthD-1, respectively. The emission of calcein was collected between 498 and 530 nm, and the emission of EthD-1 was collected between 542 and 680 nm. The total number of cells (living/dead, spread/round) was counted for each sample (composed of at least 3 images for each sample).Results and discussion
Hydrogel design
In a previous study we compared the formation of covalent cross-links via a variety of click reactions.35 Among those, dibenzocyclooctyne (DBCO) and azide grafted PEG polymers could form a gel in a few minutes in cell-friendly manner. In addition, non-covalent hydrogels formed via complexation between bisphosphonate moieties and calcium ions by grafting a bisphosphonate group on PEG polymers to bind the calcium ions have been reported by Lopez-Perez et al.29 They successfully formed a physically cross-linked gel via ionic bonds between bisphosphonates and calcium ions which can self-recover after breakage. Now, we wanted to combine these two strategies to prepare a dual cross-linked hydro-gel. To this end, we started with four-armed poly-ethylene glycol amine (Mn = 10 kDa, star-PEG-NH2) and four-armed polyethylene glycol succinimidyl glutarate (Mn = 10 kDa, star-PEG-SGA) as the polymer basis to construct our hydrogels with. We functionalized star-PEG-NH2 with either an azide or a DBCO moiety and star-PEG-SGA with alendronic acid (star-PEG-AA) respectively.35 Subsequently, we created gels via chemical cross-links between DBCO and azide grafted polymers (star-PEG-DBCO and star-PEG-N3) and physical cross-links between diphosphonates and calcium ions. We envisioned the dual cross-linked (DC) network would spontaneously form by a copper-free strain-promoted covalent click reaction (SPAAC)36 and non-covalent ionic interactions by mere mixing of all components (Fig. 1). A fully chemically cross-linked (CC) hydrogel was prepared by mixing equal amounts of functionalized PEG-DBCO and star-PEG-N3 at a concentration of 50 mg mL−1 in cell culture medium. We pre-pared fully physically cross-linked (PC) hydrogel by simply mixing equal volumes of 100 mg mL−1 star-PEG-AA in PBS (pH = 7.4) and 80 mM CaCl2 in medium to a final polymer concentration of 50 mg mL−1. For DC hydrogels, we varied the ratio of the ionic PC versus covalent CC components to obtain a set of hydrogel systems, including 20% (80p/20c), 40% (60p/40c), 50% (50p/50c), 60% (40p/60c) and 80% (20p/80c) covalent links (Table 1), all at a final total polymer concentration of 50 mg mL−1. These experiments showed us the ionic cross-links between diphosphonates and calcium ions formed immediately to instantaneously give rise to a network in case of a fully PC hydrogel (star-PEG-AA/Ca2+).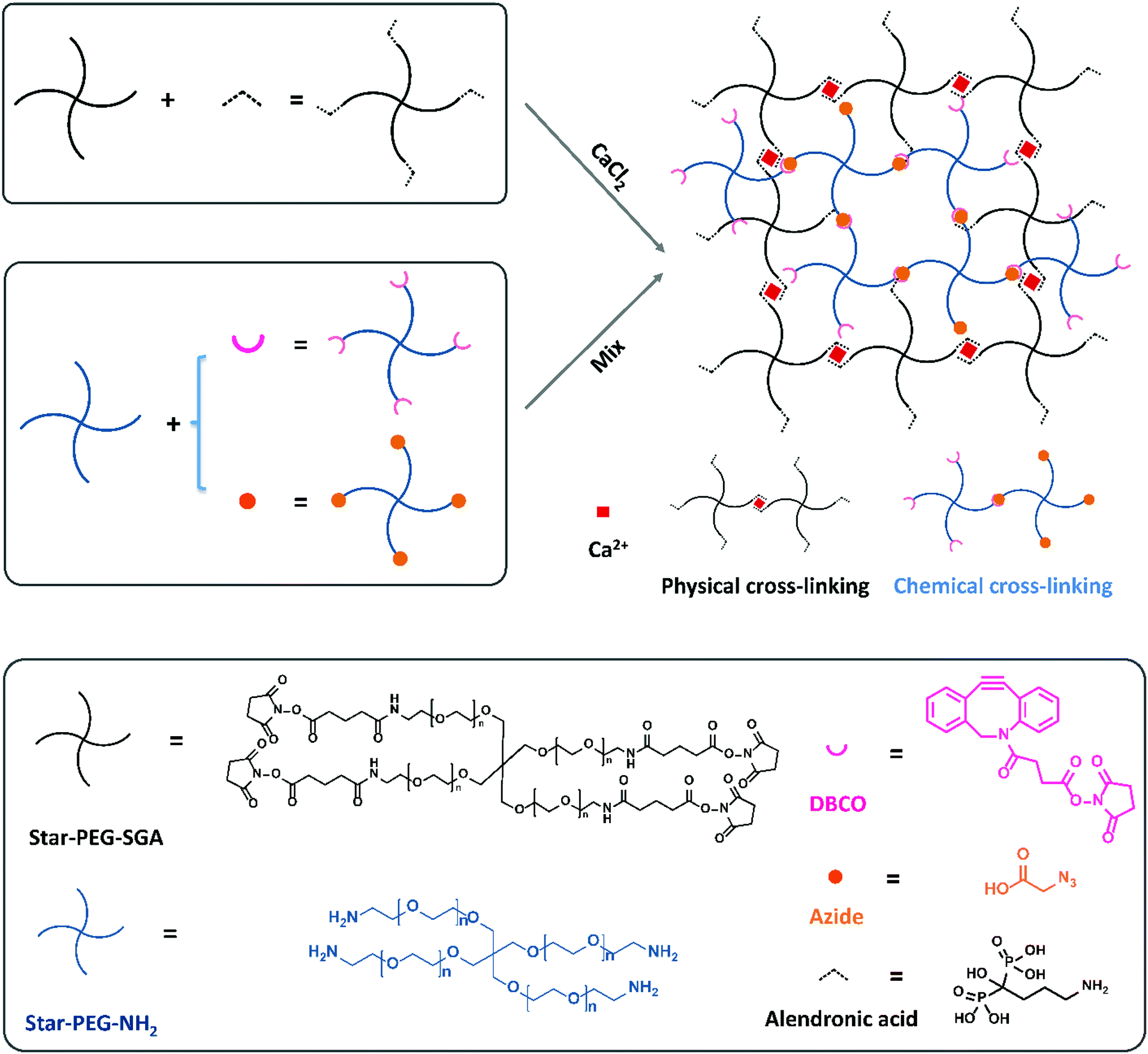 | ||
| Fig. 1 Schematic of dual cross-linked hydrogels studied and their components. Non-covalent physical cross-links form between bisphosphonates and calcium ions, and covalent chemical cross-links through a SPAAC reaction between a DBCO moiety and an azide group. | ||
Like the fully PC hydrogel, also all the DC hydrogels formed in seconds (Fig. 2C), with the observation that the more physical cross-links were incorporated in the DC hydrogels, the faster the hydrogel would form. Only the 20p/80c with the more chemical cross-links formed a stable gel in 200 s. This series of hydrogels was used for further investigation of their mechanical properties, gel morphology and swelling behavior. To be able to allow cell attachment, we also introduced an integrin-binding RGDS peptide sequence with an N-terminal azide onto star-PEG-NH2 to get a functional polymer (star-PEG-RGDS-N3) that could partly replace star-PEG-N3 to form DC hydrogels for cell encapsulation and subsequent biocompatibility investigations.36,37
 | ||
| Fig. 2 Mechanical properties of dual cross-linked hydrogels with different ratios. (A) Stress relaxation tests. (B) Strain sweeps from 0.1% to 1000%. (C) Self-recovering tests where gel failure was induced by an increasing the strain to 1000% twice. “●” represents for storage modulus value (G′), “○” represents for loss modulus value (G′′). | ||
Mechanical properties
Recent studies reported elastic cellular forces can support cell interactions, i.e., gels with faster relaxation enhance the activity of cell spreading, proliferation and differentiation.38 In similar fashion, we tested the stress relaxation properties of all our DC hydrogels. The relaxation time, defined as the time it takes upon application of a constant strain for the initial stress of the gels to decline to half its value, ranged from a few seconds to minutes (Fig. 2A). The greater the amount of PC the faster relaxation time of the DC hydrogels was. Only the DC gel containing 80% covalent cross-links took more than 15 minutes to relax which exceeded the time scale over which we measured the DC gel systems.Considering the fact that in biological environments the materials not only need to withstand the strains placed on the whole material but also the forces cells exert on the local matrix,39 we further studied the mechanical properties of our DC hydrogels, and observed the behavior of the DC gels under increasing strains. All DC hydrogels retained their original mechanical properties – in the linear region – at the first stage after which their storage modulus decreased significantly when the strain exceeded 30% (Fig. 2B). The more the CC dominated inside the DC gel systems, the longer the gel would remain in the linear region. The 20p/80c hydrogel did not lose its mechanical integrity up to a strain of 30%, while the 80p/20c gel started to yield at lower strains (5%). As hypothesized, the covalent links help to reinforce the ionic interactions and maintain the gel's mechanical properties when exposed to a higher strain. The 20p/80c and 40p/60c DC hydrogels could even retain over 80% of their initial storage modulus at a strain of 50% (Fig. S1, ESI†).
Due to the nature of the ionic cross-links of our hydrogels we expected self-recovering behaviour, as these non-covalent cross-links can reform after breaking. Here, we defined the cross over point in Fig. 2C as the strain at which the gels fail and become more viscous than elastic which is when the loss modulus (G′′ value) exceeds the storage modulus (G′ value). We hypothesized the DC hydrogels, like the PC ones,29 could quickly recover their mechanical properties after the failure at high strains. To verify this, we performed two strain sweeps from 0.1% to 1000% to ensure the strain surpasses the failure point of the DC hydrogels, followed by 10 min recovery periods at “normal” strain (1%) while monitoring the mechanical parameters by oscillatory rheology. (We also tried to monitor the self-recovering behavior with compressions tests but, unfortunately, it was not possible to perform these tests with our DC hydrogels as they are too soft.) We observed a fast recovery for each of the DC hydrogels, even for the gel mainly consisting of chemical cross-links (20p/80c). The storage modulus, immediately recovered to 50–87% and 41–96% after the first and second strain failure respectively, as depicted in Fig. S2 (ESI†). Because the 100p gel regained most of its initial mechanical properties while the 100c gel ruptured just after the first strain failure and did not recover to its initial stiffness, we think it is the PC constituents that help the DC hydrogels to regain most of their original storage modulus value after the 10 min recovery period.
Swelling test
Next, we examined our gels in a swelling test by soaking the set of hydrogels in PBS for 24 h to investigate the hydrogel stability.25 Fig. S3 (ESI†) shows the equilibrium weight swelling ratio of the different hydrogels at 1 h, 3 h, 7 h and 24 h. Moreover, not surprisingly the swelling ratio among the different hydrogel systems, going from the 100c hydrogel to the 100p hydrogel (Fig. S3, ESI†), the DC gels had a comparable swelling ratio (from 14.4 ± 0.7 to 16.9 ± 0.4 w/w) after one hour. We found that the 100c gel is more stable than the 100p gel. A significant difference of the swelling ratio was observed after one hour between the 100p (18.3 ± 0.5 w/w) and the 100c (15.2 ± 0.3 w/w) hydrogel, as well as for the 100p and 20p/80c hydrogels (14.4 ± 0.7 w/w) which contained a greater portion CC. However, the DC hydrogels swelling ratio decreased over time, especially for the gel which is dominated by physical cross-links (80p/20c). The swelling ratio at 24 h revealed the ratio of the 100p gel (24 h) was less than 80% while the 100c gel still reached 98% compared to the initial value at 1 hour (Fig. 3). As the proportion of physical cross-links increased inside the DC hydrogels, the swelling ratio decreased along with certain degree of gel disappearance after 24 hours. In combination with the dry mass of the hydrogels we determined after 1 hour and 24 hours (Fig. S4, ESI†), these observations indicate hydrogel degradation is marginal over a period of 24 hours. These results led us to believe the purely PC hydrogel has a poorer ability to entrap water possibly caused by the dissociation of the physical cross-links, leading to the unstable integrity of bulk gel materials. In contrast, the more stable 100c gel is able to confine the water during a one-day swelling period. Hence, we can conclude that the more PC parts are present in a DC hydrogel, the weaker the water absorption of the gel will get.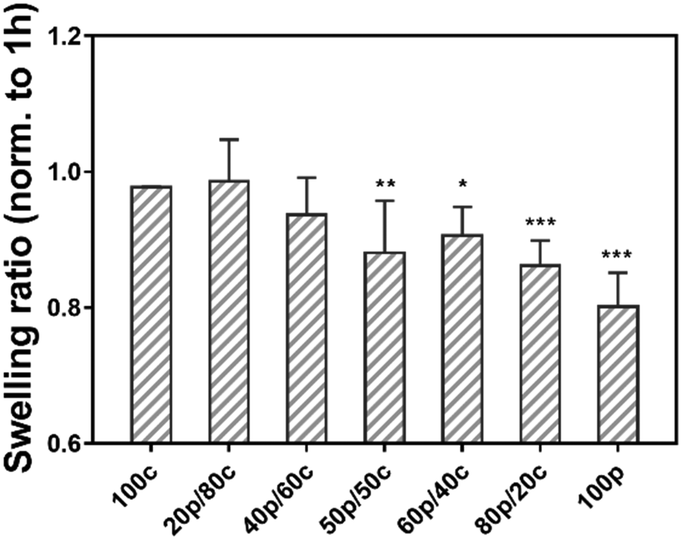 | ||
| Fig. 3 Equilibrium swelling ratio of different hydrogel combinations (total polymer concentration is 50 mg mL−1) after 24 h incubation at 37 °C. Swelling ratio normalized to 1 h. (n = 3; *p < 0.033, **p < 0.002, ***p < 0.001, two-way Anova Sidak). | ||
Cryo-scanning electron microscopy (cryo-SEM)
As has been reported for both ionically40 and covalently41 cross-linked hydrogels, we visualized the morphology of the gels (composed as shown in Table 1) via cryo-SEM microscopy. The fully CC gel displays a smaller mesh size than the fully PC gel in Fig. 4 and Fig. S6 (ESI†). In addition, as the proportion of physical cross-links increased, the mesh size inside the hydrogels became bigger and the diameter distribution got wider changing from 0.3–4.3 μm to 1.8–13.0 μm (Fig. S6, ESI†). To further substantiate this is not an artefact, we prepared and examined fully physically cross-linked hydrogel structures at different polymer concentrations (Fig. S7, ESI†) and different Ca2+ concentration (Fig. S8, ESI†). Here it can be seen that the mesh size becomes smaller and smaller upon increasing either the polymer or the Ca2+ concentration. In our opinion this corroborates that the cooling process hardly affects the network structure and that the tendency that higher concentrations of either polymer or calcium ions leads to a smaller mesh size (Fig. S7 and S8, ESI†).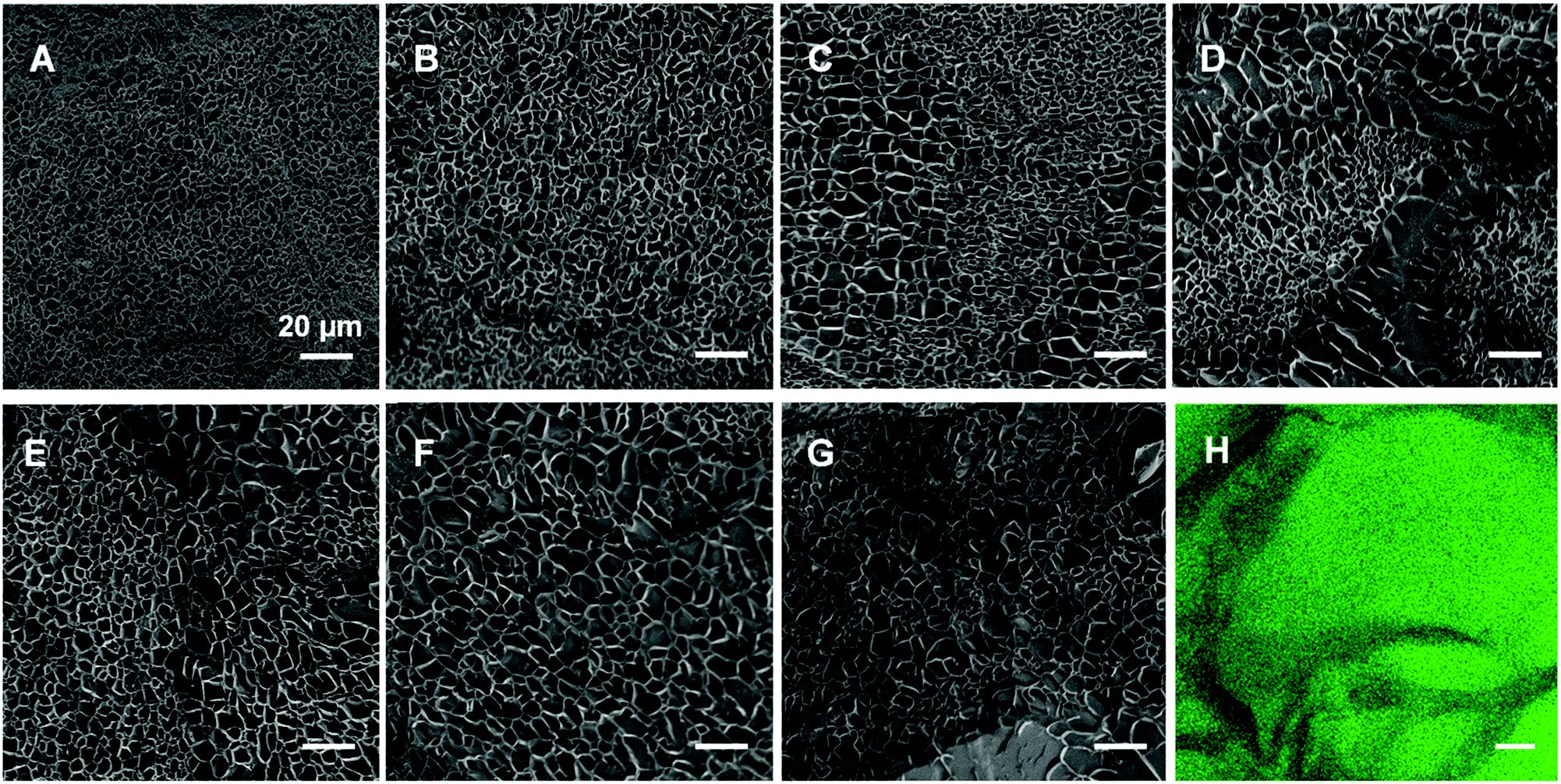 | ||
| Fig. 4 Cryo-SEM images of the different (dual) cross-linked hydrogel systems (A) 100c, (B) 20p/80c, (C) 40p/60, (D) 50p/50c, (E) 60p/40c, (F) 80p/20c, (G) 100p at a concentration of 50 mg mL−1. (H) A confocal microscopy image of 50p/50c hydrogel. The scale bar represents 20 μm. | ||
Interestingly, we observed phase separation in the DC hydrogels with equal or nearly equal amounts of covalent and non-covalent crosslinks (Fig. 4C–E). This was not as apparent in the hydrogels with a more extreme ratio of physical or chemical cross-links (Fig. 4B and F). A possible reason for this phenomenon, could be that the dominating physically or chemically cross-linked gel may impede the other constituent from its own cross-linking process. Another explanation could be that the cross-links of the minor part are just scattered over the entire gel network and are thus not distinguishable anymore. We further investigated this apparent phase separation by trying to establish the identity of the observed different mesh sizes. To this end we also incorporated carboxyfluorescein into star-PEG-N3 (star-PEG-CF-N3) and prepared a DC gel (50p/50c) containing 1% of fluorescent probe. We found the chemical cross-linked part represented by green fluorescence was surrounded by dark bands (Fig. 4H), which resembled the pattern found in the cryo-SEM samples, where the bands were made up of areas of bigger and smaller mesh sized throughout the gels, corroborating a phase separated microstructure.
Cell viability
To explore the bio-compatibility of our DC gels, we cultured human mesenchymal stem cells (HMSCs) for 5 days inside the DC hydrogel materials with the purely PC or CC hydrogels as control groups. In order to facilitate cell attachment, we introduced an RGDS-peptide motif onto the star-PEG-NH2 polymer and kept the RGDS-peptide content in all hydrogel systems the same except for the 100p gel in which we were not able to incorporate the peptide sequence as a result of the way we prepare them. As exhibited in Fig. 5 the cells were viable after 1-day culture in all DC hydrogels, with about 80% of the cells still alive. More viable cells were observed in the DC gels than in the 100c gel (only 20% cells were alive) and the 100p gel (60% cells were alive) suggesting the DC hydrogel systems are more biocompatible. Furthermore, cells started to have ragged edges in Fig. 5D gels and a few cells had become elongated in both the Fig. 5E and F gels. This substantiates our hypothesis that DC hydrogels with more physical cross-links, which exhibit faster mechanical relaxation, can enhance cell spreading behavior. After 5 days of cell culture, most cells had died in the 100c and 20p/80c gels (Fig. 5H and Fig. S9, ESI†). Furthermore, significant differences in cell viability showed up between day 1 and day 5 in the 20p/80c, 40p/60c, 50p/50c and 100p hydrogel systems. Among the DC gels, the 60p/40c and 80p/20c gels kept most cells alive (73% and 65% respectively). According to Fig. S9 (ESI†), less cells were observed after 5-day culture especially in case of physical cross-links dominating gels (60p/40c, 80p/20c and 100p). From the swelling test we know these gel structures were unstable and could lose their integrity over time and in this way, leading to cell loss to a certain degree. Hence, further optimization will be necessary to compose a DC gel that is able to trap cells efficiently and is bio-compatible at the same time over a longer period.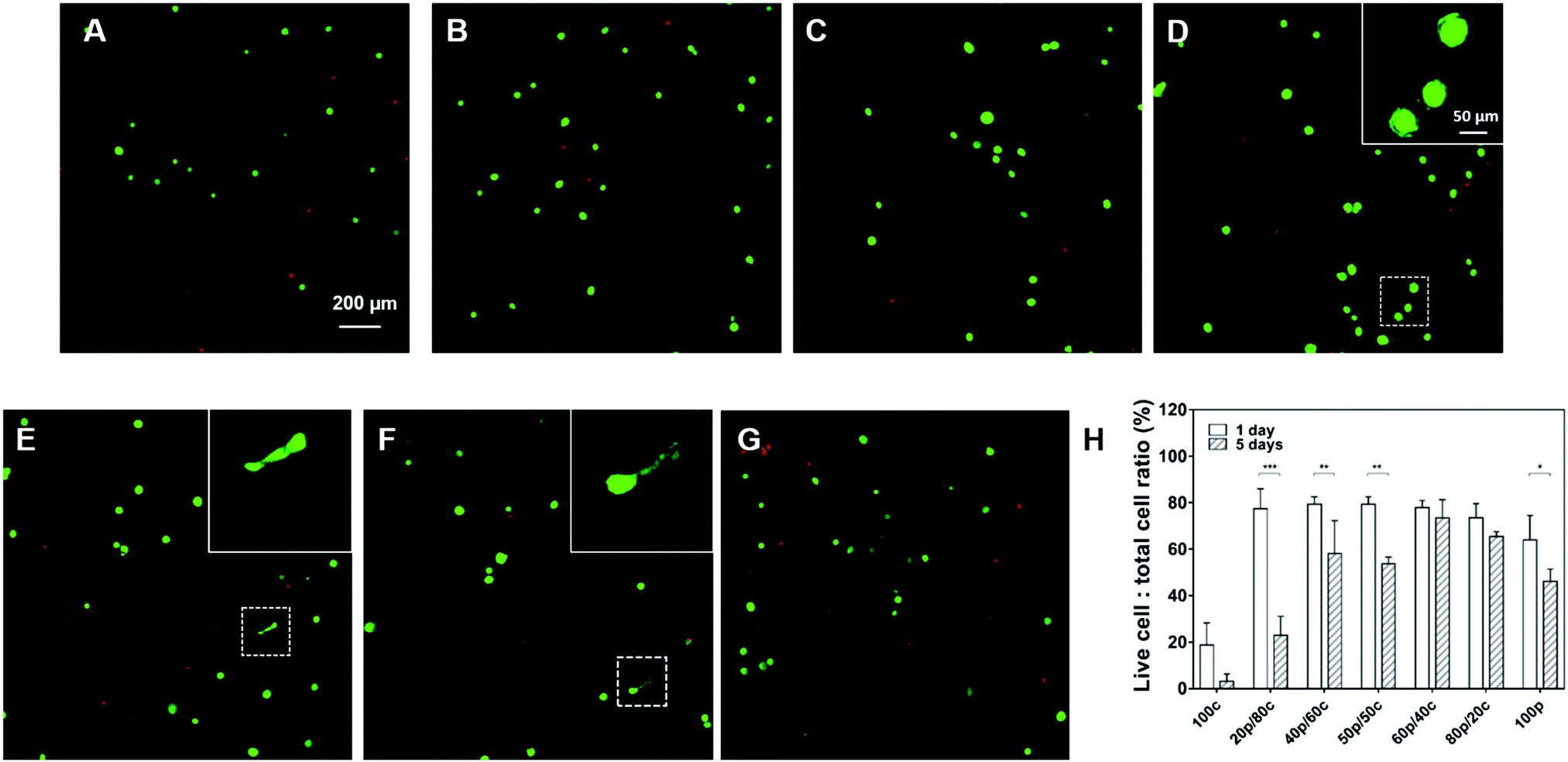 | ||
| Fig. 5 Confocal microscopy images of HMSCs encapsulated inside (A) 100c, (B) 20p/80c, (C) 40p/60, (D) 50p/50c, (E) 60p/40c, (F) 80p/20c, (G) 100p hydrogels after 24 h. Images are composed from overlays of the calcein (green) and EthD-1 (red) channels indicating live and dead cells respectively. Scale bars represent 100 μm (inset) and 200 μm respectively. (H) The live cell to total cell ratios of different hydrogel systems. (n = 3; *p < 0.033, **p < 0.002, ***p < 0.001, two-way Anova Sidak). | ||
Conclusions
We utilized ionic interactions between bisphosphonate groups and Ca2+ ions to create physical cross-links and a strain promoted azide–alkyne cycloaddition (SPAAC) reaction to introduce covalent chemical cross-links to successfully create dual-cross-linked (DC) hydrogel networks. In all cases a gel formed within seconds up to a minute upon straightforward mixing of the various components. Rheology measurements showed us that DC hydrogels containing more physical cross-links, displayed a faster relaxation time, varying from a few seconds to over 1000 s, while DC gels containing more covalent cross-links yielded at a higher strain than gels more with non-covalent linkages. At the same time, all of the DC hydrogels exhibited a striking self-recovery of mechanical properties (over 80%) after two complete ruptures regardless of their composition. Next, a swelling test revealed that the incorporation of increasing amounts of covalently cross-linked polymer significantly enhanced the capability of trapping water and led to a more stable gel network. Subsequent electron microscopy investigation of DC gel morphology revealed phase separation, which was most apparent when the PC and CC components were present in more or less equal amounts (i.e. 40p/60c, 50p/50c and 60p/40c). Furthermore, the mesh size distribution became narrower as the proportion of the chemical cross-links got larger. Finally, we encapsulated human mesenchymal stem cells (HMSCs) in our DC hydrogel networks to examine their biocompatibility. During a 5-day cell encapsulation experiment, the 60p/40c and 80p/20c gels showed the best properties with respect to cell viability without any significant differences. However, also considering the swelling test results that revealed loss of structural integrity for some gels, further optimization is still required to improve the stability of the DC hydrogels. Nevertheless, in our opinion these synthetic DC hydrogels composed of both chemical and physical cross-links are very capable to mimic the natural extracellular microenvironment. And these self-recovering, biocompatible, rapidly in situ formed hydrogel networks bear a strong potential as a 3D biomimetic matrix for investigation of cell behaviour. Especially, as similar materials have been shown to beneficial for the differentiation of encapsulated stem cells.34 We aim to further develop these materials as stem cell delivery vehicles for applications like bone tissue regeneration and replacement of damaged tissue.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The General Instrumentation department of the Radboud University is acknowledged for providing microscopy services and the Chinese Scholarship Council (CSC) and the Institute of Molecules and Materials (IMM) for the funding.References
- N. A. Peppas and R. Langer, Science, 1994, 263, 1715–1720 CrossRef CAS PubMed
.
- J. L. Drury and D. J. Mooney, Biomaterials, 2003, 24, 4337–4351 CrossRef CAS PubMed
- M. P. Lutolf and J. A. Hubbell, Nat. Biotechnol., 2005, 23, 47–55 CrossRef CAS PubMed
- K. Y. Lee and D. J. Mooney, Chem. Rev., 2001, 101, 1869–1880 CrossRef CAS PubMed
- G. Chan and D. J. Mooney, Trends Biotechnol., 2008, 26, 382–392 CrossRef CAS PubMed
- Y. Jin, C. Yu, R. J. Denman and W. Zhan, Chem. Soc. Rev., 2013, 42, 6634–6654 RSC
- Y. Qiu, J. J. Lim, L. Scott Jr, R. C. Adams, H. T. Bui and J. S. Temenoff, Acta Biomater., 2011, 7, 959–966 CrossRef CAS PubMed
- S. P. Zustiak and J. B. Leach, Biomacromolecules, 2010, 11, 1348–1357 CrossRef CAS PubMed
- A. T. Metters, K. S. Anseth and C. N. Bowman, Polymer, 2000, 41, 3993–4004 CrossRef CAS
- M. P. Lutolf, J. L. Lauer-Fields, H. G. Schmoekel, A. T. Metters, F. E. Weber, G. B. Fields and J. A. Hubbell, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 5413–5418 CrossRef CAS PubMed
- B. P. Purcell, D. Lobb, M. B. Charati, S. M. Dorsey, R. J. Wade, K. N. Zellars, H. Doviak, S. Pettaway, C. B. Logdon, J. A. Shuman, P. D. Freels, J. H. Gorman III, R. C. Gorman, F. G. Spinale and J. A. Burdick, Nat. Mater., 2014, 13, 653–661 CrossRef CAS PubMed
- H. Wang and S. C. Heilshorn, Adv. Mater., 2015, 27, 3717–3736 CrossRef CAS PubMed
- P. Lu, V. M. Weaver and Z. J. Werb, Cell Biol., 2012, 196, 395–406 CrossRef CAS PubMed
- A. M. Rosales and K. S. Anseth, Nat. Rev. Mater., 2016, 1, 15012 CrossRef CAS PubMed
- L. Saunders and P. X. Ma, Macromol. Biosci., 2019, 19, 1800313 CrossRef PubMed
- S. Ahadian, R. B. Sadeghian, S. Salehi, S. Ostrovidov, H. Bae, M. Ramalingam and A. Khademhosseini, Bioconjugate Chem., 2015, 26, 1984–2001 CrossRef CAS PubMed
- N. A. Peppas, Y. Huang, M. Torres-Lugo, J. H. Ward and J. Zhang, Annu. Rev. Biomed. Eng., 2000, 2, 9–29 CrossRef CAS PubMed
- J. Lou, R. Stowers, S. Nam, Y. Xia and O. Chaudhuri, Biomaterials, 2018, 154, 213–222 CrossRef CAS PubMed
- B. J. Adzima, C. J. Kloxin and C. N. Bowman, Adv. Mater., 2010, 22, 2784–2787 CrossRef CAS PubMed
- R. Zhang, M. Tang, A. Bowyer, R. Eisenthal and J. Hubble, Biomaterials, 2005, 26, 4677–4683 CrossRef CAS PubMed
- T. F. Scott, A. D. Schneider, W. D. Cook and C. N. Bowman, Science, 2005, 308, 1615–1617 CrossRef CAS PubMed
- C. K. Kuo and P. X. Ma, Biomaterials, 2001, 22, 511–521 CrossRef CAS PubMed
- P. Gacesa, Carbohydr. Polym., 1988, 8, 161–182 CrossRef CAS
- J. Y. Sun, X. Zhao, W. R. Illeperuma, O. Chaudhuri, K. H. Oh, D. J. Mooney, J. J. Vlassak and Z. Suo, Nature, 2012, 489, 133 CrossRef CAS PubMed
- M. H. Ghanian, H. Mirzadeh and H. Baharvand, Biomacromolecules, 2018, 19, 1646–1662 CrossRef CAS PubMed
-
H. Fleisch, A. Reszka, G. Rodan and M. Rogers, Principles of bone biology, Academic Press, 2002 Search PubMed
- M. T. Drake, B. L. Clarke and S. Khosla, Mayo Clin. Proc., 2008, 83, 1032–1045 CrossRef CAS PubMed
- M. J. Rogers, J. C. Crockett, F. P. Coxon and J. Mönkkönen, Bone, 2011, 49, 34–41 CrossRef CAS PubMed
- P. M. Lopez-Perez, R. M. da Silva, I. Strehin, P. H. Kouwer, S. C. Leeuwenburgh and P. B. Messersmith, Macromolecules, 2017, 50, 8698–8706 CrossRef CAS PubMed
-
M. J. Sánchez-Fernández, M. R. Immers, R. P. Félix-Lanao, F. Yang, J. C. Bender, J. Mecinović, S. C. Leeuwenburgh and J. C. Hest, Biomacromolecules, 2019, 20, 2913–2921 Search PubMed
- E. Kaiser, R. L. Colescott, C. D. Bossinger and P. I. Cook, Anal. Biochem., 1970, 34, 595–598 CrossRef CAS PubMed
- Z. A. Schnepp, R. Gonzalez-McQuire and S. Mann, Adv. Mater., 2006, 18, 1869–1872 CrossRef CAS
- L. J. Macdougall, V. X. Truong and A. P. Dove, ACS Macro Lett., 2017, 93–97 CrossRef CAS
- K. Zhang, Q. Feng, J. Xu, X. Xu, F. Tian, K. W. Yeung and L. Bian, Adv. Funct. Mater., 2017, 27, 1701642 CrossRef
- H. Zhan, H. de Jong and D. W. Löwik, ACS Appl. Bio Mater., 2019, 2, 2862–2871 CrossRef CAS
- A. M. Jonker, S. A. Bode, A. H. Kusters, J. C. van Hest and D. W. Löwik, Macromol. Biosci., 2015, 15, 1338–1347 CrossRef CAS PubMed
- E. Ruoslahti and M. D. Pierschbacher, Science, 1987, 238, 491–497 CrossRef CAS PubMed
- O. Chaudhuri, L. Gu, D. Klumpers, M. Darnell, S. A. Bencherif, J. C. Weaver, N. Huebsch, H. Evi Lippens, G. N. Duda and D. J. Mooney, Nat. Mater., 2016, 15, 326–334 CrossRef CAS PubMed
- D. E. Clarke, E. T. Pashuck, S. Bertazzo, J. V. Weaver and M. M. Stevens, J. Am. Chem. Soc., 2017, 139, 7250–7255 CrossRef CAS PubMed
- Z. A. Schnepp, R. Gonzalez-McQuire and S. Mann, Adv. Mater., 2006, 18, 1869–1872 CrossRef CAS
- L. J. Macdougall, V. X. Truong and A. P. Dove, ACS Macro Lett., 2017, 93–97 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0tb01042a |
| This journal is © The Royal Society of Chemistry 2020 |