
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
pH-Responsive nanocomposite fibres allowing MRI monitoring of drug release†
Ziwei
Zhang
ab,
Connor J. R.
Wells
 b,
Aaron M.
King
b,
Joseph C.
Bear
c,
Gemma-Louise
Davies
*b and
Gareth R.
Williams
*a
b,
Aaron M.
King
b,
Joseph C.
Bear
c,
Gemma-Louise
Davies
*b and
Gareth R.
Williams
*a
aUCL School of Pharmacy, University College London, 29–39 Brunswick Square, London WC1N 1AX, UK. E-mail: g.williams@ucl.ac.uk; Tel: +44(0) 207 753 5868
bUCL Department of Chemistry, University College London, 20 Gordon St, London WC1H 0AJ, UK. E-mail: gemma-louise.davies@ucl.ac.uk; Tel: +44(0) 207 679 7524
cDepartment of Chemical and Pharmaceutical Sciences, Kingston University, Penrhyn Rd, Kingston upon Thames, KT1 2EE, UK
First published on 30th June 2020
Abstract
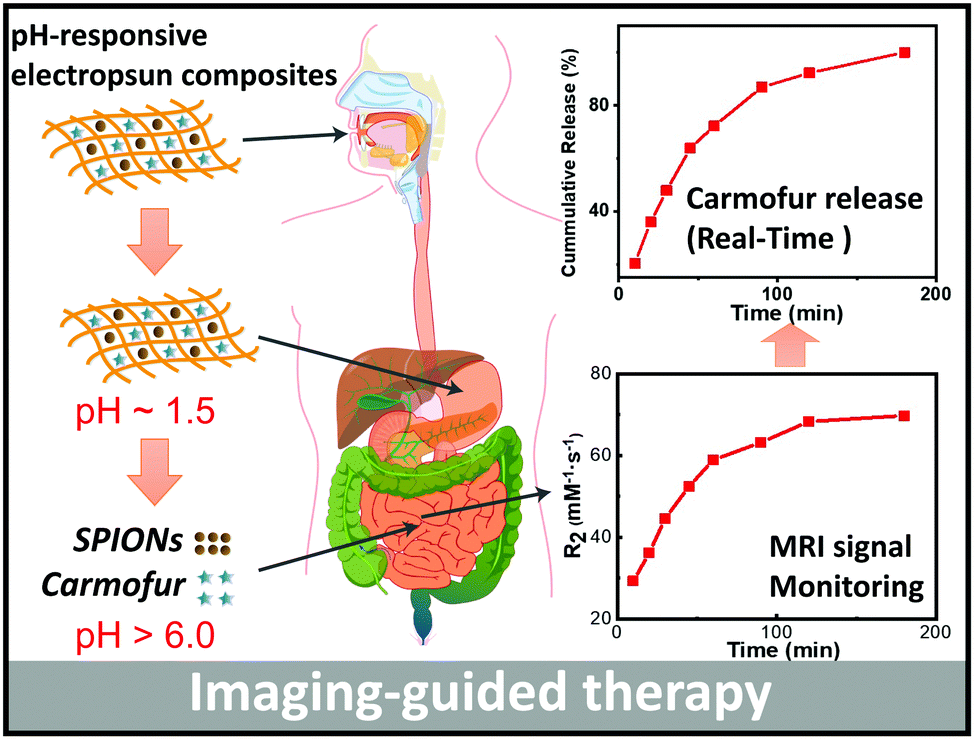
Magnetic resonance imaging (MRI) is one of the most widely-used non-invasive clinical imaging tools, producing detailed anatomical images whilst avoiding side effects such as trauma or X-ray radiation exposure. In this article, a new approach to non-invasive monitoring of drug release from a delivery vehicle via MRI was developed, using pH-responsive Eudragit L100 and S100 fibres encapsulating superparamagnetic iron oxide nanoparticles (SPIONs) and carmofur (a drug used in the treatment of colon cancer). Fibres were prepared by electrospinning, and found to be smooth and cylindrical with diameters of 645 ± 225 nm for L100 and 454 ± 133 nm for S100. The fibres exhibited pH responsive dissolution behaviour. Around the physiological pH range, clear pH-responsive proton relaxation rate changes due to matrix swelling/dissolution can be observed: r2 values of L100 fibres increase from 29.3 ± 8.3 to 69.8 ± 2.5 mM−1 s−1 over 3 h immersion in a pH 7.4 medium, and from 13.5 ± 2.0 mM−1 s−1 to 42.1 ± 3.0 mM−1 s−1 at pH 6.5. The r2 values of S100 fibres grow from 30.4 ± 4.4 to 64.7 ± 1.0 mM−1 s−1 at pH 7.4, but at pH 6.5, where the S100 fibres are not soluble, r2 remains very low (< 4 mM−1 s−1). These dramatic changes in relaxivity demonstrate that pH-responsive dissolution results in SPION release. In vitro drug release studies showed the formulations gave rapid release of carmofur at physiological pH values (pH 6.5 and 7.4), and acid stability studies revealed that they can protect the SPIONs from digestion in acid environments, giving the fibres potential for oral administration. Exploration of the relationship between relaxivity and carmofur release suggests a linear correlation (R2 > 0.94) between the two. Mathematical equations were developed to predict carmofur release in vitro, with very similar experimental and predicted release profiles obtained. Therefore, the formulations developed herein have the potential to be used for non-invasive monitoring of drug release in vivo, and could ultimately result in dramatic reductions to off-target side effects from interventions such as chemotherapy.
Introduction
Among a wide variety of available clinical imaging techniques, magnetic resonance imaging (MRI) stands out for its ability to achieve high spatial and temporal resolution without the use of ionizing radiation.1,2 In order to provide early and precise diagnosis, contrast agents (CAs) are generally employed to enhance resolution and contrast in MRI.3 Superparamagnetic iron oxide nanoparticles (SPIONs) represent an important class of MRI CA because of their size-dependent magnetic behaviour.4 By creating local magnetic field gradients, SPIONs can significantly decrease proton transverse relaxation times (T2), boosting signal contrast.5,6 Their relatively low cytotoxicity and ability to be metabolized by normal biochemical pathways, alongside their unique magnetic properties, also makes SPIONs useful for a range of biological applications, including hyperthermia and magnetic targeting.7–10 For example, SPIONs have been explored for the targeted delivery of chemotherapeutics or for local temperature-induced apoptosis.11 Recent studies into the co-delivery of drugs and SPIONs showed excellent targeting efficiency and MRI contrast coupled with minimal toxicity.12,13 However, few SPION-based systems have received regulatory approval (e.g., GastroMARK®, Feridex®, Resovist® and Feraheme®), and of those that have been approved a number have subsequently been withdrawn from the market.14,15SPIONs are highly sensitive to external conditions: for instance, they can be oxidised in an acidic environment, and they have a tendency to aggregate.16 Hence, for biological applications, SPIONs are often coated or surface modified with a biocompatible polymer, such as polyethylene glycol (PEG), polylactic-co-glycolic acid (PLGA) or polyvinyl alcohol (PVA), or natural materials such as dextran, heparin, gelatin or chitosan.17,18 This approach provides protection from aggregation or degradation, and also offers the opportunity for multifunctionality, such as the loading of a therapeutic active ingredient or specific location targeting.19
Electrospinning is a straightforward technique which can produce polymer-based nanoscale fibres via the application of an electrical field to a polymer solution.20 It has been widely explored to produce materials for a variety of fields, including tissue engineering, biosensors, wound dressings, and drug delivery.21–23 Only a few studies have probed the incorporation of SPIONs into electrospun fibres for biological applications, but there is clear promise: Huang et al. reported polystyrene fibres with a high loading capacity of SPIONs, with the resultant formulation effective in killing cancer cells via magnetic hyperthermia.24 In other work, Wang et al. revealed that drug-loaded dehydroxypropyl methyl cellulose phthalate and cellulose acetate fibres encapsulating SPIONs demonstrated superparamagnetism at room temperature, indicating the feasibility of magnetic-field induced release.25 Exploitation of the MRI activity of SPIONs for monitoring drug release from electrospun fibres, on the other hand, has never been previously reported, despite its clinical utility and promise. The environment surrounding SPIONs is crucial to their MRI signal boosting capabilities, with diffusive water access to nearby particle surfaces providing strong signal enhancement. SPIONs encapsulated within electrospun fibres are therefore expected to have significantly lower proton relaxation rate enhancement compared to non-encapsulated particles. This difference in signal could be exploited as a mechanism for monitoring the dissolution of the fibres, and quantifying both SPION and loaded active ingredient release.
Zhu et al. recently reported PLGA nanoparticles loaded with the anti-cancer drug doxorubicin and SPIONs, and found these to be potent for non-invasive MRI monitoring of drug delivery both in vitro and in vivo after intratumoral injection.26 We were interested here to develop a formulation which could be given orally, rather than requiring an invasive injection for delivery. To this end, SPIONs and carmofur (an adjuvant chemotherapy for colon cancer, employed here as a model anticancer drug) were loaded into pH-responsive fibres prepared via electrospinning, to permit both pH-responsive drug delivery and concurrent MRI-based monitoring of drug release for application in the small intestine and colon. Two pH responsive polymers (Eudragit L100 and S100, methacrylic acid/methyl methacrylate copolymers which are only soluble in water at pH > 6.0 or >7.0, respectively) were used to form the fibre filaments. These polymers can protect the SPIONs from the acidic conditions encountered in the gastric fluid following oral administration, and later dissolve to release carmofur and expose the SPIONs (Scheme 1). The fibres were fully characterised, and their drug release and proton relaxivities investigated in detail.
 | ||
| Scheme 1 The strategy underpinning nanoplatform design in this work. | ||
Results and discussion
Physical and structural properties
SPIONs were initially synthesised using co-precipitation and stabilised by polyvinylpyrrolidone (PVP) to prevent aggregation. The as-prepared PVP-stabilised SPIONs (PVP-SPIONs) possessed a mean size of 8.5 ± 2.7 nm, as determined by transmission electron microscopy (TEM; Fig. 1a and b). The X-ray diffraction (XRD) patterns of the SPIONs and PVP-stabilised SPIONs (Fig. 1c) match with the cubic cell of iron oxide: the reflections at 30.2, 35.5, 43.7, 53.6, 57.1 and 62.9° can be readily indexed to the (022), (311), (004), (333), (115), and (044) planes of the cubic inverse spinel Fe3O4 (ICSD entry 77592). TGA of the PVP-SPIONs (Fig. S1, ESI†) revealed weight loss of 3.4 wt% between 40 and 170 °C, due to the removal of physisorbed water, and a weight loss of 6.2 wt% between 170 and 500 °C which can be attributed to the degradation of the PVP stabiliser. SPIONs therefore comprise around 90 wt% of the PVP-SPIONs mass.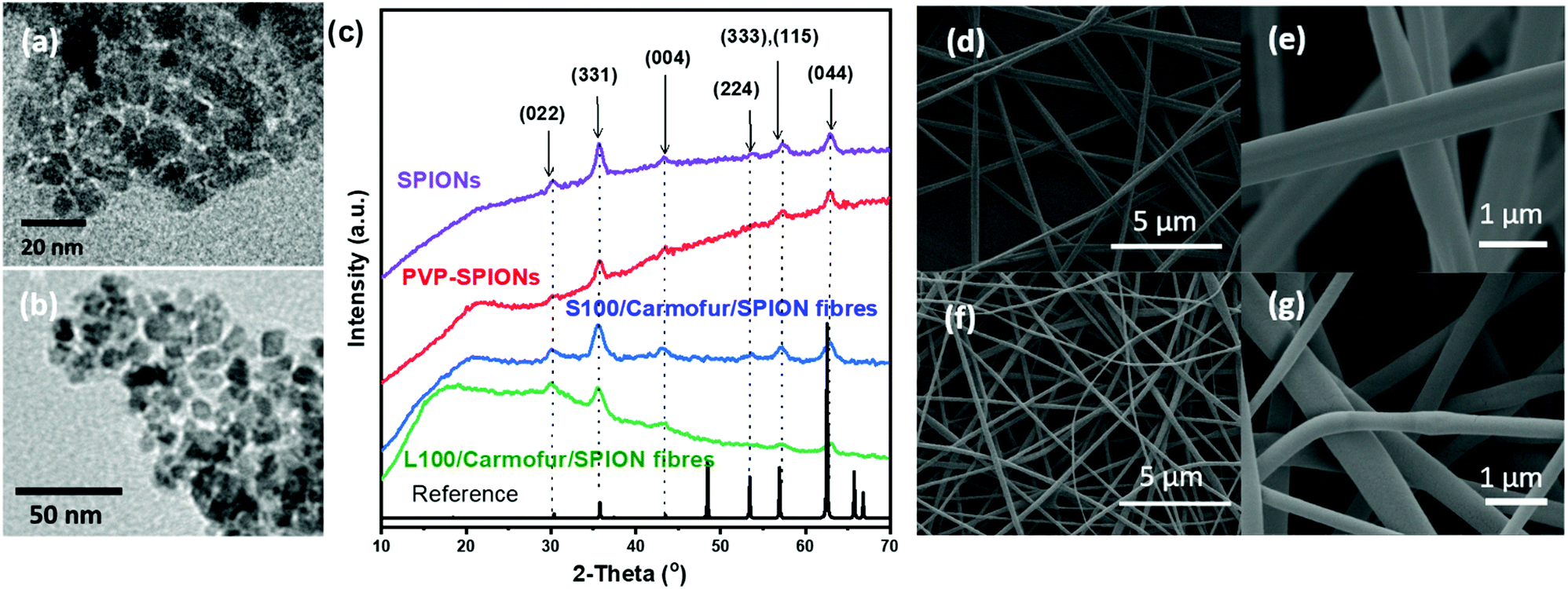 | ||
| Fig. 1 (a and b) TEM images of PVP-SPIONs (8.5 ± 2.7 nm); (c) XRD patterns of SPIONs, PVP-SPIONs and L100/Carmofur/SPION and S100/Carmofur/SPION fibres, with ICSD entry 77592 representing cubic spinel iron oxide (black and dashed lines); SEM images of (d) and (e) L100/Carmofur/SPION (645 ± 225 nm) and (f) and (g) S100/Carmofur/SPION (454 ± 133 nm) fibres. | ||
PVP-SPIONs and carmofur were encapsulated within pH-responsive Eudragit L100 or S100 nanofibres via electrospinning. Carmofur is a clinically approved antineoplastic agent used to treat breast and colorectal cancer. It is an oral derivative of fluorouracil and will be metabolised to 5-fluorodeoxyuridine monophosphate in vivo, causing interference with RNA and DNA synthesis.27,28 L100 and S100, anionic copolymers based on methacrylic acid and methyl methacrylate, are only soluble in water at pH > 6.0 or > 7.0, respectively. The resulting L100/Carmofur/SPION and S100/Carmofur/SPION composite fibres have uniform linear morphologies and smooth surfaces (Fig. 1d–g), with mean fibre diameters of 645 ± 225 and 454 ± 133 nm respectively (Fig. S2a, ESI† and Fig. 2b). The smaller diameter of the S100/Carmofur/SPION composites is likely due to the lower polymer concentration used for electrospinning (10% and 12% w/v for Eudragit S100 and Eudragit L100, respectively). This difference in solution concentration was necessary due to the gel-like consistency of Eudragit S100 at high concentrations, which can clog the needle.
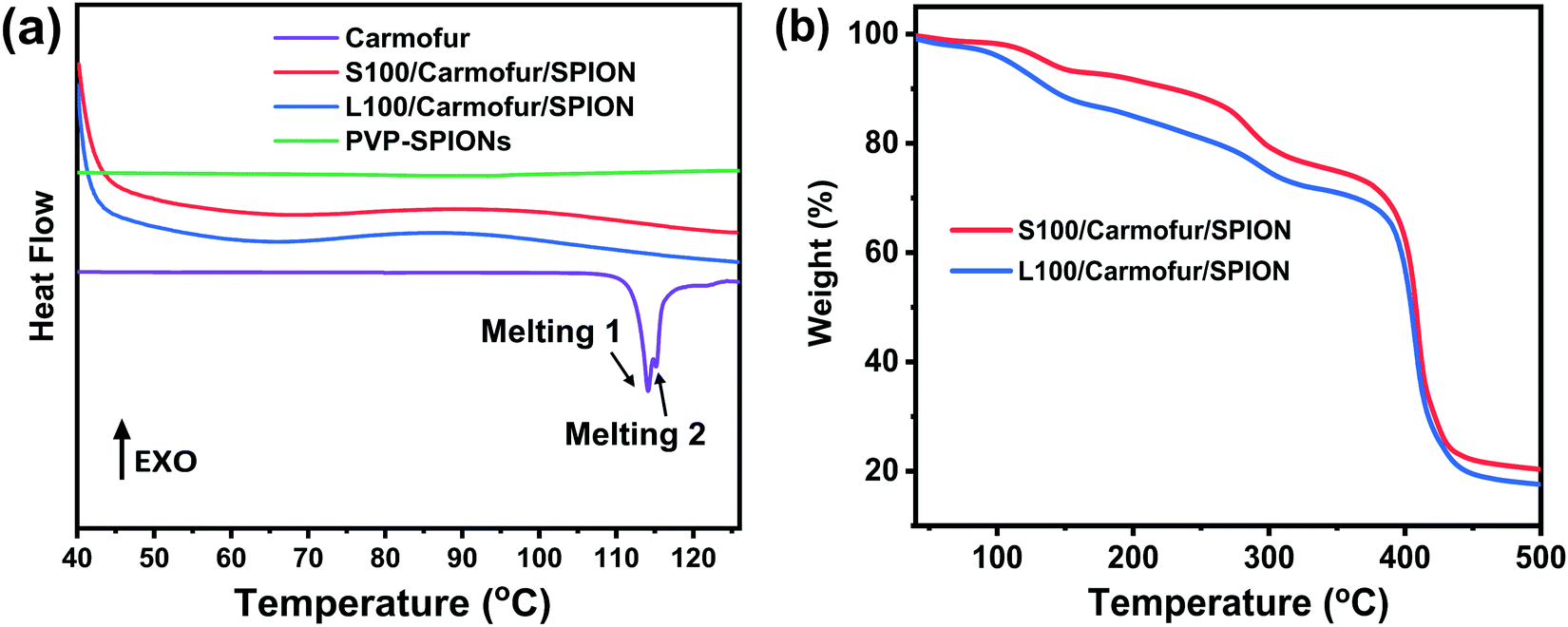 | ||
| Fig. 2 (a) DSC profiles of crystalline carmofur and S100/Carmofur/SPION and L100/Carmofur/SPION fibres; (b) TGA curves of the fibres. | ||
XRD patterns (Fig. 1c) of the fibres show the characteristic reflections of cubic iron oxide, suggesting successful incorporation of PVP-SPIONs. The polymer raw materials are amorphous, displaying only a broad background between 10–30° (see Fig. S3, ESI†). The characteristic reflections of carmofur (Fig. S3, ESI†) are not observed in the fibres’ XRD patterns, demonstrating that it is likely present in the amorphous form in the electrospun composite, owing to the very rapid drying which occurs during electrospinning.29
The XRD findings are supported by differential scanning calorimetry (DSC) analysis (Fig. 2a), where pure carmofur clearly exists as a crystalline material with two sharp endothermic melting peaks visible at ca. 114 and 115 °C, consistent with the literature.30 The raw Eudragit polymers display a broad endothermic peak between 60 and 120 °C (Fig. S4, ESI†), attributed to the loss of adsorbed water. No events can be observed in the DSC profile for PVP-SPIONs (Fig. 2a). S100/Carmofur/SPION and L100/Carmofur/SPION fibres show broad shallow endotherms between 40 to 80 °C, which can be ascribed to a loss of solvent (ethanol or DMAc, employed during preparation, or adsorbed water). The absence of the carmofur melting endotherm in the DSC curves of the drug and PVP-SPION loaded fibres confirms that it is present in the form of an amorphous solid dispersion.30
TGA curves indicate the presence of both the SPIONs and carmofur within the fibres. S100/Carmofur/SPION and L100/Carmofur/SPION fibres display multistage decomposition steps (Fig. 2b). The small mass loss of about 3% before 110 °C for both composite fibres can be attributed to solvent loss (e.g., physisorbed water). Two subsequent decomposition steps, between 100–160 °C and 220–300 °C, can be attributed primarily to loss of the loaded carmofur (TGA data for pure carmofur are shown in Fig. S5a, ESI†). A small mass loss between 170 to 500 °C is caused by removal of PVP from the loaded PVP-SPIONs (it is coincident with the mass loss in the PVP-SPIONs TGA trace in Fig. S1a, ESI†). The final stage of decomposition between 330 to 460 °C causes a weight loss of around 54%, and mainly arises due to degradation of Eudragit (see Fig. S5a for the raw Eudragit TGA data).
From the TGA of the fibres from three independent measurements (Fig. 2b and Fig. S5b, c, ESI†), we can calculate that the iron oxide content is around 20% w/w (17.3 ± 0.34% w/w for L100/Carmofur/SPION and 20.6 ± 0.26% for S100/Carmofur/SPION; mean ± S.D., n = 3). This is slightly higher than the theoretical loading (15.4% w/w for L100 and 17.9% w/w for S100 fibres). This discrepancy arises because there is a small proportion of residual decomposition products (expected to comprise carbon; around 3%) which remain at 500 °C. The carmofur loadings of the L100/Carmofur/SPION and S100/Carmofur/SPION fibres were measured by UV-vis spectroscopy and calculated to be 7.5 ± 0.4% and 8.0 ± 0.4% (mean ± S.D., n = 3), with encapsulation efficiencies of 96.1 ± 4.9% and 100.1 ± 5.4% respectively (mean ± S.D., n = 3).
Fourier-transform infrared (FTIR) spectra of the L100/Carmofur/SPION and S100/Carmofur/SPION fibres, as well as the raw materials, are given in Fig. S6 (ESI†). Raw carmofur displays bands at 1660–1720 cm−1 and at 1495 cm−1 arising from C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching vibrations. Eudragit L100 and S100 are both co-polymers of methacrylic acid and methyl methacrylate, and share similar spectra, with a characteristic stretch at 1727 or 1726 cm−1 from CO vibrations of esterified carboxylic groups, as well as stretches corresponding to ester groups between 1148 to 1251
O stretching vibrations. Eudragit L100 and S100 are both co-polymers of methacrylic acid and methyl methacrylate, and share similar spectra, with a characteristic stretch at 1727 or 1726 cm−1 from CO vibrations of esterified carboxylic groups, as well as stretches corresponding to ester groups between 1148 to 1251![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) cm−1. The presence of bands between 2900 and 2990cm−1 can be attributed to the stretching vibrations of methylene groups. For the S100/Carmofur/SPION and L100/Carmofur/SPION fibres, the characteristic stretches of the polymer can be clearly identified, but with shifts in their positions. The L100 and S100 CO stretching vibrations at 1726 or 1727 cm−1 in the raw polymer move to 1721 or 1718 cm−1 respectively in the fibres. These bands also become broader, as they incorporate the stretches of carmofur between 1666–1720 cm−1. This suggests the successful incorporation of carmofur in these fibres.29
cm−1. The presence of bands between 2900 and 2990cm−1 can be attributed to the stretching vibrations of methylene groups. For the S100/Carmofur/SPION and L100/Carmofur/SPION fibres, the characteristic stretches of the polymer can be clearly identified, but with shifts in their positions. The L100 and S100 CO stretching vibrations at 1726 or 1727 cm−1 in the raw polymer move to 1721 or 1718 cm−1 respectively in the fibres. These bands also become broader, as they incorporate the stretches of carmofur between 1666–1720 cm−1. This suggests the successful incorporation of carmofur in these fibres.29
pH responsive properties
Non-encapsulated SPIONs are sensitive to highly acidic environments such as those found in the stomach, resulting in their oxidation, eventual dissolution, and loss of magnetic properties. Their encapsulation in Eudragit-based fibres is expected to overcome this issue, with the polymers providing protection from acidic environments due to their lack of solubility at low pH.31 In order to test their stability, 10 mg of S100/Carmofur/SPION or L100/Carmofur/SPION fibres were incubated in 25 mL of pH 1.5 aqueous HCl solution, similar to the pH of gastric liquids.32 To compare their acid stability with bare nanoparticles, 2 mg of PVP-SPIONs was subjected to the same treatment. The release of Fe ions was measured using an o-phenanthroline colorimetric assay after incubation at 37 °C for 2 h, which mimics the gastric transit time. The λmax of the ferrous tris-o-phenanthroline product of dissolved iron and o-phenanthroline is around 512 nm at neutral pH. Fig. 3a shows that the [Fe] released from both types of fibres (green and blue line) after 2 h was below that of a control FeCl3 aqueous solution ([Fe] 1 mg L−1, black line), indicating less than 2 wt% of the total SPION content was degraded in the loaded fibres. This concentration is significantly lower than the [Fe] released from bare PVP-SPIONs at equivalent concentrations (between 1–10 mg L−1, equating to up to 17 wt% degradation, purple line). These results show that the Eudragit coating can protect the SPIONs from degradation in an acidic environment. Photographs (Fig. S7, ESI†) and SEM images (Fig. 3b–e) of the loaded fibres following acid incubation demonstrate the stability of the formulations: the morphology of the fibres appears largely unaffected after exposure to the acidic conditions, and the fibre size also remains similar, at 612 ± 227 nm for the L100/Carmofur/SPION fibres and 521 ± 166 nm for S100/Carmofur/SPION fibres (Fig. S2c and d, ESI†).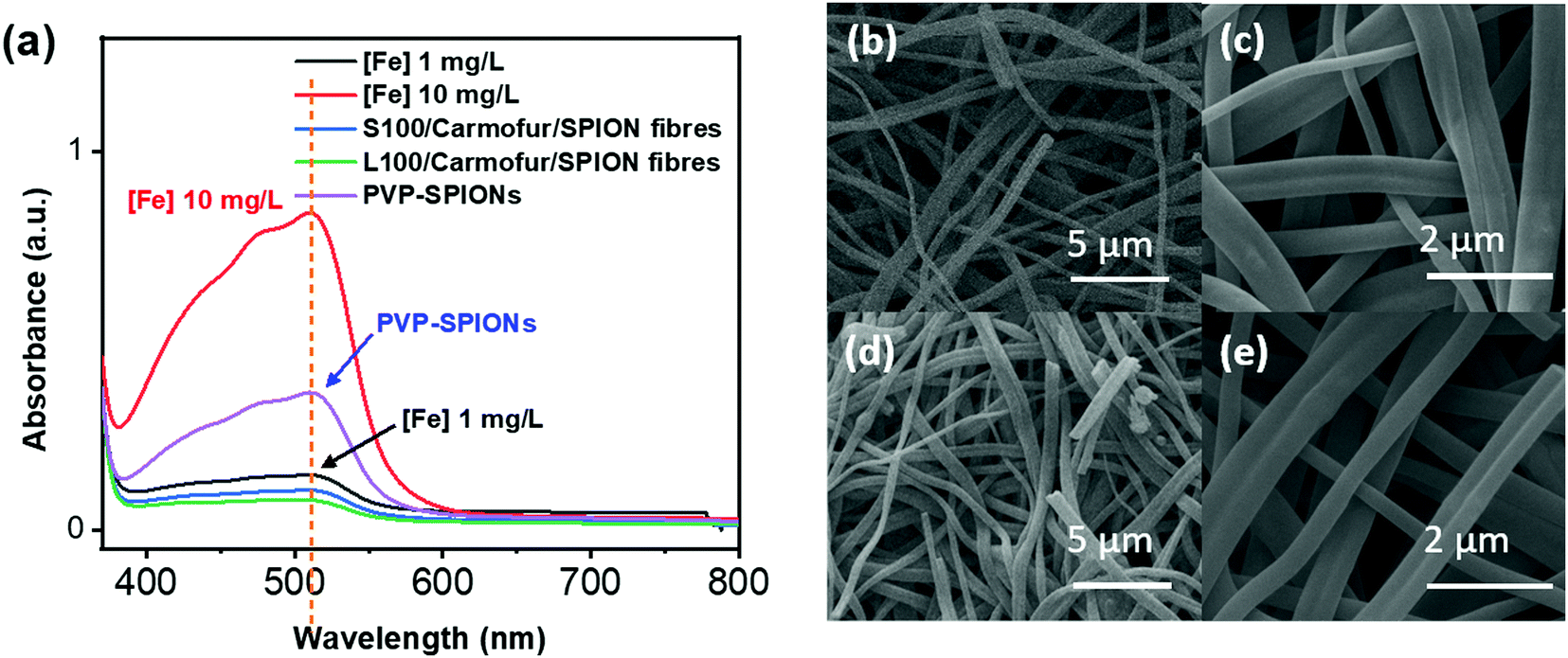 | ||
| Fig. 3 The stability of PVP-SPIONs and the fibres after immersion at pH 1.5 for 2 h. (a) The results of colorimetric assays to determine [Fe] in solution for PVP-SPIONs (purple), S100/Carmofur/SPION (blue) or L100/Carmofur/SPION (green) fibres with two control FeCl3 aqueous solutions (black and red, 1 and 10 mg L−1 respectively); SEM images of (b), (c) S100/Carmofur/SPION and (d), (e) L100/Carmofur/SPION fibres after immersion in pH 1.5 aqueous HCl for 2 h. | ||
In order to mimic the conditions encountered during oral delivery, where materials are likely to encounter a range of pH environments (gastric pH is highly acidic (pH 1.0–2.5), while the mean pH values in the proximal small intestine, colon and terminal ileum are 6.6, 7.0 and 7.5),33 drug release experiments were carried out at different pHs. Initially, S100/Carmofur/SPION and L100/Carmofur/SPION fibres were incubated at pH 1.5 (aqueous HCl solution) for 1 h, and then subsequently transferred to pH 6.5 or 7.4 PBS buffer. Carmofur release was monitored by UV-vis measurement of the supernatant after incubation (λ = 262 nm, Fig. 4). Both sets of fibres demonstrated release of around 35% of the drug content at pH 1.5 within the first hour. After introduction to PBS buffer, the L100/Carmofur/SPION fibres exhibit similar release patterns at both pH 6.5 and 7.4, with more than 90% release by 3 h and 100% release after 24 h. For the S100/Carmofur/SPION fibres, carmofur release reaches 80% after 3 h after at pH 7.4, while it takes longer (approximately 5 h) to reach this point at pH 6.5. This is due to the different pH at which L100 and S100 become water soluble. The polymer matrix in the S100 formulation remains insoluble at pH 6.5, and the drug can only reach the dissolution medium through diffusion or swelling of the polymer and the permeation of water into the centre of the fibres. At pH 7.4, S100 is water soluble and hence drug release is mediated by polymer dissolution. In contrast, L100 is water soluble at both pH 6.5 and 7.4 and therefore the release profiles are essentially identical at both pH values.
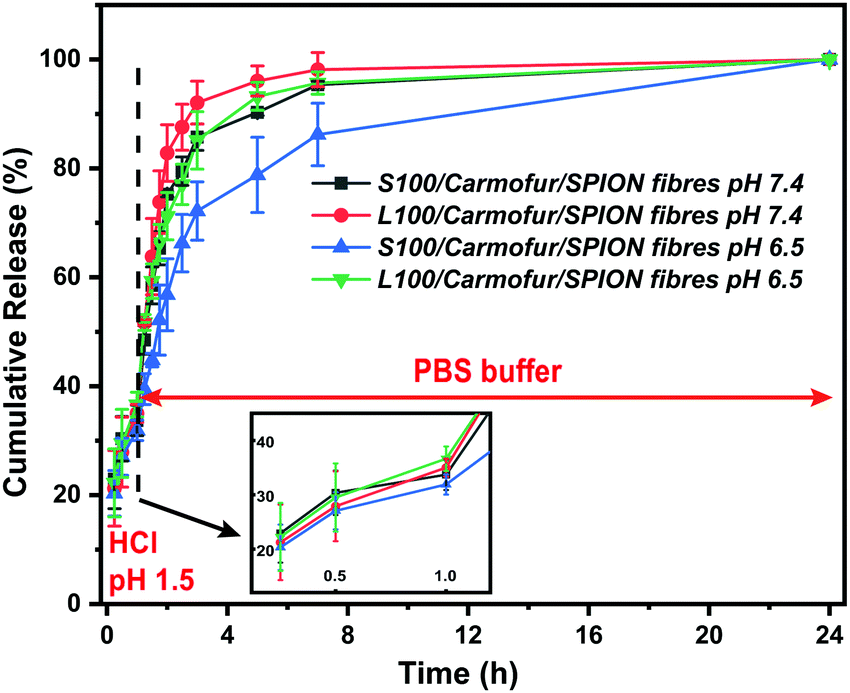 | ||
| Fig. 4 Plots showing the release of carmofur from the S100/Carmofur/SPION and L100/Carmofur/SPION fibres, as measured by UV-vis spectroscopy (λ = 262 nm). Data are given from three independent experiments as mean ± S.D. | ||
Dynamic light scattering (DLS) size distribution profiles further support the pH-sensitive dissolution properties of the fibres. After the 24 h drug release study, the particle size of the solid material in the dissolution medium was measured, and the results are shown in Fig. S8 and Table S1 (ESI†). At both pH 6.5 or 7.4, the L100/Carmofur/SPION fibres fully dissolve after 24 h and yield a dispersion with a mean particle size of 591 ± 32 nm and 380 ± 11 nm (n = 3), suggesting the amphiphilic copolymer might form micellar structures composed of a hydrophobic core and a shell with ionized carboxylate units.34 In contrast, the mean particle size of L100/Carmofur/SPION fibres dispersed at pH 5.5 for 24 h was significantly higher (3049 ± 41 nm), suggesting the polymer fibres remain intact and do not dissolve. S100/Carmofur/SPION composites displayed analogous results in DLS, with a mean size of 458 ± 31 nm at pH 7.4, indicating dissolution, and 2959 ± 176 nm at pH 6.5, suggesting that no dissolution is taking place (n = 3).
To probe the mechanism of drug release, the Peppas model (eqn (1)) was fitted to the drug release data.35Mt/M∞ (Qt) represents the extent of drug release, t is the elapsed time, k is a rate constant, and m gives information related to the mechanism of release.35
| Mt/M∞ = ktm | (1) |
The first 60% of the release data fit well with the model, as shown in Table 1 and Fig. S9 (ESI†). All the exponents at both pH values are in the range 0.45 to 0.89, indicating drug release occurs through a combination of matrix swelling and drug diffusion.35
| Fibres | pH | Peppas model | R 2 |
|---|---|---|---|
| L100/Carmofur/SPION | 7.4 | Q t = 73.8t0.75 | 0.99 |
| 6.5 | Q t = 52.7t0.53 | 0.98 | |
| S100/Carmofur/SPION | 7.4 | Q t = 61.6t0.75 | 0.99 |
| 6.5 | Q t = 35.6t0.77 | 0.91 |
SPIONs are typically T2 contrast agents, providing negative contrast by decreasing the transverse relaxation time of local protons. To explore the efficiency of their contrast behaviour when encapsulated within the composite fibres, the relaxivity (r2) of L100/Carmofur/SPION and S100/Carmofur/SPION fibres was initially measured in pH 7.4 PBS with 0.1% w/v xanthan gum and calculated according to eqn (2).
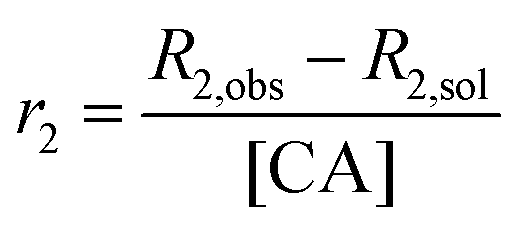 | (2) |
When fibres were immersed in the buffer, the initial relaxivity r2 value (measured after 10 min suspension at pH 7.4) was low (10.6 ± 1.9 mM−1 s−1 for L100/Carmofur/SPION, 12.0 ± 3.6 mM−1 s−1 for S100/Carmofur/SPION). The r2 values increased with time as the fibres remained suspended in buffer. This was due to the dynamic process of matrix dissolution/swelling, allowing water molecules access to the SPIONs, boosting diffusive water access and hence enhancing their relaxation rates and relaxivities. Therefore, instead of measuring a single r2 value, the fibres' r2 relaxivity was monitored as a function of time during incubation in different pH PBS (6.5 or 7.4) with 0.1% w/v xanthan gum at 37 °C. The resultant r2 relaxivity profiles can help to determine whether MRI signal could be utilised as a mechanism of monitoring fibre dissolution/swelling, and hence carmofur and SPION release.
Fig. 5a displays the transverse relaxivity (r2) profiles as a function of immersion time at different pH. Due to the very low initial relaxivity values, profiles were monitored from 10 min immersion onwards. For L100/Carmofur/SPION fibres, the transverse relaxivity increased rapidly and displayed pH-responsive behaviour. At pH 7.4, r2 rose from 29.3 ± 8.3 mM−1 s−1 after 10 min to 66.9 ± 2.7 mM−1 s−1 over 40 min and subsequently reached 69.8 ± 2.5 mM−1 s−1 after 3 h, due to dissolution/swelling and concomitant water access boosting relaxation rates as previous described. At pH 6.5, the starting relaxivity value at 10 min (13.5 ± 2.0 mM−1 s−1) was much lower than that measured at pH 7.4, which is possibly because the L100 polymer is more hydrophilic at elevated pH owing to the ionisation of carboxylic acid groups. This facilitates diffusive water access to SPIONs and thus promotes transverse relaxation in the suspension.34 The relaxivity increase is also slower at pH 6.5, with r2 reaching 42.1 ± 3.0 mM−1 s−1 after 3 h. After 3 h of immersion, all the L100/Carmofur/SPION fibres had dissolved at both pH values, and the resultant solutions were clear (Fig. S10, ESI†). However, the relaxivity value at pH 6.5 is notably lower than at pH 7.4 after 3 h. This can be attributed to the larger particle size of the dissolved L100/Carmofur/SPION formulation at pH 6.5 (Fig. S8, ESI†). The amphiphilic polymer might form micelle structures containing the SPIONs at pH 6.5, with reduced diffusive water access meaning lessened relaxivitiy enhancement, whereas at pH 7.4 the smaller particle size results in a higher surface area to volume ratio and a greater surface area for SPION–water interactions.
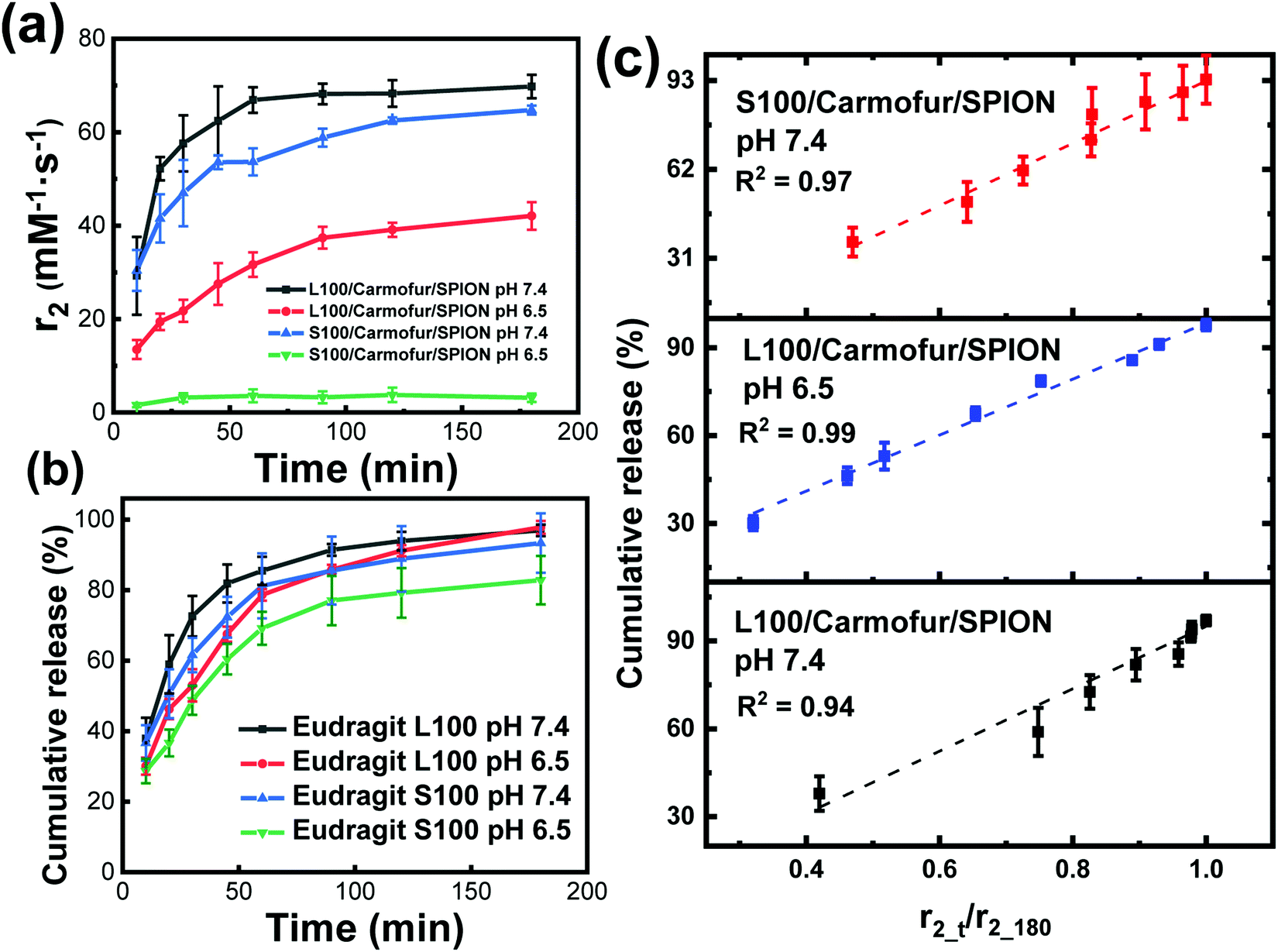 | ||
| Fig. 5 (a) The change in transverse relaxation (r2) and (b) carmofur release from the S100/Carmofur/SPION and L100/Carmofur/SPION fibres as a function of incubation time in PBS buffers with 0.1% w/v xanthan gum. (c) The relationship between relaxation behaviour (r2_t/r2_max) and cumulative carmofur release. | ||
The S100/Carmofur/SPION fibres displayed a similar relaxivity profile with respect to time at pH 7.4, with r2 values of 30.4 ± 4.4 mM−1 s−1 after 10 mins and 64.7 ± 1.0 mM−1 s−1 after 3 h. The r2 values are overall slightly lower than those observed for L100/Carmofur/SPION at the same pH, which again can be attributed to the hydrodynamic size in the suspension following dissolution. By plotting the r2 values against the hydrodynamic diameter measured earlier (Fig. S8 and S11, ESI†), a clear inverse correlation is revealed. This is consistent with the literature, where smaller particles with increased surface areas allow improved water access to the magnetic components, leading to boosted relaxivities.17 As previously noted, the S100-based fibres are insoluble at pH 6.5 (see Fig. S8 and S10, ESI†), and thus the r2 value remains low throughout the experimental time as the SPIONs remain encapsulated, preventing their effective magnetic interaction with diffusive water protons.
According to the relaxivity profiles, it is clear that L100/Carmofur/SPION composite fibres dissolve at pH 6.5 and 7.4 and S100/Carmofur/SPION fibres dissolve at pH 7.4 only, resulting in the release and potential micellisation of SPIONs, and hence increasing relaxivity due to increased water diffusive access to magnetic centres. On the other hand, the S100/Carmofur/SPION fibres do not demonstrate dissolution at pH 6.5 (but show some evidence of swelling) and extended immersion at this pH has little effect on the relaxivity. To prove that SPIONs were released from the fibres as proposed, the Fe concentration in solution was determined (Fig. S12, ESI†). The solution [Fe] concentration vs. time plot mirrors the shape of the r2 recovery profile, providing evidence that r2 is related to the release of SPIONs from the fibres. Clear linear correlations also can be observed between the concentration of Fe in the supernatant and the relaxivity (Fig. S13, ESI†). Thus, the recovery of r2 can be regarded as a kinetic process which is proportional to the dissolution of the Eudragit fibres.
In order to directly compare drug release with the relaxivity data, carmofur release from the fibres was quantified in PBS with 0.1% xanthan gum, using UV-vis spectroscopy alongside relaxivity changes (Fig. 5b). The presence of xanthan gum causes the release milieu to have a gel-like consistency, which makes it impossible to transfer between different pH values; thus, experiments were performed only at pH 6.5 or 7.4, with no initial acid stage. All the fibres displayed a rapid release of carmofur in 3 hours, consistent with the in vitro drug release tests performed without xanthan (Fig. 4). When the pH is above that at which the Eudragit dissolves, the carmofur release profile closely resembles the change in r2 with time. To explore the relationship between carmofur release and relaxivity at these pH values, plots of cumulative carmofur release vs. r2 were constructed. These reveal a clear linear correlation between the two parameters, with R2 ranging from 0.94 to 0.99 (Fig. S14 and Table S2, ESI†). In contrast, for the S100/Carmofur/SPION fibres at pH 6.5, a poor linear correlation (R2 = 0.83; Fig. S14, ESI†) was observed. This arises because the fibres are insoluble at this pH, so SPION release is minimal and the r2 value remains low throughout the experimental period. This indicates that changes in r2 signal directly correspond to carmofur release when the pH is above that at which the Eudragit dissolves, meaning that MRI could be exploited as a non-invasive means of monitoring in situ drug release from such fibres in environments such as the small intestine and colon. It should be noted that in the gastrointestinal tract the presence of bile salts, or potentially strong osmolarity in the colon, could affect the Eudragit dissolution process.36 However, it is clear from our data that there is a strong correlation between the extent of carmofur release and the r2 signal in pH conditions where the polymer is soluble, regardless of the rate of dissolution. Thus, these additional complexities in vivo are not expected to confound the findings presented here. Our approach could hence provide a non-invasive route to quantification of drug release at a site of interest, and could prove particularly helpful in treatments using highly toxic chemotherapy.
The MR signal intensity is related both to the relaxivity properties of the CA and also to its local concentration. However, the equations built considering r2_t (r2 at time t) and cumulative carmofur release (Table S2, ESI†) only take the relaxivity into consideration. In the clinic, the local CA concentration might differ as a result of varied dosages, body volumes or other pathological conditions. Hence, the r2 values in each system were normalised by calculating r2_t/r2_max. The r2_max is the maximum relaxivity value possible with the formulation, which manifests in our experiments as the relaxivity after 180 min, r2_180. Plots of drug release percentage vs. r2_t/r2_180 (Fig. 5c and Fig. S15, ESI†) reveal direct proportionality (except with the S100/Carmofur/SPION fibres at pH 6.5), and the normalised equations are given in Table 2. In the clinic, r2_t could be regarded as the local MR signal intensity at a certain time point, and r2_max as the theoretical maximum signal intensity, a constant related to the specific formulation, dose and individual. Compared to the equations built with only r2_t and cumulative carmofur release (Table S2, ESI†), these normalised equations also show good correlation coefficients but can be more universally applied.
| Formulation | pH | Fitting equation | R 2 |
|---|---|---|---|
| Eudragit L100 | 6.5 |
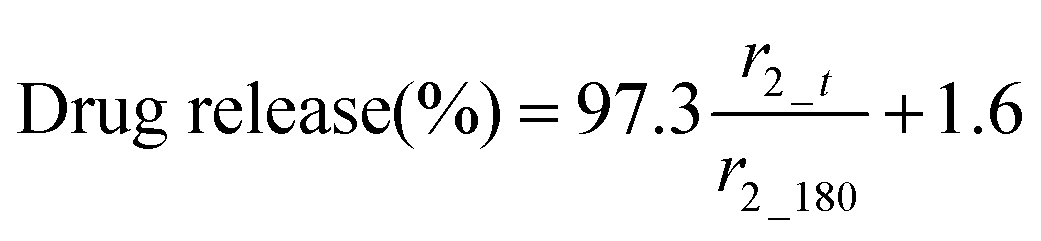
|
0.99 |
| 7.4 |
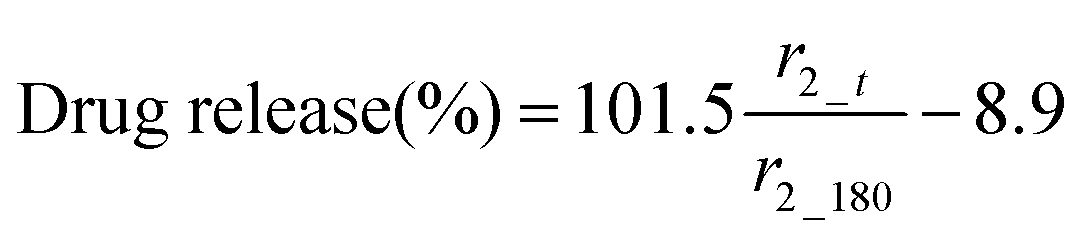
|
0.94 | |
| Eudragit S100 | 7.4 |
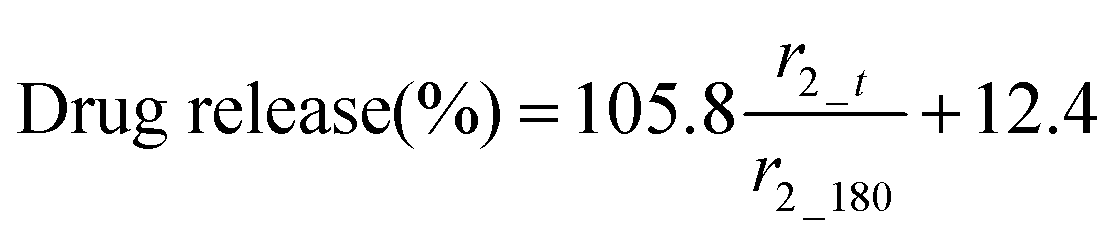
|
0.97 |
To further validate the predictive ability of the r2 data, the equations correlating carmofur release (%) with relaxation behaviour (Table 2) were applied to predict carmofur release in a new series of experiments. Relaxation behaviour changes were determined for L100/Carmofur/SPION and S100/Carmofur/SPION fibres at different concentrations (n = 3) and used to predict the extent of carmofur release. The latter was then quantified by UV-vis spectroscopy and compared with r2 predictions. The results are presented in Fig. 6. The predicted drug release curves based on r2 are very similar to the data obtained by UV-vis spectroscopy, indicating the potency of our theranostic approach. Two fit factors F1 (the difference factor) and F2 (the similarity factor) (eqn (3) and (4), respectively) were applied to statistically compare the experimental dissolution profiles determined by UV-vis measurement and those calculated based on relaxivity.37
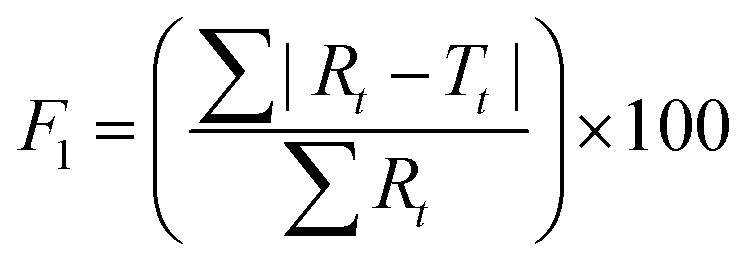 | (3) |
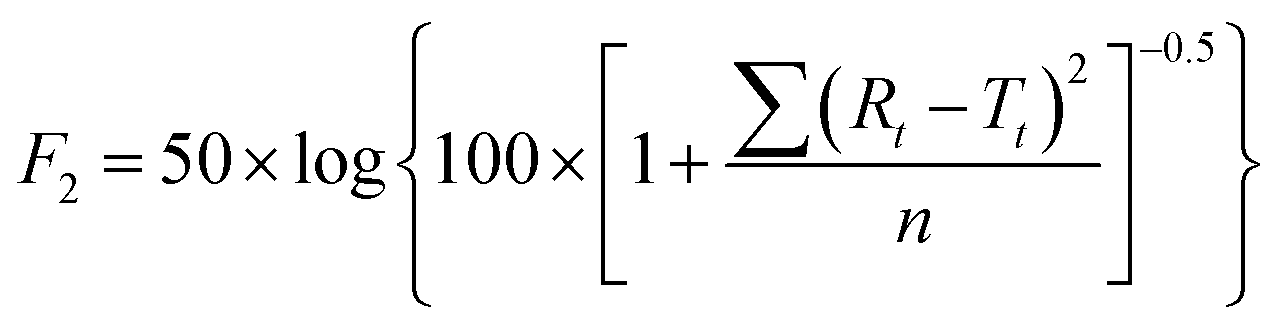 | (4) |
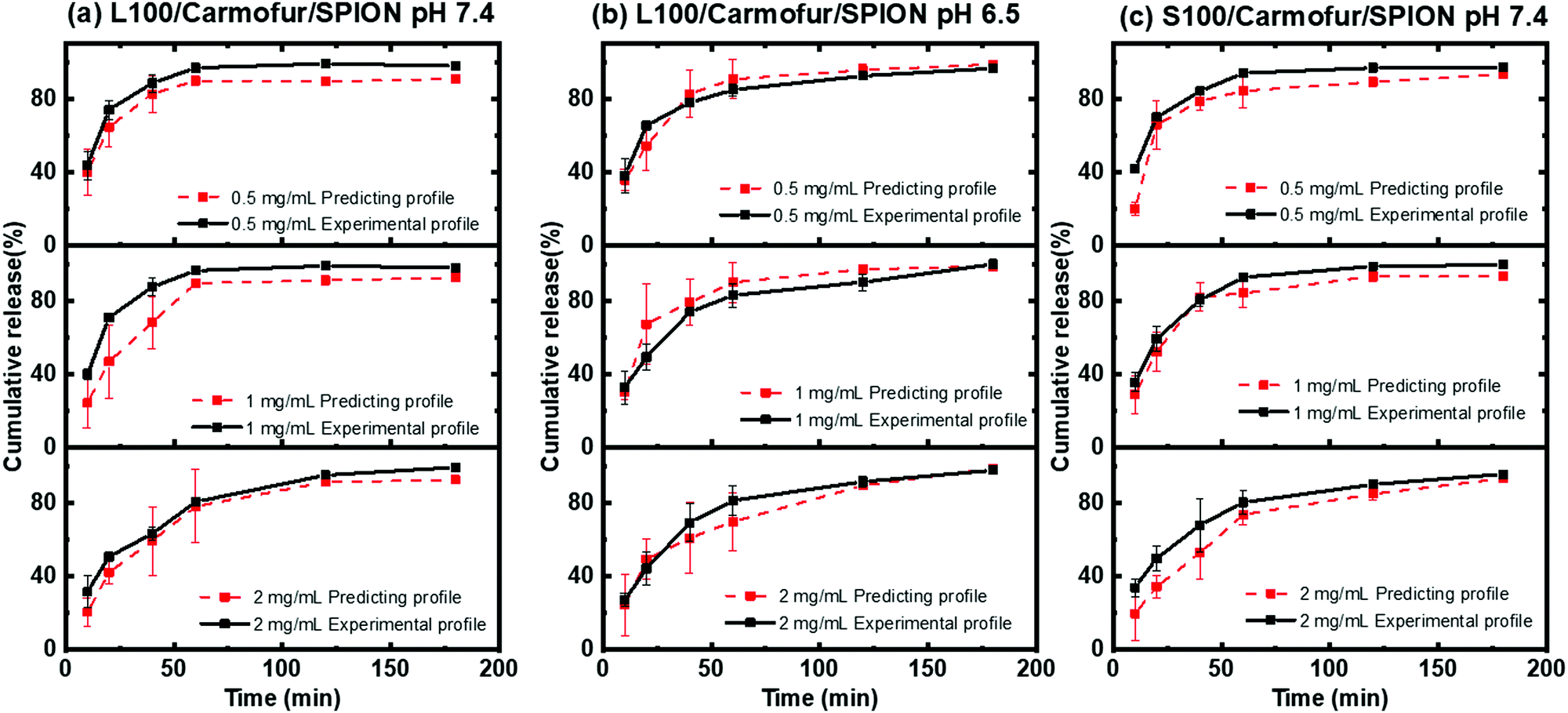 | ||
| Fig. 6 Plots of experimental (black) and predicted (red) carmofur release curves: (a) L100/Carmofur/SPION fibres at pH 7.4, (b) L100/Carmofur/SPION fibres at pH 6.5 and (c) S100/Carmofur/SPION fibres at pH 7.4. The predicted release data were calculated based on the relaxivity and the corresponding experimental release data were obtained by UV-vis measurement. Experiments were performed in PBS at 37 °C with different fibre concentrations (as labelled). | ||
Here we use the experimental release data obtained by UV-vis spectroscopy as the reference (Rt), and the predicted data as the test (Tt). The results are shown in Table 3. The F1 values of L100/Carmofur/SPION fibres at pH 6.5 and S100 Carmofur/SPION fibres at pH 7.4 lie in the range of “equivalent”, while for the L100 fibres at pH 7.4 most of the F1 values are also consistent with equivalent release patterns. In terms of similarity factor F2, the majority of the values are greater than 50. All these results suggest the reliability of our predicted curves.
| Sample | Concentration (mg mL−1) | F 1 | F 2 |
|---|---|---|---|
| L100/Carmofur/SPION pH 7.4 | 2 | 8.71 | 47.6 |
| 1 | 16.1 | 59.6 | |
| 0.5 | 8.58 | 48.3 | |
| L100/Carmofur/SPION pH 6.5 | 2 | 7.49 | 59.6 |
| 1 | 9.54 | 52.8 | |
| 0.5 | 6.48 | 61.6 | |
| S100/Carmofur/SPION pH 7.4 | 2 | 14.1 | 58.1 |
| 1 | 7.69 | 41.2 | |
| 0.5 | 10.9 | 56.2 |
Novel pH-responsive fibres loaded with SPIONs and carmofur were fabricated in this work to permit oral delivery of a drug cargo to the intestine and colon. Predictive curves were established to correlate MRI signal intensity and drug release, thereby ultimately allowing non-invasive monitoring of local drug release. Drug delivery for the treatment of colon cancer remains a very significant challenge owing to the complicated colon physiology and environmental barriers.40 The release and absorption of chemotherapeutics can be markedly affected by the changeable local environments and variable colonic residence time, making it difficult ensure that effective and safe doses are provided.40 Hence, the fact that our formulations provide local release information offers great potential benefits to control the delivered dose and provide bespoke and personalised therapy in the clinic. This can ensure that patients are given an appropriate dose to treat their disease without experiencing dangerous or unpleasant side effects. Unlike previous work,26,41 our fibres display pH-responsive relaxation behaviour around the physiological pH, potentially allowing them to be used to image abnormal local microenvironments in intestinal and colon cancer.
Conclusions
Composite pH responsive nanofibres have been prepared through electrospinning in this work. SPIONs (a negative MRI contrast agent) and carmofur (a model drug) were incorporated into polymer fibres composed of pH-responsive and biocompatible Eudragit polymers. Fibres with smooth cylindrical morphologies were generated, with an amorphous dispersion of carmofur. The encapsulation of SPIONs in the fibres led to effective protection from digestion in the acid environment of the stomach, and in vitro drug release studies reveal rapid release of carmofur at the pH values typical of the small intestine and colon. These results make our formulations promising as oral-delivery systems for colonic cancer. The fibres also exhibit pH responsive relaxation behaviour around the physiological pH range, making them ideal candidates for the development of ultra-sensitive reporters to detect abnormal microenvironments in the small intestine and colon.Further investigation of the fibres’ relaxivity behaviour showed them to have pH responsive r2 profiles closely correlated to the extent of drug release. On that basis, a novel quantification method allowing drug release to be monitored via proton relaxation changes was established and used to predict with a high degree of accuracy the carmofur release profiles in a new series of experiments. This offers the exciting possibility to non-invasively monitor the extent of drug release in situ. As most chemotherapeutic agents are cytotoxic and non-specific, their safety remains a critical issue and hence our formulations potentially open up a new route to dramatically decrease off-target side effects in chemotherapy.
Experimental
Chemicals were purchased as follows: sodium hydroxide and hydrous ethanol (Fisher Scientific Ltd); sodium chloride, N,N-dimethylacetamide (DMAc), acetone, anhydrous ethanol, polyvinylpyrrolidone (PVP; 40 kDa), hydroxylamine hydrochloride, o-phenanthroline, xanthan gum, FeCl3·6H2O and FeCl2·4H2O (Sigma-Aldrich); Eudragit L100 and S100 (Röhm GmbH); and, Carmofur (ChemCruz™). Ultrapure water was collected from a Millipore MilliQ system operated at 18.2 MΩ.FeCl3·6H2O (6.5 g, 0.024 mol) and FeCl2·4H2O (2.48 g, 0.012 mol) were dissolved in 25 mL of deoxygenated ultrapure water. This solution was added dropwise into 250 mL of an aqueous NaOH solution (0.5 M) at 40 °C, and stirred for 1 h at this temperature. The resultant SPIONs were washed by centrifugation with DI water until the supernatant was pH neutral, and the resultant black precipitate dried under vacuum. For PVP stabilisation, SPIONs (100 mL, 10 mg mL−1 in water) were mixed with 2 mL of an aqueous PVP 40k solution (25.6 g L−1, 0.64 mM), and the suspension was shaken (100 rpm) at room temperature. After 24 h, the suspension was mixed with 500 mL of aqueous acetone (H2O/acetone, 1:10 v/v) and centrifuged at 13200 rpm for 20 min. The supernatant was removed, and the resultant black precipitate washed with ethanol and dried in an oven at 50 °C for 24 h.
A 12% (w/v) solution of Eudragit L100 and 10% (w/v) solution of Eudragit S100 were prepared in a mixture of DMAc and ethanol (2:8 v/v) and stirred vigorously for 24 h. Carmofur (to give final concentrations of 12 or 10 mg mL−1) and PVP-SPIONs (at final concentrations of 24 or 20 mg mL−1) were then added to the Eudragit L100 or S100 solutions respectively, with sonication for 20 min. An HCP 35–35000 power supply (FuG Elektronik GmbH) was used to generate an electric field. A 5 mL plastic syringe fitted with a narrow-bore stainless-steel needle (18 G, with outer and inner diameter of 1.25 and 0.838 mm, respectively) was filled with the required working solution. The spinneret was connected to the positive electrode of the power supply via an alligator clip and a flat plate aluminium collector attached to the grounded electrode. The working solution was dispensed with the aid of a syringe pump (KDS 100, Cole-Parmer) under ambient conditions (22 ± 3 °C and relative humidity 40 ± 5%), with a flow rate of 1.0 mL/h. The applied voltage and the distance from the spinneret to collector were set to 16 kV and 15 cm, respectively.
A MiniFlex 600 diffractometer (Rigaku) supplied with Cu-Kα radiation was used to collect XRD patterns (λ = 0.15418 nm, 40 kV, 15 mA). Patterns were recorded over the 2θ range from 3 to 70° (step = 0.01°). The morphology of the fibres was analysed with a field emission scanning electron microscope (FEI Quanta 200F) connected to a secondary electron detector (Everheart-Thornley Detector-ETD). Samples were coated with a 20 nm gold sputter (using a Quorum Q150T coater) before measurement. The size distribution of the fibres was determined from the SEM micrographs by measuring the fibres at >100 points in the images, with the aid of the ImageJ software (version 1.52s, National Institutes of Health).
TEM images were obtained on a JEOL JEM-1200 microscope operated at 120 kV with a beam current of ca. 80 mA. A Gatan Orius 11-megapixel camera was used to take images. TEM samples were prepared by depositing a drop of PVP-stabilised SPIONs in aqueous suspension on a formvar-coated 300-mesh copper grid, and then drying the grids in the oven (45 °C). Average particle size was measured with the ImageJ software (version 1.52s, National Institutes of Health).
Thermogravimetric analysis was undertaken on a Discovery instrument (TA Instruments, Waters LLC). Ca. 3 mg of each sample was loaded into an aluminium pan and heated from 40 to 500 °C at 10 °C min−1 under a nitrogen flow of 25 mL min−1. Data were recorded using the Trios software and analysed with TA Universal Analysis. A Q2000 DSC (TA Instruments, Waters LLC) was used for calorimetry. A small amount of sample (approximately 3 mg) was loaded in a non-hermetically sealed aluminium pan (T130425, TA instruments) and DSC experiments carried out from 40 to 126 °C, with a temperature ramp of 10 °C min−1 and nitrogen purge of 25 mL min−1. DSC data were recorded with the TA Advantage software package and analysed using TA Universal Analysis.
For ICP-MS, samples (containing ca. 0.06 mg SPIONs) were digested using hot HNO3 digestion and then diluted to 10 mL with DI water. The iron concentrations (mM) were quantified on an Agilent 7500cx spectrometer.
An MQC+ benchtop NMR analyser (Oxford Instruments) was used to measure transverse relaxation times (T2) of protons at 37 °C and 23 MHz. The Carr–Purcell–Meiboom–Gill (CPMG) method was used to measure T2, with 4 scans per experiment. The water relaxation rate enhancement per mmol of contrast agent (relaxivity) is defined by eqn (2).
Stability studies were carried out by dispersing 10 mg of the nanofibres or 2 mg of PVP-SPIONs in 25 mL of an aqueous HCl solution (pH 1.5). Experiments were carried out in a shaking incubator (100 rpm) at 37 °C for 2 h. The resulting solutions were centrifuged for 15 min (13200 rpm) to sediment any undissolved fibres or particles. 5 mL samples of the supernatants were removed and neutralised with a few drops of aqueous 0.2 M NaOH. 0.9 mL of the neutralised sample was added to 0.3 mL of a 10% (w/v) hydroxylamine hydrochloride solution in water and 0.6 mL of an aqueous 0.2% (w/v) o-phenanthroline solution. Finally, UV-vis spectra were recorded on an Agilent Cary 100 spectrophotometer over the wavelength range 370 to 800 nm.
The loading capacity of carmofur (LC%) can be calculated as the amount of entrapped drug divided by the total fibre weight. Encapsulation efficiency (EE) is the percentage of the drug present that is successfully entrapped into the fibres. To calculate the loading and encapsulation efficiency of carmofur, 10 mg of the fibres (n = 5) was added into 10 mL of ethanol and sonicated until the polymer was fully dissolved. A PVDF-type syringe filter (0.22 μm) was used to filter the resultant solutions, and the filtrates centrifuged for 10 min (13200 rpm) to remove the SPIONs. The supernatants were analysed with UV spectroscopy at 262 nm (Cary 100 instrument, Agilent), and the LC and EE calculated based on a pre-determined calibration curve.
The carmofur release study was undertaken using a 50 mL suspension of fibres (∼0.5 mg mL−1). Samples were incubated in a pH 1.5 HCl solution for 1 h, and then transferred to pH 6.5 or 7.4 PBS. Experiments were undertaken in a shaking incubator (100 rpm) at 37 °C. 1 mL aliquots were withdrawn from the dissolution medium at predetermined time points and filtered through a PVDF-type syringe filter (0.22 μm). To maintain a constant volume, 1 mL of fresh pre-heated buffer was added to the dissolution vessel. The filtrates were centrifuged for 15 min (13200 rpm) to remove any SPIONs, and then analysed with an Agilent Cary 100 spectrophotometer. Carmofur quantifications were performed at λmax of 262 nm. Dilutions were undertaken when necessary to bring concentrations into the linear range of the calibration curve. Experiments were performed in triplicate and the results are reported as mean ± standard deviation (S.D.).
In a separate set of experiments, 5 mg of L100/Carmofur/SPION fibres was incubated in 10 mL of pH 5.5 acetate buffer under the same conditions as used for drug release (100 rpm, 37 °C). After 24 h, 2 mL aliquots were taken from each of the pH 7.4, 6.5 or 5.5 experiments for dynamic light scattering (DLS) measurement. For the L100/Carmofur/SPION system at pH 5.5 and S100/Carmofur/SPION at pH 6.5, where the fibres were aggregated in the form of mats, sonication was applied to ensure a homogenous suspension was obtained before taking aliquots and performing DLS measurements. To obtain the particle size data, a ZetaSizer ZS instrument (Malvern) fitted with a 4 mW He–Ne 633 nm laser module was used, and scattered light measured at 173° (back scattering). The attenuator position was selected automatically by the instrument and particle sizes are reported as the mean of 5 measurements. Each sample was analysed three times.
To monitor changes in proton relaxivity, a dispersion of approximately 10 mg of each fibre formulation in 8 mL of a 0.1% (w/v) aqueous xanthan gum solution was placed into a 10 mm-diameter NMR tube, which was held at 37 °C. The transverse relaxation time (T2) was directly monitored over 3 h. At predetermined time points, 0.3 mL of suspension was taken from the NMR tube, diluted, and filtered through a PVDF-type syringe filter (0.22 μm). To measure the free iron concentration, suspensions were analysed by ICP-MS after hot nitric acid digestion. To measure the carmofur concentration, centrifugation (13200 rpm for 10 min) was conducted to remove the SPIONs, and the supernatant analysed on an Agilent Cary 100 spectrophotometer. All experiments were performed in triplicate and the results are reported as mean ± S.D. In a set of experiments to predict drug release, dispersions of fibre samples at different concentration (0.5, 1.0 and 2 mg mL−1) in 0.1% w/v xanthan gum buffer (n = 3) were placed into 10 mm NMR tubes. The transverse relaxation time was monitored at 37 °C for 3 h. At selected time points, 0.3 mL aliquots were taken from the NMR tube and the [Fe] and carmofur content in each aliquot quantified as above.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors gratefully thank Dr Andrew Weston for assistance with electron microscopy experiments. The authors further acknowledge financial support from the EPSRC (grants EP/N509577/1, supporting AMK; and the EPSRC Centre for Doctoral Training in the Advanced Characterisation of Materials (EP/L015277/1), supporting CJRW).References
- E. Haacke and J. Reichenbach, Susceptibility weighted imaging in MRI: basic concepts and clinical applications. John Wiley & Sons, 2014 Search PubMed.
- G.-L. Davies, I. Kramberger and J. J. Davis, Chem. Commun., 2013, 49, 9704–9721 RSC.
- D. H. Carr, J. Brown, G. M. Bydder, R. E. Steiner, H. J. Weinmann, U. Speck, A. S. Hall and I. R. Young, Am. J. Roentgenol., 1984, 143, 215–224 CrossRef CAS PubMed.
- M. Mahmoudi, S. Sant, B. Wang, S. Laurent and T. Sen, Adv. Drug Delivery Rev., 2011, 63, 24–46 CrossRef CAS PubMed.
- A. Neuwelt, N. Sidhu, C.-A. Hu, G. Mlady, S. C. Eberhardt and L. O. Sillerud, Am. J. Roentgenol., 2015, 204, 302–W313 CrossRef PubMed.
- R. Jin, B. Lin, D. Li and H. Ai, Curr. Opin. Pharmacol., 2014, 18, 18–27 CrossRef CAS PubMed.
- A. S. Perera, S. Zhang, S. Homer-Vanniasinkam, M.-O. Coppens and M. Edirisinghe, ACS Appl. Mater. Interfaces, 2018, 10, 15524–15531 CrossRef CAS PubMed.
- T. Kang, F. Li, S. Baik, W. Shao, D. Ling and T. Hyeon, Biomaterials, 2017, 136, 98–114 CrossRef CAS PubMed.
- S. Laurent, S. Dutz, U. O. Häfeli and M. Mahmoudi, Adv. Colloid Interface Sci., 2011, 166, 8–23 CrossRef CAS PubMed.
- A. Jordan, R. Scholz, P. Wust, H. Fähling and R. Felix, J. Magn. Magn. Mater., 1999, 201, 413–419 CrossRef CAS.
- A. Hervault and N. T. K. Thanh, Nanoscale, 2014, 6, 11553–11573 RSC.
- A. R. Chowdhuri, T. Singh, S. K. Ghosh and S. K. Sahu, ACS Appl. Mater. Interfaces, 2016, 8, 16573–16583 CrossRef CAS PubMed.
- J. Zeng, L. Jing, Y. Hou, M. Jiao, R. Qiao, Q. Jia, C. Liu, F. Lei and M. Gao, Adv. Mater., 2014, 26, 2694–2698 CrossRef CAS PubMed.
- D. Bobo, K. J. Robinson, J. Islam, K. J. Thurecht and S. R. Corrie, Pharm. Res., 2016, 33, 2373–2387 CrossRef CAS PubMed.
- Y. X. Wang and J. M. Idee, Quant. Imaging Med. Surg., 2017, 7, 88–122 CrossRef PubMed.
- S. Laurent, D. Forge, M. Port, A. Roch, C. Robic, L. Vander Elst and R.-N. Muller, Chem. Rev., 2008, 108, 2064–2110 CrossRef CAS PubMed.
- L. Ternent, D. A. Mayoh, M. R. Lees and G.-L. Davies, J. Mater. Chem. B, 2016, 4, 3065–3074 RSC.
- N. Ž. Knežević, E. Ruiz-Hernández, W. E. Hennink and M. Vallet-Regí, RSC Adv., 2013, 3, 9584–9593 RSC.
- X. Guo, Z. Wu, W. Li, Z. Wang, Q. Li, F. Kong, H. Zhang, X. Zhu, Y.-P. Du, Y. Jin, Y. Du and J. You, ACS Appl. Mater. Interfaces, 2016, 8, 3092–3106 CrossRef CAS PubMed.
- J. Doshi and D. H. J. Reneker, J. Electrost., 1995, 35, 151–160 CrossRef CAS.
- D.-G. Yu, X.-Y. Li, X. Wang, J.-H. Yang, S. A. Bligh, G. R. Williams and D.-G. Yu, ACS Appl. Mater. Interfaces, 2015, 7, 18891–18897 CrossRef CAS PubMed.
- M. Jin, D.-G. Yu, X. Wang, C.-F. Geraldes, G. R. Williams and S.-A. Bligh, Adv. Healthcare Mater., 2016, 5, 977–985 CrossRef CAS PubMed.
- G. R. Williams, B. T. Raimi-Abraham and C. Luo, Nanofibres in Drug Delivery, UCL Press, 2018 Search PubMed.
- C. Huang, S. J. Soenen, J. Rejman, J. Trekker, L. Chengxun, L. Lagae, W. Ceelen, C. Wilhelm, J. Demeester and S. C. De Smedt, Adv. Funct. Mater., 2012, 22, 2479–2486 CrossRef CAS.
- L. Wang, M. Wang, P.-D. Topham and Y. Huang, RSC Adv., 2012, 2, 2433–2438 RSC.
- X. Zhu, J. Li, P. Peng, N. H. Nassab and B. R. Smith, Nano Lett., 2019, 19, 6725–6733 CrossRef CAS PubMed.
- Y. Koyama and Y. Koyama, Cancer Treat Rep., 1980, 64, 861–867 CAS.
- Y. Koyama, K. Inokuchi and Y. Koyama, Jpn. J. Clin. Oncol., 1980, 10, 83–92 CrossRef.
- D.-G. Yu, J.-J. Li, G. R. Williams and M. Zhao, J. Controlled Release, 2018, 292, 91–110 CrossRef CAS PubMed.
- B. Sanchez-Vazquez, A. J. Amaral, D. G. Yu, G. Pasparakis and G. R. Williams, AAPS PharmSciTech, 2017, 18, 1460–1468 CrossRef CAS PubMed.
- H. Li, B. Sanchez-Vazqueza, R. P. Trindadea, Q. Zoua, Y. Maia, L. Dou, L. Zhu and G. R. Williams, Colloids Surf., B, 2019, 183, 110411 CrossRef CAS PubMed.
- L. C. Rose, J. C. Bear, P. Southern, P. D. McNaughter, R. B. Piggott, I. P. Parkin, S. Qi, B. P. Hills and A. G. Mayes, J. Mater. Chem. B, 2016, 4, 1704–1711 RSC.
- D. F. Evans, G. Pye, R. Bramley, A. G. Clark, T.-J. Dyson and J. D. Hardcastle, Gut, 1988, 29, 1035–1041 CrossRef CAS PubMed.
- Y. Zhao, Y.-W. Luo, B.-G. Li and S. Zhu, Langmuir, 2011, 27, 11306–11315 CrossRef CAS PubMed.
- P. L. Ritger and N. A. Peppas, J. Controlled Release, 1987, 5, 23–36 CrossRef CAS.
- S. Matoori, Y. Bao, A. Schmidt, E. J. Fischer, R. Ocho-Sanchez, M. Tremblay, M. M. Oliveira, C. F. Rose and J.-C. Leroux, Small, 2019, 15, 1902347 CrossRef CAS PubMed.
- H. Flanner and J. W. Moore, Pharm. Tech., 1996, 20, 64–74 Search PubMed.
- Food and Drug Administration. Guidance for Industry: Dissolution Testing of Immediate Release Solid Oral Dosage Forms. US Department of Health and Human Services, Food and Drug Administration, Center for Drug Evaluation and Research, Rockville, MD, 1997 Search PubMed.
- G. Freitag, Drug Inf. J., 2001, 35, 865–874 CrossRef.
- C. G. Wilson and P. J. Crowley, Controlled Release in Oral Drug Deliver, Springer, 2011 Search PubMed.
- S. Matoori, M. Roveri1, P. Tiefenboeck, A. Romagna, O. Wuerthinger, O. Kolokythas and J. M. Froehlich1, Eur. Radiol., 2019, 3, 11 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0tb01033b |
| This journal is © The Royal Society of Chemistry 2020 |