
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMechanically adaptive implants fabricated with poly(2-hydroxyethyl methacrylate)-based negative photoresists†
Baptiste
Monney
 a,
Allison E.
Hess-Dunning
bc,
Paul
Gloth
bc,
Jeffrey R.
Capadona
bc and
Christoph
Weder
*a
a,
Allison E.
Hess-Dunning
bc,
Paul
Gloth
bc,
Jeffrey R.
Capadona
bc and
Christoph
Weder
*a
aAdolphe Merkle Institute, University of Fribourg, Chemin des Verdiers 4, CH-1700 Fribourg, Switzerland. E-mail: christoph.weder@unifr.ch
bAdvanced Platform Technology Center, Louis Stokes Cleveland Veterans Affairs Medical Center, Cleveland, OH 44106, USA
cDepartment of Biomedical Engineering, Case Western Reserve University, 2071 Martin Luther King Jr. Drive, Cleveland, OH 44106, USA
First published on 8th June 2020
Abstract
Neural implants that are based on mechanically adaptive polymers (MAPs) and soften upon insertion into the body have previously been demonstrated to elicit a reduced chronic tissue response than more rigid devices fabricated from silicon or metals, but their processability has been limited. Here we report a negative photoresist approach towards physiologically responsive MAPs. We exploited this framework to create cross-linked terpolymers of 2-hydroxyethyl methacrylate, 2-hydroxyethyl acrylate and 2-ethylhexyl methacrylate by photolithographic processes. Our systematic investigation of this platform afforded an optimized composition that exhibits a storage modulus E′ of 1.8 GPa in the dry state. Upon exposure to simulated physiological conditions the material swells slightly (21% w/w) leading to a reduction of E′ to 2 MPa. The large modulus change is mainly caused by plasticization, which shifts the glass transition from above to below 37 °C. Single shank probes fabricated by photolithography could readily be implanted into a brain-mimicking gel without buckling and viability studies with microglial cells show that the materials display excellent biocompatibility.
Introduction
Mechanically adaptive polymers (MAPs) represent the subset of stimuli-responsive materials that feature stimuli-responsive mechanical properties.1 Polymers that soften in response to physiological conditions form part of this class of materials and are considered useful in the context of implantable devices, notably intracortical electrodes.2–4 These devices represent the central element of brain–machine interfaces, but their lifetime has shown to be limited by the chronic inflammatory response of the brain. Several factors have been implicated in this process, including implantation- and motion-induced injuries and the materials used to manufacture the devices.5,6 The reduction of the stiffness and/or size of the electrode has been shown to reduce the tissue response but this renders the devices more fragile and thus complicates the insertion procedure.7–9 The implantation of soft electrodes can be achieved by coating the electrode with a stiff, water-soluble polymer that dissolves once in the brain,10–13 by using electrode shuttles,8,14 or other auxiliary tools, such as a mosquito-inspired insertion guide.15 Another approach involves building the electrodes on a mechanically adaptive substrate that can undergo a stiff-to-soft transition when placed in physiological conditions.16–23 Cellulose nanocrystal nanocomposites and photopolymerizable (meth)acrylate and thiol–ene polymers have been investigated in this context.16–26 The water-induced mechanical switching in these materials is based on a plasticization of the polymer matrix and, in the case of the nanocomposites, the weakening/disassembly of the hydrogen-bonded nanocellulose network upon interaction of the cellulose with water. The available materials display modulus changes of up to three orders of magnitude and excellent biocompatibility, but so far these materials were typically shaped by laser ablation, which complicates the fabrication process and limits their adoption by the field.2–4,9,10,26–29 The ability to process and shape MAPs via conventional photolithographic techniques would simplify their integration into standard microfabrication processes for neuroprosthetic devices.30,31Some of us recently explored the mechanically adaptive behavior of networks made by the photopolymerization of 2-hydroxyethyl methacrylate (HEMA) and ethylene glycol dimethacrylate (EGDMA) and demonstrated the possibility to prepare such MAPs in a photolithographic process.23 Optimized compositions exhibited a 500-fold drop of the storage modulus E′ from 2.5 GPa to 5 MPa when exposed to simulated physiological conditions, on account of modest swelling (30% w/w). The materials could be processed into shapes with feature sizes in the 10–100 μm range using soft- or contact lithography. However, as for all materials made from liquid photo-resins, the potential presence and leaching of unreacted monomers is of concern.32–34 The problem of leachable residues can be significantly reduced by introducing the cross-links in a post-polymerization process, i.e., after purification of the polymerization product. In the case of p(HEMA) this can be achieved with external cross-linkers35 or alternatively via the acid-catalyzed transesterification of the HEMA's hydroxyl groups with the methacrylate esters.36–39 We have utilized this latter approach to create a new family of physiologically responsive HEMA-based MAPs. These materials can be accessed from their precursors (HEMA-based copolymers and a photoacid generator) through a conventional negative photoresist approach that involves exposure, post-exposure bake (PEB), and development. A systematic investigation afforded a composition whose storage modulus E′ changes by three orders of magnitude upon exposure to simulated physiological conditions and which shows modest swelling. We show that photolithographically produced shanks can readily be implanted into a tissue mimic and demonstrate that the new materials display excellent biocompatibility towards microglial cells.
Results and discussion
P(HEMA) can be cross-linked through the photoacid-catalyzed transesterification of the HEMA repeating units (Scheme 1a), as reported previously.36–39 Thus, we first explored the cross-linking capability of a p(HEMA)-based photoresist composed of p(HEMA) and 0.5 wt% of the photoacid generator (PAG) (4-phenylthiophenyl)diphenylsulfonium triflate (PR1, Scheme 1b–d). This PAG is suitable for i-line irradiation, displays a low absorption coefficient at 365 nm (ca. 100 mol L−1 cm−1 in methanol, ESI,† Fig. S1a) and is thus useful for applications in thick films. The PAG also offers excellent thermal stability (Fig. S1b, ESI†). The concentration of the PAG in PR1 was kept low, in order to avoid optical gradient effects during exposure, i.e., the formation of a cross-link density gradient, which would lead to inhomogeneous swelling and thus bending of the samples (Fig. S2, ESI†). A low PAG content further limits the quantity of leachable compound, and reduces the reaction rate and consequentially the influence of thermal gradients at the beginning of the post-exposure bake (PEB). Homogeneous films of PR1 of a thickness of 150–250 μm (depending on the requirements of the respective experiments) were produced by solution casting mixtures of p(HEMA) and the PAG, drying, and compression molding at 140 °C. The thickness of the films, at least in the range studied here, had no significant influence on the final properties. Dynamic mechanical analysis (DMA) was used to investigate the effect of the cross-linking induced by this process. The DMA traces of the neat p(HEMA) (Fig. S3, ESI†) and p(HEMA) which for comparative purposes was exposed to UV light (365 nm, 4500 mJ cm−2, Fig. 1a, black) are comparable and show the typical features of an amorphous thermoplastic polymer, i.e., a glassy plateau, and a drop of the storage modulus E′ around the glass transition (Tg = 144 °C) followed by sample failure. By contrast, the UV-irradiated p(HEMA) photoresist (PR1)shows an increase of E′ above Tg, indicating that the dissociated PAG indeed catalyzes the cross-linking reaction in this temperature range (Fig. 1a, blue). An isothermal DMA trace of a non-irradiated PR1 film at 120 °C shows that E′ remains constant for hours and reflects the absence of any cross-linking reaction (Fig. 1b, green). By contrast, the corresponding DMA trace of an irradiated p(HEMA) photoresist (PR1) displays a rapid increase of E′ on account of cross-linking, before the E′ increase becomes asymptotic. This behavior is related to the fact that Tg increases upon cross-linking and eventually reaches the curing temperature so that the reaction rate drops (Fig. 1b, blue, Fig. S4, ESI†).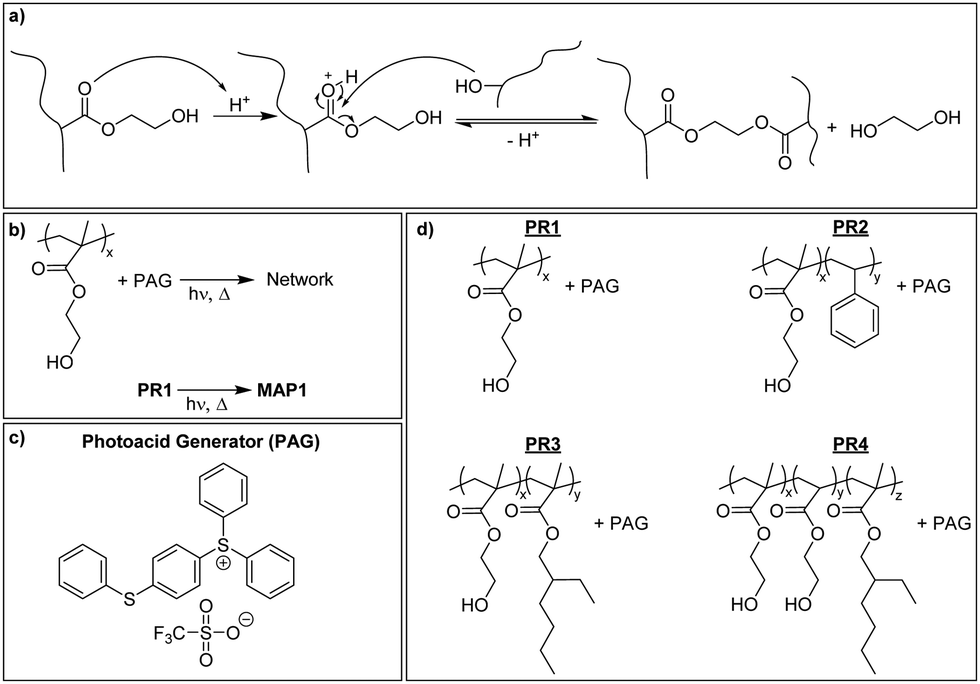 | ||
| Scheme 1 (a) Transesterification reaction involving the 2-hydroxyethyl moieties. (b) General reaction scheme to create the mechanically adaptive polymers (MAP) from the parent photoresists (PR). (c) Chemical structure of (4-phenylthiophenyl)diphenylsulfonium triflate, the photoacid generator (PAG) employed. (d) Chemical composition of PR1, PR2, PR3 and PR4. | ||
 | ||
| Fig. 1 (a) Dynamic mechanical analysis (DMA) traces showing (a) the storage modulus of a UV-light exposed, neat p(HEMA) film (black curve) and of an exposed, PR1 film (blue curve). (b) Evolution of the storage modulus of a non-irradiated (solid, green) and an irradiated (solid, blue) PR1 sample during an isothermal experiment at 120 °C. UV Exposure conditions: exposure @365 nm, 4500 mJ cm−2. | ||
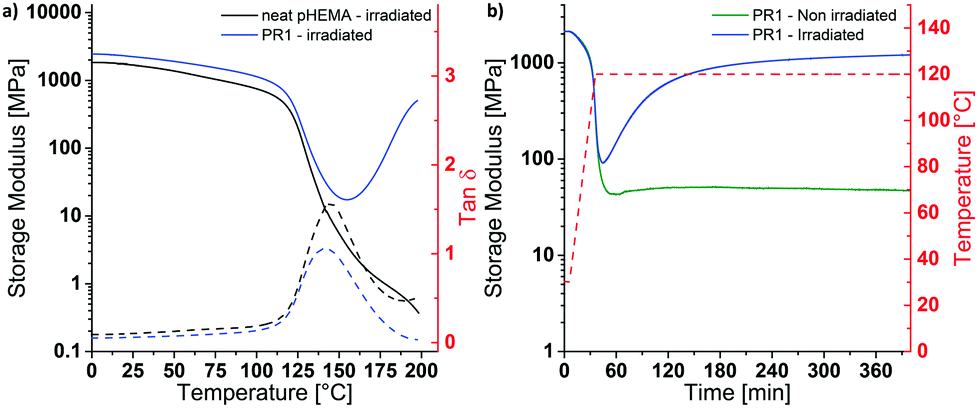
Having established by DMA experiments that the combination of UV exposure and post-exposure bake (but neither process alone) induces cross-linking of PR1 films, we explored how the properties of the MAPs made from this photoresist are influenced by the post-exposure bake time. In explorative experiments, samples were thus exposed to UV light (365 nm, 4500 mJ cm−2) and then cured at 110 °C for 30, 60, or 120 min and developed in a methanol/aqueous NaOH bath, followed by rinsing in methanol:water mixtures. The swelling behavior in artificial cerebrospinal fluid (ACSF) at 37 °C and the dynamic mechanical properties in ACSF were measured to assess the influence of exposure to simulated physiological conditions (Fig. S5 and Table S1, ESI†). In that respect, a post exposure bake of 60 min at 110 °C was found to give the best compromise between softening performance (low swelling and low swollen E′) and processing time, although we note that it may be possible to optimize this further. The material made by exposing, curing and developing PR1 under these conditions (denoted MAP1) exhibits a Tg of 137 °C and a storage modulus E′ of 2.5 ± 0.1 GPa at 25 °C (Fig. 2a). When exposed to simulated physiological conditions (ACSF at 37 °C), the network swells by 43% w/w, and this induces a drop of the Tg to ca. 4 °C and a reduction of the storage modulus E′ at 37 °C to 4 MPa (Fig. 2b and d).
 | ||
| Fig. 2 DMA traces showing (a) The storage modulus (solid) and loss tangent (dashed) of dry MAP1 (black) and dry MAP4 (red). (b) The storage modulus (solid) and loss tangent (dashed) of ACSF-swollen MAP1 (black) and ACSF-swollen MAP4 (red). The samples were pre-swollen in ACSF at 37 °C for 24 hours prior to measurement. (c) Stress–strain curves of dry MAP1 (black) and MAP4 (red) at 25 °C. (d) Swelling of MAP1 (black) and MAP4 in ACSF at 37 °C. Curing conditions: MAP1: exposure @365 nm 4500 mJ cm−2, PEB 60 min at 110 °C. MAP4: exposure @365 nm 4500 mJ cm−2, PEB 60 min at 90 °C. | ||
While the mechanical contrast that can be realized by exposing dry MAP1 to (simulated) physiological conditions is significant, the softening comes at the expense of a relatively high degree of swelling, which is undesirable, because it can lead to delamination of the multilayered structure of the neural electrode.4 The extent of swelling can be reduced by either decreasing the hydrophilicity or by increasing the cross-link density. Increasing the cross-link density would increase the rubbery plateau modulus and thus reduce the mechanical contrast, which is undesirable. Tuning of the hydrophilicity of the material can be achieved through the incorporation of co-monomers without affecting, in principle, the rubbery plateau modulus. In order to reduce the swelling without affecting the cross-link density, two copolymers with hydrophobic monomers were prepared. HEMA was thus copolymerized with either styrene, which keeps the Tg of the copolymer high, or 2-ethylhexyl methacrylate (EHMA), which lowers the Tg. Initial experiments were carried out with copolymers containing about 15 mol% of the hydrophobic monomers. A comparison of the differential scanning calorimetry traces of PR1 and the copolymers p(HEMA84-co-styrene16) (PR2) and p(HEMA85-co-EHMA15) (PR3) reveals Tg values of 109, 105 and 92 °C (Table S2, ESI†), respectively, and confirms that the introduction of styrene and EHMA affects the Tg as expected. Photoresist films consisting of 99.5 wt% of polymer and 0.5 wt% of PAG (PR2 or PR3) and were processed into MAP2 and MAP3 following the procedure described for PR1/MAP1. The swelling behavior in ACSF at 37 °C and the dynamic mechanical properties of the ACSF-swollen MAPs were measured to assess the mechanical switching. MAP2 and MAP3 made using similar processing conditions as applied for MAP1 (exposure @365 nm 4500 mJ cm−2, PEB 60 min at 110 °C) show indeed reduced swelling levels of 20 and 12% w/w, respectively (Table S3, ESI†). The DMA traces of the ACSF-swollen MAP2 and MAP3 show Tgs of ca. 47 °C and 42 °C, and E′ (at 37 °C) values of ca. 124 MPa and ca. 83 MPa, respectively (Table S3, ESI†). Thus, while the incorporation of the hydrophobic monomers indeed reduces the swelling, it also limits the extent of plasticization, to the extent that neither MAP2 nor MAP3 are fully softened at 37 °C. Because increasing the EHMA fraction not only lowers Tg, but also the swelling, we sought to reduce the dry Tg of the PR by replacing a part of the HEMA by 2-hydroxyethyl acrylate (HEA). Thus, p(HEMA57-co-HEA27-co-EHMA16) was synthesized and photoresist films (PR4) were fabricated following the procedure described for PR1/MAP1. While PR4 exhibits indeed a much lower dry Tg (63 °C, DSC, Table S2, ESI†) than PR1-PR3, the properties in the ACSF-swollen state of MAP4 that had been cured for 60 min at 110 °C are not very different from those of MAP1–MAP3 (Tg = ca. 30 °C, E′ at 37 °C = ca. 33 MPa, Table S3 and Fig. S6, ESI†), arguably because the lower Tg leads to faster curing and hence a higher cross-link density, if cured under similar conditions. Indeed, when the post-exposure bake was performed at a lower temperature (90 °C, 60 min), the targeted property matrix could be accessed. MAP4 produced under these conditions exhibits a dry Tg of 90 °C (DMA) and a storage modulus E′ of 1.8 ± 0.3 GPa at 25 °C (Fig. 2a and Table 1). When placed under simulated physiological conditions (ACSF at 37 °C), the network swells by 21% w/w and its storage modulus E′ at 37 °C drops below 2 MPa, with a Tg of ca. 4 °C (Fig. 2b, d and Table 1). Thus, the optimization of the polymer composition allowed us to reduce the swelling to one half in comparison to MAP1, while excellent mechanical contrast was maintained. The kinetics of the softening process were studied by adding ACSF that had been pre-heated to 37 °C into the DMA submersion bath during a measurement that started with the sample in the dry state. Upon addition of ACSF, MAP4 swells rapidly and reaches the rubbery plateau in less than 5 min (Fig. S7b, ESI†). Despite exhibiting a lower dry storage modulus at 25 °C than MAP1 (1.8 ± 0.3 GPa vs. 2.5 ± 0.1 GPa), MAP4 shows a higher toughness (662 ± 134 kJ mm−3vs. 454 ± 102 kJ mm−3, Fig. 2c and Table 1), which is advantageous as this reduces the risk of catastrophic failure upon buckling.
| MAP1 | MAP4 | |
|---|---|---|
| Dry MAP | ||
| E′ (at 25 °C) [MPa] | 2517 ± 128 | 1760 ± 290 |
| T g (DMA) [°C] | 135 ± 3.5 | 92 ± 2.5 |
| E (at 25 °C) [MPa] | 1756 ± 250 | 1639 ± 98 |
| Elongation at break [%] | 2.5 ± 0.6 | 3.1 ± 0.4 |
| Stress at break [MPa] | 32 ± 6 | 36 ± 2 |
| Toughness [kJ mm−3] | 454 ± 102 | 662 ± 134 |
| ACSF-swollen MAP | ||
| Swelling in ACSF (at 37 °C) [% w/w] | 43 ± 2.5 | 21 ± 0.8 |
| E′ (at 37 °C) [MPa] | 4.5 ± 2.7 | 1.4 ± 0.4 |
| T g (DMA) [°C] | 6 ± 2 | 4 ± 1 |
With the ultimate aim of fabricating complex devices, we explored the patterning of the material via a photolithographic process. Initial experiments revealed that PR4 samples exposed with a dose of 4500 mJ cm−2 showed poor patternability, thus indicating an overexposure of the photoresist. The photopatternability was improved by reducing the exposure dose to 480 mJ cm−2, with no significant change of the mechanical properties of the final material (Fig. S8, ESI†). Thus, a film of PR4 was exposed through a photomask using a mask aligner (365 nm, 60 s, 8 mW cm−2) and after a post-exposure bake (PEB) at 90 °C for 60 min, the photoresist was developed by immersion in a 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of methanol and aqueous sodium hydroxide (pH 10). Using the procedure described above, a MAP4 shank was obtained (Fig. 3a). The object shows a good patterning fidelity, as shown by the pictures of the probe (top view: Fig. 3a, cross-sectional view: Fig. S9, ESI†). The probe can be divided through the thickness into 3 sections. The top 85 μm of the probe shows vertical (90°) sidewalls, while the middle (35 μm) and bottom section (15 μm) show sidewalls with a 98° and 125° angle relative to the bottom surface of the probe, respectively. The width of the top section (202.4 ± 0.3 μm) slightly deviates from the width of the photomask used (197.9 ± 0.6 μm).
:1 mixture of methanol and aqueous sodium hydroxide (pH 10). Using the procedure described above, a MAP4 shank was obtained (Fig. 3a). The object shows a good patterning fidelity, as shown by the pictures of the probe (top view: Fig. 3a, cross-sectional view: Fig. S9, ESI†). The probe can be divided through the thickness into 3 sections. The top 85 μm of the probe shows vertical (90°) sidewalls, while the middle (35 μm) and bottom section (15 μm) show sidewalls with a 98° and 125° angle relative to the bottom surface of the probe, respectively. The width of the top section (202.4 ± 0.3 μm) slightly deviates from the width of the photomask used (197.9 ± 0.6 μm).
 | ||
| Fig. 3 Pictures of the insertion experiments in agarose gel. (A) Picture of the MAP4 probe used for the insertion experiments. (B) The dry probe is approaching the brain-mimicking agarose gel. (C) The probe is inserted in the agarose gel without buckling. (D) The probe is fully inserted in the agarose gel. (E) After 3 hours, the probe is successfully retracted. (F) The retracted, swollen probe is moved to another location. (G) When attempting reinsertion, the swollen probe buckles. The experiments were performed at room temperature. | ||
The suitability of MAP4 as mechanically adaptive substrate was further explored by inserting the MAP4 probe in a brain-mimicking agarose gel. The MAP4 probe could be successfully inserted in the agarose gel, without buckling. After 3 h in the agarose gel, the probe was retracted and reinserted at another location. Upon reinsertion, the probe buckled due to the water-induced softening of the MAP4 network (Fig. 3). Note that the probe did not bend upon softening.
Extraction experiments, coupled with GC-MS analysis were performed to verify that no extractable, low-molecular weight compounds leach from the material over time. Thus, MAP4 samples were placed in methanol for 24 h and the extracts were analyzed by GC-MS. The chromatograms of the extracts were comparable to the methanol reference and show none of the peaks associated to relevant compounds involved during the fabrication of the materials (monomers, UV-exposed PAG and ethylene glycol) (Fig. S10, ESI†). These results confirm that all soluble compounds are removed during the development step.
Cell viability assays were performed by exposing HAPI rat microglial cells to MAP4 using an established procedure.40 The cytotoxicity of the material was evaluated after 24 h and 48 h, using a live/dead test kit. After 24 h and 48 h, the microglial cells exposed to the photoresist remained viable and showed no significant cytotoxicity compared to the control cells cultured on glass substrates (Fig. 4).
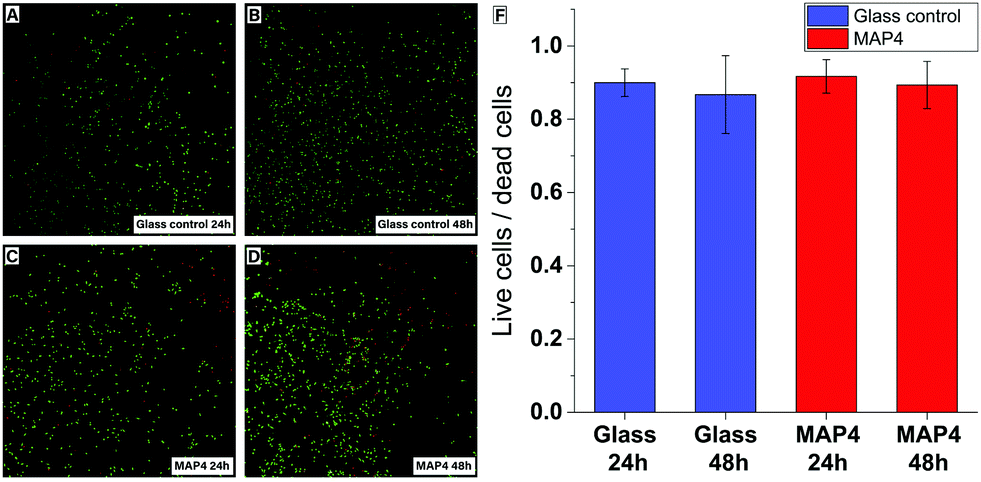 | ||
| Fig. 4 Viability assay of MAP4. Fluorescence microscopy images of the microglial cells exposed to glass (A) and (B) or MAP4 (C) and (D). Green indicates live cells and red indicates dead cells. (F) Live/dead cell ratio for the glial cells exposed to glass or MAP4. | ||
Conclusion
We demonstrated a negative photoresist approach to create mechanically adaptive polymers based on p(HEMA) that are initially stiff when dry and that soften when exposed to a physiological environment. The properties of the new MAPS can be tailored in a wide range by incorporating co-monomers and/or by modifying the processing conditions. When the composition and processing conditions were optimized to achieve a maximal mechanical contrast with minimum swelling in physiological conditions, a material that shows a reduction of the storage modulus E′ from 1.8 GPa to 2 MPa at a swelling level (21% w/w) that is much lower than that of previously studied MAPs for which the adaptive behavior was shown to lead to improved neural integration (70% w/w).28 A dummy neural shank fabricated by a conventional photolithographic process could be readily inserted into a brain-mimicking gel without buckling. Moreover, the mechanically adaptive polymer network exhibits excellent initial in vitro biocompatibility towards microglial cells. The new materials thus combine mechanically adaptive properties, biocompatibility and advanced microfabrication capabilities which makes it useful as a substrate for neural electrodes. Due to the water-absorbing nature of the substrate, the fabrication of functional neural probes would require the use of an insulating/moisture barrier such as Parylene to isolate the electronic components.29 Additional studies are underway to explore the potential of the new MAPs to be loaded with drugs that further reduce the inflammatory response,40 and to be surface-functionalized for improved control over the in vivo tissue response.Experimental
Materials
2-Hydroxyethyl methacrylate (HEMA, >99%, Sigma-Aldrich), 2-hydroxyethyl acrylate (HEA, 96%, Sigma-Aldrich), 2-ethylhexyl methacrylate (EHMA, >99%, TCI), styrene (>99%, Sigma-Aldrich), 2,2′-azobis(2-methylpropionitrile) (AIBN, >98%, Sigma-Aldrich), dodecanethiol (99%, Sigma-Aldrich), hydroquinone (>99%, Fluka), (4-phenylthiophenyl)diphenylsulfonium triflate (Sigma-Aldrich), potassium chloride (anhydrous, >99%, Sigma-Aldrich), sodium phosphate monobasic monohydrate (>99%, Sigma-Aldrich), sodium phosphate dibasic heptahydrate (98–102%, Sigma-Aldrich), calcium chloride dihydrate (>99%, Sigma-Aldrich), magnesium chloride hexahydrate (99–102%, Sigma-Aldrich), sodium chloride (>99%, Sigma-Aldrich) were used without further purification, except for AIBN, which was recrystallized twice from methanol and styrene which was passed through a basic alumina column before use. ACSF was prepared as reported elsewhere.41Dynamic mechanical analysis
Dynamic mechanical analyses (DMA) were performed on a TA Instruments Q800 DMA using a frequency strain analysis temperature ramp (0–180 °C, 3 °C min−1, 8 minutes equilibration time at 0 °C) or an isothermal step (120 °C, 6 h) at an amplitude of 15 μm and a frequency of 1 Hz. Submersion DMA in ACSF was performed on the same instrument using a multi-frequency strain analysis temperature ramp (5–75 °C, 3 °C min−1) with an amplitude of 30 μm and a frequency of 1 Hz. Before submersion experiments, samples were conditioned by immersion in ACSF at 37 °C for 24 h. All DMA data reported are averages and standard deviation from triplicate measurements and the graphs presented show representative experiments, unless otherwise specified.The softening kinetic experiments were performed on the same instrument using a multi-frequency strain analysis in submersion mode with an open furnace. The sample was mounted on the submersion clamp and the test was started at room temperature, in dry conditions. After ∼5 min, pre-heated ACSF (∼42 °C) was added to the submersion bath to trigger the softening. The following parameters were modified in order to measure the rapid change of modulus: amplitude = 20 μm, static force = 0.005 N, stabilization cycle = 2, average cycle = 2, sampling interval = 0.1.
Tensile tests
Tensile tests were performed on a Zwick/Roell Z010 tensile tester equipped with a 200 N load cell. The strain rate was 1 mm min−1.Size exclusion chromatography
SEC experiments were performed on an Agilent 1260 Infinity II system equipped with one Agilent PolarGel M guard column (particle size = 8 μm) and two Agilent PolarGel M columns (ID = 7.5 mm, L = 300 mm, particle size = 8 μm). Signals were recorded by an interferometric refractometer (Agilent 1260 series). Measurements were conducted using DMF + 0.05 M LiBr as the eluent, at a temperature of 60 °C, and with a flow rate of 1.0 mL min−1. Molecular weights were determined based on narrow molecular weight poly(ethylene oxide) calibration standards.Differential scanning calorimetry
DSC was conducted on a Mettler Toledo DSC 2 STAR system under nitrogen from −25 to 180 °C with heating and cooling rates of 20 °C min−1. The glass transition temperature was determined by the midpoint method from data collected in the second heating cycle.Thermogravimetric analysis (TGA)
TGA was conducted on a Mettler Toledo TGA/DSC 1 STAR system under nitrogen with a temperature ramp that had a heating rate of 10 °C min−1 and a maximum temperature of up to 550 °C.UV-Vis spectrometry
UV-Vis spectroscopy was carried out with a Shimadzu UV-2401PC spectrophotometer. Quartz cuvettes (path length = 1 cm) were used to measure the absorption from 320 to 420 nm.NMR spectroscopy
1H NMR spectroscopy was carried out at 297.2 K on a Bruker Avance DPX 400 spectrometer (400.19 MHz) or a Bruker Avance III 300 (300.13 MHz). Spectra were calibrated to the residual solvent peak of DMSO-d6 (2.50 ppm). The relaxation time D1 was 5 s and the number of scans was 128.Gas chromatography-mass spectrometry (GC-MS)
GC-MS was measured on a Thermo Scientific Trace GC Ultra equipped with a Zebron ZB-5MS column. A mass range of 40–540 m/z, and ion source of 250 °C and a temperature ramp from 50 to 280 °C at 20 °C min−1 were selected.Synthesis of p(HEMA)
2-Hydroxyethyl methacrylate (412 mmol, 53.65 g, 50.0 mL), dodecanethiol (0.5 mol%, 2.00 mmol, 417 mg, 493 μL), and ethanol (240 mL) were placed in a 500 mL 2-necked round bottomed flask equipped with a reflux condenser and a magnetic stir bar. The solution was purged with nitrogen for 90 min at room temperature. The mixture was then preheated for 30 min in an oil bath set to 60 °C. A solution of AIBN (3 mol%, 12.4 mmol, 2.00 g) in dioxane (15 mL) was added and the reaction mixture was stirred at 60 °C for 3 hours under a constant flow of nitrogen. The polymerization was quenched by the addition of hydroquinone (ca. 0.5 g) and the polymer was precipitated twice in diethyl ether and subsequently dried under vacuum at 60 °C overnight. p(HEMA) (38 g, 71%) was obtained as a white powder. Tg (DSC): 103 °C. SEC in DMF: Mn = 22 kDa, D = 1.53. 1H NMR (400 MHz, DMSO-d6) δ 4.80 (s, 1H, –OH), 3.91 (s, 2H, –CH2CH2OH), 3.59 (s, 2H, –CH2CH2OH), 2.18–1.28 (m, backbone), 1.23–0.56 (m, backbone) (Fig. S11, ESI†).Synthesis of p(HEMA-co-styrene)
2-Hydroxyethyl methacrylate (90 mol%, 165 mmol, 21.5 g, 20.0 mL), styrene (10 mol%, 18.3 mmol, 1.9 g, 2.10 mL), dodecanethiol (0.1 mol%, 0.18 mmol, 37.0 mg, 44 μL) and ethanol (102 mL) were placed in a 250 mL 2-necked round bottomed flask equipped with a reflux condenser and a magnetic stir bar. The solution was purged with nitrogen for 60 min at room temperature. The mixture was then preheated for 30 min in an oil bath set to 60 °C. A solution of AIBN (2 mol%, 3.66 mmol, 602 mg) in dioxane (5 mL) was added and the reaction was stirred at 60 °C for 3 hours under a constant flow of nitrogen. The polymerization was quenched by the addition of hydroquinone (ca. 0.5 g). The polymer was then precipitated twice in diethyl ether and subsequently dried under vacuum at 60 °C overnight. p(HEMA-co-styrene) (11.6 g, 50%) was obtained as a white material. Tg (DSC): 105 °C SEC in DMF: Mn = 31 kDa, D = 1.55. Polymer composition (NMR): 84% HEMA, 16% styrene 1H NMR (300 MHz, DMSO-d6) δ 7.37–6.74 (m, 5H, aromatics), 4.91–4.50 (m, 1H, –OH), 3.89 (s, 2H, –CH2CH2OH), 3.57 (2H, –CH2CH2OH), 2.15–1.26 (m, backbone), 1.17–0.24 (m, backbone) (Fig. S12, ESI†).Synthesis of p(HEMA-co-EHMA)
2-Hydroxyethyl methacrylate (85 mol%, 165 mmol, 21.5 g, 20.0 mL), 2-ethylhexyl methacrylate (15 mol%, 29.1 mmol, 5.77 g, 6.50 mL), dodecanethiol (0.1 mol%, 0.19 mmol, 39.2 mg, 46 μL) and ethanol (102 mL) were placed in a 500 mL 2-necked round bottomed flask equipped with a reflux condenser and a magnetic stir bar. The solution was purged with nitrogen for 60 min at room temperature. The mixture was then preheated for 30 min in an oil bath set to 60 °C. A solution of AIBN (2 mol%, 3.88 mmol, 637 mg) in dioxane (5 mL) was added and the reaction was stirred at 60 °C for 3 hours under a constant flow of nitrogen. The polymerization was quenched by the addition of hydroquinone (ca. 0.5 g). The polymer was then precipitated twice in a diethyl ether:hexane mixture (9:1) and subsequently dried under vacuum at 60 °C overnight. p(HEMA-co-EHMA) (15.2 g, 56%) was obtained as a white material. Tg (DSC): 62 °C SEC in DMF: Mn = 51 kDa, D = 1.76. Polymer composition (NMR): 85% HEMA, 15% EHMA (Fig. S5, ESI†). 1H NMR (300 MHz, DMSO-d6) δ 4.85–4.70 (s, 1H, –OH), 4.03–3.81 (s, 2H, –CH2CH2OH), 3.81–3.7 (br, 2H, –CH2CH(C2H5)C4H9), 3.67–3.48 (s, 2H, –CH2CH2OH), 2.20–1.20 (m, backbone), 1.05–0.53 (m, backbone) (Fig. S13, ESI†).
Synthesis of p(HEMA-co-HEA-co-EHMA)
2-Hydroxyethyl methacrylate (60 mol%, 272 mmol, 35.4 g, 33.0 mL), 2-hydroxyethyl acrylate (25 mol%, 113 mmol, 13.2 g, 13.0 mL), 2-ethylhexyl methacrylate (15 mol%, 68.0 mmol, 13.5 g, 15.2 mL), dodecanethiol (0.1 mol%, 0.45 mmol, 91.8 mg, 109 μL) and ethanol (220 mL) were placed in a 500 mL 2-necked round bottomed flask equipped with a reflux condenser and a magnetic stir bar. The solution was purged with nitrogen for 90 min at room temperature. The mixture was then preheated for 30 min in an oil bath set to 60 °C. A solution of AIBN (2 mol%, 9.07 mmol, 1.49 g) in dioxane (15 mL) was added and the reaction was stirred at 60 °C for 3 hours under a constant flow of nitrogen. The polymerization was quenched by the addition of hydroquinone (ca. 0.5 g). The polymer was then precipitated twice in a diethyl ether:hexane mixture (9:1) and subsequently dried under vacuum at 60 °C overnight. p(HEMA-co-HEA-co-EHMA) (37 g, 60%) was obtained as a white material. Tg (DSC): 62 °C. SEC in DMF: Mn = 32 kDa, D = 1.97. Polymer composition (NMR): 57% HEMA, 27% HEA, 16% EHMA. 1H NMR (400 MHz, DMSO-d6) δ 4.90–4.61 (m, 1H), 4.17–3.68 (m, 2H, –CH2CH2OH and –CH2CH(C2H5)C4H9), 3.67–3.48 (m, 2H, –CH2CH2OH), 2.30–1.13 (m, backbone), 1.04–0.57 (m, backbone) (Fig. S14, ESI†).
Photoresist film preparation
P(HEMA), p(HEMA-co-styrene), p(HEMA-co-EHMA), or p(HEMA-co-HEA-co-EHMA) (1.00 g) and 4-(phenylthiophenyl)diphenylsulfonium triflate (5 mg, 0.5 wt%) were dissolved in methanol (8 mL). The solution was stirred at room temperature until dissolution was complete. The solution was then filtered through a 0.45 μm disk filter and poured into a poly(tetrafluoroethylene) Petri dish (6 cm diameter). After letting most of the solvent evaporate during 24 h at room temperature in the dark, a photoresist film had formed that was removed from the mold and dried under vacuum (in the dark, r.t. → 100 °C). The dry films were then re-processed by compression-molding at 140 °C (p(HEMA)) or 125 °C (p(HEMA-co-HEA-co-EHMA)). Films with a thickness of between 100 and 250 μm were thus produced.DMA sample preparation
Rectangular samples (25 × 5 mm) for DMA experiments were cut from the dry photoresist films at 70 °C to prevent the film from shattering and were then placed into a PTFE Petri dish and irradiated at 365 nm at 25 mW cm−2 for 180 s in a Hönle LED Cube 100 IC. The light intensity was measured using a Hönle UV Meter BASIC equipped with a FS VIS D1 probe. The post-exposure bake was then performed for 60 min in a pre-heated oven at 110 °C (p(HEMA)) or 90 °C (p(HEMA-co-HEA-co-EHMA)). The samples were then developed in a 4:1 v/v mixture of methanol and aqueous NaOH (pH 10) for 20 min at room temperature. The films were then immersed in methanol:water mixtures in which the water content was gradually increased from 3:2 to 2:3 to 1:4 v/v. The gradual solvent change caused the probed to slowly de-swell. Samples were left in each bath for 20 min. Finally, the samples were dried in an oven at 100 °C overnight (normal pressure).
Tensile test sample preparation
Dog-bone shaped samples were prepared by exposing the dry p(HEMA) and p(HEMA-co-HEA-co-EHMA) photoresist films of through a photomask-on-film at 365 nm at 20 mW cm−2 for 100 s in a Hönle LED Cube 100 IC. Stray light effects were reduced by covering the mirrors of the LED Cube's irradiation chamber with black cardboard. The irradiation intensity was measured through the photomask. The films were then processed using the procedure described above, affording dog-bone shaped samples (design and dimensions in Fig. S15, ESI†).Determination of the extent of swelling
Precisely weighed amounts (10–15 mg) of the dry polymer samples were immersed in ACSF (4–5 mL) for 24 h at 37 °C. The samples were then wiped dry with a paper tissue and immediately weighed. The extent of swelling was calculated using the following formula:where mD is the dry sample mass and mS is the swollen sample mass. Values quoted are averages of 3 samples, unless otherwise specified. Swelling kinetics were established by measuring weights over the course of 72 h using the above procedure.
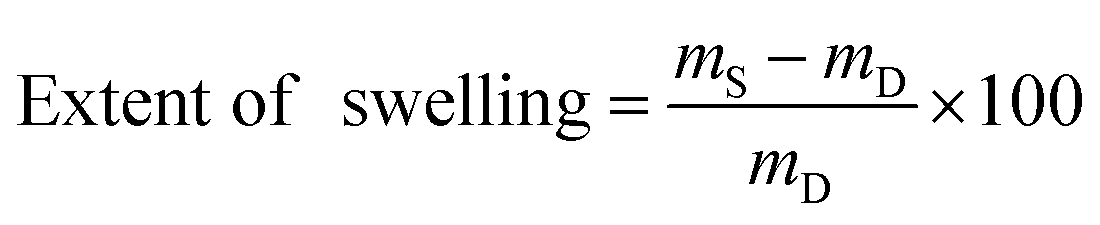
Sample design
Planar neural probe-like structures were designed to have a 200 μm wide and 4 mm long shank and a 2 mm wide and 3.5 mm long tab. The design was drafted in CAD software, then printed onto a transparent film using a laser photoplotter (Fineline Imaging, Colorado Springs, CO USA). The transparency mask was adhered to a blank glass photomask to ensure compatibility with the exposure equipment.Photolithography
Neural probe structures based on the new MAPs were fabricated by first mounting pre-made photoresist films (thickness ∼135 μm) onto partial silicon wafers. The films were placed onto the wafers and placed in an oven at 90 °C for 15 min to remove any absorbed moisture. Light pressure was applied to lightly adhere the films to the silicon. The samples were exposed to i-line (365 nm) UV light (60 s, 8 mW cm−2) through the photomask using a Karl Suss MA6/BA6 alignment tool in “Hard Contact” mode. The samples were post-exposure baked in an oven at 90 °C for 60 min and developed in a 3:1 v/v mixture of methanol and aqueous NaOH (pH 10) for 90 min. The swelling of the probes during the development process caused delamination and release from the silicon substrates. The probes were transferred into a 3:1 v/v mixture of methanol and deionized water and left for 30 min. Deionized water was then added gradually (approximately 1 part every 15 min) until a 1:4 v/v methanol:water ratio was reached. The gradual solvent change caused the probe to slowly de-swell. Probes were then removed from the solution and placed onto a Teflon film. The probes were dried under ambient conditions for ca. 2 h and then in an oven at 100 °C overnight.
Probe insertion experiment
To demonstrate functionality, probes were inserted into a 0.6% aqueous agarose gel that mimics the physical properties of brain tissue.42 The MAP probe was mounted on an acrylic rod and secured with super glue (Loctite Super Glue Gel Control). A motorized insertion tool advanced the probe into the agarose gel at a rate of 850 μm s−1, at room temperature. The probe was left in place for 3 h before extracting it, and attempting to re-insert it in a new location.Extraction experiment
MAP4 films (1 cm × 1 cm × 0.2 cm) were stirred in 2 mL of methanol for 24 hours. The methanol extract was then analyzed by GC-MS. Solutions of HEMA, HEA, EHMA and ethylene glycol at a concentration of 1 or 100 ppm (depending on the intensity of the GC-MS signal) were prepared in methanol and analyzed by GC-MS. A solution of the PAG (1 ppm) was prepared in methanol, exposed to UV light (365 nm, 4500 mJ cm−2) and subsequently analyzed by GC-MS.Cell culture
HAPI cells were maintained in DMEM Dulbecco's Modified Eagle Medium supplemented with 5% fetal bovine serum and 1% penicillin–streptomycin. HAPI cells were used between passages 5–25 for all experiments. For all experiments, cells were seeded at a density of 8000 cells per cm2. All representative images from the assays detailed below were acquired by fluorescence microscopy using an inverted AxioObserver Z1 (Zeiss) equipped with an AxioCam MRm camera. Exposure times were held constant between conditions for each assay. All samples were tested three times per assay, and a minimum of three assays were performed to ensure consistency and repeatability of the measurements.Cell viability
To analyze the cytotoxic effect of these materials the live/dead viability/cytotoxicity Kit (Life Technologies) was used. HAPI cells were seeded on ethylene oxide sterilized films of MAP4 (1 cm × 1 cm × 100 μm) or glass coverslips in 24-well plates. For 48 h time points, media was changed at the 24 h mark. At the end of each time point cells were incubated in 8 μM ethidium homodimer-1 (EthD-1) and 0.1 mM calcein-AM in Dulbecco's phosphate buffered saline (DPBS). The samples were then washed in DPBS once. Fluorescent images were taken (488 nm green/live, 594 nm red/dead) to quantitatively determine the cell viability and cytotoxicity of the films compared to known substrates. Cells were counted using Neurolucida cell counting software.Author contributions
B. M. and C. W. conceived the idea and designed the study. B. M. designed, synthesized, and characterized the materials and developed the photolithography procedure. P. G. performed the cytotoxic assay. A. H. D. and P. G. performed the photolithography and the insertion in the artificial brain. All authors discussed the results and contributed to the interpretation of the data. B. M. and C. W. wrote the paper. All authors contributed to editing of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors are grateful for the financial support received from the Swiss National Science Foundation (Grant No. 200020_172619 and IZPIP0_177995, PIRE “Bioinspired Materials and Systems”), the Adolphe Merkle Foundation, the US National Science Foundation (PIRE “Bioinspired Materials and Systems”, grant OISE 1844463), the Department of Veterans Affairs Rehabilitation Research and Development Service Merit Review Awards (GRANT1264735 and GRANT12418820), Research Career Scientist Award (GRANT12635707) and the Advanced Platform Technology Center (I50 RX001871). The contents do not represent the views of the U.S. Department of Veterans Affairs or the United States Government.Notes and references
- M. W. Urban, Stimuli-Responsive Materials, The Royal Society of Chemistry, Cambridge, 2016 Search PubMed.
- J. P. Harris, J. R. Capadona, R. H. Miller, B. C. Healy, K. Shanmuganathan, S. J. Rowan, C. Weder and D. J. Tyler, J. Neural Eng., 2011, 8, 066011 CrossRef CAS PubMed.
- J. P. Harris, A. E. Hess, S. J. Rowan, C. Weder, C. A. Zorman, D. J. Tyler and J. R. Capadona, J. Neural Eng., 2011, 8, 46010 CrossRef CAS PubMed.
- J. R. Capadona, D. J. Tyler, C. A. Zorman, S. J. Rowan and C. Weder, MRS Bull., 2012, 37, 581–589 CrossRef CAS.
- M. Jorfi, J. L. Skousen, C. Weder and J. R. Capadona, J. Neural Eng., 2015, 12, 011001 CrossRef PubMed.
- S. M. Wellman and T. D. Y. Y. Kozai, ACS Chem. Neurosci., 2017, 8, 2578–2582 CrossRef CAS PubMed.
- T. D. Y. Kozai, A. S. Jaquins-Gerstl, A. L. Vazquez, A. C. Michael and X. T. Cui, ACS Chem. Neurosci., 2015, 6, 48–67 CrossRef CAS PubMed.
- T. D. Y. Kozai and D. R. Kipke, J. Neurosci. Methods, 2009, 184, 199–205 CrossRef CAS PubMed.
- A. Lecomte, E. Descamps and C. Bergaud, J. Neural Eng., 2018, 15, 031001 CrossRef PubMed.
- H. C. Lee, F. Ejserholm, J. Gaire, S. Currlin, J. Schouenborg, L. Wallman, M. Bengtsson, K. Park and K. J. Otto, J. Neural Eng., 2017, 14, 036026 CrossRef PubMed.
- M. Lo, S. Wang, S. Singh, V. B. Damodaran, H. M. Kaplan, J. Kohn, D. I. Shreiber and J. D. Zahn, Biomed. Microdevices, 2015, 17, 34 CrossRef PubMed.
- F. Wu, L. W. Tien, F. Chen, J. D. Berke, D. L. Kaplan and E. Yoon, J. Microelectromech. Syst., 2015, 24, 62–69 CAS.
- Z. Xiang, S.-C. C. Yen, N. Xue, T. Sun, W. M. Tsang, S. Zhang, L.-D. De Liao, N. V. Thakor and C. Lee, J. Micromech. Microeng., 2014, 24, 065015 CrossRef.
- F. Vitale, S. R. Summerson, B. Aazhang, C. Kemere and M. Pasquali, ACS Nano, 2015, 9, 4465–4474 CrossRef CAS PubMed.
- A. J. Shoffstall, S. Srinivasan, M. Willis, A. M. Stiller, M. Ecker, W. E. Voit, J. J. Pancrazio and J. R. Capadona, Sci. Rep., 2018, 8, 122 CrossRef PubMed.
- J. R. Capadona, K. Shanmuganathan, D. J. Tyler, S. J. Rowan and C. Weder, Science, 2008, 319, 1370–1374 CrossRef CAS PubMed.
- K. Shanmuganathan, J. R. Capadona, S. J. Rowan and C. Weder, Prog. Polym. Sci., 2010, 35, 212–222 CrossRef CAS.
- L. Montero De Espinosa, W. Meesorn, D. Moatsou and C. Weder, Chem. Rev., 2017, 117, 12851–12892 CrossRef CAS PubMed.
- L. Hsu, C. Weder and S. J. Rowan, J. Mater. Chem., 2011, 21, 2812–2822 RSC.
- J. Mendez, P. K. Annamalai, S. J. Eichhorn, R. Rusli, S. J. Rowan, E. J. Foster and C. Weder, Macromolecules, 2011, 44, 6827–6835 CrossRef CAS.
- K. Shanmuganathan, J. R. Capadona, S. J. Rowan and C. Weder, ACS Appl. Mater. Interfaces, 2010, 2, 165–174 CrossRef CAS PubMed.
- M. Mao, J. He, X. Li, B. Zhang, Q. Lei, Y. Liu and D. Li, Micromachines, 2017, 8, 113 CrossRef.
- B. Monney, A. G. Dibble, D. Moatsou and C. Weder, ACS Omega, 2020, 5, 3090–3097 CrossRef CAS.
- T. Ware, D. Simon, D. E. Arreaga-Salas, J. Reeder, R. Rennaker, E. W. Keefer and W. Voit, Adv. Funct. Mater., 2012, 22, 3470–3479 CrossRef CAS.
- T. Ware, D. Simon, K. Hearon, T. H. Kang, D. J. Maitland and W. Voit, Macromol. Biosci., 2013, 13, 1640–1647 CrossRef CAS PubMed.
- T. Ware, D. Simon, C. Liu, T. Musa, S. Vasudevan, A. Sloan, E. W. Keefer, R. L. Rennaker and W. Voit, J. Biomed. Mater. Res., Part B, 2014, 102, 1–11 CrossRef PubMed.
- A. Sridharan, J. K. Nguyen, J. R. Capadona and J. Muthuswamy, J. Neural Eng., 2015, 12, 036002 CrossRef PubMed.
- J. K. Nguyen, D. J. Park, J. L. Skousen, A. E. Hess-Dunning, D. J. Tyler, S. J. Rowan, C. Weder and J. R. Capadona, J. Neural Eng., 2014, 11, 056014 CrossRef PubMed.
- A. E. Hess, J. R. Capadona, K. Shanmuganathan, L. Hsu, S. J. Rowan, C. Weder, D. J. Tyler and C. A. Zorman, J. Micromech. Microeng., 2011, 21, 054009 CrossRef.
- A. del Campo and E. Arzt, Chem. Rev., 2008, 108, 911–945 CrossRef CAS PubMed.
- D. Simon, T. Ware, R. Marcotte, B. R. Lund, D. W. Smith, M. Di Prima, R. L. Rennaker and W. Voit, Biomed. Microdevices, 2013, 15, 925–939 CrossRef CAS PubMed.
- M. Goldberg, Clin. Oral Investig., 2008, 12, 1–8 CrossRef PubMed.
- T. E. Schafer, C. A. Lapp, C. M. Hanes, J. B. Lewis, J. C. Wataha and G. S. Schuster, J. Biomed. Mater. Res., 1999, 45, 192–197 CrossRef CAS.
- G. Lodiene, E. Morisbak, E. Bruzell and D. Ørstavik, Int. Endod. J., 2008, 41, 72–77 CAS.
- S. Yang, J. Ford, C. Ruengruglikit, Q. Huang and J. Aizenberg, J. Mater. Chem., 2005, 15, 4200–4202 RSC.
- T. Aoai, J. S. Lee, H. Watanabe, S. Kondo, N. Miyagawa, S. Takahara and T. Yamaoka, J. Photopolym. Sci. Technol., 1999, 12, 303–306 CrossRef CAS.
- M. Vasilopoulou, S. Boyatzis, I. Raptis, D. Dimotikalli and P. Argitis, J. Mater. Chem., 2004, 14, 3312–3320 RSC.
- C. D. Diakoumakos, I. Raptis, A. Tserepi and P. Argitis, Polymer, 2002, 43, 1103–1113 CrossRef CAS.
- J. Lee, T. Aoai, S. N. I. Kondo, N. Miyagawa, S. Takahara and T. Yamaoka, J. Polym. Sci., Part A: Polym. Chem., 2002, 40, 1858–1867 CrossRef CAS.
- J. K. Nguyen, M. Jorfi, K. L. Buchanan, D. J. Park, E. J. Foster, D. J. Tyler, S. J. Rowan, C. Weder and J. R. Capadona, Acta Biomater., 2016, 29, 81–93 CrossRef CAS PubMed.
- Preparation of Artificial CSF, http://www.alzet.com/products/guide_to_use/cfs_preparation.html, (accessed 8 July 2019).
- Z.-J. Chen, G. T. Gillies, W. C. Broaddus, S. S. Prabhu, H. Fillmore, R. M. Mitchell, F. D. Corwin and P. P. Fatouros, J. Neurosurg., 2004, 101, 314–322 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: UV-VIS spectrum and TGA of the PAG, Swelling kinetics of the photoresist, additional mechanical and swelling data, DSCs and NMRs of the polymers. See DOI: 10.1039/d0tb00980f |
| This journal is © The Royal Society of Chemistry 2020 |