
Surface-fragmenting hyperbranched copolymers with hydrolysis-generating zwitterions for antifouling coatings†
 *a
and
Guangzhao
Zhang
a
*a
and
Guangzhao
Zhang
a
Abstract
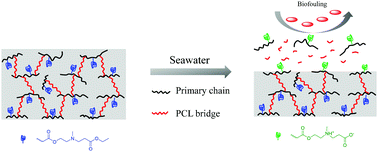
Zwitterionic polymers have attracted increasing attention due to their excellent fouling resistance ability and eco-friendliness. Yet, their non-degradability and hydrophilic nature limit their applications. In this study, we have prepared a novel surface-fragmenting hyperbranched copolymer with tertiary carboxybetaine ester (TCB) primary chains and poly(ε-caprolactone) (PCL) bridged chains, where the former and the latter can hydrolyze and degrade in marine environments, continuously generating zwitterions, so the polymer coating has a fouling resistant and renewable surface. Our study demonstrates that the degradation rate of the polymer is well controlled by the content of PCL bridges. Protein resistance and antibacterial assays show that the coating can inhibit the adhesion of protein and marine bacteria (Pseudomonas sp.). This new surface-fragmenting, self-regenerating hyperbranched zwitterionic copolymer has multiple applications in antifouling coatings.
 Please wait while we load your content...
Please wait while we load your content...

