
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Nanoparticle-based biosensors for detection of extracellular vesicles in liquid biopsies
Beatriz
Martín-Gracia†
 ab,
Alba
Martín-Barreiro†
ab,
Carlos
Cuestas-Ayllón
a,
Valeria
Grazú
ab,
Aija
Line
c,
Alicia
Llorente
d,
Jesús
M. de la Fuente
*ab and
María
Moros
*ab
ab,
Alba
Martín-Barreiro†
ab,
Carlos
Cuestas-Ayllón
a,
Valeria
Grazú
ab,
Aija
Line
c,
Alicia
Llorente
d,
Jesús
M. de la Fuente
*ab and
María
Moros
*ab
aAragón Materials Science Institute (ICMA), CSIC/University of Zaragoza, Zaragoza, Spain. E-mail: m.moros@csic.es; j.m.fuente@csic.es
bBiomedical Research Networking Center in Bioengineering, Biomaterials and Nanomedicine (CIBER-BBN), Spain
cLatvian Biomedical Research and Study Centre, Riga, Latvia
dDepartment of Molecular Cell Biology, Institute for Cancer Research, Oslo University Hospital, Oslo, Norway
First published on 4th June 2020
Abstract
Tumor-derived extracellular vesicles have emerged as an alternative source of cancer biomarkers in liquid biopsies. Despite their clinical potential, traditional methods for isolation and analysis have hampered their translation into the clinic. The use of nanomaterial-based biosensors can speed up the development of analytical methods for quantifying extracellular vesicles in a specific, highly reproducible, robust, fast and inexpensive way. Here we review the utility of extracellular vesicles as a novel type of liquid biopsies and the recent advances in nanoparticle-based biosensors for their analysis. We aim to emphasise the limitations and challenges that hinder extracellular vesicle analysis using these biosensors and point out potential solutions.
 María Moros | Dr María Moros is a researcher at the Aragón Materials Science Institute (ICMA), Zaragoza, Spain. She obtained her PhD in Chemistry (PhD extraordinary award) from the Institute of Nanoscience of Aragon (Zaragoza), under the supervision of Dr J. M. de la Fuente and Dr V. Grazú. After postdoctoral work in this group from 2012 to 2015, she held a Marie Curie Fellowship at the Institute of Applied Sciences and Intelligent Systems in Naples. She joined the ICMA with a Juan de la Cierva Fellowship and she obtained an ERC Starting Grant in 2019. Her research interests involve using nanoparticles to develop more sensitive biosensors and mechanotransduction tools for regenerative medicine, and to understand how the grafting molecules can affect their biological fate. |
1. Introduction
Liquid biopsies are considered a very promising alternative to conventional tissue biopsies for cancer detection, monitoring tumor progression and tracking tumor evolution.1 Recently, tumor-derived extracellular vesicles (EVs) have emerged as an alternative source of biomarkers in liquid biopsies. Although EVs were initially proposed to be cellular waste, to date it is known that they mediate intercellular communication and play a major role in a variety of normal and pathological processes, including cancer.2 As the cargoes that EVs carry largely depend on their parent cells, EVs hold great promise as prognostic elements.3 Despite their clinical potential, the use of complex and time-consuming traditional methods for isolation and analysis has limited their clinical translation.4 Furthermore, characterization of EVs can be challenging because of the high heterogeneity of the isolates, which generally contain a mix of EVs of different origin, with diverse sizes and cargo content.5,6 In this context, the development of new analytical platforms to perform high-throughput analyses in an easy and sensitive way without sample pre-treatment could speed up their clinical translation. Ideally, point of care (POC) biosensors will allow for a sensitive, selective and fast detection of EVs while remaining easy to use and inexpensive. In recent years great efforts have been devoted to develop novel biosensors for EV analysis based on microfluidics, nanomaterials or plasmonics to name a few. However the majority of these platforms are only proof of concept works that have not entered into the market.In this review we present the recent progress in the detection of EVs and describe the state-of-the-art in nanomaterial-based biosensors. Although several reviews have focused on some isolation and detection techniques,7–14 this review concentrates on the advantages of using nanomaterials, mainly nanoparticles (NPs) to develop biosensors. We also provide a comprehensive overview of the potential use of EVs as a novel type of liquid biopsies and the techniques that are currently used to analyze EV biomarkers in biofluids. Then, we focus on the advantages of using NPs to design and develop innovative biosensors, and how the correct selection of biomolecules and nanostructures and the way to combine both could improve their analytical performance. The main aim of this review is to present in a critical way the state-of–the-art in NP-based biosensors and finally address current challenges in the detection of EVs, considering the advantages and limitations of each technique.
2. Liquid biopsies and extracellular vesicles
2.1. Liquid biopsies
Liquid biopsies of cancer are samples of biofluids such as blood or urine that are used for the analysis of cancer cells or cancer tissue-derived molecules.15,16 Liquid biopsies have emerged as a very promising alternative to conventional tissue biopsies since they can be obtained in a noninvasive or minimally invasive way, thus avoiding the risks related to tissue sampling and allowing serial sampling during the course of disease. Importantly, they have been shown to reflect intratumoral heterogeneity better than tissue biopsies, and are suitable for longitudinal monitoring of cancer evolution and detection of resistance-conferring tumor cell subclones.17 Hence, liquid biopsies have a potential utility for cancer diagnosis, detection of minimal residual disease, tracking tumor progression and predicting the emergence of chemoresistance.18 The most common types of liquid biopsies are circulating tumor cells (CTCs) and circulating cell-free DNA (cfDNA) or RNA (cfRNA) (Table 1).19–21 CTC analyses range from the enumeration and immunophenotyping of CTCs to the single cell genomic, transcriptomic or proteomic profiling and tumor growth assays.19,22 cfDNA can be exploited for the detection and quantification of tumor mutations, copy number variations and methylation markers,21 whereas cfRNA can be used for the profiling of mRNAs and non-coding RNAs, and the identification of tumor-specific fusion transcripts and splice variants.22 Some of such tests, for example the enumeration of CTCs using the CELLSEARCH® CTC Test23 or the detection of epidermal growth factor receptor (EGFR) mutations using the cobas® EGFR Mutation Test,24 have been approved by the Food and Drug Administration and are available for routine clinical use. More recently, tumor-educated platelets and tumor-derived EVs have emerged as an alternative source of cancer tissue-derived biomarkers in liquid biopsies.18,22| Types of liquid biopsies | Main source | Detection | Example | |
|---|---|---|---|---|
| CA: cancer antigen; Ct: circulating tumor; CTC: circulating tumor cell; dPCR: digital PCR; FISH: fluorescence in situ hybridization; hTERT: human telomerase reverse transcriptase; ICH: immunocytochemistry; NGS: next-generation sequencing; NSCLC: non-small cell lung cancer; lncRNA: long noncoding RNA; PCA3: prostate cancer antigen 3; PSA: prostate-specific antigen; qPCR: quantitative PCR; RNA-Seq: RNA-sequencing | ||||
| Circulating tumor cells | Cell count | Blood | Several approaches based on the biological, physical and functional properties of CTCs. After CTC isolation, different molecular analysis including NGS, FISH and ICH or testing for drug sensitivity can be done | Enumeration of CTCs by CellSearch for metastatic breast, prostate or colorectal cancer |
| Single cell analysis: | ||||
| – genomics | ||||
| – transcriptomics | ||||
| – proteomics | ||||
| – cytogenetics | ||||
| Tumour-derived cell-free molecules | Proteins | Blood | Immunoassays | – PSA, prostate cancer |
| – CA 15–3, breast cancer | ||||
| ctDNA: | Any biofluid in contact with cancer | PCR (qPCR, dPCR) | – EGFR mutation test, NSCLC | |
| mutations | ||||
| amplifications | ||||
| deletions | ||||
| translocations | NGS | – Epi proColon based on gene methylation, colorectal cancer | ||
| methylation | ||||
| ctRNA: | PCR (qPCR, dPCR) | – hTERT, prostate cancer | ||
| – expression profiles (i.e. of mRNAs, miRNAs, lncRNAs) | ||||
| – mRNA splicing | RNA-Seq | – PCA3, prostate cancer | ||
| Tumor-derived EVs | EV count | Any biofluid in contact with cancer | Nanoparticle tracking analysis, tunable resistive pulse sensing. Other platforms are under development | Increased in pancreatic cancer |
| Proteins: expression and modifications | Mass spectrometry, ELISA | Many examples such as Del-1 for breast cancer | ||
| RNA: expression profiles, i.e. of miRNA, lncRNA, mRNA | PCR, RNA-Seq | Many examples such as | ||
| – AR-V7, prostate cancer | ||||
| – ExoDx Prostate test | ||||
| – miR-196a and miR-1246 for pancreatic cancer | ||||
| DNA: presence and modifications | PCR, NGS | Mutations in KRAS and TP53 in pancreatic cancer | ||
| Lipids and metabolites | Mass spectrometry | See ref. 26 and 27 for examples in prostate cancer | ||
| – exosomes | ||||
| – microvesicles | ||||
| – apoptotic bodies | ||||
2.2. Extracellular vesicles
The term “EV” refers to all types of particles released from cells that are enclosed by a lipid bilayer and cannot replicate.28 EVs contain a large variety of molecules, including proteins, nucleic acids, lipids and metabolites. According to their biogenesis, three main subtypes of EVs have been defined: exosomes, microvesicles (also called ectosomes, shedding vesicles or microparticles) and apoptotic bodies.29 Exosomes correspond to intraluminal vesicles of multivesicular bodies that are released from cells after fusion of the limiting membrane of these organelles with the plasma membrane. The majority of them range between 30 and 150 nm in diameter.30,31 Microvesicles are generated by budding from the plasma membrane and range between 50 and 1000 nm in diameter.31 Apoptotic bodies are highly heterogeneous EVs formed during apoptotic cell death, and the majority of them have a diameter ranging between 1 and 5 μm. However, the release of smaller EVs (<1 μm in diameter) during the progression of apoptosis has also been reported.32 Some specific types of cancer cells have been found to release unusually large EVs (1–10 μm in diameter) referred to as large oncosomes or large EVs,33,34 but their biogenesis is not fully understood so far. As the current methods for EV isolation do not allow accurate separation of EV subtypes, the International Society for Extracellular Vesicles (ISEV) in the recently updated position paper recommends using the term “extracellular vesicle” instead of terms like “exosome” or “microvesicle”, unless the biogenesis pathway of the studied vesicles is clearly established.28 In this review, however, in order to not modify the terminology of the revised manuscripts, we will utilize the terms employed in the original works.Although initially considered a waste disposal mechanism,35 both live cell and apoptotic cell-derived EVs have turned out to be important mediators of intercellular communication acting in a paracrine and systemic manner.32,36 Overwhelming evidence suggests that cancer-derived EVs promote cancer progression in various ways. For instance, EVs released by highly aggressive, drug resistant or hypoxia-experienced cancer cells transfer their phenotypic traits to other cancer cells.37–39 Cancer-derived EVs can also be taken up by various cell types constituting the tumor microenvironment leading to stromal activation, induction of angiogenesis, lymphangiogenesis and immune suppression.40,41 Furthermore, cancer-derived EVs can act systemically by helping to establish pre-metastatic niches in lymph nodes and organ-specific distal sites.42
2.3. Extracellular vesicles as a novel type of liquid biopsies
EVs are released by the vast majority, if not all, cell types in the body; hence blood and other biofluids contain a mixture of EVs released by various cells. Multiple studies have shown that the levels of EVs in biofluids of cancer patients are higher than in healthy controls.43–50 Furthermore, some studies suggest that elevated levels of EVs are associated with the presence of minimal residual disease, therapy failure and disease progression, and that the level of EVs significantly drops after surgery.51,52 These findings support the idea that the presence of cancer stimulates the release of EVs; however, whether these EVs are produced by cancer cells themselves or represent a systemic response to the disease or treatment is still a matter of debate.18,53 Moreover, increased levels of EVs have been found in the blood of patients with various non-cancer diseases and physiological stress conditions, suggesting that the release of EVs is a common response to various stress cues.18 Thus, the EV level per se does not appear to be a highly specific biomarker of cancer. On the other hand, EVs isolated from plasma and other biofluids of cancer patients have been shown to contain cancer cell-derived molecules such as truncated epidermal growth factor receptor EGFRvIII,54 mutated DNA and mRNA fragments and cancer-specific splice variants and fusion transcripts,55–57 as well as cancer-associated mRNA, protein and miRNA signatures.55,58 These findings have raised the idea that cancer-derived EVs may serve as a source of RNA, protein, lipid, DNA and metabolite-based biomarkers for early detection of cancer, monitoring cancer progression and tracking tumor evolution (Table 1 and Fig. 1).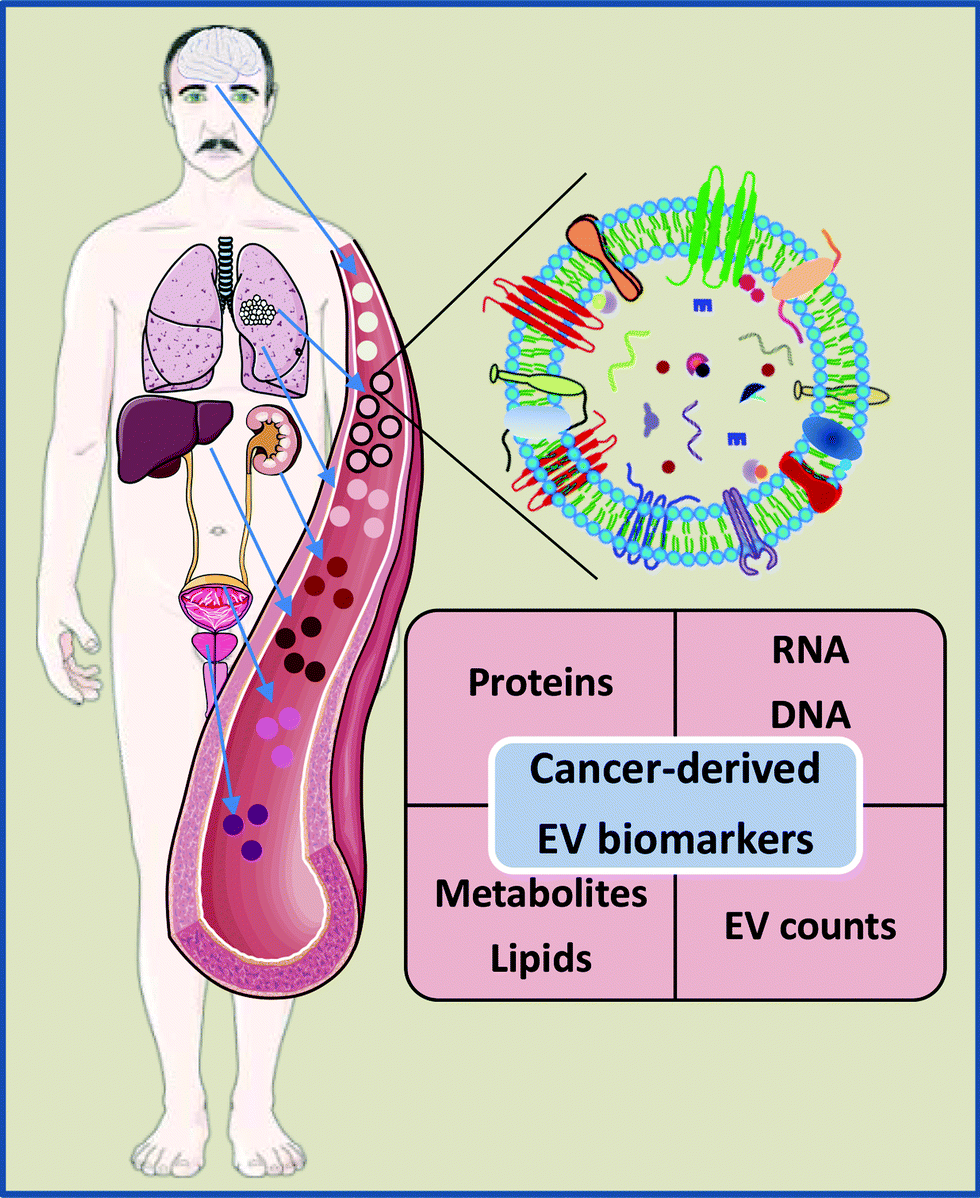 | ||
| Fig. 1 EV-based biomarkers in liquid biopsies. Human blood and other biofluids contain a mixture of EVs released by various cell types. Importantly, EVs isolated from cancer patients’ biofluids contain various cancer-derived molecules. This has raised the idea that EVs may serve as a source of protein, RNA, DNA, lipid and metabolite based cancer biomarkers. Moreover, several studies indicate that the levels of EVs are increased in cancer patients. Therefore, the count of specific EV subpopulations in biofluids may be used as a biomarker on its own. Some elements of the figure originate from Servier Medical Art image bank. | ||
EVs may have several advantages over CTC, cfDNA or cfRNA-based liquid biopsies. At first, they are more abundant than CTCs and therefore may reflect intratumor heterogeneity better than CTCs and/or be easier to detect at earlier stages of cancer, in particular for tumors that release few CTCs, such as tumors of the central nervous system.18,59,60 Secondly, in contrast to vesicle-free cfDNA and cfRNA, EVs contain molecular signatures reminiscent of their parental cells and protect their cargo from degradation.18,59 Indeed, several studies have shown that EV-enclosed DNA yields higher sensitivity and specificity for detecting KRAS, EGFR and BRAF mutations than total circulating cfDNA.56,61–63 Our recent study demonstrated that some miRNA biomarkers show better diagnostic performance if tested in EV-enclosed RNA as compared to total circulating cfRNA.64
Currently, one of the main biological challenges in developing EV-based blood tests is that the proportion of cancer-derived EVs in the total pool of EVs present in blood is low and highly variable among patients leading to high variability in the assay performance. One of the possible solutions is to isolate specific EV subpopulations that may serve as biomarkers by themselves or may be enriched with cancer-derived molecules. For instance, glypican-1 (GPC1) has been identified as a highly specific marker of pancreatic cancer-derived EVs and the levels of GPC1-positive EVs have been shown to have diagnostic and prognostic value in pancreatic cancer.65,66 Similarly, prostate-specific membrane antigen (PSMA) has been used for the isolation of prostate-specific EVs. Although PSMA is a prostate-specific, not prostate cancer-specific, protein, the plasma levels of PSMA-positive EVs could discriminate prostate cancer from benign prostatic hyperplasia and correlate with the aggressiveness of the disease.67,68 Conceivably, these EV subpopulations are also enriched in cancer-derived molecules. Furthermore, EVs have been detected in various other biofluids including lymph, saliva, urine, cerebrospinal fluid, breast milk and bronchoalveolar lavage fluid;56,69–71 hence these biofluids may serve as organ-specific liquid biopsies for cancers that are in contact with the given biofluid.
2.4. State-of-the-art techniques to analyze EV biomarkers in biofluids
The use of omics methodologies such as mass spectrometry for protein, lipid and metabolite quantification, and next generation sequencing for quantification of nucleic acids, has led to the discovery of numerous EV-based biomarkers for several diseases.18,72,73 In the biomarker development pipeline, the validation phase follows the discovery phase. In this phase, the ability of the biomarker to separate specific patient groups using larger and independent patient cohorts is verified, and the best analytical platform to quantify the biomarker is established. In fact, the quantitative verification of the biomarkers identified in the discovery phase studies is considered a bottleneck in biomarker development. The analytical method should be able to quantify the biomarker(s) in a specific, highly reproducible, robust, fast and cheap way in order to facilitate the implementation of the biomarker test in the clinic. Conventional techniques to analyse EVs include methods such as nanoparticle tracking analysis (NTA), flow cytometry, dynamic light scattering (DLS), western blotting (WB) or immunoassays among others (Table 2). These techniques can provide valuable information about EV size, concentration or specific markers, as summarized in excellent reviews where readers are referred to for detailed information.10,11,74,75 On the other hand, mass spectrometry and next generation sequencing, although very useful for the discovery of biomarkers, are currently not widely used in clinical laboratories. Instead, PCR-based tests, for the quantification of nucleic acids, and immunological methods, mainly enzyme linked immunosorbent assays (ELISA), for the quantification of proteins, are part of clinical lab routines and are commonly used for diagnostic purposes (Table 1). These methods have high sensitivity and do not require complex equipment. In terms of EVs, a polymerase chain reaction (PCR)-based test for the detection of RNAs in urinary EVs that can reduce the number of unnecessary biopsies has already been commercialized.76 The potential of lateral flow immunoassays (LFIAs) for detection of protein biomarkers in EVs has also been shown since a LFIA using the membrane proteins tetraspanins as targets is able to detect purified EVs from human plasma and urine.77 LFIAs are based on similar principles as ELISA tests, in which a capture antibody is immobilized on a solid phase, and are ideal POC tests because they require few resources. Interestingly, immunoisolation is particularly suitable for liquid biopsies of specific cancer types and several microfluidic devices based on this principle are being developed.78| WB | ELISA | Flow cytometry | NTA | DLS | |
|---|---|---|---|---|---|
| What is measured | Presence and molecular weight of proteins | Quantification of surface protein markers | Concentration of EVs, phenotype | Concentration of EVs, size | Size |
| Signal | Colorimetric, chemiluminescence, fluorescence | Colorimetric, chemiluminescence, fluorescence | Light scattering, fluorescence | Light scattering | Light scattering |
| LOD (EVs per mL) | 1011–1012 | 109–1010 | 107–109 | 107–109 | 108–1012 |
| Specificity | High (specific antigen-Ab) | High (specific antigen-Ab) | High if Abs are used | Low | Low |
| Ref. | 79,80 | 79–81 | 82,83 | 75,80 | 84 |
In addition to the analytical approaches, the standardization of the preanalytical procedures is essential for robust biomarker quantification and data interpretation.85 It is critical that the collection and storage of biofluids follows specific guidelines.86 Moreover, when working with EVs, the way in which vesicles are isolated/purified should be carefully investigated. The main goal of EV isolation is to concentrate the molecular signal contained in the EVs and/or remove contaminants that may mask the signal or perturb the analytical measurement. At the moment, several methods to isolate/purify EVs exist, including ultracentrifugation, size-exclusion chromatography, precipitation, immunoisolation, microfluidics or filtration.78,87–89 These methods have advantages and disadvantages that have to be considered in relation with the specific biofluid, the biomolecule that is going to be measured and the analytical method that is going to be used. In fact, it has been shown that the yield and purity of EVs significantly vary between different isolation methods.90,91 Therefore, the isolation of EVs may introduce a potential error in the quantification of biomarkers. Thus, ideally, a routine test should not require a prior isolation of EVs from the biofluids. This could be possible if the EV-associated biomarker is very abundant and/or the quantification method is very sensitive and not affected by other materials found in the biofluid. This strategy has already given promising results. For example, Duijvesz et al. have developed a highly sensitive time-resolved fluorescence immunoassay (TR-FIA) for capture/detection of prostate cancer derived EVs that allows the quantification of EV proteins directly from urine.46 Other example is the ExoScreen assay, which is able to detect EVs directly in serum using two antibodies to capture the vesicles that are then detected using photosensitizer-beads.92
3. Development of nanoparticle-based biosensors
Currently, a nanomaterial can be defined as “a natural, incidental or manufactured material containing particles, in an unbound state or as an aggregate or as an agglomerate and where, for 50% or more of the particles in the number size distribution, one or more external dimensions is in the size range 1–100 nm”.93 On the other hand, the International Organization for Standardization defines a NP as “a material with all external dimensions in the nanoscale, where the lengths of the longest and the shortest axes of the nano-object do not differ significantly”. In this review we will mainly focus on the use of NPs for the development of biosensors with enhanced performance. Materials on the nanometre scale have unique optical, electronic, and magnetic properties that are different from the bulk material. Of particular interest is the possibility of changing their physicochemical properties by tuning the shape and size of many NPs. For instance, important features that can be tailored on demand are the surface plasmon resonance (SPR) of gold NPs (AuNPs), the emission wavelength of carbon nanotubes (CNTs) or Quantum Dots (QDs), or the magnetic properties of magnetic nanoparticles (MNPs). One general advantage of NPs is their large surface area to volume ratio, enabling the attachment of an enhanced number of biomolecules.94,95 In addition, as NPs and biomolecules have similar sizes, they can interact more effectively. NPs therefore hold huge interest in biomedical applications. Indeed, NP-based biosensors have become one of the major topics in the field of diagnostics.96,97 The use of NPs allows the development of devices with increased sensitivity and lower limits of detection (LODs), features with growing interest in the sensing field. Furthermore, lab-on-a-chip based assays allow the rapid analysis of low amounts of samples, thus reducing clinical care costs.77In the field of EVs, several detection platforms have been reported based on diverse sensing techniques, such as colorimetry, fluorescence, SPR, Surface-enhanced Raman spectroscopy (SERS), electrochemistry or nuclear magnetic resonance, showing the potential of NPs in diagnostics.79,98,99 These methods, along with their advantages and disadvantages, will be discussed in the next sections.
3.1. Functionalization of NPs with antibodies and aptamers
When developing biosensors, antibodies (Abs) and aptamers are two of the most commonly used molecular recognition biomolecules. These molecules are generally coupled to NPs in order to selectively recognize EVs. The most common targets to identify and detect EVs are protein markers such as tetraspanins (CD63, CD9, CD81), ALIX or TSG101.3 Moreover, EVs also contain specific markers directly related with their cellular origin; the analysis of these antigens could be used to detect EVs derived from cancer cells for instance.2 Aside from proteins, EVs also contain RNAs; among all RNA species, miRNAs are in general very abundant, and can be used as biomarkers,100 although aptamers and Abs are not commonly used to target them.Different types of Abs or Ab-derived fragments can be used to functionalize NPs to detect EV antigens.101 The most widely used is Immunoglobulin G (IgG). IgG consists of four polypeptide chains linked together by disulphide bonds, forming a Y-shaped structure (Fig. 2a and b). The IgG structure can be subdivided into two parts, the antigen binding fragment (Fab) and the constant fragment (Fc) (Fig. 2b). The Fab region (arms of the Ab) contains the antigen-binding site, which confers antigen specificity. A single IgG has two antigen-binding sites that are found at the extremity of the arms. In order to improve the biosensor performance, it is highly important to leave these antigen binding sites available, so that they can interact with their antigens. One of the key factors to develop a reliable biosensor, therefore, is the technique used to immobilize the Abs on the NP surface, as the selected methodology can impact the Ab structure and activity, and ultimately the biosensor sensitivity.102,103 The methodologies used to functionalize Abs on NPs are based on physical adsorption, covalent binding or the use of specific adaptor molecules (Fig. 2c–g).104–106 Generally, strategies that provide a better orientation of the Ab on the NP surface and without involving the antigen-binding sites will result in a better outcome than strategies providing a random orientation of the Ab.107 Here we present some of these strategies, which could be implemented to build biosensors to detect EVs.
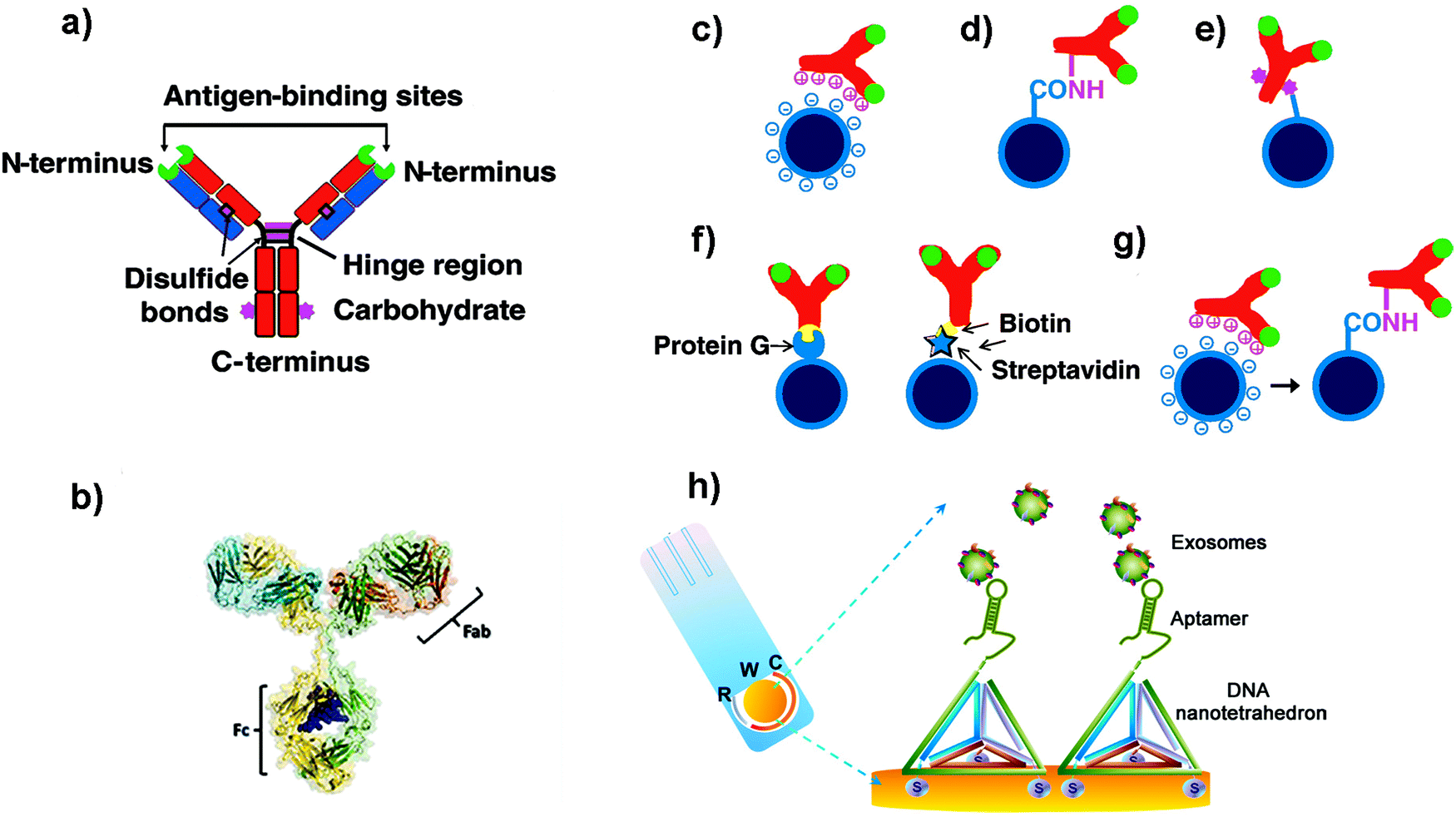 | ||
| Fig. 2 (a and b) Schematic cartoon showing (a) the Y-shaped structure of an Ab (which is the ligand to be attached to the NP); the two light chains (variable regions) and the heavy chains (constant regions) are coloured in blue and red respectively. The green colour represents the antigen binding sites. The purple colour represents groups which can be used for attachment to NPs. (b) Three-dimensional model of an Ab from X-ray crystallography studies. (c–g) Schematic representation of different strategies used to functionalize NPs with Abs: (c) electrostatic adsorption; (d) covalent binding via amine groups on the Ab; (e) covalent binding via carbohydrate groups on the Ab; (f) use of adaptor biomolecules (streptavidin–biotin, Protein G); (g) ionic adsorption plus covalent binding. Adapted from ref. 105 with permission from Elsevier. (h) Schematic illustration of a nanotetrahedron-assisted electrochemical aptasensor. Aptamer-containing nanotetrahedra were immobilized via three thiol groups onto the gold electrodes for direct capture of exosomes in suspension. R: reference electrode area; W: working electrode area, with a diameter of 4 mm; C: counter electrode area. Adapted with permission from ref. 108. Copyright (2017) American Chemical Society. | ||
One of the most common strategies to covalently link Abs to NPs is to use the amine groups from the Abs (Fig. 2d).105 However, using this approach, Abs will be immobilized with a random orientation, as some Abs will be well oriented, while others will not have their antigen binding sites available. Moreover, taking into account the different pKa values of the amine groups of the Ab, at the pH conventionally used for this kind of reaction, the most reactive amine groups are the terminal ones. Unfortunately, these moieties are located in the antigen binding area, and their use could reduce the recognition efficacy of the Abs. In order to overcome this limitation, other strategies have been developed to improve the orientation of the Abs. For instance, Puertas et al. demonstrated that by binding the Ab on MNPs through its sugar moieties (located in the Fc region, Fig. 2e), by their partial oxidation and formation of a Schiff base, the LOD of a LFIA could be greatly improved.109 Other works suggested the use of bioorthogonal click chemistry to functionalize the surface of NPs with Abs. The cycloaddition between 1,2,4,5-tetrazines (Tz) and trans-cyclooctene (TCO) is a straightforward method to bind Abs on the NP surface.110 Furthermore, it is fast, catalyst free and chemoselective. Abs modified with TCO have been used to detect cells and pathogens before being coupled with MNPs functionalized with Tz.110,111 When compared with direct Ab binding, the use of bioorthogonal click chemistry yielded higher sensitivity. This strategy could enhance the signal to a greater extent than other two-step labelling strategies that are routinely used, such as the coupling of avidin-modified Abs with biotin-conjugated MNPs. In addition, consecutive steps of orthogonal chemistry can further amplify the signal and increase the sensitivity.112
However, all these examples imply the chemical modification of the Abs. This modification can ultimately affect Ab structure and activity. To overcome this concern, other strategies have been proposed. Some groups have demonstrated that unspecific reversible interactions between the Ab and the NPs can be used to orient the Ab before performing a covalent coupling (Fig. 2g).113,114 In these cases, the incubation pH can be selected to orient the Ab, as the net protein surface charge depends on the isoelectric point of the Ab.115 Puertas et al. described this strategy to bind different types of Abs to MNPs, demonstrating that in all cases the activity was higher than when using a random conjugation.12 This approach has been also used for the binding of Abs to AuNPs116 and multiwalled carbon nanotubes (MWCNTs),117 showing an improvement of the analytical performance of the biosensors when using this approach.
Aside from the orientation, the Ab density is also an important factor to take into account.114,118 Although the use of adaptor molecules (Fig. 2f) could provide worse Ab coating, the improvement on the presentation is able to enhance its activity.119 On the other side, Van der Heide et al. described that using protein A as an adaptor molecule resulted in higher Ab per AuNP and higher binding efficacy when compared to random immobilization through the most reactive amine groups.120
Aptamers are synthetic single-stranded oligonucleotides (DNA or RNA) that bind to target molecules, such as proteins or nucleic acids, with high affinity and selectivity. Thus, they have been used in many biomedical applications, including biosensing. Aptamers are selected from an oligonucleotide library by Systematic Evolution of Ligands by Exponential Enrichment (SELEX), a process that can be automatized.121,122 Target recognition and subsequent binding is based on electrostatic and hydrophobic interactions and the three-dimensional structure that they adopt. Compared to Abs, aptamers are thermally stable, have a smaller size, are more flexible and lack immunogenicity.123,124 When developing biosensors, the density of the aptamers on the surface of the support can be a critical factor. For instance, a dense coating could end with a high steric crowding and aptamer entanglement, thus resulting in poorer accessibility.125 It has been suggested that immobilization through the 3′ end or the addition of a linker can improve the target binding, presumably due to a decreased steric hindrance or an improved folding.126 In this sense, the use of DNA nanotetrahedron structures to functionalize the aptamers can greatly improve the accessibility and binding ability of aptamers to their targets.127,128 For instance, Wang et al. developed an electrochemical biosensor to detect EVs where the aptamers were oriented using a tetrahedron structure (Fig. 2h).108 The sensitivity of the aptasensor was increased 100-fold when compared with that obtained using single-stranded aptamers.
3.2. NP-based biosensors for EV analysis
In a typical LFIA sandwich assay, the sample is added to the sample-pad and it migrates by capillarity to the conjugate-pad. There, the analyte finds a conjugate composed of the detection Ab conjugated to particles such as latex beads or AuNPs (Fig. 3a).129,130 This complex flows through the nitrocellulose membrane, where the analyte is now recognised by the capture Ab immobilized at the test line (TL). Finally, the excess of conjugate reaches the control line (CL) where a secondary Ab is located. This Ab recognizes the detection Ab, to indicate that the test worked properly. The excess of sample migrates to the absorbent pad (Fig. 3a).131,132 In summary, if the target analyte is present, both the CL and the TL should appear and be detected visually. If the target analyte is not present, only the CL appears. The best well-known example of this bioanalytical method is the human pregnancy test, based on the detection of human Chorionic Gonadotropin (hCG) in urine.
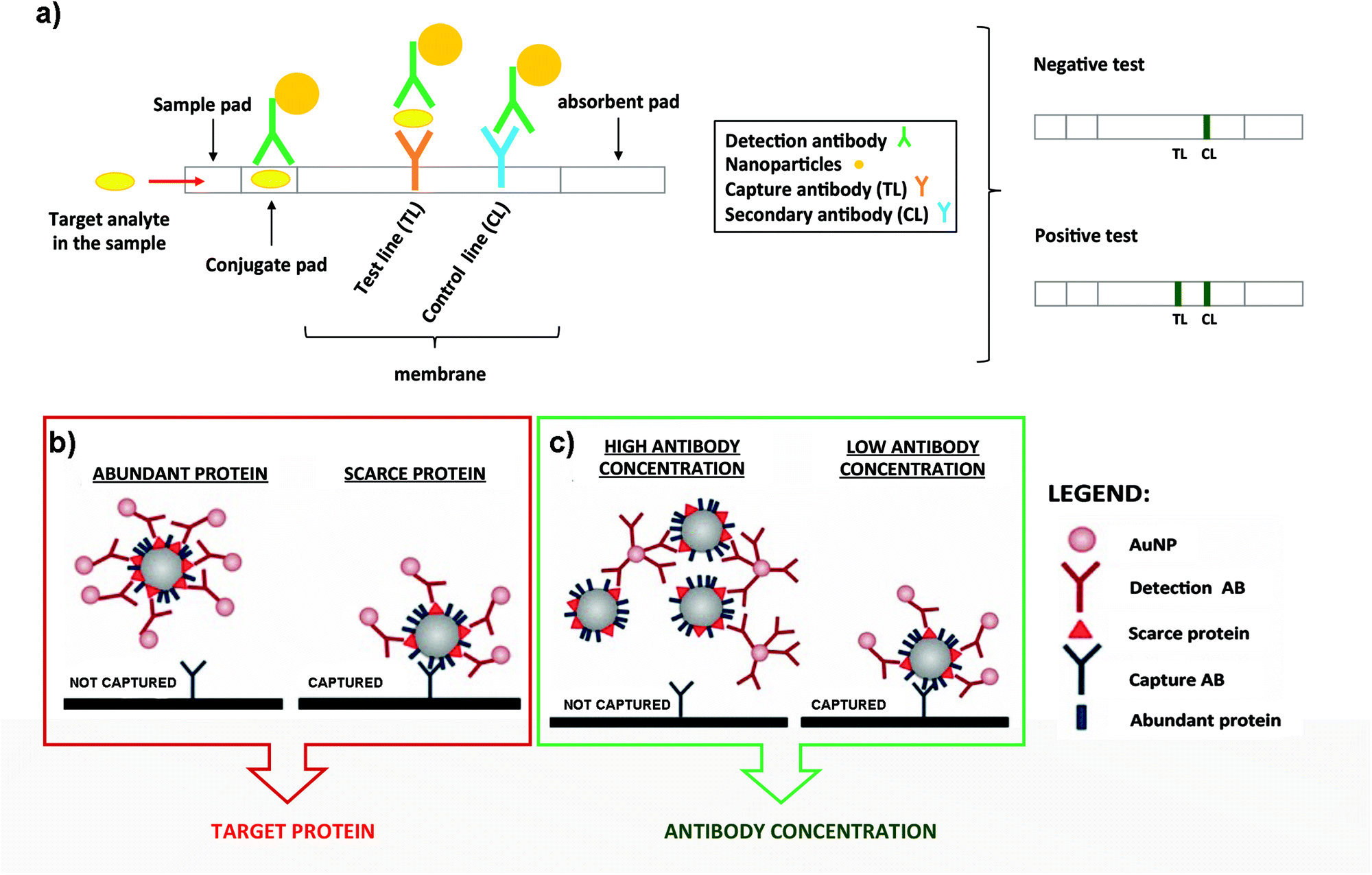 | ||
| Fig. 3 (a) Schematic representation of a typical LFIA when the target analyte is present (left panel). Naked eye detection of a negative and positive test (rigth panel). (b) Steric hindrance model for exosomal detection using LFIA: if a marker is abundant, the exosome is completely covered by the conjugate, composed of the detection Ab functionalized on NPs. This reduces the availability of epitopes for the capture antibody present at the TL (left panel). However, if the detection marker is scarcer, the exosome can be captured (right panel). (c) Model of the effect of the Ab concentration coupled to AuNPs. When AuNPs are conjugated using a high concentration of the Ab, bigger aggregates are generated due to the crosslinking of Abs and exosomes. This results in an impaired flow of the mixture on the strip and lower capture capacity (left panel). However, if the detection Ab is conjugated at lower concentration, these complexes are not formed and each exosome can bind to several AuNPs, resulting in a better capture at the TL (right panel). Adapted from ref. 133. Copyright (2018), with permission from Springer Nature. | ||
LFIA test strips are therefore ideal candidates for EV detection when onsite analysis and simplicity are needed. Oliveira-Rodríguez et al. developed a LFIA using tetraspanins as targets to detect purified exosomes from cell culture supernatants of Ma-Mel-86c melanoma cells.77 In this work the authors were able to detect 8.54 × 108 exosomes per mL when combining anti-CD9 and anti-CD81 as capture Abs and anti-CD63 as the detection Ab. The detection Ab was labelled with 40 nm AuNPs. In addition, and as a proof of concept, these authors managed to visually detect 5 μg and 20 μg of plasma and urine-derived exosomes respectively (commercial exosomes from healthy donors) when different combinations of capture and detection Abs against CD9, CD81 and CD63 were used. The Ab pair was selected case-by-case depending on the different protein composition (localization and density of tetraspanins) present on the exosomal surface.
Some studies have been carried out to evaluate the performance of different NPs as labels, including AuNPs, carbon black nanoparticles (CB) and MNPs. To select the best option, the following parameters were compared: simplicity for the bioconjugate formation, stability over time and ease of visualization on the strips.134 NPs were functionalized with anti-CD63 and tested to detect EVs purified from plasma of healthy donors on strips containing anti-CD9 as the capture antibody. Conjugates made using AuNPs provided the best performance providing similar sensitivity results as CB (≈109 EVs per mL), but providing a better fitting in the linear range. Moreover, AuNPs are easier to get functionalized with Abs. In contrast, MNPs provided low sensitivity and generated a retention-like line at the end of the sample pad during the test. This study also explored a multiple-targeted approach, incorporating the anti-CD81 Ab in an additional capture line. Although single-targeted and multiple-targeted detection provided similar LODs when using a reflectance reader, the multiple-targeted detection had a broader detection range. Since EVs from different origin express diverse proteins on the surface, the incorporation of several test lines may allow the detection of a broader range of EVs, opening the possibility to study a concrete disease marker.
LFIA immunoassays using AuNPs as labels have been successfully employed for the detection of the endogenously expressed tumour-derived antigen MICA (MHC class I chain-related protein A) in exosomes.133 In this case it was very important to consider potential competition events and steric effects in LFIA assays, highlighting the importance of targeting scarce proteins with the detection Ab present on the NPs (Fig. 3b). In fact, targeting abundant proteins on the exosomal surface could lead to steric impediments, impairing the subsequent binding of the EVs with the capture Ab present at the TL.77 Furthermore, controlling the density of the detection Ab on the AuNP surface allowed an improvement of the LOD, mainly driven by the decrease of the aggregates because of the crosslinking of Abs and exosomes (Fig. 3c). Taking into account all these considerations, MICA-containing exosomes purified from metastatic melanoma cell lines were detected at a concentration of 5 × 1010 exosomes per mL, using anti-CD9 and anti-MICA Abs as capture and detection Abs respectively. This was the first time that canonical exosome markers as well as an endogenously expressed tumour-derived antigen were detected using a LFIA.
In order to increase the sensitivity of LFIA sensors, an amplification step could be carried out. Wu et al. recently reported a LFIA system using two different AuNP bioconjugates.135 The first one included a monoclonal anti-CD9 Ab, while the second bioconjugate was labelled with an anti-BSA Ab. The first bioconjugate recognized the exosomes and could be retained at the TL, where a polyclonal anti-CD9 Ab was immobilized. The second bioconjugate was then added, enhancing the staining, as the first bioconjugate was blocked with BSA. This enhancement improved the optical intensity of the red band formed by the AuNPs on the TL. This system was used to detect isolated exosomes from MCF-7 human breast cancer cells, showing a LOD of 1.3 × 106 particles per mL, improving the sensitivity by two orders of magnitude when compared to conventional LFIAs. This LFIA was also successfully used for detecting MCF-7 exosomes diluted in ultracentrifuged foetal bovine serum, proving its potential application in practical diagnostics.
Besides, it is also possible to detect exosomes through the phospholipids present within their lipid bilayer. Dong et al. studied this possibility using biotin-tagged 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-poly(ethylene glycol) (DSE-PEG-biotin) to label exosomes through the strong hydrophobic interaction of the fatty acid tails and the phospholipid membrane.136 Based on the high affinity between streptavidin and biotin, fluorescent nanospheres conjugated to streptavidin (FNs-SA) were used as the detection system. On the other hand, streptavidin was deposited at the TL in the LFIA test strips. In this complex system, the biotin-EVs formed a complex with the FNs-SA and with the streptavidin present at the TL. With this approach, ultracentrifuged exosomes from human epithelial Cal 27 cells were tested in the test strips and the minimal detectable concentration using a portable UV lamp was 2.0 × 106 particles per mL. To collect the exosomes, however, two rounds of ultracentrifugation were performed, consequently increasing the time: (i) to isolate the exosomes from cell culture media, and (ii) to remove the excess of reagents after labelling with DSPE–PEG–biotin.
As an alternative to standard LFIA performed using Abs, a lateral flow aptamer assay (LFAA) was proposed by Yu et al.137 This LFAA system was based on a competitive format, where in the presence of exosomes, a CD63 aptamer functionalized on AuNPs could interact with the CD63 exosomal proteins. On the other hand, a CD63 aptamer complementary strand was deposited at the TL. In the presence of exosomes, the aptamers present on the AuNPs will interact with them, avoiding the subsequent binding of the AuNPs to the TL. However, in the absence of exosomes, the AuNPs@aptamer could interact with the complementary aptamer present at the TL, and give a positive signal. Some parameters such as the concentration of the blocking buffer to pre-treat the strips, the streptavidin ratio to AuNP aptamer and the optimal incubation time were optimized, reaching a LOD of 6.4 × 108 particles per mL for exosomes derived from lung carcinoma A459 cells. Although the idea of using aptamers instead of Abs could be advantageous, further work should be conducted to validate this LFAA: the addition of a CL on the test strips, the control of AuNP aggregation and the possibility to assemble the strips in a more reproducible way could all help to improve the system.
For instance, Chen et al. used positively charged MNPs to isolate exosomes directly from plasma.138 The exosomes were thereafter eluted using a high concentration of sodium chloride. By performing this anion exchange-based isolation, exosomes were recovered with high efficiency and high purity in a fast way. Once isolated, aptamer-coated iron oxide nanoparticles were added for the visual detection of exosomes (Fig. 4). The rationale behind this experiment is that iron oxide nanoparticles have weak intrinsic HRP-like activity, catalyzing a change of color when 3,3,5,5′-tetramethylbenzidine (TMB-substrate of peroxidase) and H2O2 are present. Interestingly, the presence of aptamers on the MNP surface increased the HRP-like activity when compared with naked MNPs. In the presence of exosomes, these aptamers could bind to them by molecular recognition, getting desorbed from the MNP surface. Once the aptamers were desorbed, a decrease in the catalytic activity of those NPs was achieved, followed by a decrease in the colour change when TMB and H2O2 were present. This color change could be detected by UV-vis spectroscopy, and a linear correlation between the absorbance and the concentration of exosomes was found. The LOD of this aptasensor was 7.0 × 106 particles per mL for exosomes isolated from plasma (healthy donors) and 3.58 × 106 particles per mL for exosomes isolated from simulated prostate cancer (PCa) plasma samples. Similarly, single-walled carbon nanotubes (s-SWCNTs) coated with CD63 aptamers were used to detect exosomes due to their HRP-like intrinsic activity.139 The detection limit was 5.2 × 108 particles per mL and the whole detection process took 40 minutes. Interestingly, no complex technologies to enhance the signal were needed. Using graphitic nitride nanosheets and a similar detection approach, a LOD of 13.52 × 108 particles per mL was reported.140
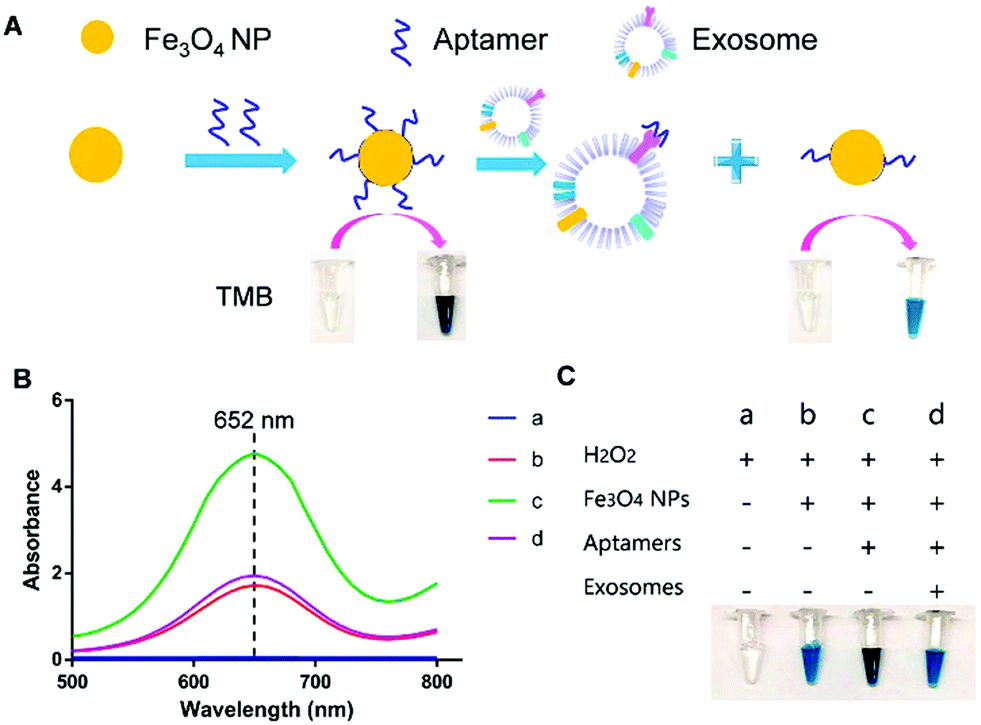 | ||
| Fig. 4 Visible detection of exosomes. (A) Schematic representation of the detection mechanism for the visible detection of exosomes. (B) UV-vis-absorption spectra of TMB–H2O2 (curve a); TMB–H2O2 and iron oxide NPs (curve b); TMB–H2O2 and aptamer–iron oxide NPs (curve c); and TMB–H2O2, aptamer–iron oxide NPs, and exosomes (curve d). (C) Digital images of TMB–H2O2 (image a); TMB–H2O2 and iron oxide NPs (image b); TMB–H2O2 and aptamer–iron oxide NPs (image c); and TMB–H2O2, aptamer–iron oxide NPs, and exosomes (image d). Reprinted with permission from ref. 138. Copyright (2018) American Chemical Society. | ||
AuNPs are commonly used to design colorimetric-based biosensors as their optical properties depend on the NP separation. Aggregation causes a shift in the extinction coefficient that can be appreciated with a colour change. Taking advantage of this phenomenon, Jiang et al. described a biosensor composed of aptamers and AuNPs to detect and profile exosomes.141 Complexation of aptamers with AuNPs protected them from aggregation at high ionic strength, as aptamers stabilized the AuNPs by steric repulsion. When exosomes were present, these could bind to the aptamers, resulting in the displacement of these ligands from the AuNP surface, destabilizing the AuNPs under high ionic strength conditions. This led to AuNP aggregation, resulting in a red-to-blue color change that could be measured by UV-vis spectroscopy. This sensor was used to differentiate exosomes derived from different cancer cell lines depending on the CD63 expression level. Furthermore, the authors were able to detect exosomal proteins restricted to a unique cell line by using an aptamer that could bind to protein tyrosine kinase-7, overexpressed in human acute lymphoblastic leukemia cells.
The aggregation of AuNPs in combination with other steps to amplify the signal has also been reported. For instance, Liu et al. designed a pair of DNA-labelled Abs that could bind to the same target biomarker present on exosomes.142 Upon synchronous recognition of the protein on the exosome surface, the DNA strands could hybridize, generating a unique DNA signal. This double-stranded DNA signal was amplified twice by recombinase polymerase amplification combined with transcription-mediated amplification to produce RNA strands. The RNA products were proportional to the initial concentration of the biomarker and could be detected by using oligonucleotide-coated AuNPs, complementary to the RNA. RNA recognition by the AuNPs promoted their aggregation and a red-to-blue color change that could be measured by absorption spectroscopy. With this technique, the authors demonstrated the possibility to detect EGFR and Epstein–Barr virus latent membrane protein 1 (LMP1)-positive exosomes derived from nasopharyngeal carcinoma cells. Despite being more complex than other colorimetric methods, the developed biosensor was highly sensitive, and was able to detect 100 particles per mL.
DNA hybridization chain reaction (HCR) has also been used to enhance the signal in colorimetric biosensors.143,144 Zhang et al. used aptamer-conjugated MNPs to isolate exosomes from cell culture media.144 Thereafter, a bivalent-cholesterol-labelled DNA probe was incorporated into the exosome membrane by hydrophobic interactions. The sticky end of this anchor triggered an enzyme-linked HCR, where alkaline phosphatase (ALP) was introduced. ALP enables the removal of the phosphate group from ascorbic acid 2-phosphate to produce ascorbic acid, which can further reduce Ag+. This reaction led to the formation of silver shells on gold nanorods (AuNRs), and gave rise to a change of colour that could be distinguished with the naked eye. Interestingly, the colour changed from pink to brown, green or purple with increasing concentration of exosomes. This allowed an easier detection with the naked eye when compared with the development of a unique colour. The reported LOD was 1.6 × 105 particles per mL by UV-vis spectroscopy. The sensor was tested with plasma from donors, obtaining similar concentration values as standard techniques, that is, ultracentrifugation followed by NTA.
Traditional assays for the identification of EVs by fluorescence techniques are based on immunoassays, such as ELISA and western blot, where Abs are coupled to organic fluorophores.147 These methods have excellent analytical performances, but sometimes the stability of the fluorophore is compromised, thus limiting their application. Fluorescent NPs have advantageous optical properties, such as high photostability and quantum yields, low photobleaching, size-tunable emission, extremely broad excitation range and narrow emission which allow large Stokes shift, that can overcome the limitations of current fluorophores.148 These properties allow improving the LOD and even enabling single molecule detection, making fluorescence-based nanobiosensor devices more sensitive and reliable when compared to the classic fluorescence detection methodologies. Fluorescent NPs include NPs made with silica and organically modified silica,149 metals,150 metal oxides,151 metal nanoclusters,152,153 upconversion NPs (UCNPs),154,155 organic polymers,156 quantum dots (QDs),157,158 silicon quantum dots159 and different carbonaceous nanomaterials such as carbon dots, carbon nanotubes, carbon nanoclusters and nanodiamonds.160,161
Lanthanide-doped UCNPs undergo a non-linear photophysical process whereby low-energy radiation, usually in the near infrared (NIR) range, is converted to higher-energy radiation, for example, visible light (anti-Stokes shift). Thus, the fluorescence emission of UCNPs takes place at shorter wavelengths than the absorbed light. This feature makes them especially attractive for biological applications and nanobiosensor development, since it avoids the use of ultraviolet (UV) light, therefore minimizing the autofluorescence of biological samples.162,163 UCNPs coupled to aptamers have been used as energy donors in the development of aptasensors based on Luminescence Resonance Energy Transfer (LRET) in combination with other fluorophores or NPs that act as acceptors. LRET and Fluorescence Resonance Energy Transfer (FRET) are mechanisms that occur due to the very short distance interaction between the energy levels of two luminescent/fluorescent molecules in which the emission wavelength of the donor molecule coincides with the excitation wavelength of the acceptor molecule. In this way, the excited donor transfers its energy to the acceptor, which emits a photon. Wang et al. used tetramethyl rhodamine (TAMRA) and UCNPs functionalized with two DNA aptamers to target the epithelial cell adhesion molecule (EpCAM) present on the exosome membrane of some cell lines.164 After the aptamer–exosome binding, both DNA strands got closer and the distance between the energy donor (UCNPs) and the acceptor (TAMRA) was reduced, promoting the LRET process (Fig. 5). Due to the coincidence between the emission wavelength of the donor and the excitation spectrum of the acceptor, the excitation of UCNPs by IR light produces a UV emission that excites the TAMRA molecule, leading to a yellow emission (585 nm) that is linearly correlated with the exosome concentration. This LRET sensor reached a LOD of 8 × 104 particles per mL.
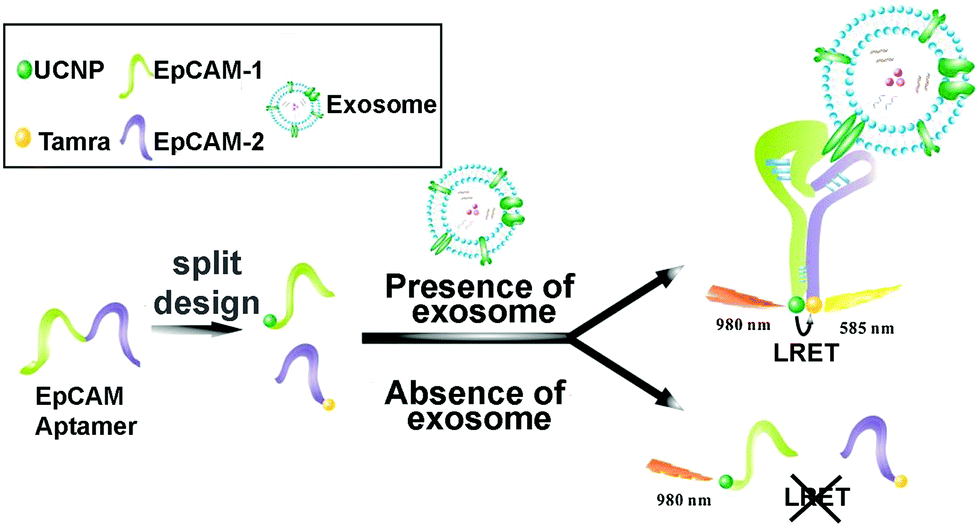 | ||
| Fig. 5 Aptasensor based on LRET between UCNP donor and TAMRA acceptor for highly sensitive detection of exosomes. The two DNA strands from the EpCAM aptamer are labeled with UCNPs and TAMRA, which get closer to each other when recognition of EpCAM exosomes occurs. The LRET fluorescence response enables the quantitative detection of exosomes. Reprinted with permission from ref. 164. Copyright (2019) American Chemical Society. | ||
The possibility of immobilizing this type of sensors in a user-friendly POC format makes them even more interesting. This is the case of an exosomal aptasensor based on a LRET system that used AuNRs as the acceptor and UCNPs immobilized on a paper support as the donor.165 As before, the sequence of the CD63 aptamer was split into two different fragments, and AuNRs and UCNPs were decorated with only one of the fragments. In the presence of exosomes, the CD63 protein present on their surface was combined with both strands of the aptamer bound to AuNR and UCNP-paper respectively; this reduced the distance between acceptor and donor, allowing the LRET to take place. The analytical signal was the quenching effect produced by AuNRs over the green luminescence of UCNPs under IR excitation, reporting a LOD of 1.1 × 106 particles per mL.
QDs are also widely used in fluorescence-based biosensing, due to their high emission quantum yield, size tunable emission profiles with a narrow spectral band and unique photophysical properties.166 Bai et al. used QDs to build a bead-based exosome microfluidic chip for exosome isolation and multiplexed detection.167 Anti-CD9-labeled magnetic beads were used to isolate exosomes, while three QD probes labelled with Abs to detect tumoral markers were used for multiplexed detection of exosome surface markers (carcinoma embryonic antigen, CEA; Cytokeratin 19; Progastrin-releasing peptide, proGRP). With this novel microfluidic immunoassay system and using an inverted fluorescence microscope, it was possible to discriminate between plasma-derived exosomes from lung cancer patients and healthy controls. Further, minimal differences were found between these experimental results and clinical data obtained using traditional methods for the detection of CEA in real samples. Although CdSe-based QDs are the most used QDs, works that use other kind of QDs as fluorescent labels for EV detection combined with fluorescence microscopy have been reported: InP/ZnS QDs,168 silicon QDs169 and gold–carbon dots170 among others.
Although metallic NPs can be used as fluorescent labels because of their intrinsic fluorescence, not many works have employed them to detect EVs. He et al. used CuO NPs modified with CD63 aptamers to form sandwich complexes with exosomes previously isolated with magnetic beads.171 These complexes were subsequently dissolved in acidic medium to obtain copper(II) ions which were thereafter reduced to fluorescent CuNPs (copper nanoparticles) in the presence of sodium ascorbate and poly(thymine) (Fig. 6a). CuNPs’ concentration and fluorescence were proportional to the exosome content, obtaining a LOD of 4.8 × 107 particles per mL. Many times the fluorescence of these metallic NPs is not intrinsic, but comes from ligands placed on their surface during their synthesis. For instance, Gao et al. designed a complex exosome detection method using a rolling circle amplification (RCA) reaction.172 A series of long DNA hairpin structures with components such as CD63 aptamer, linker and spacer sequences were obtained using the RCA reaction. In the presence of exosomes, the RCA product could attach to the exosomal membrane, opening its hairpin structure and exposing a linker sequence. A fluorescent AuNP-linker/complementor bioconjugate (AuNP-L/cL) could then pair with this linker sequence by toehold-mediated strand displacement, releasing the fluorescent cL probe. The fluorescence signal was proportional to exosome concentration, reaching detection limits in the order of 1 × 108 particles per mL.
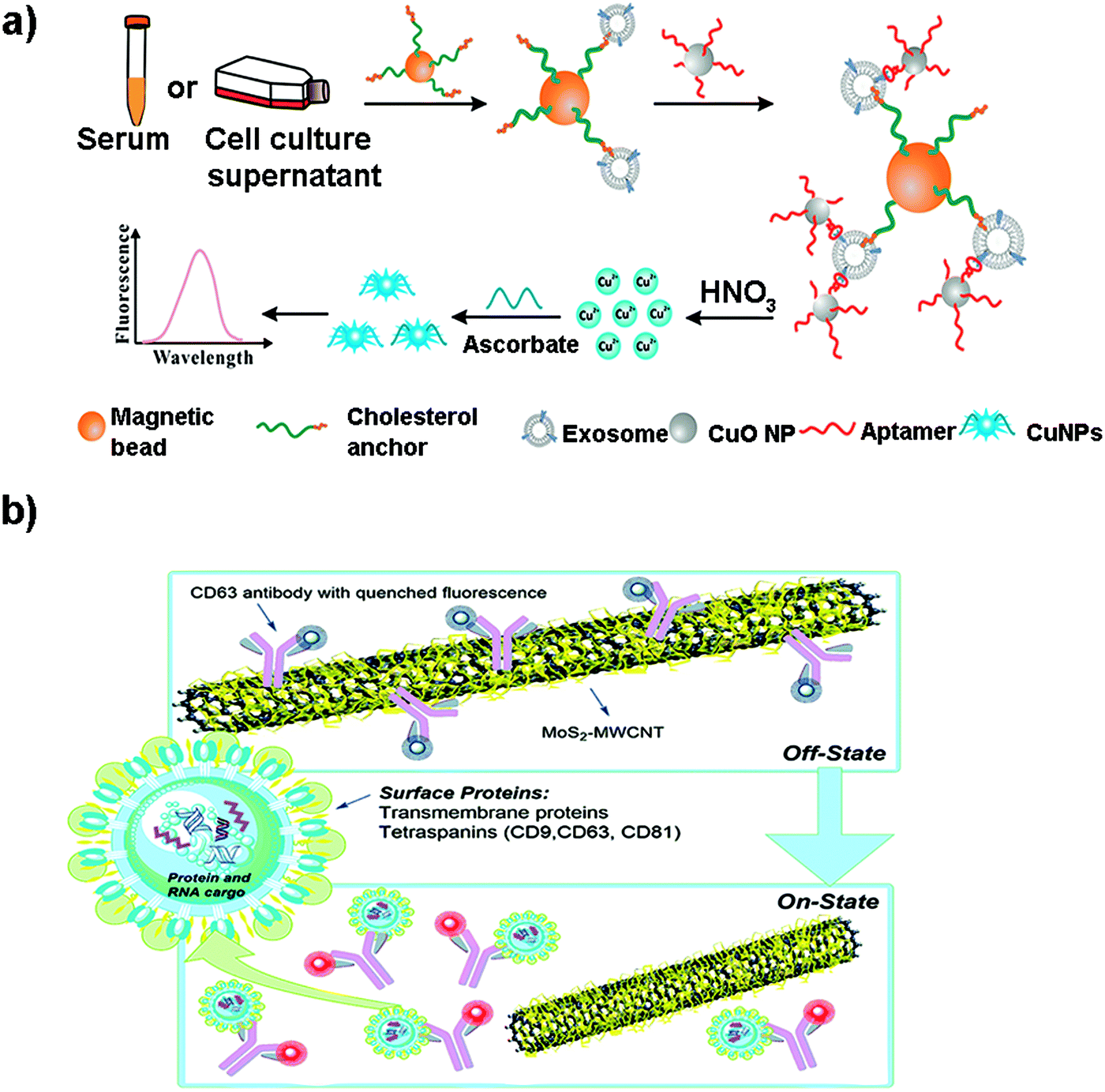 | ||
| Fig. 6 (a) Direct capture and rapid detection of exosomes using a copper-mediated signal amplification strategy. After the formation of sandwich complexes (Magnetic Bead–exosome–CuO NP), the unbound CuO NP probes are separated by a magnet and dissolved by acidolysis to convert CuO NPs into copper(II) ions (Cu2+) and reduced to fluorescent copper nanoparticles (CuNPs) by sodium ascorbate in the presence of poly(thymine). The fluorescence intensity of CuNPs increases with the increase of Cu2+ concentration, which is directly proportional to the concentration of exosomes. Reprinted with permission from ref. 173. Copyright (2018) American Chemical Society. (b) A MoS2–MWCNT fluorescence nanosensor based on FRET in which the fluorophore-CD63 (donor) transfers energy to the nano-quencher MoS2–MWCNT (acceptor) and provides an “on–off” sensor to detect the exosome–antibody recognition. Reprinted with permission from ref. 173. Published by the Royal Society of Chemistry. | ||
Other metallic materials reported in the literature are MXenes, new nanosheet structures that combine transition metal carbides and thus metallic conductivity with hydrophilic nature due to their oxygen or hydroxyl terminated surfaces. This property facilitates their interaction with biomolecules, positioning them as highly interesting nanobiointerfaces for the development of biosensors, where they are used as nano-quenchers.174 The detection mechanism is based on a FRET phenomenon in which a fluorophore (donor) transfers energy to the nano-quencher (acceptor) by distance-dependent fluorescence quenching coupling. When this distance increases, the fluorescence of the donor is recovered, as the nano-quencher does not act anymore. This phenomenon has been exploited to detect EVs using a fluorescent-labelled Cyanine (Cy3)-CD63 aptamer and Ti3C2 MXenes as the 2D nano-quencher interface.175 MXenes could absorb the aptamer by chelation interaction of the hydrogen–metal bond, turning off the Cy3 fluorescence. When exosomes were added, the Cy3 fluorescence was recovered because of the release of the aptamer from the nanosheet, as a consequence of its higher affinity with the surface of the exosome. A LOD of 1.4 × 103 particles per mL was obtained. This FRET mechanism has been also exploited to detect EVs using s-SWCNTs, MoS2–multiwall carbon nanotubes (MWCNT), graphene oxide (GO) or MoS2 nanosheets (Fig. 6b).139,173,176–178
Finally, polymeric NPs such as lanthanide chelate-doped polystyrene beads are also being used as sensing platforms. Islam et al. used these NPs with long-lifetime fluorescence when compared with the fluorescence of highly effective markers, such as europium, to develop a NP-based time resolved fluorescence immunoassay (NP-TRFIA).179 The polystyrene beads were conjugated with Abs or lectins against tetraspanins present on the exosome surface. Anti-CD9 Abs were immobilized on the surface of a microwell plate so that they could capture the exosomes. Thereafter, polystyrene beads were added and TRF detection was performed. It was demonstrated that beads coated with lectins showed a 2–10 fold higher signal when compared to Eu-chelates. EVs from minimally processed urine samples were detected with a LOD of 0.03 ng mL−1. Another example for the development of a fluorescence-based immunosensor was the use of polydiacetylene liposomes conjugated to anti-CD63 Abs.180 Based on the optical properties of polydiacetylene, the interaction with exosomes isolated from human plasma led to changes in the fluorescence signal, achieving a detection limit of 3 × 108 particles per mL.
For instance, Boriachek et al. used QDs and a voltammetric immunoassay for the electrochemical detection of exosomes.186 To this end, biotinylated CdSe QDs were functionalized with Abs against HER-2 and FAM134B, as potential markers of breast and colon cancer respectively. After acid dissolution of the CdSe QDs and anodic stripping voltammetric quantification of Cd2+, a sensitive detection of 105 exosomes per mL was achieved in exosomes derived from serum samples of patients with colorectal adenocarcinoma. The same authors developed a more sophisticated dual isolation and electrochemical detection using gold-loaded ferric oxide nanocubes (Au-NPFe2O3NCs) functionalized with CD63 Abs. Exosomes derived from BeWo placental choriocarcinoma cells were first isolated using these magnetic nanocubes (Fig. 7a and b).187 The exosomes were subsequently transferred to screen-printed electrodes previously functionalized with an anti-placental exosome Ab. The oxidation of TMB in the presence of H2O2 was accomplished because of the HRP-like activity of the Au-NPFe2O3NCs. Subsequent addition of stop solution produced diimine, a product that is electroactive and stable. Naked eye detection along with electrochemical quantification reported a LOD of 103 exosomes per mL. In addition, electrochemical biosensors based on metallic NPs have been successfully applied for multiplexing. For instance, Zhou et al. designed an electrochemical sensor for exosome and microsome detection by direct electro-oxidation of AgNPs and CuNPs labelled with anti-EpCAM and anti-PSMA respectively. The platform required only 25 μL of sample and exhibited a LOD of 50 exosomes per sensor.188
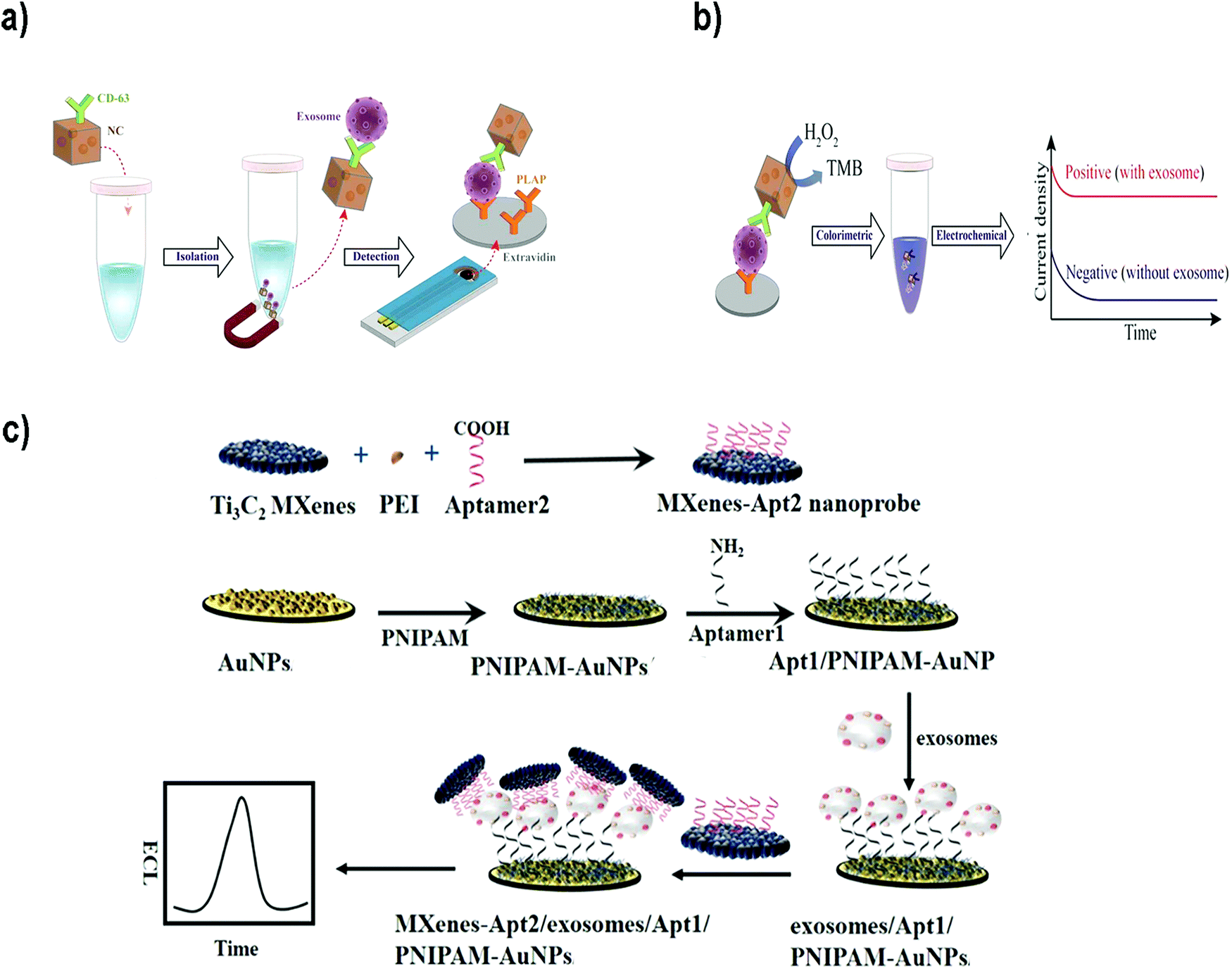 | ||
| Fig. 7 (a and b) Electrochemical biosensor based on dual isolation and electrochemical detection using Au-NPFe2O3NCs. (a) Schematic representation of the assay for direct exosome isolation and detection from cell culture media. In this method, the Au-NPFe2O3NCs were initially functionalized with a generic antibody (CD63) and dispersed in cell culture media to capture bulk exosomes. After magnetic capture and purification, exosome-bound Au-NPFe2O3NCs were transferred to specific Ab-modified, screen-printed electrodes. (b) The HRP-like activity of Au-NPFe2O3NCs was then used to achieve naked-eye detection along with electrochemical quantification of specific exosomes present in cell culture media. Reproduced from ref. 187 with permission from The Royal Society of Chemistry. (c) The principle of an ECL biosensor for exosome detection using Ti3C2 MXene nanosheets to enhance the signal. Reprinted from ref. 189. Copyright (2018), with permission from Elsevier. | ||
Electrochemiluminescent (ECL) biosensors are based on the emission of light (chemiluminescence) when an electrochemical reaction takes place, commonly on an electrode. Therefore, this methodology combines electrochemical and luminescence techniques, and merges the advantages of both methods.190 The use of NPs to build this type of sensors offers advantages such as amplification of the ECL signal, or the possibility to use NPs as the sensor nucleus (ECL comes directly from the NP) or as resonance energy transfer acceptors (NPs and fluorophores-combined ECL). Zhanga et al.189 developed a sensor using Ti3C2 MXene nanosheets to enhance the ECL signals of luminol. Exosomes were first captured onto an electrode surface functionalized with an aptamer to recognize the exosomal EpCAM protein. On the other side, Ti3C2 MXene nanosheets were modified with CD63 aptamers to recognize the exosomes that were already captured (Fig. 7c). The catalytic and conductivity characteristics of the nanosheets together with the possibility of improving the electron transfer on the electrode interface led to a luminol amplification signal. A LOD of 1.25 × 105 particles per mL for EVs derived from MCF-7 cells was reported. Similarly, black phosphorus quantum dots (BPQDs) functionalized with MXenes have also been used to develop a ECL biosensor to detect EVs with a LOD of 3.7 × 104 exosomes per mL.191 Other examples could be found in the literature reporting ECL aptasensors for exosome detection using mercaptopropionic acid (MPA)-modified Eu3+-doped CdS nanocrystals (MPA-CdS:Eu NCs) functionalized with CD63 aptamers.192 MPA-CdS:Eu NCs were immobilized on glassy carbon electrodes and they behaved as ECL emitters in the presence of H2O2. Exosomes were added and the ECL intensity recorded. Subsequently, a DNA sequence that could fold into a G-quadruplex/hemin DNAzyme was introduced. This structure had HRP-like activity, and could catalyze the reduction of H2O2. This decomposition of H2O2 resulted in a decrease of the ECL signal of the MPA-CdS:Eu NCs. A LOD of 7.41 × 104 exosomes per mL for MCF-7 breast tumor cells was achieved. The platform was applied to detect exosomes in serum, showing potential application in real sample diagnosis.
The presence of metals can solve this weakness, as they can enhance the signal up to 1015 times, depending of the geometry (thickness, size and shape) and the composition of the metal nanostructures.195,196 This technique is referred to as SERS (Fig. 8).197 SERS is a plasmonic-based spectroscopic technique that combines a laser with the optical properties of metallic nanostructures to obtain detailed chemical information of molecules adsorbed or attached to them.198 The technique is supported by the formation of regions of intense field enhancement, caused by local surface plasmon resonances (LSPR) at the metal–dielectric interface.199 LSPR excitation will induce an enhanced electromagnetic field, increasing the number of scattered photons in the presence of a Raman-active molecule. Therefore, SERS is a powerful technique that can provide particular signals in complex environments, offering at the same time high sensitivity and multiplexing ability. Furthermore, it allows the analysis of small sample concentrations with short acquisition times, which is ideal for the measurement of biological samples in the clinic and medical research. Because of these advantages, SERS biosensors comprising NPs are currently being used to detect and identify the molecular differences between different groups of EVs.
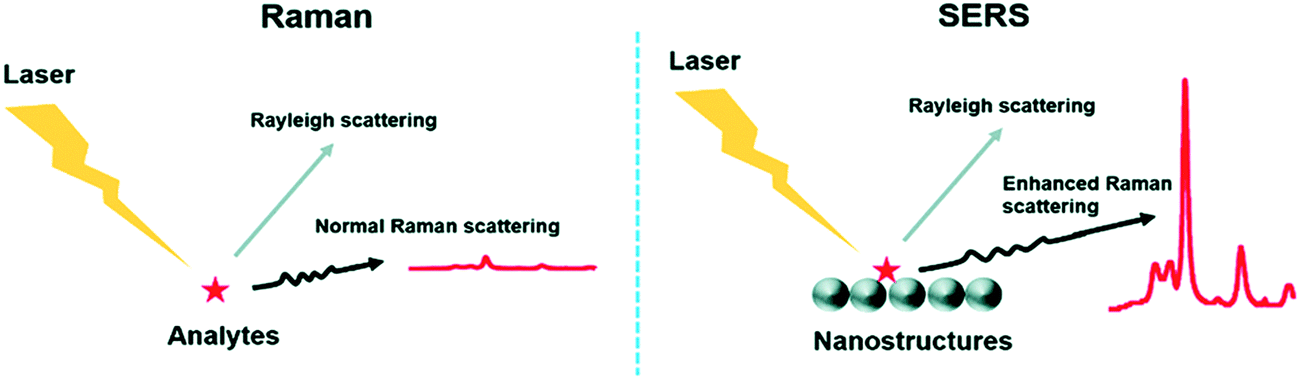 | ||
| Fig. 8 Comparison between the Raman technique and the SERS technique. The main difference lies in the enhanced Raman scattering from the nanostructures used as substrates in SERS. Reproduced from ref. 197 with permission from The Royal Society of Chemistry. | ||
SERS biosensors can be divided into two types:8 (i) solution-based, where an EV is generally captured between 2 NPs and afterwards deposited on a substrate to perform the measurement (Fig. 9a and b); (ii) solid-based, where an EV is directly captured on a substrate where the measurement will be done. Glass slides non-coated or coated with a metal layer or NPs (to increase the sensitivity) can be used as the substrate (Fig. 9c and d).
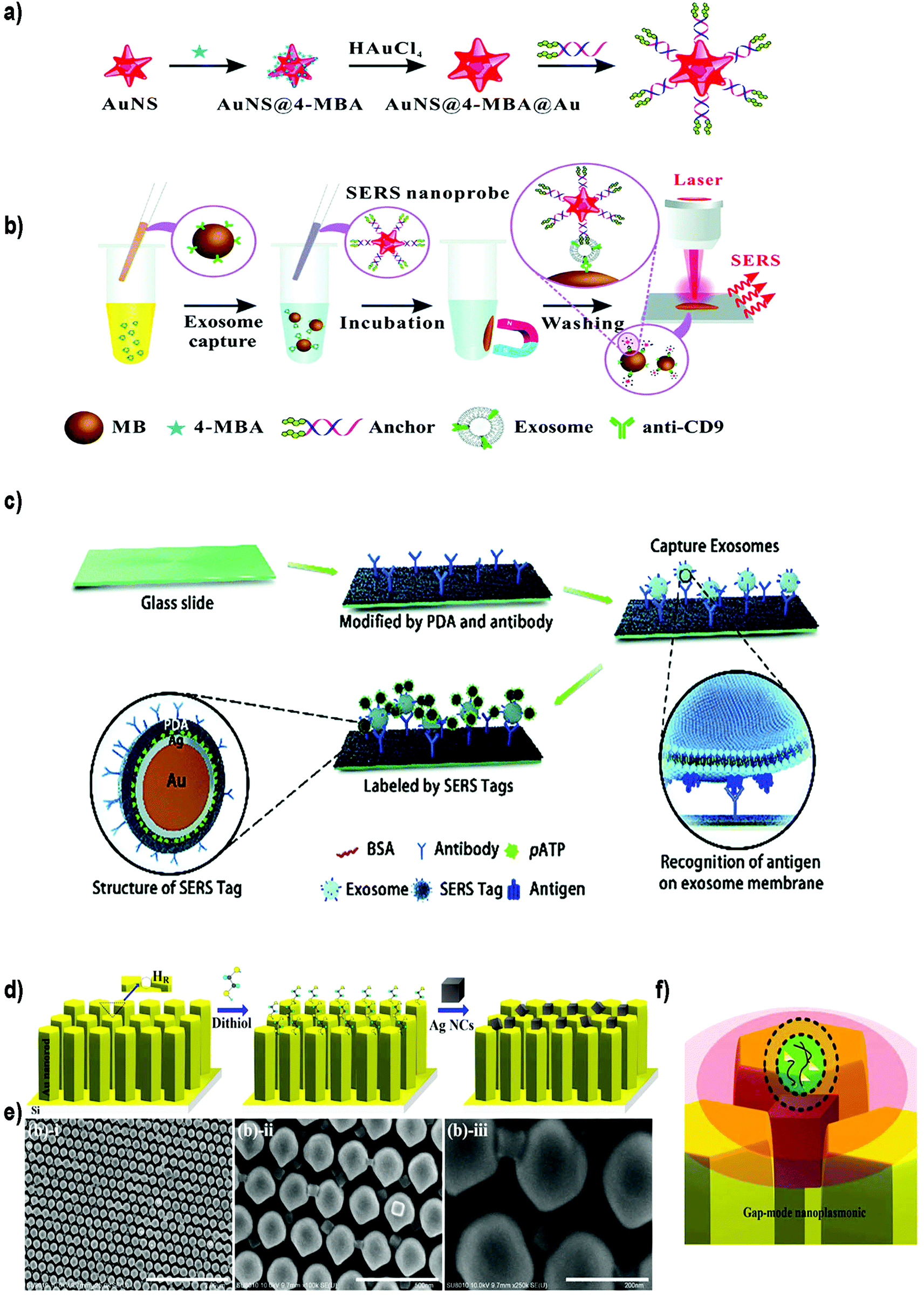 | ||
| Fig. 9 Different SERS-based biosensors for the detection of exosomes. (a) Fabrication of SERS probes: Au nanostars containing reporter molecules (4-MBA) and modified with a bivalent cholesterol-labelled DNA anchor. (b) SERS sensing strategy for exosome detection. Reproduced from ref. 200 with permission from The Royal Society of Chemistry. (c) A schematic view of a PDA chip and Au@Ag@PDA SERS tag-based exosome sensors. Reproduced from ref. 201 with permission from The Royal Society of Chemistry. (d) Schematic illustration of the experimental process of AgNCs on a specified high-density hot-ring diameter area Au NR array substrate. (e) Field-emission scanning electron microscopy images of as-fabricated samples and (f) schematic illustration of electromagnetic enhancement. Adapted from ref. 202 with permission from Elsevier. | ||
Zong et al.203 were the first to report a SERS-based strategy to detect exosomes from tumor cell lines. The methodology used magnetic beads and SERS nanoprobes (Au@Ag NRs@SERS reporter) functionalized with specific Abs able to recognize CD63 and the tumoral marker HER2 (epidermal growth factor receptor-2). When exosomes were present, both the magnetic beads and SERS nanoprobes attached to the exosome, forming a sandwich type immunocomplex. Magnetic beads were then used to separate the exosomes from the cell media and SERS signals were detected in the isolated vesicles. Exosomes derived from a breast cancer cell line were specifically detected, reaching a LOD of 6 × 104 exosomes per mL. Going a step forward, Wang et al. used aptamers and the multiplexing ability of SERS to simultaneously detect multiple exosomes.204 Magnetic beads coated with a layer of silica and a layer of Au (MB@SiO2@Au) were functionalized with CD63 aptamers to capture extracellular vesicles. On the other hand, AuNPs decorated with a SERS reporter and with a specific aptamer were fabricated (AuNP@aptamer) as SERS probes. To allow multiplexing, three kinds of SERS probes were synthesized using different SERS reporters. Breast cancer (SKBR3), prostate cancer (LNCaP) and colorectal cancer (T84) cells were selected as model cells, and the aptamers designed accordingly to target proteins overexpressed in exosomes derived from these cancer cell lines. If only one type of exosomes is present, its specific probe will recognize it, forming a sandwich-type apta-immunocomplex containing MB@SiO2@Au, the target exosomes and the SERS probes. After magnetic separation, SERS signals of the supernatant are measured, finding a decreased signal of this probe when compared with the other SERS probes. If three populations of exosomes are present, the signal of the three types of probes will decrease. The possibility to detect and distinguish different types of exosomes at the same time was demonstrated experimentally. The methodology was tested using exosomes already purified from the aforementioned cell lines, obtaining LODs of 32, 73 and 203 × 103 exosomes per mL for SKBR3, T84 and LNCaP, respectively. The biosensor was also used to detect exosomes from patients suffering from breast, colorectal and prostate cancer, matching in all cases their corresponding type of exosomes.
Other groups have reported the possibility to detect EVs in a non-specific way, that is, without adding Abs or aptamers to recognize the EVs. For instance, Tian et al. reported the use of SERS probes composed of gold nanostars and reporter molecules, further modified with a bivalent cholesterol-labelled DNA anchor (Fig. 9a and b).200 Target exosomes were captured using magnetic beads functionalized with anti CD9 Abs. The captured exosomes were then labelled with the SERS nanoprobes via hydrophobic interactions between the cholesterol moieties and the exosomal lipid membranes, resulting in a sandwich complex. This complex could be magnetically captured and deposited on a silica slide for detection using a Raman spectrometer. Exosomes derived from HepG2 hepatocellular carcinoma cells were used as a model for liver cancer diagnosis. A LOD of 27 × 103 exosomes per mL was reported, with a linear relation between exosome concentration and corresponding SERS signal ranging from 40 to 4 × 107 exosomes per μL. As a proof of concept, serum samples from healthy and liver cancer patients were tested. As already reported, the number of exosomes was elevated in cancer patients, and the results were comparable with those obtained using state-of-the-art technologies (purification using the ExoEasy kit and quantification with qNano). Meanwhile, Stremersch et al. used cationic AuNPs to ionically adsorb exosome-like vesicles (ELVs), demonstrating the possibility to discriminate ELVs isolated from B16F10 melanoma cell cultures and red blood cells (RBCs) with SERS.205 The same group subsequently improved the system by applying an extra silver layer in situ to remove interfering signals generated by the AuNP coating.206 The Au@AgNPs core–shell system resulted in SERS signals with improved signal-to-noise ratio, and consequently, more vibrational modes could be identified. Furthermore, the improved system decreased the acquisition time by a factor of 20. Lee et al. took advantage of α3β1 integrin overexpression in exosomes derived from some cancerous cell lines to detect exosomes isolated and purified from human ovarian carcinoma cell lines (SKOV-3).207 In this case, SERS probes were prepared using AgNPs functionalized with the LXY30 peptide, able to selectively label the aforementioned integrin, and allowed a specific detection of exosomes derived from SKOV-3 cells.
Recently, Pang et al. developed a smart system to test the expression of programmed death-ligand 1 (PD-L1) on the exosome membrane using magnetite@TiO2 particles to capture the exosomes.208 In recent years, several reports have revealed the correlation of exosomal PD-L1 expression and anti-PD-L1/PD-1 therapy in order to treat tumours.209,210 The possibility to predict if a patient will respond to anti-PD-L1/PD-1 therapy is of utmost importance, considering the high price and side effects of immune drugs. To study PD-L1 expression, exosomes purified from A549 non-small cell lung cancer (NSCLC) cells and BEAS-2B normal human bronchial epithelial cells were incubated with magnetite@TiO2 particles to enrich and separate exosomes from cell culture media or serum samples. No aptamers or Abs were needed, as TiO2 can specifically interact with the phosphate groups present on the lipid bilayer of exosomes.211 Once separated, exosomes were labelled with the SERS tags, consisting of Ag@Au@SERS reporter nanoprobes functionalized with anti PD-L1 Abs. With this methodology exosomes containing PD-L1 were detected with a LOD of 1000 exosomes per mL. Interestingly, the authors tested the possibility to detect the individual level of exosomal PD-L1 in NSCLC patients at different stages, using undiluted serum. Differences in exosomal PD-L1 expression could be found among healthy patients and both early and late NSCLC groups. Moreover, only 4 μL of serum sample and 40 minutes were needed to complete the assay.
SERS biosensors can also be built by directly immobilizing the EVs on a substrate before adding the SERS tag. For instance, Li et al. created a SERS immunosensor for clinical pancreatic cancer diagnosis using a polydopamine (PDA) polymerized substrate to encapsulate Abs in order to capture exosomes isolated from real samples or from culture media (Fig. 9c).201 Au@Ag@Raman reporter@PDA NPs modified with an Ab were subsequently used as SERS tags. PDA substrates were used to capture exosomes and Au@Ag@PDA SERS tags. Both the substrate and the NPs were functionalized with diverse Abs to detect common exosomal proteins (CD63 and CD9) or specific pancreatic cancer-derived exosomal proteins (GPC1, EGFR, EpCAM and Macrophage migration inhibitory factor). Importantly, this immunosensor was able to distinguish healthy from pancreatic cancer patients, as well as metastatic from non-metastatic tumors.
On the other side, efforts have been also devoted to enhance the SERS signal by improving the SERS substrate. Kwizera et al. combined SERS technology with a 3D printing methodology to develop a chip platform functionalized with Abs to detect exosomes.212 In this case, 35 nm AuNRs functionalized with a Raman reporter were used as SERS tags. This platform had microliter sample requirement and allowed the detection of 2 × 106 exosomes per mL from breast cancer cell lines in 2 hours. Further, they showed that exosomes derived from different cancer cells gave different protein profiles when compared to exosomes derived from healthy cells. Other authors have developed a strong plasmonic gap-mode SERS substrate by assembling silver nanocubes (AgNCs) on an AuNR pillar array surface, in order to enhance the SERS signal (Fig. 9d–f).202 The Raman reporter molecule was directly deposited on this substrate. Using this approach, the possibility to distinguish exosomes derived from lung normal and cancer cell lines was demonstrated. Exosomes could be detected at concentrations 104–105 times lower than that found in general blood samples (1011 exosomes per mL) in a short time.
Remarkably, SERS can also serve as a valuable tool to characterize subpopulations of exosomes from different origin if combined with a multivariate method able to condense the SERS data. For instance, Carmicheal et al. used principal component and differential function analyses (PC-DFA) in conjunction with SERS data in order to classify exosomes from various cellular origins.213 Using this method, the authors were able to differentiate serum-derived exosomes isolated from healthy and pancreatic cancer patients with high sensitivity and specificity. Meanwhile, Choi et al. used PCA to classify SERS spectra (Fig. 10).214,215 By using this approach, NSCLC derived exosomes were distinguished from normal alveolar cell-derived exosomes with 95.3% sensitivity and 97.3% specificity. Thereafter, the same group identified the unique SERS profiles of NSCLC derived exosomes and correlated the unique peaks with the Raman profiles of different exosomal protein markers that could contribute to them (Fig. 10).215 In these approaches exosomes were deposited onto a slide containing AuNPs.
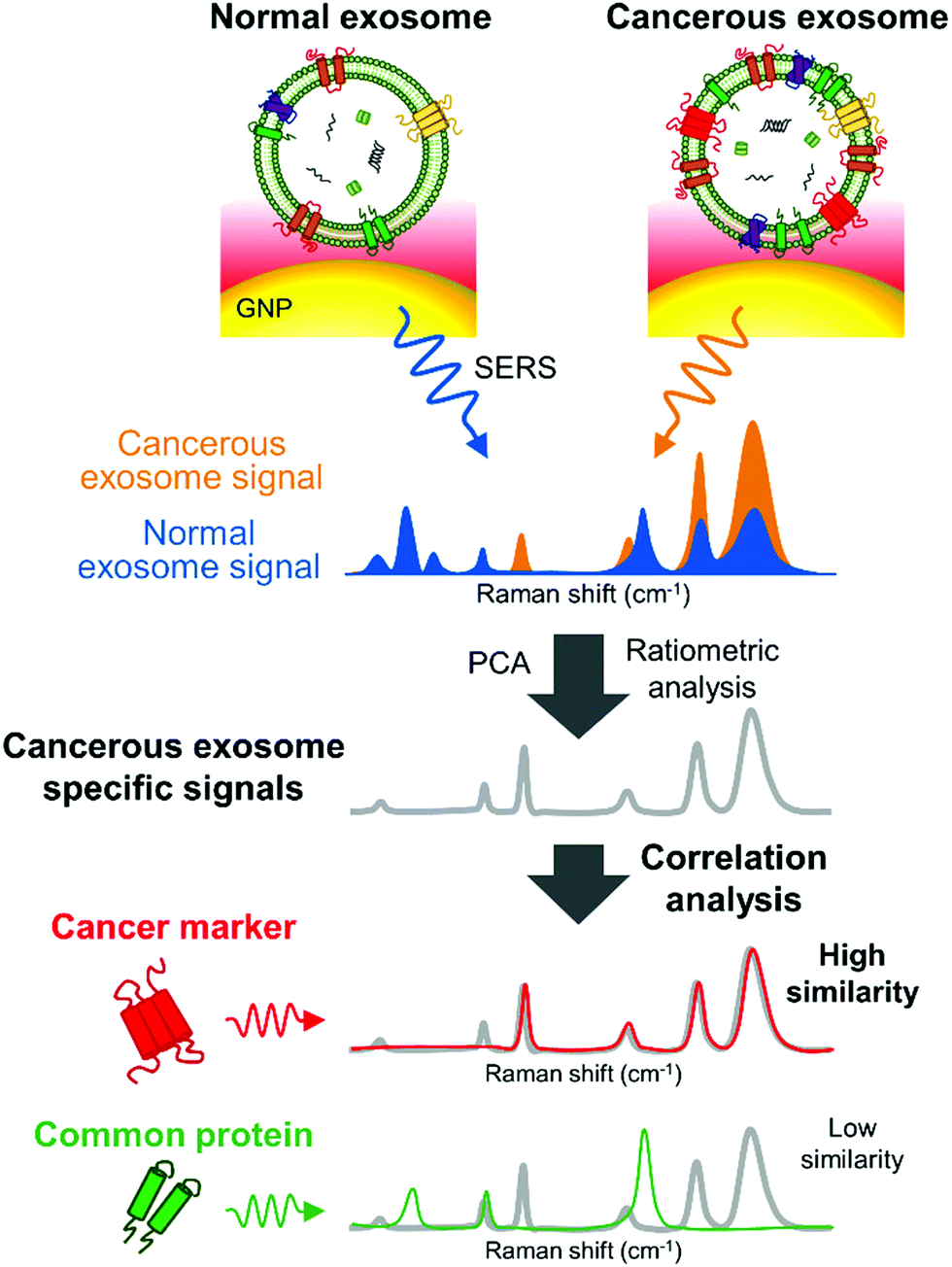 | ||
| Fig. 10 Detection of unique Raman scattering profiles of lung cancer cell-derived exosomes and comparison to the profiles of their potential surface protein markers. Cancerous exosome-specific protein markers are associated in terms of signal similarity. Reprinted with permission from ref. 215. Copyright (2018) American Chemical Society. | ||
Lastly, SERS-based biosensors have been also used for the detection of microRNAs in EVs. In this context, exosomal microRNAs have attracted attention in recent years due to their significant role in regulating cancer progression.216 Pang et al. proposed a one-step and one-pot assay for exosomal microRNA detection, based on a duplex-specific nuclease (DSN) assisted SERS biosensor.217 To this end, Fe3O4@Ag-DNA NPs were used to capture the exosomes and as SERS substrate, while Au@Ag@SERS reporter structures were used as SERS tags. The Fe3O4@Ag–Au@Ag SERS tag conjugates were formed through the DNA linking between both types of structures. DNA probes were specifically designed to recognize miRNA from plasma-derived exosomes from pancreatic ductal adenocarcinoma (PDAC) and chronic pancreatitis (CP) patients. The Raman signal was induced by the “hot spots” between the Au@Ag of SERS tag and the Ag shell of the Fe3O4@Ag substrate, especially after magnetic isolation. When exosomal microRNA was present, DNA could hybridize with it. DSN nuclease was added to selectively cleave the DNA of the DNA/microRNA duplex, so that the SERS tags could be separated from the Fe3O4@Ag substrate. This induced SERS signal quenching which could be correlated with the original concentration of microRNA. Using this technology, a detection limit of 1 aM (microRNA 10b) was achieved. It was also reported that microRNA 10b levels were higher in exosomes derived from PDAC patients when compared to those derived from CP or normal patients. Similarly, Ma et al. proposed the use of MNPs linked via DNA to Au@SERS reporter@AgAu nanoparticles (SERS tags).218 DNA could specifically bind with the complementary exosomal miRNA, while MNPs were used to separate exosomes from serum and plasma media. Once again miRNA/DNA duplexes could be specifically cleaved by DSN, releasing SERS tags while miRNA was involved in other rounds of signal amplification. This technique could detect a concentration of 5 fM of miRNA21.
A remarkable example of a SPR-based platform was reported by Im et al.79 The nano-plasmonic exosome (nPLEX) sensor comprised arrays of periodic nanoholes patterned in a gold film. Nanoholes were designed for matching their dimensions (<200 nm) to exosome size, enhancing the detection sensitivity. Three-dimensional simulation studies were performed to optimize the geometry of the nanoholes, finding enhanced electromagnetic fields tightly confined within the exosome size range. A multichannel multifluidics system was integrated to perform high-throughput analyses. The surface was passivated using PEG, and anti CD63 Abs were functionalized onto PEG chains. As a proof of concept, exosomes isolated from a human ovarian cancer cell line (CaOV3) were tested. Upon binding of exosomes, a change in the local refractive index took place, shifting the spectral peaks. The magnitude of spectral shift correlated with the molecular mass density covering the sensor surface and thus enabled quantitative analysis of EV proteins. The LOD was ∼3000 exosomes, 102-fold higher than that of an ELISA performed using the same exosomes. Importantly, the signal could be significantly increased by using AuNPs as secondary labels. 10 nm spherical AuNPs showed a 20% increase, while the signal could be enhanced by 300% when using 50 nm Au nanostars. Subsequently, the platform was functionalized with Abs against various exosomal targets and tested using samples from ovarian cancer patients and healthy patients as controls. It was found that the levels of EpCAM and CD24 were elevated in the ovarian cancer patient samples. An intrinsic diagnosis accuracy of 97% was reported, although a larger cohort of patients should be used to validate the platform. Importantly, the whole analysis could be accomplished in less than 30 min, and ascites samples from patients were used with minimal processing, as they were only membrane filtered to remove cells and debris.
4. Conclusions
In recent years, much effort has been put into the development of POC devices for the isolation and analysis of EVs.223 For instance, the Exochip and Exosearch are smart microfluidic platforms able to capture and quantify EVs, using external fluorometers.224,225 Going a step forward, other groups have developed microfluidic devices where the detection system is also included,222,226 or have demonstrated the possibility to detect EVs using smartphones.227,228 The combination of these platforms with NPs could enhance their performance, speeding up the translation of EVs into clinical settings. Indeed, NPs offer many advantages when compared to bulk materials, such as unique optical properties or a high surface area that can be exploited to design innovative biosensors. Among the different sensing methodologies that can be used to detect EVs, each technique has inherent advantages and drawbacks that need to be taken into account (Table 4). For instance, LFIA tests are easy to use and do not require sophisticated equipment, but their sensitivity is lower than that reported using more complex methodologies. Similarly, colorimetric biosensors using NPs could be implemented as POC assays, as detection can be done by the naked eye; however, an amplification method is routinely needed to enhance the sensitivity, hampering in many cases this portability. Electrochemical and fluorescence-based biosensors are promising candidates for POC analysis as they can be portable and sensitive at the same time,227,228 but the use of NPs in these completely portable systems has not been reported. Other methodologies such as SPR in combination with NPs, while promising, have not been extensively explored to date. For instance, although AuNPs have been shown to increase the sensitivity of SPR platforms, their use is still limited.79 Similarly, MNPs are widely used to separate EVs, but their use to analyse them using RMN is limited because of the small size of EVs.80Currently, the majority of the devices reported to detect EVs (Table 3) use Abs or aptamers to recognize them. Although the chemistry to functionalize these biomolecules onto the NPs is crucial, almost any work takes this point into account. As can be seen in Table 3, these NP-based devices can provide higher sensitivities than the ones obtained using standard techniques for EV analyses, such as NTA (107–109 EVs per mL), flow cytometry (107–109 EVs per mL), ELISA (109–1010 EVs per mL) or WB (1011–1012 EVs per mL) (Table 2).79–81,229,230 Selecting the appropriate chemistry to couple the biomolecule could further enhance it.
| Type of biosensor | Nanomaterial | Target | Recognition molecule | EVs’ origin | Previous isolation | LOD | Vol EVs (μL) | POC* | Ref. |
|---|---|---|---|---|---|---|---|---|---|
| *POC devices: biosensors described in the original reference as fully portable or automatized. Ab = antibody; Apt = aptamer; UC = ultracentrifugation; RBC = red blood cell; R6G = rhodamine 6 G; TEIR = total exosome isolation reagent; Y = yes; N = no; NA = not applicable; NP = not provided | |||||||||
| LFIA | AuNPs | CD9, CD81 and CD63 | Ab | Metastatic melanoma (Ma-Mel-86c) | Yes (UC) | 8.54 × 108 exosomes per mL | <100 | Y | 77 |
| LFIA | AuNPs | CD9, CD81 and CD63 | Ab | Plasma (healthy donors) | Yes (ExoQuickTM) | 3.4 × 109 EVs per mL | <100 | Y | 134 |
| LFIA | AuNPs | CD9 and MICA | Ab | Metastatic melanoma (Ma-Mel-55) | Yes (UC) | 5 × 1010 exosomes per mL | <100 | Y | 133 |
| LFIA | AuNPs | CD9 | Ab | Breast cancer (MCF-7) | Yes (TEIR) | 1.3 × 106 particles per mL | <100 | Y | 135 |
| LFAA | AuNPs | CD63 | Apt | Lung carcinoma (A549) | Yes (UC) | 6.4 × 108 particles per mL | NP | Y | 137 |
| LFIA | CdSe/ZnS QDs into FNs | Phospholipid membrane | Phospholipid-biotin | Epithelial (Cal 27) | Yes (UC) | 2.0 × 106 particles per mL | <100 | Y | 136 |
| Colorimetric | Fe3O4 NPs | EpCAM | Apt | Plasma from healthy patients | Yes (magnetic isolation) | 7.0 × 106 particles per mL | <100 | N | 138 |
| Colorimetric | s-SWCNTs | CD63 | Apt | Breast cancer & normal (MCF-7, MCF-10A) | Yes (UC) | 5.2 × 108 particles per mL | NP | N | 139 |
| Colorimetric | Graphitic carbon nitride nanosheets | CD63 | Apt | Breast cancer & normal (MCF-7, MCF-10A) | Yes (UC) | 13.52 × 108 particles per mL | <100 | N | 140 |
| Colorimetric | AuNPs | LMP1 | DNA-Ab | Nasopharyngeal carcinoma | No | 1 × 102 particles per mL | 100 | N | 142 |
| Colorimetric | AuNR | CD63 and phospholipid membrane | Apt and cholesterol-DNA | Breast cancer (MCF-7) | Yes (UC) | 1 × 105 particles per mL | NP | N | 144 |
| Fluorescence | CuO NPs | CD63 | Cholesterol anchor and Apt | Liver cancer (HepG2) | Yes (magnetic isolation) (exoEasy Maxi Kit) | 5 × 107 exosomes per mL | NP | N | 171 |
| Human serum (healthy and cancer patients) | No | 4.8 × 107 exosomes per mL | NP | N | |||||
| Fluorescence | AuNP-linker/complementor | CD63 | Apt | Liver cancer (HepG2) | No | 108 exosomes per mL | 30 | N | 172 |
| Fluorescence | Lanthanide chelate-doped polystyrene beads | CD9, CD81, and CD63 | Abs and lectins | Human prostate cancer (LNcaP) | Yes (UC) | 0.03 ng mL−1 | 200 | N | 179 |
| Fluorescence | Polydiacetylene liposomes | CD63 | Ab | Human plasma (HansaBioMed Life Science) | Yes (UC) | 3 × 108 exosomes per mL | 30 | N | 180 |
| Fluorescence | QDs | CEA, Cyfra21–1, Pro-GRP | Ab | Lung carcinoma (A549 and H226), SCLC (H446) and umbilical vein endothelial (HUVEC) | Yes (UC) | N/A | 80 | N | 167 |
| Plasma (lung cancer patients) | 80 | N | |||||||
| Fluorescence | QDs | CD63 | Ab | Monocyte-like (THP-1) | Yes (UC) | N/A | NP | N | 168 |
| Fluorescence | QDs | CD63 | Apt | Breast cancer (SKBR3) | Yes (ExoQuick™) | N/A | 500 | N | 169 |
| FRET (quenching fluorescence) | MoS2-MWCNT | CD63 | Ab | Breast cancer (MCF-7) | Yes (TEIR and UC) | 1.48 × 106 exosomes per mL | 50 | N | 173 |
| FRET (quenching fluorescence) | GO | CD63 and EpCAM | Apt | Colorectal cancer (SW480) | Yes (UC) | 2.1 × 107 exosomes per mL | 5 | N | 177 |
| FRET (quenching fluorescence) | GO | CD63, EpCAM, PTK7, PSMA, PDGF, CEA, AFP | Apt | Breast cancer (MCF-7), cervical cancer (HeLa), gastric cancer (SGC7901), liver cancer (HepG2), mammary epithelial (MCF-10A) | Yes (UC) | 1.6 × 105 exosomes per mL | 10 | N | 178 |
| FRET (quenching fluorescence) | MXene nanosheets | CD63 | Apt | Melanoma (B16), breast cancer (MCF-7), ovarian carcinoma (OVAR-3) and liver cancer (Hep G2) | Yes (UC) | 1.4 × 103 exosomes per mL | 30 | N | 175 |
| LRET | AuNRs-UCNPS (paper) | CD63 | Apt | Liver cancer (HepG2) | Yes (UC) | 1.1 × 106 exosomes per mL | 10 | N | 165 |
| LRET | UCNPs | EpCAM | Apt | Breast cancer (MDA-MB-231) | Yes (UC) | 8 × 104 exosomes per mL | 10 | N | 164 |
| Electrochemical (voltammetric) | Au-NPFe2O3NC | CD63 | Ab | Placental choriocarcinoma (BeWo) | No (magnetic isolation) | 103 exosomes per mL | 50 | Y (qualitative) | 187 |
| Electrochemical (voltammetric) | CdSeQD | HER-2 and FAM134B | Ab | Serum samples from patients with colorectal carcinoma | Yes (TEIR and UC) | 105 exosomes per mL | 10 | N | 186 |
| Electrochemical (voltammetric) | AgNPs and CuNPs | EpCAM and PSMA | Ab | Vertebral-Cancer of the Prostate (VCaP) | Yes (UC) | 2 × 103 exosomes per mL | 25 | N | 188 |
| Blood from PCa patients | N/A | 25 | N | ||||||
| Electrochemiluminescence | Ti3C2MXenes | EpCAM | Apt | Melanoma (B16), breast cancer (MCF-7), and liver cancer (HepG2) | Yes (UC) | 1.25 × 105 exosomes per mL | 100 | N | 189 |
| Electrochemiluminescence | BPQDs and MXenes | EpCAM and CD63 | Apt | Breast cancer (MCF-7), cervical cancer (HeLa) | Yes (UC) | 3.7 × 104 exosomes per mL | NP | N | 191 |
| Electrochemiluminescence | MPA-CdS:Eu NCs | CD63 | Apt | Breast cancer (MCF-7) | Yes (UC) | 7.41 × 104 exosomes per mL | 50 | N | 192 |
| Plasma and serum samples from breast cancer patients | N/A | 50 | N | ||||||
| SERS | Au@Ag NRs | CD63, HER2 | Ab | Breast cancer (SKBR3), lung fibroblasts (MRC5) | Yes (ExoQuickTM) | 6 × 104 exosomes per mL | 1 | N | 203 |
| Plasma (breast cancer patients) | No | N/A | |||||||
| SERS | AuNPs | CEA, H2 and PSMA | Apt | Breast cancer (SKBR3, LNCaP, T84) | Yes (UC and ExoQuickTM) | 32 × 103 exosomes per mL | 2 | N | 204 |
| Plasma (breast cancer patients) | No | N/A | |||||||
| SERS | AuNPs | Phospholipid membrane | No | Melanoma cancer (B16F10), RBCs | Yes (UC) | N/A | 200 | N | 205 |
| SERS | Au@Ag NPs | Phospholipid membrane | No | Melanoma cancer (B16F10), RBCs | Yes (UC) | 1 exosome | NP | N | 206 |
| SERS | Au NPs | CD 9 | Ab | Liver cancer (HepG2) | Yes (UC) | 2.7 × 10 4 exosomes per mL | 60 | N | 200 |
| Plasma (carcinoma patients) | Yes (UC and ExoQuickTM) | N/A | |||||||
| SERS | Au NPs | α3β1 integrin | Peptide | Ovarian carcinoma (SKOV-3) & T lymphocyte (Jurkat) | Yes (UC) | N/A | NP | N | 207 |
| SERS | Ag(shell) Au(core) @PDA particles | CD9, CD63, MIF, GPC1 | Ab | Pancreatic (PANC-01 and HPDE6-C7) | Yes (UC) | 5.44 × 102 exosomes per mL | 2 | N | 201 |
| Plasma (pancreatic cancer patients) | No | N/A | |||||||
| SERS | Surface with Au pillars and Ag NCs | No | No | Lung normal (NL-20, BEAS and L929) and cancer (PC-9, H1975, HCC827) cells | Yes (UC) | N/A | 5 | N | 202 |
| SERS | Au@Ag@MBA NPs Fe3O4@TiO2 particles | PD-L1 | Ab | NSCLC (A549), bronchial epithelial (BEAS-2B) | Yes (UC) | 1 × 103 exosomes per mL | NP | N | 208 |
| Plasma (lung cancer patients) | No | N/A | 4 | ||||||
| SERS | Au NRs | EpCAM, CD44, HER2, EGFR, IGFR, CD81, CD63, CD9 | Ab | Breast cancer (MMM231, MM468, SKBR3) & breast normal (MCF12A) | Yes (UC) | 2 × 106 exosomes per mL | 15 | N | 212 |
| Plasma (breast cancer patients) | Yes (UC) | N/A | |||||||
| SERS | Au NPs | No | No | Pancreatic (CD 18/HPAF, MiaPaCa and HPDE) | Yes (UC) | N/A | NP | N | 213 |
| SERS | Surfaces coated with AuNPs | No | No | Lung carcinoma, pulmonary alveolar epithelial (H1299, H522) | Yes (UC) | 10 9exosomes per mL | 50 | N | 214 |
| Plasma (lung cancer patients) | No | N/A | |||||||
| SERS | Au@Ag NPs | Exosomal miRNA | DNA | Pancreatic cancer (PANC-01), normal pancreatic (HPDE6-C7), breast cancer (MCF-7), normal breast (MCF-10) | Yes (TEIR) | 1 a M (miRNA) | NP | N | 217 |
| Plasma (pancreatic and breast cancer patients) | Yes (UC) | N/A | |||||||
| SERS | Au@R6GAgAu NPs | Exosomal miRNA | DNA | Cervical cancer (HeLa), lung cancer (A549), normal renal (293) | Yes (UC) | 5 × 10−18 mol exosomes per mL or 5 fM miRNA | NP | N | 218 |
| Plasma (cervical cancer and lung cancer patients) | Yes (UC) | N/A | |||||||
| SPR | Nanoholes patterned in a Au film + Au nanostars | CD63 | Ab | Ovarian cancer (CaOV3) | No | 1 × 107 particles per mL | 0.3 | Y | 79 |
| Ovarian cancer and control patients | N/A | ||||||||
| μNMR | MNPs | CD63 | Ab | Glioblastoma multiforme (GBM20/3) | Yes (UC) | 1 × 107 particles per mL | 1 | Y | 80 |
| Plasma (glioblastoma multiforme and healthy patients) | N/A | ||||||||
| LFIA | Colorimetric | SERS | Fluorescence | Electrochemical | SPR | μNMR | |
|---|---|---|---|---|---|---|---|
| Principle | Optical properties (coloured particles) | Colour changes | Optical properties (light dispersion) | Fluorescence intensity, FRET or LRET, quenching | Voltammetric and electrochemiluminescence | Optical properties (light reflection) | Magnetic properties (relaxation time of water molecules) |
| Portability | Always (qualitative measurements) | Yes for qualitative/quantitative measurements (naked eye/smartphone) | Portable devices have been described but integrated platforms are not common | Possible (compact fluorescence detectors) | Possible (miniaturized electrodes integrated into microfluidic devices) | Possible (i.e. fiber-optic sensors for smartphone231) | Possible (microcoils integrated in a platform) |
| Naked eye detection | Yes | Yes | No | No | Sometimes | No | No |
| Simple (technique) | Yes | Yes | No | No | No | No | No |
| Expensive equipment | $ | $-$$ | $$$ | $$$ | $$ | $$$ | $$$ |
| Sensitivity | Low | Low | High | High | High | High | High |
| Unskilled operators | Yes | Possible | No | No | No | No | No |
| Other advantages | Long shelf life, batches can be prepared in advance, no extra equipment needed, high potential for commercialization | Cost-effective, no additional equipment (for semi-quantitative or qualitative read out) | Can distinguish subpopulations, multiple detection and quantification, analysis can be performed in low concentrated samples, statistical methods can improve the results | Simplicity and targeted bioimaging | Reproducibility, real-time and fast answer, possibility of automation and robustness | Fast, label free, high throughout analysis | High penetration (can be performed in turbid samples), low background |
| Other disadvantages | Sample pre-treatment is needed depending on the sample, qualitative or semi-quantitative read-out | Low sensitivity if an amplification method is not included | SERS nanoprobes need to be prepared in advance, sample pre-treatment is necessary, total analysis time | Background fluorescence and spectral interference | Sensitive to the surrounding environment, short or limited shelf life, cross-sensitivity | Portability hindered when using bulk SPR sensors, complicated optics | Difficult to be engineered to detect EVs because of their size |
| Examples | 77,133–137,232 | 138,142,144,228 | 201,204,206,208,214,217,233 | 225,227,234 | 99,108,235 | 79 | 80 |
Despite progressive advances in the field, the majority of the devices where NPs are used to analyse EVs require their previous isolation using traditional methods (Table 3). Methodologies such as ultracentrifugation, density gradient centrifugation or size exclusion chromatography are generally time consuming, should be performed by skilled personal and can incur sample loss. On top of that, more than 1000 different protocols to retrieve EVs from biological fluids have been reported.236 Thus, without standard isolation procedures it is difficult to establish detailed comparisons to evaluate the performance of these detection platforms. These complications are exacerbated by the fact that many reports lack minimal information about the EVs, or important analytical information of the device such as limit of quantification, precision, accuracy or LOD. Even when the LOD is reported, as the units of quantification differ between studies, it is difficult to establish a real comparison. To overcome this issue, recommendations for EV analysis and reporting can be followed.28,237
Lastly, the majority of the studies listed in Table 3 are at the level of proof-of-concept, having being performed with EVs isolated from cell lines. Although some of them included patient samples, statistics should be improved by using large patient cohorts. Advances in these fields will enable the acceleration of EVs incorporation into the clinic.
5. Future outlook
Nowadays the potential of EVs as a source of biomarkers is widely accepted and we believe that liquid biopsies will open a new avenue for early detection of cancer. Proof of this is the fact that the first EV-based biopsies have started to reach the market. One example is the ExoDx Prostate Test, a test based on measurement of specific EV-RNA in urine by PCR that can be used to predict high-grade prostate cancer.238 We expect that during the next years additional tests will be generated for other cancer types and diseases and that the first EV-protein tests will be developed.In the last few years, lab on a chip platforms have been attracting more attention as they can improve portability, efficiency, automation and miniaturization among others. The combination of NPs with these platforms could enhance their performance and speed up the translation of liquid biopsies into the clinic; however, there is still a long way to reach this goal as the field faces several important challenges. For instance, EV-based markers should be detectable with a few and simple processing steps, preferentially directly in the cell-free biofluid. Further technology advancements to specifically detect tumor-derived subpopulations of EVs in a complex mixture are also necessary, as human biofluids contain a mixture of EVs released from diverse cell types. Lastly, high-throughput EV separation along with high purity recovery and device standardization should be also pursued.
In conclusion, although nanomaterials are playing a key role in developing novel biosensors with enhanced sensitivity for the detection of EVs, major efforts should be devoted to enhance the portability and reproducibility of the devices to be used in clinical settings. For future prospective, prototypes where EVs isolation and analysis are integrated into the same device are highly promising towards automated and user-friendly POC devices. We also believe that orientation of molecules on NPs could greatly improve the sensitivity, and that by controlling the functionalization processes, many platforms could enhance their performance. We expect that in the next few decades, by addressing the aforementioned challenges, the nanobiosensor field will result in the development of easy platforms with the capacity to test clinical samples with high selectivity and accuracy.
Abbreviations
| Abs | Antibodies |
| Ag NCs | Silver nanocubes |
| AgNPs | Silver nanoparticles |
| ALP | Alkaline phosphatase |
| aM | Attomolar |
| Au-NPFe2O3NCs | Gold-loaded ferric oxide nanocubes |
| AuNPs | Gold nanoparticles |
| AuNRs | Gold nanorods |
| BPQDs | Black phosphorus quantum dots |
| BSA | Bovine serum albumin |
| CB | Carbon black nanoparticles |
| CEA | Carcinoma embryonic antigen |
| cfDNA | Circulating cell-free DNA |
| cfRNA | Circulating cell-free RNA |
| CL | Control line |
| CNTs | Carbon nanotubes |
| CP | Pancreatic cancer |
| CTCs | Circulating tumor cells |
| CuNP | Copper nanoparticles |
| CuO NPs | Copper oxide nanoparticles |
| Cy3 | Cyanine |
| DNA | Deoxyribonucleic acid |
| DSN | Duplex-specific nuclease |
| ECL | Electrochemiluminescence |
| EGFR | Epidermal growth factor receptor |
| ELISA | Enzyme linked immunosorbent assay |
| EM | Electron microscope |
| EpCAM | Epithelial cell adhesion molecule |
| EVs | Extracellular vesicles |
| Fab | Antigen binding fragment |
| FBS | Fetal bovine serum |
| Fc | Constant fragment |
| fM | Femtomolar |
| FRET | Fluorescence resonance energy transfer |
| GO | Graphene oxide |
| GPC1 | Glypican-1 |
| HCR | Hybridization chain reaction |
| HER2 | Human epidermal growth factor receptor-2 |
| HRP | Horseradish peroxidase |
| IgG | Immunoglobulin G |
| ISEV | International Society for Extracellular Vesicles |
| LFAA | Lateral flow aptamer assay |
| LFIA | Lateral flow immunoassay |
| LMP-1 | Epstein–Barr virus latent membrane protein 1 |
| LOD | Limit of detection |
| LRET | Luminescence resonance energy transfer |
| LSPR | Local surface plasmon resonance |
| MICA | MHC class I chain-related protein A |
| MIF | Macrophage migration inhibitory factor |
| miRNAs | micro RNAs |
| MNPs | Magnetic nanoparticles |
| MPA-CdS:Eu NCs | Mercaptopropionic acid-modified Eu3+-doped CdS nanocrystals |
| MWCNTs | Multiwalled carbon nanotubes |
| NCs | Nanocubes |
| NIR | Near infrared |
| NP-TRFIA | NP-based time resolved fluorescence immunoassay |
| nPLEX | Nano-plasmonic exosome |
| NPs | Nanoparticles |
| NRs | Nanorods |
| NSCLC | Non-small cell lung cancer |
| NTA | Nanoparticle tracking analysis |
| PC-DFA | Principal component differential function analysis |
| PCa | Prostate cancer |
| PCA | Principal component analysis |
| PCR | Polymerase chain reaction |
| PD-1 | Programmed cell death-1 |
| PD-L1 | Programed cell death-ligand 1 |
| PDA | Polydopamine |
| PDAC | Pancreatic ductal adenocarcinoma |
| PEG | Poly ethylene glycol |
| POC | Point-of-care |
| proGRP | Progastrin-releasing peptide |
| PSMA | Prostate-specific membrane antigen |
| QDs | Quantum dots |
| RCA | Rolling circle amplification reaction |
| RNA | Ribonucleic acid |
| s-SWCNTs | Single-walled carbon nanotubes |
| SELEX | Systematic evolution of ligands by exponential enrichment |
| SERS | Surface-enhanced Raman spectroscopy |
| SPR | Surface plasmon resonance |
| TAMRA | Tetramethyl rhodamine |
| TCO | trans-Cyclooctene |
| TL | Test line |
| TMB | 3,3′,5,5′-Tetramethylbenzidine |
| TR-FIA | Time-revolved fluorescence immunoassay |
| Tz | Tetrazines |
| UC FBS | Ultracentrifuged fetal bovine serum |
| UCNPs | Upconversion nanoparticles |
| UV-vis | Ultraviolet-visible spectroscopy |
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the TRANSCAN2-JTC2016 call (Project PROSCANEXO), Spanish AEI funds (BIO2017-84246-C2-1-R project and Juan de la Cierva fellowship (IJCI-2016-30520) to M. M.) and Fondo Social de la DGA (grupos DGA). B. M. G. thanks the DGA Fellowship program for her PhD position. The Research Council of Norway, the Norwegian Cancer Society, and the South-Eastern Norway Regional Health Authority are also acknowledged. We acknowledge support of the publication fee by the CSIC Open Access Publication Support Initiative through its Unit of Information Resources for Research (URICI).Notes and references
- F. Castro-Giner, S. Gkountela, C. Donato, I. Alborelli, L. Quagliata, C. Ng, S. Piscuoglio and N. Aceto, Diagnostics, 2018, 8, 31 CrossRef PubMed.
- M. Cordonnier, G. Chanteloup, N. Isambert, R. Seigneuric, P. Fumoleau, C. Garrido and J. Gobbo, Cell Adhes. Migr., 2017, 11, 151–163 CrossRef CAS.
- Y. Zhang, Y. Liu, H. Liu and W. H. Tang, Cell Biosci., 2019, 9, 19 CrossRef PubMed.
- Z. Zhao, J. Fan, Y.-M. S. Hsu, C. J. Lyon, B. Ning and T. Y. Hu, Lab Chip, 2019, 19, 1114–1140 RSC.
- C. Z. J. Lim, L. Zhang, Y. Zhang, N. R. Sundah and H. Shao, ACS Sens., 2020, 5, 4–12 CrossRef CAS PubMed.
- X. Li, A. L. Corbett, E. Taatizadeh, N. Tasnim, J. P. Little, C. Garnis, M. Daugaard, E. Guns, M. Hoorfar and I. T. S. Li, APL Bioeng., 2019, 3, 011503 CrossRef PubMed.
- H. Shao, H. Im, C. M. Castro, X. Breakefield, R. Weissleder and H. Lee, Chem. Rev., 2018, 118, 1917–1950 CrossRef CAS PubMed.
- N. Cheng, D. Du, X. Wang, D. Liu, W. Xu, Y. Luo and Y. Lin, Trends Biotechnol., 2019, 37, 1236–1254 CrossRef CAS PubMed.
- T. Rojalin, B. Phong, H. J. Koster and R. P. Carney, Front. Chem., 2019, 7, 279 CrossRef CAS PubMed.
- B. S. Chia, Y. P. Low, Q. Wang, P. Li and Z. Gao, TrAC, Trends Anal. Chem., 2017, 86, 93–106 CrossRef CAS.
- K. Boriachek, M. N. Islam, A. Möller, C. Salomon, N.-T. Nguyen, M. S. A. Hossain, Y. Yamauchi and M. J. A. Shiddiky, Small, 2018, 14, 1702153 CrossRef PubMed.
- D. L. M. Rupert, V. Claudio, C. Lässer and M. Bally, Biochim. Biophys. Acta, Gen. Subj., 1861, 2017, 3164–3179 Search PubMed.
- V. Shpacovitch and R. Hergenröder, Anal. Chim. Acta, 2018, 1005, 1–15 CrossRef CAS PubMed.
- W. Wang, J. Luo and S. Wang, Adv. Healthcare Mater., 2018, 7, 1800484 CrossRef.
- P. G. Febbo, A. M. Martin, H. I. Scher, J. C. Barrett and J. A. Beaver, et al. , Clin. Pharmacol. Ther., 2020, 1–5 Search PubMed.
- A. Babayan and K. Pantel, Genome Med., 2018, 10, 21 CrossRef PubMed.
- A. R. Parikh, I. Leshchiner, L. Elagina, L. Goyal and C. Levovitz, et al. , Nat. Med., 2019, 25, 1415–1421 CrossRef CAS PubMed.
- M. H. Vasconcelos, H. R. Caires, A. Ābols, C. P. R. Xavier and A. Linē, Drug Resist. Updates, 2019, 47, 100647 CrossRef PubMed.
- K. Pantel and C. Alix-Panabières, Nat. Rev. Clin. Oncol., 2019, 16, 409–424 CrossRef CAS PubMed.
- L. Sorber, K. Zwaenepoel, V. Deschoolmeester, P. E. Y. Van Schil, J. Van Meerbeeck, F. Lardon, C. Rolfo and P. Pauwels, Lung Cancer, 2017, 107, 100–107 CrossRef CAS PubMed.
- G. Siravegna, B. Mussolin, T. Venesio, S. Marsoni, J. Seoane, C. Dive, N. Papadopoulos, S. Kopetz, R. B. Corcoran, L. L. Siu and A. Bardelli, Ann. Oncol., 2019, 30, 1580–1590 CrossRef CAS PubMed.
- G. De Rubis, S. Rajeev Krishnan and M. Bebawy, Trends Pharmacol. Sci., 2019, 40, 172–186 CrossRef CAS.
- V. Müller, S. Riethdorf, B. Rack, W. Janni, P. A. Fasching, E. Solomayer, B. Aktas, S. Kasimir-Bauer, K. Pantel and T. Fehm, Breast Cancer Res., 2012, 14, R118 CrossRef.
- M. Oellerich, E. Schütz, J. Beck and P. D. Walson, Ther. Drug Monit., 2019, 41, 115–120 CrossRef CAS PubMed.
- G. De Rubis, S. Rajeev Krishnan and M. Bebawy, Trends Pharmacol. Sci., 2019, 40, 172–186 CrossRef CAS PubMed.
- M. Clos-Garcia, A. Loizaga-Iriarte, P. Zuñiga-Garcia, P. Sánchez-Mosquera, A. Rosa Cortazar, E. González, V. Torrano, C. Alonso, M. Pérez-Cormenzana, A. Ugalde-Olano, I. Lacasa-Viscasillas, A. Castro, F. Royo, M. Unda, A. Carracedo and J. M. Falcón-Pérez, J. Extracell. Vesicles, 2018, 7, 1470442 CrossRef PubMed.
- T. Skotland, K. Ekroos, D. Kauhanen, H. Simolin, T. Seierstad, V. Berge, K. Sandvig and A. Llorente, Eur. J. Cancer, 2017, 70, 122–132 CrossRef CAS PubMed.
- C. Théry, K. W. Witwer, E. Aikawa, M. J. Alcaraz and J. D. Anderson, et al. , J. Extracell. Vesicles, 2018, 7, 1535750 CrossRef PubMed.
- M. Yáñez-Mó, P. R. M. Siljander, Z. Andreu, A. B. Zavec and F. E. Borràs, et al. , J. Extracell. Vesicles, 2015, 4, 27066 CrossRef PubMed.
- B. Vestad, A. Llorente, A. Neurauter, S. Phuyal, B. Kierulf, P. Kierulf, T. Skotland, K. Sandvig, K. B. F. Haug and R. Øvstebø, J. Extracell. Vesicles, 2017, 6, 1344087 CrossRef PubMed.
- M. Colombo, G. Raposo and C. Théry, Annu. Rev. Cell Dev. Biol., 2014, 30, 255–289 CrossRef CAS PubMed.
- S. Caruso and I. K. H. Poon, Front. Immunol., 2018, 9, 1486 CrossRef PubMed.
- D. Di Vizio, M. Morello, A. C. Dudley, P. W. Schow and R. M. Adam, et al. , Am. J. Pathol., 2012, 181, 1573–1584 CrossRef CAS PubMed.
- T. Vagner, C. Spinelli, V. R. Minciacchi, L. Balaj, M. Zandian, A. Conley, A. Zijlstra, M. R. Freeman, F. Demichelis, S. De, E. M. Posadas, H. Tanaka and D. Di Vizio, J. Extracell. Vesicles, 2018, 7, 1505403 CrossRef CAS PubMed.
- R. M. Johnstone, M. Adam, J. R. Hammond, L. Orr and C. Turbide, J. Biol. Chem., 1987, 262, 9412–9420 CAS.
- M. Tkach and C. Théry, Cell, 2016, 164, 1226–1232 CrossRef CAS.
- N. Milman, L. Ginini and Z. Gil, Drug Resist. Updates, 2019, 45, 1–12 CrossRef PubMed.
- M. I. Zonneveld, T. G. H. Keulers and K. M. A. Rouschop, Cancers, 2019, 11, 154 CrossRef CAS PubMed.
- L. Sadovska, C. B. Santos, Z. Kalniņa and A. Linē, J. Circ. Biomarkers, 2015, 4, 2 CrossRef PubMed.
- S. Maacha, A. A. Bhat, L. Jimenez, A. Raza, M. Haris, S. Uddin and J. C. Grivel, Mol. Cancer, 2019, 18, 55 CrossRef PubMed.
- T. L. Whiteside, Adv. Exp. Med. Biol., 2017, 1036, 81–89 CrossRef CAS PubMed.
- L. Nogués, A. Benito-Martin, M. Hergueta-Redondo and H. Peinado, Mol. Aspects Med., 2018, 60, 15–26 CrossRef PubMed.
- M. Logozzi, A. De Milito, L. Lugini, M. Borghi and L. Calabrò, et al. , PLoS One, 2009, 4, e5219 CrossRef.
- E. Alegre, L. Zubiri, J. L. Perez-Gracia, M. González-Cao, L. Soria, S. Martín-Algarra and A. González, Clin. Chim. Acta, 2016, 454, 28–32 CrossRef CAS PubMed.
- A. Caivano, I. Laurenzana, L. De Luca, F. La Rocca, V. Simeon, S. Trino, F. D’Auria, A. Traficante, M. Maietti, T. Izzo, G. D’Arena, G. Mansueto, G. Pietrantuono, L. Laurenti, P. Musto and L. Del Vecchio, Tumor Biol., 2015, 36, 9739–9752 CrossRef CAS.
- D. Duijvesz, C. Y. L. Versluis, C. A. M. Van Der Fels, M. S. Vredenbregt-Van Den Berg, J. Leivo, M. T. Peltola, C. H. Bangma, K. S. I. Pettersson and G. Jenster, Int. J. Cancer, 2015, 137, 2869–2878 CrossRef CAS PubMed.
- Y. Matsumoto, M. Kano, Y. Akutsu, N. Hanari, I. Hoshino, K. Murakami, A. Usui, H. Suito, M. Takahashi, R. Otsuka, H. Xin, A. Komatsu, K. Iida and H. Matsubara, Oncol. Rep., 2016, 36, 2535–2543 CrossRef CAS PubMed.
- M. Nawaz, G. Camussi, H. Valadi, I. Nazarenko, K. Ekström, X. Wang, S. Principe, N. Shah, N. M. Ashraf, F. Fatima, L. Neder and T. Kislinger, Nat. Rev. Urol., 2014, 11, 688–701 CrossRef PubMed.
- E. Ogorevc, V. Kralj-Iglic and P. Veranic, Radiol. Oncol., 2013, 47, 197–205 CAS.
- S. R. Zorrilla, M. Pérez-Sayans, S. Fais, M. Logozzi, M. G. Torreira and A. G. García, Cancers, 2019, 11, 429 CrossRef CAS PubMed.
- L. König, S. Kasimir-Bauer, A. K. Bittner, O. Hoffmann, B. Wagner, L. F. Santos Manvailer, R. Kimmig, P. A. Horn and V. Rebmann, Oncoimmunology, 2018, 7, e1376153 CrossRef PubMed.
- D. Osti, M. Del Bene, G. Rappa, M. Santos, V. Matafora, C. Richichi, S. Faletti, G. V. Beznoussenko, A. Mironov, A. Bachi, L. Fornasari, D. Bongetta, P. Gaetani, F. DiMeco, A. Lorico and G. Pelicci, Clin. Cancer Res., 2019, 25, 266–276 CrossRef CAS PubMed.
- F. Cappello, M. Logozzi, C. Campanella, C. C. Bavisotto, A. Marcilla, F. Properzi and S. Fais, Eur. J. Pharm. Sci., 2017, 96, 93–98 CrossRef CAS.
- K. Al-Nedawi, B. Meehan, J. Micallef, V. Lhotak, L. May, A. Guha and J. Rak, Nat. Cell Biol., 2008, 10, 619–624 CrossRef CAS PubMed.
- Q. Bao, L. Gong, J. Wang, J. Wen, Y. Shen and W. Zhang, Ann. Surg. Oncol., 2018, 25, 2642–2651 CrossRef PubMed.
- S. García-Silva, A. Benito-Martín, S. Sánchez-Redondo, A. Hernández-Barranco and P. Ximénez-Embún, et al. , J. Exp. Med., 2019, 216, 1061–1070 CrossRef PubMed.
- E. Lázaro-Ibáñez, A. Sanz-Garcia, T. Visakorpi, C. Escobedo-Lucea, P. Siljander, Á. Ayuso-Sacido and M. Yliperttula, Prostate, 2014, 74, 1379–1390 CrossRef PubMed.
- M. A. S. Broggi, L. Maillat, C. C. Clement, N. Bordry, P. Corthésy, A. Auger, M. Matter, R. Hamelin, L. Potin, D. Demurtas, E. Romano, A. Harari, D. E. Speiser, L. Santambrogio and M. A. Swartz, J. Exp. Med., 2019, 216, 1091–1107 CrossRef CAS PubMed.
- L. Sadovska, J. Eglitis and A. Line, Anticancer Res., 2015, 35, 6379–6390 CAS.
- H. Shao, J. Chung and D. Issadore, Biosci. Rep., 2016, 36, e00292 CrossRef PubMed.
- E. Castellanos-Rizaldos, D. G. Grimm, V. Tadigotla, J. Hurley, J. Healy, P. L. Neal, M. Sher, R. Venkatesan, C. Karlovich, M. Raponi, A. Krug, M. Noerholm, J. Tannous, B. A. Tannous, L. E. Raez and J. K. Skog, Clin. Cancer Res., 2018, 24, 2944–2950 CrossRef CAS.
- K. Allenson, J. Castillo, F. A. San Lucas, G. Scelo, D. U. Kim, V. Bernard, G. Davis, T. Kumar, M. Katz, M. J. Overman, L. Foretova, E. Fabianova, I. Holcatova, V. Janout, F. Meric-Bernstam, P. Gascoyne, I. Wistuba, G. Varadhachary, P. Brennan, S. Hanash, D. Li, A. Maitra and H. Alvarez, Ann. Oncol., 2017, 28, 741–747 CrossRef CAS.
- F. A. San Lucas, K. Allenson, V. Bernard, J. Castillo, D. U. Kim, K. Ellis, E. A. Ehli, G. E. Davies, J. L. Petersen, D. Li, R. Wolff, M. Katz, G. Varadhachary, I. Wistuba, A. Maitra and H. Alvarez, Ann. Oncol. Off. J. Eur. Soc. Med. Oncol., 2016, 27, 635–641 CrossRef CAS PubMed.
- E. Endzelinš, A. Berger, V. Melne, C. Bajo-Santos, K. Sobolevska, A. Abols, M. Rodriguez, D. Šantare, A. Rudnickiha, V. Lietuvietis, A. Llorente and A. Line, BMC Cancer, 2017, 17, 730 CrossRef PubMed.
- S. A. Melo, L. B. Luecke, C. Kahlert, A. F. Fernandez, S. T. Gammon, J. Kaye, V. S. LeBleu, E. A. Mittendorf, J. Weitz, N. Rahbari, C. Reissfelder, C. Pilarsky, M. F. Fraga, D. Piwnica-Worms and R. Kalluri, Nature, 2015, 523, 177–182 CrossRef CAS PubMed.
- J. Y. Qian, Y. L. Tan, Y. Zhang, Y. F. Yang and X. Q. Li, Oncol. Lett., 2018, 16, 1253–1258 Search PubMed.
- Y. H. Park, H. W. Shin, A. R. Jung, O. S. Kwon, Y. J. Choi, J. Park and J. Y. Lee, Sci. Rep., 2016, 6, 30386 CrossRef CAS PubMed.
- C. N. Biggs, K. M. Siddiqui, A. A. Al-Zahrani, S. Pardhan, S. I. Brett, Q. Q. Guo, J. Yang, P. Wolf, N. E. Power, P. N. Durfee, C. D. MacMillan, J. L. Townson, J. C. Brinker, N. E. Fleshner, J. I. Izawa, A. F. Chambers, J. L. Chin and H. S. Leong, Oncotarget, 2016, 7, 8839–8849 CrossRef PubMed.
- O. D. Murillo, W. Thistlethwaite, J. Rozowsky, S. L. Subramanian and R. Lucero, et al. , Cell, 2019, 177, 463–477 CrossRef CAS PubMed.
- C. Pisano, J. Galley, M. Elbahrawy, Y. Wang, A. Farrell, D. Brigstock and G. E. Besner, J. Clin. Pediatr. Surg., 2020, 55, 54–58 CrossRef.
- J. Y. Hur, J. S. Lee, I. A. Kim, H. J. Kim, W. S. Kim and K. Y. Lee, Transl. Lung Cancer Res., 2019, 8, 1051–1060 CrossRef PubMed.
- J. Linxweiler and K. Junker, Nat. Rev. Urol., 2020, 17, 11–27 CrossRef PubMed.
- S. Fais, L. O’Driscoll, F. E. Borras, E. Buzas and G. Camussi, et al. , ACS Nano, 2016, 10, 3886–3899 CrossRef CAS PubMed.
- U. Erdbrügger and J. Lannigan, Cytometry, Part A, 2016, 89, 123–134 CrossRef PubMed.
- P. Ziaei, C. E. Berkman and M. G. Norton, ACS Appl. Nano Mater., 2018, 1, 2004–2020 CrossRef CAS.
- J. McKiernan, M. J. Donovan, E. Margolis, A. Partin, B. Carter, G. Brown, P. Torkler, M. Noerholm, J. Skog, N. Shore, G. Andriole, I. Thompson and P. Carroll, Eur. Urol., 2018, 74, 731–738 CrossRef CAS PubMed.
- M. Oliveira-Rodríguez, S. López-Cobo, H. T. Reyburn, A. Costa-García, S. López-Martín, M. Yáñez-Mó, E. Cernuda-Morollón, A. Paschen, M. Valés-Gómez and M. C. Blanco-López, J. Extracell. Vesicles, 2016, 5, 31803 CrossRef PubMed.
- G. Li, W. Tang and F. Yang, Biotechnol. J., 2020, 1900225 CrossRef CAS PubMed.
- H. Im, H. Shao, Y. Il Park, V. M. Peterson, C. M. Castro, R. Weissleder and H. Lee, Nat. Biotechnol., 2014, 32, 490–495 CrossRef CAS PubMed.
- H. Shao, J. Chung, L. Balaj, A. Charest, D. D. Bigner, B. S. Carter, F. H. Hochberg, X. O. Breakefield, R. Weissleder and H. Lee, Nat. Med., 2012, 18, 1835–1840 CrossRef CAS PubMed.
- T. Kilic, A. T. D. S. Valinhas, I. Wall, P. Renaud and S. Carrara, Sci. Rep., 2018, 8, 9402 CrossRef PubMed.
- R. Friedrich, S. Block, M. Alizadehheidari, S. Heider, J. Fritzsche, E. K. Esbjörner, F. Westerlund and M. Bally, Lab Chip, 2017, 17, 830–841 RSC.
- M. Sáenz-Cuesta, Front. Immunol., 2015, 6, 1–12 Search PubMed.
- V. Filipe, A. Hawe and W. Jiskoot, Pharm. Res., 2010, 27, 796–810 CrossRef CAS PubMed.
- A. Clayton, D. Buschmann, J. B. Byrd, D. R. F. Carter and L. Cheng, et al. , J. Extracell. Vesicles, 2018, 7, 1473707 CrossRef PubMed.
- A. Clayton, E. Boilard, E. I. Buzas, L. Cheng and J. M. Falcón-Perez, et al. , J. Extracell. Vesicles, 2019, 8, 1647027 CrossRef PubMed.
- M. Y. Konoshenko, E. A. Lekchnov, A. V. Vlassov and P. P. Laktionov, Biomed. Res. Int., 2018, 2018, 8545347 Search PubMed.
- P. Li, M. Kaslan, S. H. Lee, J. Yao and Z. Gao, Theranostics, 2017, 7, 789–804 CrossRef CAS PubMed.
- B. Y. Chen, C. W. H. Sung, C. Chen, C. M. Cheng, D. P. C. Lin, C. Te Huang and M. Y. Hsu, Clin. Chim. Acta, 2019, 493, 14–19 CrossRef CAS PubMed.
- F. Royo, P. Zuñiga-Garcia, P. Sanchez-Mosquera, A. Egia, A. Perez, A. Loizaga, R. Arceo, I. Lacasa, A. Rabade, E. Arrieta, R. Bilbao, M. Unda, A. Carracedo and J. M. Falcon-Perez, J. Extracell. Vesicles, 2016, 5, 29497 CrossRef PubMed.
- G. K. Patel, M. A. Khan, H. Zubair, S. K. Srivastava, M. Khushman, S. Singh and A. P. Singh, Sci. Rep., 2019, 9, 5335 CrossRef PubMed.
- Y. Yoshioka, N. Kosaka, Y. Konishi, H. Ohta, H. Okamoto, H. Sonoda, R. Nonaka, H. Yamamoto, H. Ishii, M. Mori, K. Furuta, T. Nakajima, H. Hayashi, H. Sugisaki, H. Higashimoto, T. Kato, F. Takeshita and T. Ochiya, Nat. Commun., 2014, 5, 3591 CrossRef PubMed.
- E. Commission, Off. J. Eur. Union. 2011/696/EU, 2011, 38–40.
- B. A. Kairdolf, X. Qian and S. Nie, Anal. Chem., 2017, 89, 1015–1031 CrossRef CAS PubMed.
- P. Malik, V. Katyal, V. Malik, A. Asatkar, G. Inwati and T. K. Mukherjee, ISRN Nanomater., 2013, 2013, 327435 Search PubMed.
- H. Su, S. Li, Y. Jin, Z. Xian, D. Yang, W. Zhou, F. Mangaran, F. Leung, G. Sithamparanathan and K. Kerman, Adv. Health Care Technol., 2017, 3, 19–29 CrossRef.
- Z. Farka, T. Juřík, D. Kovář, L. Trnková and P. Skládal, Chem. Rev., 2017, 117, 9973–10042 CrossRef CAS PubMed.
- D. Issadore, C. Min, M. Liong, J. Chung, R. Weissleder and H. Lee, Lab Chip, 2011, 11, 2282–2287 RSC.
- S. Jeong, J. Park, D. Pathania, C. M. Castro, R. Weissleder and H. Lee, ACS Nano, 2016, 10, 1802–1809 CrossRef CAS PubMed.
- X. Huang, T. Yuan, M. Tschannen, Z. Sun, H. Jacob, M. Du, M. Liang, R. L. Dittmar, Y. Liu, M. Liang, M. Kohli, S. N. Thibodeau, L. Boardman and L. Wang, BMC Genomics, 2013, 14, 319 CrossRef CAS PubMed.
- A. K. Trilling, J. Beekwilder and H. Zuilhof, Analyst, 2013, 138, 1619 RSC.
- S. Sharma, H. Byrne and R. J. O’Kennedy, Essays Biochem., 2016, 60, 9–18 CrossRef PubMed.
- A. Makaraviciute and A. Ramanaviciene, Biosens. Bioelectron., 2013, 50, 460–471 CrossRef CAS PubMed.
- A. J. Sivaram, A. Wardiana, C. B. Howard, S. M. Mahler and K. J. Thurecht, Adv. Healthcare Mater., 2018, 7, 1700607 CrossRef PubMed.
- J.-M. Montenegro, V. Grazu, A. Sukhanova, S. Agarwal, J. M. de la Fuente, I. Nabiev, A. Greiner and W. J. Parak, Adv. Drug Delivery Rev., 2013, 65, 677–688 CrossRef CAS PubMed.
- M. A. Parracino, B. Martín and V. Grazú, Photoactive Inorganic Nanoparticles, Elsevier, 2019, pp. 211–257 Search PubMed.
- A. C. Marques, P. J. Costa, S. Velho and M. H. Amaral, J. Controlled Release, 2020, 320, 180–200 CrossRef CAS PubMed.
- S. Wang, L. Zhang, S. Wan, S. Cansiz, C. Cui, Y. Liu, R. Cai, C. Hong, I.-T. Teng, M. Shi, Y. Wu, Y. Dong and W. Tan, ACS Nano, 2017, 11, 3943–3949 CrossRef CAS PubMed.
- S. Puertas, M. Moros, R. Fernández-Pacheco, M. R. Ibarra, V. Grazú and J. M. de la Fuente, J. Phys. D: Appl. Phys., 2010, 43, 474012 CrossRef.
- J. B. Haun, N. K. Devaraj, S. A. Hilderbrand, H. Lee and R. Weissleder, Nat. Nanotechnol., 2010, 5, 660–665 CrossRef CAS PubMed.
- M. Liong, M. Fernandez-Suarez, D. Issadore, C. Min, C. Tassa, T. Reiner, S. M. Fortune, M. Toner, H. Lee and R. Weissleder, Bioconjugate Chem., 2011, 22, 2390–2394 CrossRef CAS PubMed.
- V. M. Peterson, C. M. Castro, H. Lee and R. Weissleder, ACS Nano, 2012, 6, 3506–3513 CrossRef CAS PubMed.
- E. Polo, S. Puertas, M. Moros, P. Batalla, J. M. Guisán, J. M. de la Fuente and V. Grazú, Methods Mol. Biol., 2013, 1051, 149–163 CrossRef CAS PubMed.
- S. Puertas, P. Batalla, M. Moros, E. Polo, P. Del Pino, J. M. Guisán, V. Grazú and J. M. de la Fuente, ACS Nano, 2011, 5, 4521–4528 CrossRef CAS PubMed.
- G. Ruiz, K. Tripathi, S. Okyem and J. D. Driskell, Bioconjugate Chem., 2019, 30, 1182–1191 CrossRef CAS PubMed.
- C. Parolo, A. de la Escosura-Muñiz, E. Polo, V. Grazú, J. M. de la Fuente and A. Merkoçi, ACS Appl. Mater. Interfaces, 2013, 5, 10753–10759 CrossRef CAS PubMed.
- S. Puertas, M. de Gracia Villa, E. Mendoza, C. Jiménez-Jorquera, J. M. de la Fuente, C. Fernández-Sánchez and V. Grazú, Biosens. Bioelectron., 2013, 43, 274–280 CrossRef CAS PubMed.
- B. Saha, P. Songe, T. H. Evers and M. W. J. Prins, Analyst, 2017, 142, 4247–4256 RSC.
- K. Tripathi and J. D. Driskell, ACS Omega, 2018, 3, 8253–8259 CrossRef CAS PubMed.
- S. van der Heide and D. A. Russell, J. Colloid Interface Sci., 2016, 471, 127–135 CrossRef CAS PubMed.
- C. Tuerk and L. Gold, Science, 1990, 249, 505–510 CrossRef CAS PubMed.
- A. D. Ellington and J. W. Szostak, Nature, 1990, 346, 818–822 CrossRef CAS PubMed.
- S. Y. Toh, M. Citartan, S. C. B. Gopinath and T.-H. Tang, Biosens. Bioelectron., 2015, 64, 392–403 CrossRef CAS PubMed.
- V. Crivianu-Gaita and M. Thompson, Biosens. Bioelectron., 2016, 85, 32–45 CrossRef CAS PubMed.
- Z. Ge, H. Pei, L. Wang, S. Song and C. Fan, Sci. China: Chem., 2011, 54, 1273–1276 CrossRef CAS.
- E. Baldrich, A. Restrepo and C. K. O’Sullivan, Anal. Chem., 2004, 76, 7053–7063 CrossRef CAS PubMed.
- M. Lin, P. Song, G. Zhou, X. Zuo, A. Aldalbahi, X. Lou, J. Shi and C. Fan, Nat. Protoc., 2016, 11, 1244–1263 CrossRef CAS.
- R. Schlapak, J. Danzberger, D. Armitage, D. Morgan, A. Ebner, P. Hinterdorfer, P. Pollheimer, H. J. Gruber, F. Schäffler and S. Howorka, Small, 2012, 8, 89–97 CrossRef CAS PubMed.
- NanoComposix, Lateral Flow Assay Development Guide v 1.4, 2018.
- InnovaBioscience, Guide to Lateral Flow Immunoassays, 2017.
- H. V. Hsieh, J. L. Dantzler and B. H. Weigl, Diagnostics, 2017, 7, 29 CrossRef CAS PubMed.
- K. M. Koczula and A. Gallotta, Essays Biochem., 2016, 60, 111–120 CrossRef PubMed.
- S. López-Cobo, C. Campos-Silva, A. Moyano, M. Oliveira-Rodríguez, A. Paschen, M. Yáñez-Mó, M. C. Blanco-López and M. Valés-Gómez, J. Nanobiotechnol., 2018, 16, 47 CrossRef PubMed.
- M. Oliveira-rodríguez, E. Serrano, A. C. García, S. López, M. Y. Mo and E. Cernuda-morollón, Biosens. Bioelectron., 2016, 87, 38–45 CrossRef PubMed.
- T. Wu, Y. Yang, Y. Cao, Y. Huang, L. P. Xu, X. Zhang and S. Wang, Sci. China: Chem., 2018, 61, 1423–1429 CrossRef CAS.
- D. Dong, L. Zhu, J. Hu, D. W. Pang and Z. L. Zhang, Talanta, 2019, 200, 408–414 CrossRef CAS PubMed.
- Q. Yu, Q. Zhao, S. Wang, S. Zhao, S. Zhang, Y. Yin and Y. Dong, Anal. Biochem., 2020, 594, 113591 CrossRef CAS.
- J. Chen, Y. Xu, Y. Lu and W. Xing, Anal. Chem., 2018, 90, 14207–14215 CrossRef CAS PubMed.
- Y. Xia, M. Liu, L. Wang, A. Yan, W. He, M. Chen, J. Lan, J. Xu, L. Guan and J. Chen, Biosens. Bioelectron., 2017, 92, 8–15 CrossRef CAS PubMed.
- Y.-M. Wang, J.-W. Liu, G. B. Adkins, W. Shen, M. P. Trinh, L.-Y. Duan, J.-H. Jiang and W. Zhong, Anal. Chem., 2017, 89, 12327–12333 CrossRef CAS PubMed.
- Y. Jiang, M. Shi, Y. Liu, S. Wan, C. Cui, L. Zhang and W. Tan, Angew. Chem., Int. Ed., 2017, 56, 11916–11920 CrossRef CAS PubMed.
- W. Liu, J. Li, Y. Wu, S. Xing, Y. Lai and G. Zhang, Biosens. Bioelectron., 2018, 102, 204–210 CrossRef CAS PubMed.
- F. He, H. Liu, X. Guo, B.-C. Yin and B.-C. Ye, Anal. Chem., 2017, 89, 12968–12975 CrossRef CAS PubMed.
- Y. Zhang, D. Wang, S. Yue, Y. Lu, C. Yang, J. Fang and Z. Xu, ACS Sens., 2019, 4, 3210–3218 CrossRef CAS PubMed.
- W. Nawrot, K. Drzozga, S. Baluta, J. Cabaj and K. Malecha, Sensors, 2018, 18, 1–21 CrossRef PubMed.
- D. Wu, A. C. Sedgwick, T. Gunnlaugsson, E. U. Akkaya, J. Yoon and T. D. James, Chem. Soc. Rev., 2017, 46, 7105–7123 RSC.
- B. S. Chia, Y. P. Low, Q. Wang, P. Li and Z. Gao, TrAC, Trends Anal. Chem., 2017, 86, 93–106 CrossRef CAS.
- S. M. Ng, M. Koneswaran and R. Narayanaswamy, A review on fluorescent inorganic nanoparticles for optical sensing applications, Royal Society of Chemistry, 2016, vol. 6 Search PubMed.
- L. Huang, S. Yang, J. Chen, J. Tian, Q. Huang, H. Huang, Y. Wen, F. Deng, X. Zhang and Y. Wei, Mater. Sci. Eng., C, 2019, 94, 270–278 CrossRef CAS.
- X. Miao, Z. Cheng, H. Ma, Z. Li, N. Xue and P. Wang, Anal. Chem., 2018, 90, 1098–1103 CrossRef CAS PubMed.
- S. Kailasa, B. G. Rani, M. S. Bhargava Reddy, N. Jayarambabu, P. Munindra, S. Sharma and K. Venkateswara Rao, Mater. Chem. Phys., 2020, 242, 122524 CrossRef CAS.
- A. Martín-Barreiro, S. de Marcos and J. Galbán, Microchim. Acta, 2018, 185(3), 171 CrossRef PubMed.
- A. Martín-Barreiro, S. de Marcos, J. M. de la Fuente, V. Grazú and J. Galbán, Sens. Actuators, B, 2018, 277, 261–270 CrossRef.
- M. Ali, M. Sajid, M. A. U. Khalid, S. W. Kim, J. H. Lim, D. Huh and K. H. Choi, Spectrochim. Acta, Part A, 2020, 226, 117610 CrossRef CAS PubMed.
- Y. Wang, Z. Wei, X. Luo, Q. Wan, R. Qiu and S. Wang, Talanta, 2019, 195, 33–39 CrossRef CAS PubMed.
- D. Chen, C. Liu, J. Tang, L. Luo and G. Yu, Polym. Chem., 2019, 10, 1168–1181 RSC.
- Y. Liu, P. Dong, Q. Jiang, F. Wang, D. W. Pang and X. Liu, Sens. Actuators, B, 2019, 279, 334–341 CrossRef CAS.
- X. Lu, C. Wang, J. Qian, C. Ren, K. An and K. Wang, Anal. Chim. Acta, 2019, 1047, 163–171 CrossRef CAS PubMed.
- H. Yanagawa, A. Inoue, H. Sugimoto, M. Shioi and M. Fujii, MRS Commun., 2019, 9, 1079–1086 CrossRef CAS.
- R. A. Almotiri, K. J. Ham, V. M. Vijayan and S. A. Catledge, Materials, 2019, 12(13), 2097 CrossRef CAS PubMed.
- M. O. Dekaliuk, O. Viagin, Y. V. Malyukin and A. P. Demchenko, Phys. Chem. Chem. Phys., 2014, 16, 16075–16084 RSC.
- G. Chen, H. Qiu, P. N. Prasad and X. Chen, Chem. Rev., 2014, 114, 5161–5214 CrossRef CAS.
- D. Mendez-Gonzalez, E. Lopez-Cabarcos, J. Rubio-Retama and M. Laurenti, Adv. Colloid Interface Sci., 2017, 249, 66–87 CrossRef CAS PubMed.
- Y. Wang, D. Luo, Y. Fang, W. Wu, Y. Wang, Y. Xia, F. Wu, C. Li, J. Lan and J. Chen, Sens. Actuators, B, 2019, 298, 126900 CrossRef.
- X. Chen, J. Lan, Y. Liu, L. Li, L. Yan, Y. Xia, F. Wu, C. Li, S. Li and J. Chen, Biosens. Bioelectron., 2018, 102, 582–588 CrossRef CAS.
- C. H. R, J. D. Schiffman and R. G. Balakrishna, Sens. Actuators, B, 2018, 258, 1191–1214 CrossRef.
- Y. Bai, Y. Lu, K. Wang, Z. Cheng, Y. Qu, S. Qiu, L. Zhou, Z. Wu, H. Liu, J. Zhao and H. Mao, Nano-Micro Lett., 2019, 11, 1–11 CrossRef CAS PubMed.
- G. Dobhal, D. Ayupova, G. Laufersky, Z. Ayed, T. Nann and R. V. Goreham, Sensors, 2018, 18, 1–12 CrossRef PubMed.
- S. Zong, J. Zong, C. Chen, X. Jiang, Y. Zhang, Z. Wang and Y. Cui, Nanotechnology, 2018, 29, 065705 CrossRef.
- X. Jiang, S. Zong, C. Chen, Y. Zhang, Z. Wang and Y. Cui, Nanotechnology, 2018, 29, 175701 CrossRef PubMed.
- F. He, J. Wang, B. C. Yin and B. C. Ye, Anal. Chem., 2018, 90, 8072–8079 CrossRef CAS PubMed.
- M. L. Gao, B. C. Yin and B. C. Ye, Analyst, 2019, 144, 5996–6003 RSC.
- M. Tayebi, M. Tavakkoli Yaraki, H. Y. Yang and Y. Ai, Nanoscale Adv., 2019, 1, 2866–2872 RSC.
- K. Huang, Z. Li, J. Lin, G. Han and P. Huang, Chem. Soc. Rev., 2018, 47, 5109–5124 RSC.
- Q. Zhang, F. Wang, H. Zhang, Y. Zhang, M. Liu and Y. Liu, Anal. Chem., 2018, 90, 12737–12744 CrossRef CAS PubMed.
- C. Zhu, Z. Zeng, H. Li, F. Li, C. Fan and H. Zhang, J. Am. Chem. Soc., 2013, 135, 5998–6001 CrossRef CAS PubMed.
- H. Wang, H. Chen, Z. Huang, T. Li, A. Deng and J. Kong, Talanta, 2018, 184, 219–226 CrossRef CAS PubMed.
- D. Jin, F. Yang, Y. Zhang, L. Liu, Y. Zhou, F. Wang and G. J. Zhang, Anal. Chem., 2018, 90, 14402–14411 CrossRef CAS PubMed.
- M. K. Islam, P. Syed, L. Lehtinen, J. Leivo, K. Gidwani, S. Wittfooth, K. Pettersson and U. Lamminmäki, Sci. Rep., 2019, 9, 1–9 CrossRef PubMed.
- C. Kim and K. Lee, Biomacromolecules, 2019, 20, 3392–3398 CrossRef CAS PubMed.
- H. A. Abdulbari and E. A. M. Basheer, ChemBioEng Rev., 2017, 4, 92–105 CrossRef CAS.
- S. Kaushal, S. S. Nanda, S. Samal and D. K. Yi, ChemBioChem, 2020, 21, 576–600 CrossRef CAS PubMed.
- S. Alim, J. Vejayan, M. M. Yusoff and A. K. M. Kafi, Biosens. Bioelectron., 2018, 121, 125–136 CrossRef CAS PubMed.
- S. Shrivastava, N. Jadon and R. Jain, TrAC, Trends Anal. Chem., 2016, 82, 55–67 CrossRef CAS.
- A. de la Escosura-Muñiz, C. Parolo and A. Merkoçi, Mater. Today, 2010, 13, 24–34 CrossRef.
- K. Boriachek, M. N. Islam, V. Gopalan, A. K. Lam, N. T. Nguyen and M. J. A. Shiddiky, Analyst, 2017, 142, 2211–2219 RSC.
- K. Boriachek, M. K. Masud, C. Palma, H. P. Phan, Y. Yamauchi, M. S. A. Hossain, N. T. Nguyen, C. Salomon and M. J. A. Shiddiky, Anal. Chem., 2019, 91, 3827–3834 CrossRef CAS PubMed.
- Y. G. Zhou, R. M. Mohamadi, M. Poudineh, L. Kermanshah, S. Ahmed, T. S. Safaei, J. Stojcic, R. K. Nam, E. H. Sargent and S. O. Kelley, Small, 2016, 12, 727–732 CrossRef CAS PubMed.
- H. Zhang, Z. Wang, Q. Zhang, F. Wang and Y. Liu, Biosens. Bioelectron., 2019, 124–125, 184–190 CrossRef CAS PubMed.
- Z. Shi, G. Li and Y. Hu, Chin. Chem. Lett., 2019, 30, 1600–1606 CrossRef CAS.
- D. Fang, D. Zhao, S. Zhang, Y. Huang, H. Dai and Y. Lin, Sens. Actuators, B, 2020, 305, 127544 CrossRef.
- B. Qiao, Q. Guo, J. Jiang, Y. Qi, H. Zhang, B. He, C. Cai and J. Shen, Analyst, 2019, 144, 3668–3675 RSC.
- B. Chase, Appl. Spectrosc., 1994, 48, 14A–19A CrossRef CAS.
- D. W. Shipp, F. Sinjab and I. Notingher, Adv. Opt. Photonics, 2017, 9, 315 CrossRef.
- U. K. Sur and J. Chowdhury, Curr. Sci., 2013, 105, 923 CAS.
- M. Fan, G. F. S. Andrade and A. G. Brolo, Anal. Chim. Acta, 2020, 1097, 1–29 CrossRef CAS PubMed.
- H. Guo, L. He and B. Xing, Environ. Sci.: Nano, 2017, 4, 2093–2107 RSC.
- J. Langer, D. Jimenez de Aberasturi, J. Aizpurua, R. A. Alvarez-Puebla and B. Auguié, et al. , ACS Nano, 2020, 14, 28–117 CrossRef CAS PubMed.
- M. E. Stewart, C. R. Anderton, L. B. Thompson, J. Maria, S. K. Gray, J. A. Rogers and R. G. Nuzzo, Chem. Rev., 2008, 108, 494–521 CrossRef CAS PubMed.
- Y.-F. Tian, C.-F. Ning, F. He, B.-C. Yin and B.-C. Ye, Analyst, 2018, 143, 4915–4922 RSC.
- T.-D. Li, R. Zhang, H. Chen, Z.-P. Huang, X. Ye, H. Wang, A.-M. Deng and J.-L. Kong, Chem. Sci., 2018, 9, 5372–5382 RSC.
- K. Sivashanmugan, W.-L. Huang, C.-H. Lin, J.-D. Liao, C.-C. Lin, W.-C. Su and T.-C. Wen, J. Taiwan Inst. Chem. Eng., 2017, 80, 149–155 CrossRef CAS.
- S. Zong, L. Wang, C. Chen, J. Lu, D. Zhu, Y. Zhang, Z. Wang and Y. Cui, Anal. Methods, 2016, 8, 5001–5008 RSC.
- Z. Wang, S. Zong, Y. Wang, N. Li, L. Li, J. Lu, Z. Wang, B. Chen and Y. Cui, Nanoscale, 2018, 10, 9053–9062 RSC.
- S. Stremersch, M. Marro, B.-E. Pinchasik, P. Baatsen, A. Hendrix, S. C. De Smedt, P. Loza-Alvarez, A. G. Skirtach, K. Raemdonck and K. Braeckmans, Small, 2016, 12, 3292–3301 CrossRef CAS PubMed.
- J. C. Fraire, S. Stremersch, D. Bouckaert, T. Monteyne, T. De Beer, P. Wuytens, R. De Rycke, A. G. Skirtach, K. Raemdonck, S. De Smedt and K. Braeckmans, ACS Appl. Mater. Interfaces, 2019, 11, 39424–39435 CrossRef CAS PubMed.
- C. Lee, R. Carney, K. Lam and J. W. Chan, J. Raman Spectrosc., 2017, 48, 1771–1776 CrossRef CAS.
- Y. Pang, J. Shi, X. Yang, C. Wang, Z. Sun and R. Xiao, Biosens. Bioelectron., 2020, 148, 111800 CrossRef CAS PubMed.
- G. Chen, A. C. Huang, W. Zhang, G. Zhang, M. Wu, W. Xu, Z. Yu, J. Yang, B. Wang, H. Sun, H. Xia, Q. Man, W. Zhong, L. F. Antelo, B. Wu, X. Xiong, X. Liu, L. Guan, T. Li, S. Liu, R. Yang, Y. Lu, L. Dong, S. McGettigan, R. Somasundaram, R. Radhakrishnan, G. Mills, Y. Lu, J. Kim, Y. H. Chen, H. Dong, Y. Zhao, G. C. Karakousis, T. C. Mitchell, L. M. Schuchter, M. Herlyn, E. J. Wherry, X. Xu and W. Guo, Nature, 2018, 560, 382–386 CrossRef CAS PubMed.
- F. L. Ricklefs, Q. Alayo, H. Krenzlin, A. B. Mahmoud and M. C. Speranza, et al. , Sci. Adv., 2018, 4, eaar2766 CrossRef PubMed.
- F. Gao, F. Jiao, C. Xia, Y. Zhao, W. Ying, Y. Xie, X. Guan, M. Tao, Y. Zhang, W. Qin and X. Qian, Chem. Sci., 2019, 10, 1579–1588 RSC.
- E. A. Kwizera, R. O’Connor, V. Vinduska, M. Williams, E. R. Butch, S. E. Snyder, X. Chen and X. Huang, Theranostics, 2018, 8, 2722–2738 CrossRef CAS PubMed.
- J. Carmicheal, C. Hayashi, X. Huang, L. Liu, Y. Lu, A. Krasnoslobodtsev, A. Lushnikov, P. G. Kshirsagar, A. Patel, M. Jain, Y. L. Lyubchenko, Y. Lu, S. K. Batra and S. Kaur, Nanomedicine, 2019, 16, 88–96 CrossRef CAS PubMed.
- J. Park, M. Hwang, B. Choi, H. Jeong, J. Jung, H. K. Kim, S. Hong, J. Park and Y. Choi, Anal. Chem., 2017, 89, 6695–6701 CrossRef CAS PubMed.
- H. Shin, H. Jeong, J. Park, S. Hong and Y. Choi, ACS Sens., 2018, 3, 2637–2643 CrossRef CAS.
- R. Bhome, F. Del Vecchio, G.-H. Lee, M. D. Bullock, J. N. Primrose, A. E. Sayan and A. H. Mirnezami, Cancer Lett., 2018, 420, 228–235 CrossRef CAS PubMed.
- Y. Pang, C. Wang, L. Lu, C. Wang, Z. Sun and R. Xiao, Biosens. Bioelectron., 2019, 130, 204–213 CrossRef CAS.
- D. Ma, C. Huang, J. Zheng, J. Tang, J. Li, J. Yang and R. Yang, Biosens. Bioelectron., 2018, 101, 167–173 CrossRef CAS PubMed.
- L. Grasso, R. Wyss, L. Weidenauer, A. Thampi, D. Demurtas, M. Prudent, N. Lion and H. Vogel, Anal. Bioanal. Chem., 2015, 407, 5425–5432 CrossRef CAS PubMed.
- Y. P. Chen, M. Q. Zou, C. Qi, M.-X. Xie, D.-N. Wang, Y.-F. Wang, Q. Xue, J.-F. Li and Y. Chen, Biosens. Bioelectron., 2013, 39, 112–117 CrossRef CAS PubMed.
- H. Shao, J. Chung, K. Lee, L. Balaj, C. Min, B. S. Carter, F. H. Hochberg, X. O. Breakefield, H. Lee and R. Weissleder, Nat. Commun., 2015, 6, 6999 CrossRef CAS PubMed.
- S. Jeong, J. Park, D. Pathania, C. M. Castro, R. Weissleder and H. Lee, ACS Nano, 2016, 10, 1802–1809 CrossRef CAS PubMed.
- J. C. Contreras-Naranjo, H.-J. Wu and V. M. Ugaz, Lab Chip, 2017, 17, 3558–3577 RSC.
- S. S. Kanwar, C. J. Dunlay, D. M. Simeone and S. Nagrath, Lab Chip, 2014, 14, 1891–1900 RSC.
- Z. Zhao, Y. Yang, Y. Zeng and M. He, Lab Chip, 2016, 16, 489–496 RSC.
- J. Sierra, J. Marrugo-Ramírez, R. Rodriguez-Trujillo, M. Mir and J. Samitier, Sensors, 2020, 20, 1317 CrossRef PubMed.
- J. Ko, M. A. Hemphill, D. Gabrieli, L. Wu, V. Yelleswarapu, G. Lawrence, W. Pennycooke, A. Singh, D. F. Meaney and D. Issadore, Sci. Rep., 2016, 6, 31215 CrossRef CAS PubMed.
- L.-G. Liang, M.-Q. Kong, S. Zhou, Y.-F. Sheng, P. Wang, T. Yu, F. Inci, W. P. Kuo, L.-J. Li, U. Demirci and S. Wang, Sci. Rep., 2017, 7, 46224 CrossRef PubMed.
- R. Xu, D. W. Greening, H.-J. Zhu, N. Takahashi and R. J. Simpson, J. Clin. Invest., 2016, 126, 1152–1162 CrossRef PubMed.
- F. A. W. Coumans, E. L. Gool and R. Nieuwland, Platelets, 2017, 28, 242–248 CrossRef CAS PubMed.
- Y. Liu, Q. Liu, S. Chen, F. Cheng, H. Wang and W. Peng, Sci. Rep., 2015, 5, 12864 CrossRef CAS PubMed.
- S. L. Cobo, C. C. Silva, A. Moyano, M. O. Rodríguez, A. Paschen, M. Y. Mó, M. Carmen, B. López and M. V. Gómez, J. Nanobiotechnol., 2018, 16, 47 CrossRef PubMed.
- K. Sivashanmugan, W. L. Huang, C. H. Lin, J. Der Liao, C. C. Lin, W. C. Su and T. C. Wen, J. Taiwan Inst. Chem. Eng., 2017, 80, 149–155 CrossRef CAS.
- S. D. Ibsen, J. Wright, J. M. Lewis, S. Kim, S. Y. Ko, J. Ong, S. Manouchehri, A. Vyas, J. Akers, C. C. Chen, B. S. Carter, S. C. Esener and M. J. Heller, ACS Nano, 2017, 11, 6641–6651 CrossRef CAS PubMed.
- Q. Zhou, A. Rahimian, K. Son, D. S. Shin, T. Patel and A. Revzin, Methods, 2016, 97, 88–93 CrossRef CAS PubMed.
- J. Van Deun, P. Mestdagh, P. Agostinis, Ö. Akay and S. Anand, et al. , Nat. Methods, 2017, 14, 228–232 CrossRef CAS PubMed.
- J. A. Welsh, E. Van Der Pol, G. J. A. Arkesteijn, M. Bremer and A. Brisson, et al. , J. Extracell. Vesicles, 2020, 9, 1713526 CrossRef PubMed.
- J. McKiernan, M. J. Donovan, E. Margolis, A. Partin, B. Carter, G. Brown, P. Torkler, M. Noerholm, J. Skog, N. Shore, G. Andriole, I. Thompson and P. Carroll, Eur. Urol., 2018, 74, 731–738 CrossRef CAS PubMed.
Footnote |
| † Equal contribution. |
| This journal is © The Royal Society of Chemistry 2020 |