
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceStable anchoring of bacteria-based protein nanoparticles for surface enhanced cell guidance†
Marc
Martínez-Miguel
 ab,
Adriana R.
Kyvik
ac,
Lena
M. Ernst
a,
Albert
Martínez-Moreno
a,
Olivia
Cano-Garrido
cd,
Elena
Garcia-Fruitós‡
cd,
Esther
Vazquez
cd,
Nora
Ventosa
ac,
Judith
Guasch
*abc,
Jaume
Veciana
ac,
Antonio
Villaverde
cd and
Imma
Ratera
*ac
ab,
Adriana R.
Kyvik
ac,
Lena
M. Ernst
a,
Albert
Martínez-Moreno
a,
Olivia
Cano-Garrido
cd,
Elena
Garcia-Fruitós‡
cd,
Esther
Vazquez
cd,
Nora
Ventosa
ac,
Judith
Guasch
*abc,
Jaume
Veciana
ac,
Antonio
Villaverde
cd and
Imma
Ratera
*ac
aDepartment of Molecular Nanoscience and Organic Materials, Institut de Ciència de Materials de Barcelona (ICMAB-CSIC), Campus UAB, Bellaterra 08193, Spain. E-mail: iratera@icmab.es
bDynamic Biomimetics for Cancer Immunotherapy, Max Planck Partner Group, ICMAB-CSIC, Campus UAB, Bellaterra 08193, Spain
cNetworking Research Center on Bioengineering, Biomaterials and Nanomedicine (CIBER-BBN), Spain
dInstitut de Biotecnologia i de Biomedicina and Departament de Genètica i de Microbiologia, Universitat Autònoma de Barcelona (UAB), Cerdanyola del Vallès 08193, Spain
First published on 29th April 2020
Abstract
In tissue engineering, biological, physical, and chemical inputs have to be combined to properly mimic cellular environments and successfully build artificial tissues which can be designed to fulfill different biomedical needs such as the shortage of organ donors or the development of in vitro disease models for drug testing. Inclusion body-like protein nanoparticles (pNPs) can simultaneously provide such physical and biochemical stimuli to cells when attached to surfaces. However, this attachment has only been made by physisorption. To provide a stable anchoring, a covalent binding of lactic acid bacteria (LAB) produced pNPs, which lack the innate pyrogenic impurities of Gram-negative bacteria like Escherichia coli, is presented. The reported micropatterns feature a robust nanoscale topography with an unprecedented mechanical stability. In addition, they are denser and more capable of influencing cell morphology and orientation. The increased stability and the absence of pyrogenic impurities represent a step forward towards the translation of this material to a clinical setting.
Introduction
Cell behavior is influenced by numerous factors, ranging from biological and chemical to physical cues, such as the influence of nearby cells and soluble molecules or the surface stiffness of the extracellular matrix (ECM).1 Indeed, cells are able to receive and integrate these inputs, resulting in responses such as adhesion, proliferation, migration, and apoptosis.2,3 Using this knowledge, tissue engineering aims to mimic the physiological and environmental conditions of biological systems to provide better strategies for regenerative medicine.4 Several surface engineering approaches have been used to design and produce materials able to interact with cells and study their behavior.5 For example, micro- and nano-structured surfaces produced by hard lithography techniques have been of great utility to study the influence of different surface topographies.6–8 Other approaches such as self-assembly and soft-lithography enabled versatile chemical functionalizations and immobilizations of biological materials on surfaces.9–13 Among the techniques used to fabricate these soft materials, microcontact printing (μCP) stands as a cost-effective and fast soft-lithography technique to control surface chemistry at the microscale.14 μCP allows the use of a broad range of materials to coat surfaces such as proteins,15,16 nucleic acids,17 genetically modified viruses,18 and electroactive ligands with dynamic surfaces.19,20 Recently, μCP has been used to print proteins and peptides to generate micropatterned surfaces for cell guidance and mechanotransduction studies.21–23Our group applied the same technique to pattern protein nanoparticles (pNPs) on surfaces. These pNPs consisted of bacterial inclusion bodies (IBs) of green fluorescent proteins (GFPs) or fibroblast growth factors (FGFs).27,55–58 Such pNPs were produced through biological synthesis in recombinant bacteria obtaining highly pure aggregates,24 which preserved the function of the protein of interest.25 They were roughly spherical and showed diameters ranging from 50 to a few hundred nanometers, although their sizes and geometries can be tuned.26 In these studies, negatively charged pNPs were anchored through electrostatic interactions on amino terminated self-assembled monolayers (SAMs), resulting in the preferential adhesion of fibroblasts in pNP-rich micropatterned areas.27 pNPs were thus proved to be useful nanomaterials for cell culture and for the development of biomaterials for regenerative medicine. Nevertheless, some challenges still have to be overcome in order to achieve wider applicability, such as the lack of a strong and specific binding of the pNPs to surfaces as well as their safety issues related to their production in Escherichia coli (E. coli), which are bacteria rich in endotoxins.
There are several strategies for protein attachment to surfaces that are compatible with μCP, such as affinity-based biological reactions28 and surface chemistry strategies such as the thiol–gold,15 amine-reactive crosslinking after the silanization of glass,29,30 or thiol–maleimide interfacial reactions,31 among others. The interfacial maleimide–thiol reaction is an interesting option due to its resulting covalent interaction and compatibility with many biological functional groups of proteins.31 Moreover, the natural presence of thiol groups in cysteine residues32 allows a strong binding with the maleimide of a wide range of proteins into specific micropatterns, without prior modification. To our knowledge, the covalent attachment of pNPs to surfaces has not yet been reported in contrast with the well-known anchoring of single proteins32 and the immobilization of other nano-objects, such as liposomes,33 gold nanoparticles,34 and molecularly imprinted polymeric nanoparticles.35 The covalent binding of pNPs on surfaces through the maleimide–thiol reaction is interesting to prevent the pNP displacement caused by the exerted forces of cells in long lasting cell culture studies. Such covalent bindings are also expected to enhance the life time of the pNP-decorated surfaces.
On the other hand, the production of IB-like pNPs in E. coli as a cell factory represents a major drawback due to the presence of lipopolysaccharides (LPS) in their outer cell membrane, which are also known as endotoxins. LPS are able to over-activate the immune system at only μg per kg of LPS/body mass ratios36,37 and its removal from proteins produced in Gram-negative bacteria, such as E. coli, is tedious and expensive.36,38 Among the alternatives, Gram-positive lactic acid bacteria (LAB) stand out,39 which are LPS-free and have been classified as generally recognized as safe (GRAS) microorganisms by the Food and Drug Administration (FDA),40 thus allowing their development as biologically-safe production platforms. In spite of the high solubility of LAB-protein products, fully functional IB-like pNPs composed of relevant proteins in humans and veterinary medicine were produced and purified.41 Since then, LAB-produced IB-like pNPs have continued to be developed42 and used for applications such as therapeutic protein delivery.43
In this work we aimed at covalently attaching pNPs derived from LAB to pre-functionalized surfaces, thus improving both the robustness and biocompatibility of pNP-decorated surfaces for cell guidance. Our objective was to increase the adhesion of large pNPs to their supporting substrate through covalent bonding to produce dense and well-defined pNP patterns that would promote cell adhesion with resistance to cell-applied forces. At the same time, we explored the efficacy of the non-pyrogenic pNPs produced in LAB compared to those produced in E. coli. With these objectives in mind, pNPs produced in Lactococcus lactis (L. lactis) were micropatterned onto maleimide-terminated SAMs and used to study the adhesion and spreading of human osteosarcoma U2-OS cells. Thus, we demonstrate the suitability of LAB as safe cell factories and the possibility to produce endotoxin-free nanomaterials in a cost-effective and scalable process, while still sharing many of the positive features of pNPs derived from E. coli for cell culture studies.41
Experimental
Bacterial strains, plasmids and IB-like protein nanoparticle production
IB-like pNPs were produced both in the E. coli strain MC4100 (StrpR), transformed with the expression vector pTV1GFP (ApR), and in L. lactis subsp. cremoris NZ9000, clpP−htrA− (clpP-htrA; EmR) (kindly provided by INRA, Jouy-en-Josas, France; patent Nr. EP1141337B1) strain44 transformed with the expression vector pNZ8148 (CmR) (MoBiTech). E. coli was grown in lysogeny broth (LB) rich medium at 37 °C with its respective antibiotic supplementation (ampicillin, 100 μg ml−1; and streptomycin, 30 μg ml−1) at 37 °C and 250 rpm. L. lactis was grown in M17 medium enriched with 0.5% glucose at 30 °C with antibiotic supplementation (chloramphenicol, 5 μg ml−1; and erythromycin, 2.5 μg ml−1) at 30 °C without shaking. The production of pNPs in E. coli was induced when reaching an OD550 = 0.5 with 1 mM isopropyl β-D-1-thiogalactopyranoside (IPTG), while the production of IB-like pNPs in L. lactis was induced by 12.5 ng ml−1 nisin when OD550 = 0.5. After 3 h, cell cultures were harvested for NP purification.Production of GFP IB-like pNPs in Escherichia coli
All the materials used were sterile and the procedure was carried out in a laminar flow hood under sterile conditions. The E. coli MC4100 pTV1GFP strain was grown in LB rich medium supplemented with 300 μl of ampicillin and 300 μl of streptomycin in 300 ml shake flasks at 37 °C and 250 rpm until it reached an OD550 = 0.715. The culture was induced to produce GFP pNPs using 1 mM IPTG for 3 h. Purification started when the induced culture optical density reached 4.38 units. Protease inhibitors were then added to the broth (one small complete EDTA-free pill). 3 ml of lysozyme at 0.5 mg ml−1 was also added to the medium and the culture was incubated for 2 h at 37 °C and 250 rpm. After this step, the culture was frozen overnight (ON) at −80 °C. Afterwards, 1.2 ml of Triton X-100 was added to the media when the culture defrosted. The culture was then incubated with agitation at room temperature (RT) for 1 h. A first sterility control was performed by spreading 100 μl of the culture broth in an antibiotic-free LB Petri dish and incubating it ON at 37 °C. The rest of the culture was frozen ON at −80 °C. The freezing/defrosting process was repeated by spreading 100 μl of the culture in an antibiotic-free LB plaque after each cycle, until no organism was grown in the dish after 24 h. After 3 cycles of freezing/defrosting, the broth was sterile. 75 μl of Nonidet NP-40 was then added to the mixture and kept under agitation for 1 h at 37 °C and 250 rpm. Afterwards, the culture was centrifuged at 3900 rpm for 30 min and the pellet was resuspended with 15 ml of lysis buffer and Triton X-100. The mixture was frozen ON and the culture was defrosted and centrifuged at 3900 rpm for 15 min at 4 °C. The supernatant was discarded and the pellet was resuspended with 15 ml of phosphate-buffered saline (PBS). The mixture was transferred to a 1 ml Eppendorf tube (each one containing 10 ml of the original broth). Samples were then centrifuged and the supernatant was discarded. GFP pNPs were finally purified and ready to be used. They were kept frozen at −80 °C until their use.Production of GFP IB-like pNPs in Lactococcus lactis
All the materials used were sterile and the procedure was carried out in laminar flow hoods always under sterile conditions. The L. lactis NZ9000 clpP−htrA− pNZ8148-VP1GFP strain was cultured in M17 medium enriched with 0.5% glucose and supplemented with 150 μl of erythromycin and 75 μl of chloramphenicol in 300 ml shake flasks at 30 °C and 250 rpm until it reached an OD550 = 0.37. Recombinant gene expression was induced to produce GFP pNPs using 12.5 ng ml−1 nisin for 3 h. Purification started when the optical density reached 3.8 units. The culture was distributed in 50 ml sterile falcons and centrifuged at 3900 rpm for 30 min. The supernatant was then discarded and the pellet was resuspended in 30 ml PBS/falcon, which was frozen ON at −80 °C. Afterwards, the sample was defrosted and disrupted for three rounds using a French Press at 15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 PSI. The resulting solution was frozen ON at −80 °C and 600 μl of lysozyme/falcon was added to the sample and kept under agitation for 2 h at 37 °C and 250 rpm when defrosted. Then, the mixture was frozen ON at −80 °C and 120 μl of Triton X-100/falcon was added after defrosting. Falcons were agitated for 1 h at RT. After, a first sterility control was performed by spreading 100 μl of the culture broth in an antibiotic-free 0.5% glucose M17 broth Petri dish and leaving it ON at 30 °C. The rest of the culture was frozen at −80 °C ON. A freezing/defrosting process was repeated spreading 100 μl of culture in an antibiotic-free M17 broth plaque after each freezing cycle, until no organism was grown in the media. After 3 cycles of freezing/defrosting, the broth was sterile. Then, 75 μl of NP-40 was added to the mixture and kept under agitation for 1 h at 4 °C. 18 μl of 1 M MgSO4 and 18 μl of 1 mg ml−1 DNAse were added to each falcon and kept under agitation for 1 h at 37 °C and 250 rpm. The mixture was centrifuged at 3900 rpm for 30 min and the supernatant was discarded. The pellet was resuspended with 5 ml lysis buffer and 0.5% Triton X-100. Falcons were then frozen ON at −80 °C. Tubes were centrifuged at 3900 rpm for 30 min after defrosting and the bacterial pellet was resuspended with 5 ml of sterile PBS. The resulting solution was transferred to 1 ml Eppendorf tubes (each one containing 10 ml of the original broth). Aliquots were centrifuged at 1300 rpm for 15 min and the supernatant was discarded. GFP pNPs were finally purified and ready to be used. They were kept frozen at −80 °C and defrosted for immediate use.
000 PSI. The resulting solution was frozen ON at −80 °C and 600 μl of lysozyme/falcon was added to the sample and kept under agitation for 2 h at 37 °C and 250 rpm when defrosted. Then, the mixture was frozen ON at −80 °C and 120 μl of Triton X-100/falcon was added after defrosting. Falcons were agitated for 1 h at RT. After, a first sterility control was performed by spreading 100 μl of the culture broth in an antibiotic-free 0.5% glucose M17 broth Petri dish and leaving it ON at 30 °C. The rest of the culture was frozen at −80 °C ON. A freezing/defrosting process was repeated spreading 100 μl of culture in an antibiotic-free M17 broth plaque after each freezing cycle, until no organism was grown in the media. After 3 cycles of freezing/defrosting, the broth was sterile. Then, 75 μl of NP-40 was added to the mixture and kept under agitation for 1 h at 4 °C. 18 μl of 1 M MgSO4 and 18 μl of 1 mg ml−1 DNAse were added to each falcon and kept under agitation for 1 h at 37 °C and 250 rpm. The mixture was centrifuged at 3900 rpm for 30 min and the supernatant was discarded. The pellet was resuspended with 5 ml lysis buffer and 0.5% Triton X-100. Falcons were then frozen ON at −80 °C. Tubes were centrifuged at 3900 rpm for 30 min after defrosting and the bacterial pellet was resuspended with 5 ml of sterile PBS. The resulting solution was transferred to 1 ml Eppendorf tubes (each one containing 10 ml of the original broth). Aliquots were centrifuged at 1300 rpm for 15 min and the supernatant was discarded. GFP pNPs were finally purified and ready to be used. They were kept frozen at −80 °C and defrosted for immediate use.
High resolution scanning electron microscopy of GFP IB-like pNPs
IB-like pNPs were resuspended in ultrapure water and sonicated for 10 min before the deposition of a few microdrops on silicon chips. Substrates were left to air dry for 2 min and then, were dried completely with filter paper. Samples were observed in a FEI Magellan 400L XHR SEM (Nanolab technologies, Milpitas, CA) operating at 2 kV. Images were obtained using a vCD back-scattered electron detector.Colorimetric quantification of accessible cysteine residues of GFP IB-like pNPs
The comparative quantification of accessible cysteine residues between the different IB-like pNPs produced was carried out by following a previously described protocol using 4′4-dithiopyridine (DTDP), which interacts with accessible thiol groups of cysteines and yields a product that strongly absorbs light at 324 nm.45 Briefly, IB-like pNP samples were diluted in PBS up to a concentration of 4 μg ml−1 and sonicated 10 min before the addition of the DTDP reagent. The progress of the reaction was monitored on a spectrophotometer (JASCO V-780) for 5 min at 324 nm. The increment of absorbance was compared in each sample after 5 min.Preparation of functional terminated SAMs
Glass substrates were placed in piranha solution [concentrated H2SO4, aqueous H2O2, 3:1] for 40 min, rinsed with ultrapure water and dried under a nitrogen stream. A 3 nm layer of titanium was deposited on the glass substrates in order to improve the gold adhesion using a vapor deposition equipment (Edwards Auto 306; Edwards, Crawley, UK). On top of that, an 8 nm layer of gold was deposited. Substrates were cleaned through 5 min sonication consecutively in dichloromethane, acetone, and ethanol, and then dried under a nitrogen stream. Prior to the SAM formation, substrates were activated in an ozone cleaner (UVO-cleaner: model 42 series; Jelight company, USA) for 20 min. Substrates were then immersed in 1 mM ethanolic solutions containing either an HO-terminated molecule [HO-EG3-C11-S-S-C11-EG3-OH, where EGx is poly(ethylene)glycol, and Cx is –(CH2)x–] or a maleimide-terminated molecule [a 98:2 mix of HO-EG3-C11-S-S-C11-EG3-OH and S-S-C4CONH-C3-EG3-C-NHCO-C2-Mal (lipoic acid–poly(ethylene)glycol-maleimide) (Mal = maleimide) molecules] (Prochimia Surfaces, Poland) in a controlled argon atmosphere for 24 h (Scheme 1).
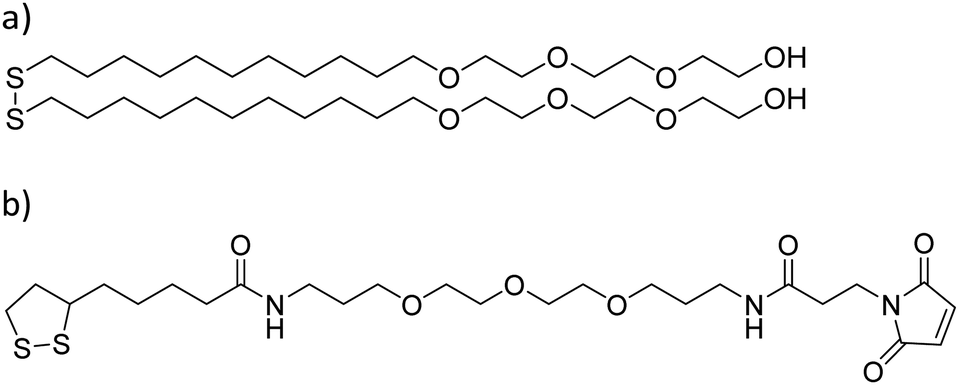 | ||
| Scheme 1 Molecular structures used in the formation of –OH and maleimide terminated SAMs. (a) HO-EG3-C11-S-S-C11-EG3-OH and (b) S-S-C4CONH-C3-EG3-C-NHCO-C2-Mal. EGx = polyethylene glycol, Cx = –(CH2)x–, and Mal = maleimide. | ||
Contact angle measurements of deposited IB-like pNPs
IB-like pNPs were resuspended in PBS up to a final concentration of 20 μg ml−1. Freshly prepared HO- and maleimide-terminated SAMs were immersed in pNP suspensions for 2 h and gently rinsed with ultrapure water before measuring the wettability of the substrates with a Drop Shape Analyzer – a DSA100 (Krüss, Germany) contact angle goniometer. Three sets of static contact angles at different positions in each sample were measured and the average values were obtained.Microcontact printing
Polydimethylsiloxane (PDMS) stamps were fabricated using a commercial kit (Sylgard 184 Silicone Elastomer kit; Sigma-Aldrich, USA), mixing the PDMS precursor and the curing agent together in a 10:1 (w/w) ratio, and degassing the mixture to remove bubbles in a vacuum desiccator for 20 min. The mixture was then poured into a previously silanized photolithographically patterned silicon master, degassed again, cured for 2 h at 60 °C, and released. Before soaking the clean stamps in the pNP suspension, the stamps were sonicated for 5 min in acetone and dried in vacuum for 10 min. Then, the stamps were sonicated for 5 min in a sodium dodecyl sulfate (SDS) solution [10% in ultrapure water, w/w] and conditioned in the same media for 5 min. Afterwards, stamps were dipped in ultrapure water to remove the excess of SDS and dried under a nitrogen stream. After the latter pre-treatment, stamps were immersed in previously sonicated E. coli- or L. lactis-pNP solutions in PBS for 1 h. Stamps were then gently rinsed with ultrapure water, dried under a nitrogen stream, and placed on the SAM-coated substrates under a 13 g weight. After 1 h of contact, stamps were carefully removed in the presence of PBS.
Cell culture, seeding, and immunostaining
Human osteosarcoma cells (U2-OS) were obtained from the American Type Culture Collection (ATCC; Manassas, VA, USA). Cells were routinely cultured in Dulbecco's Modified Eagle's Medium (DMEM) (Thermo Fisher Scientific, USA) supplemented with 10% fetal bovine serum (FBS) and 1% penicillin/streptomycin in a humidified atmosphere containing 10% CO2 at 37 °C. U2-OS cells were seeded on IB-like pNP micropatterned surfaces at a concentration of 5000 cells per cm2. Plates were incubated at 37 °C and under 10% CO2 for 24 h. Thereafter, cells were fixed by the addition of 4% paraformaldehyde in PBS for 20 min. After fixation, cells were permeabilized by adding 0.1% Triton in PBS and treated with a blocking solution (1% bovine serum albumin in PBS) for 30 min to prevent non-specific binding. After blocking, substrates were incubated for 1 h at RT with a mouse monoclonal anti-paxillin antibody (Sigma-Aldrich, USA) diluted at a ratio of 1:400. After incubation with the primary antibody, samples were washed with PBS using a shaker at 50 rpm for 10 min and then they were incubated for 45 min at RT with the secondary antibody Alexa Fluor 647-labeled goat anti-mouse IgG (1:100; Thermo Fisher Scientific, USA) and with Hoechst (1:1000; Thermo Fisher Scientific, USA). Primary and secondary antibodies were diluted in the blocking solution. Finally, samples were washed with PBS using a shaker for 10 min and mounted with ProLong Gold Antifading Mountant (Thermo Fisher Scientific, USA).
Optical and confocal microscopy
pNP-printed SAMs were observed using a fluorescence microscope (Olympus BX51 with a U-RFL-T reflected fluorescence system; Olympus, Japan) in order to assess the result of the pNP deposition by μCP. To study cell adhesion and morphology, confocal microscopy images were obtained using a Leica TCS SP5 AOBS spectral confocal microscope (Leica Microsystems, Mannheim, Germany) with a HCX PL APO lambda blue 63× objective.Image analysis
Focal adhesion (FA) quantification as well as cell orientation and morphology data were extracted from microscopy images and treated with the ImageJ software (National Institute of Health, USA). The treatment of the obtained data and the statistical Kruskal–Wallis ANOVA test were performed using the Origin software (OriginLab, USA). To determine the FA mean area, images were treated with contrast enhancement and a mean filter to improve structure resolution. Afterwards, captures were turned into binary using the threshold tool, and the “Analyze Particles” function was used to measure the area of the FAs in the cell. The mean FA area per cell was determined and plotted using Origin. The “Analyze Particles” function was also used to approximate the whole cell area into ellipses to determine cell orientation. The angle between the long axis of the ellipses and the stripped pattern was calculated. Then, orientation data were classified into 10° segments, ranging from 0 to 180°, and represented using radial plots. Values between 180° and 360° are a symmetric repetition of the data to improve the plot visualization. Similarly, upon orientation analysis, images were turned into binary using the threshold tool to calculate the cell area, which were approximated into ellipses, and their elongation was calculated as their aspect ratios minus one.Results and discussion
GFP IB-like pNPs produced by E. coli and L. lactis were covalently attached to maleimide-terminated SAMs through their cysteine aminoacids by means of a Michael addition interfacial reaction using the μCP technique, as recently shown for other nano-objects.46–48More specifically, the exposed thiol groups of the cysteine residues of GFP at the surface of the pNPs reacted with the maleimide groups of the SAM forming covalent bonds, thus providing a more stable binding in comparison with a simple physisorption procedure (Fig. 1).
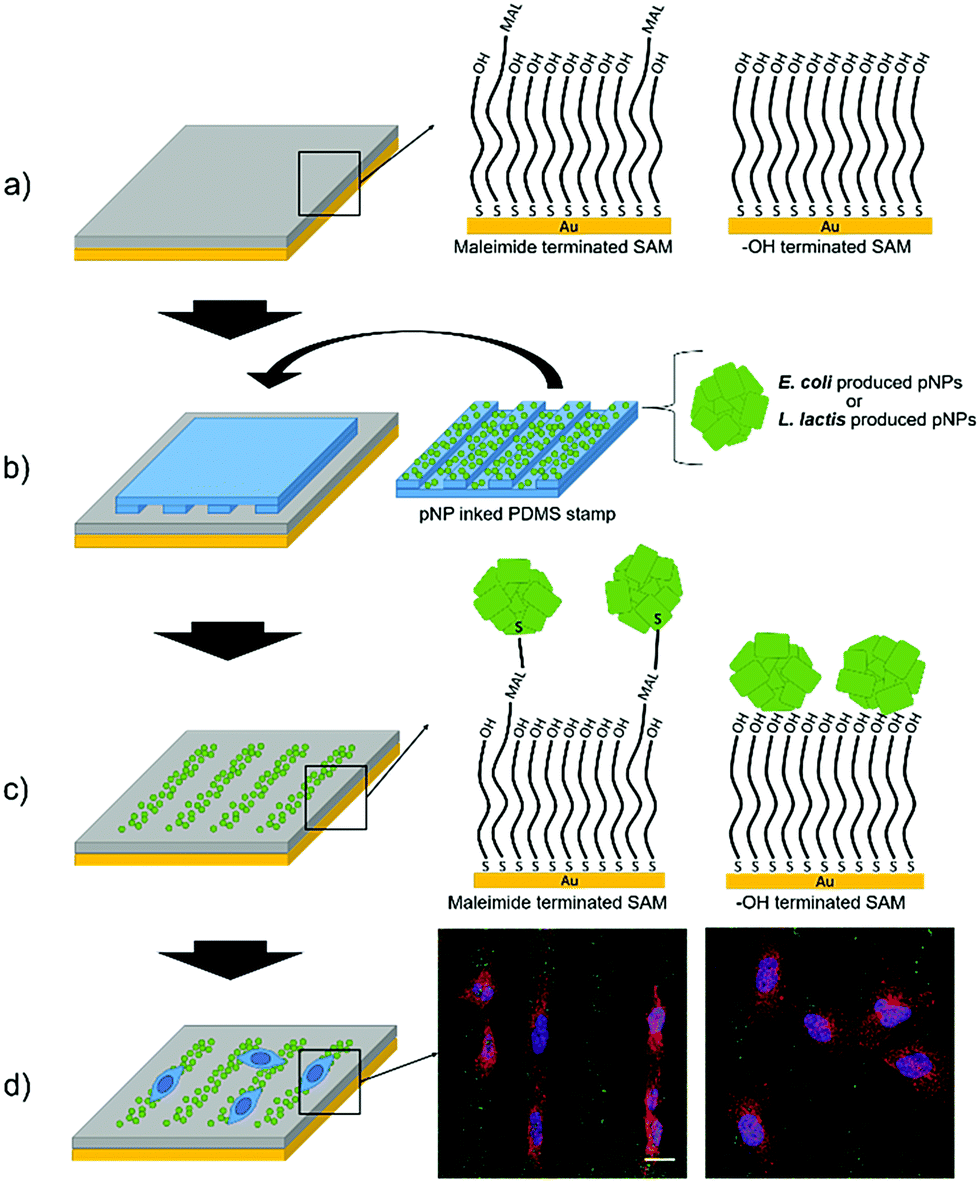 | ||
| Fig. 1 Scheme of the strategy for culturing cells on micropatterns of pNPs. (a) HO- and maleimide-terminated SAMs were prepared. (b) A micropatterned PDMS stamp was inked in a pNP suspension, produced either in E. coli or in L. lactis, and put gently in contact with the SAM to form the stripped pattern of pNPs. (c) In the maleimide terminated SAM, a covalent bond was formed between the maleimide and the exposed cysteine groups of proteins, while in the HO-terminated SAM, pNPs were merely physisorbed on the surface. (d) Cells were seeded on the surfaces and studied using confocal microscopy. Scale bar = 20 μm. | ||
In order to assess the effect of the covalent bond, the same pNPs were also printed on HO-terminated SAMs, in which pNPs are expected to be only physisorbed. Moreover, to compare the viability of LAB produced pNPs (Lactis-pNPs) with E. coli produced pNPs (Coli-pNPs), both are produced and printed onto the different substrates using suspensions with the same mass concentrations. Thus, the seeding of cells on these substrates and the study of their morphology and orientation allowed us to measure the effect of the covalently attached pNPs and the influence of the host organism in which these particles were produced (Fig. 1). Finally, we also explored mechanotransduction by the formation of focal adhesions (FAs), given that stronger applied forces were expected from cells seeded on the covalently attached pNPs compared to the physisorbed ones. Indeed, FAs are protein complexes that are formed on the cell membrane, which communicate cells with their ECM and are responsible for cell attachment and migration.
Interactions between IB-like pNPs and functionalized surfaces
First, we analyzed the surface coverage of the pNPs attained upon μCP of Coli-pNP and Lactis-pNP suspensions with similar mass concentrations on the two functional terminated SAMs, HO-SAM and maleimide-SAM, without any further treatment. Thus, 20 μm wide stripped patterns of pNPs were printed on the SAMs and analyzed without rinsing using fluorescence microscopy to determine the pNP surface coverage and to compare the number of pNPs adhered on the different terminated surfaces (Fig. 2a and b). The quantitative surface coverage analysis was performed by the integration of the GFP fluorescence intensity of microscopy images in 30 random fields per sample. Total integrated intensity was weighted against the total area of the field to obtain the average percentage of coverage. This analysis indicates that surfaces decorated with Lactis-pNPs show a higher surface coverage in comparison with those decorated with Coli-pNPs. Furthermore, the maleimide-terminated SAMs with Lactis-pNPs exhibit the highest surface coverage. These results were further validated by water contact angle measurements of the surfaces decorated with pNPs. A higher contact angle was detected on the SAMs decorated with Lactis-pNPs in comparison with those decorated with Coli-pNPs (Fig. S2, ESI†) in agreement with the higher quantity of deposited material on the surface.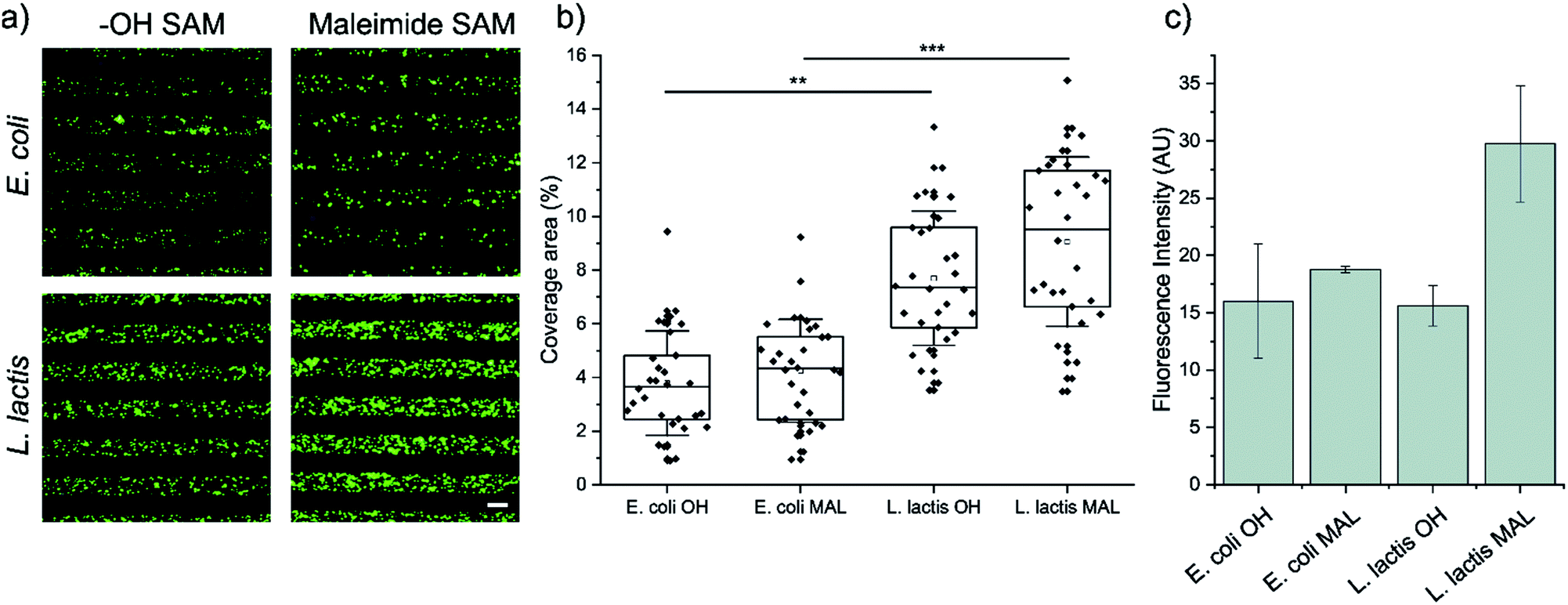 | ||
| Fig. 2 (a) Representative fluorescence microscopy images of 20 μm striped μCP patterns of E. coli (up) and L. lactis (down) GFP pNPs on functionalized SAMs with HO- (left) or maleimide (right) terminated groups. Scale bar = 20 μm. Images were taken immediately after the removal of the PDMS stamp. (b) Percentage of E. coli and L. lactis pNPs coverage area after being adhered to maleimide or HO-terminated SAMs through μCP. Higher coverage area relates to higher pNP density in the pattern. The information was extracted from fluorescence microscopy images. Nexperiments = 3; and Nimages = 30. Statistical analysis was performed using a Kruskal–Wallis ANOVA test. **p < 0.001, and ***p < 0.0001. (c) Fluorescence intensity retention after rinsing with ultrapure water on HO- and maleimide-terminated surfaces with deposited GFP pNPs produced in E. coli and L. lactis. | ||
Afterwards, the pNP retention was assessed by the integration of the GFP fluorescence intensity of microscopy images after rinsing the surfaces with ultrapure water, thus eliminating any interference from non-covalent interactions (Fig. 2c). The maleimide-terminated surface decorated with Lactis-pNPs showed a higher fluorescence intensity than the HO-terminated surface, indicating that covalent bonds between maleimide groups on the surface and the cysteine residues of GFP were properly formed. Thus, the density of attached pNPs and their resistance to being washed away increased. Even though there is a slight increase of fluorescence for the Coli-pNPs decorated maleimide-terminated SAM with respect to the HO-terminated SAM, there is no statistically significant difference between them. To compare the remaining density of attached Coli-pNPs and Lactis-pNPs after washing with water on a given surface, it should be taken into account that the specific fluorescence of GFP Coli-pNPs is approximately 200% higher than that of Lactis-pNPs (see the ESI,† Section S4). According to this result, the higher integrated fluorescence intensities based on the images of surfaces decorated with Lactis-pNPs can be correlated with a relevant increase in the interaction of the particulate material with the surface in comparison with that of Coli-pNPs.
To understand if the physicochemical characteristics of the pNPs influence the differential formation of covalent bonds between the external cysteine residues of GFP-pNPs and the maleimide groups of the surface, we compared the average sizes and Z-potential of the pNPs produced in either E. coli or L. lactis. The Z-potential of both particles is very similar (−37 mV) (see Section S1 of the ESI†), while the average sizes of Coli-pNPs and Lactis-pNPs are 350 and 520 nm (Fig. S1, ESI†), respectively. The observed differences in the size of the two types of pNPs could be at least partially responsible for the larger formation of covalent bonds on the Lactis-pNPs.41 In addition, the number of accessible thiol groups of the cysteine residues on the pNP surfaces was investigated. Specifically, we performed a colorimetric assay in which pNPs were incubated with 4,4′-dithiopyridine (DTDP). This reagent interacts with thiol groups and yields a product that absorbs light at 324 nm, allowing the quantification of cysteine residues of proteins in a fast and simple manner.45 To comparatively quantify the amount of thiol groups in each sample, we diluted the pNPs to equal concentrations and monitored the reaction with DTDP over 5 min. Lactis-pNPs showed an approximately 25% higher amount of accessible thiol groups than Coli-pNPs (Fig. S3, ESI†). Therefore, enhanced accessibility to thiol groups in Lactis-pNPs could also be another explanation for the improved interactions.
It is important to note that the proposed interfacial pNP-SAM reaction requires the presence of accessible thiol groups. Although the tendency of the thiols to dimerize, no reducing agents were applied during the printing process, which indicates that the possible dimerization of thiols on the pNPs did not impede the interaction with the surface. Another indication of the absence of dimerization is that size distributions, as measured by DLS,41 showed clearly defined intensity peaks, as well as a negative Z-potential which supports its colloidal stability by the action of the electrostatic repulsion between NPs. Furthermore, the DTDP assay confirmed the presence of accessible thiol groups without scattering for as long as 2 h after the resuspension of the pNP samples. Moreover, the resulting patterns are stable and well formed, and in SAMs featuring maleimide groups these patterns are denser and more resilient to washes, reinforcing the hypothesis that the interfacial maleimide–thiol reaction successfully occurred.
Interaction between cells and pNP-decorated surfaces
U2-OS cells were seeded on substrates micropatterned with 20 μm wide stripes made of either Coli-pNPs or Lactis-pNPs using hydrophilic HO-terminated or maleimide-terminated SAMs. The interaction of cells with the four types of pNP-decorated surfaces was studied using an immunofluorescence staining technique after 24 h of U2-OS cell seeding (Fig. 3a). The quantification of the averaged FAs, cell elongation, and cell orientation was performed.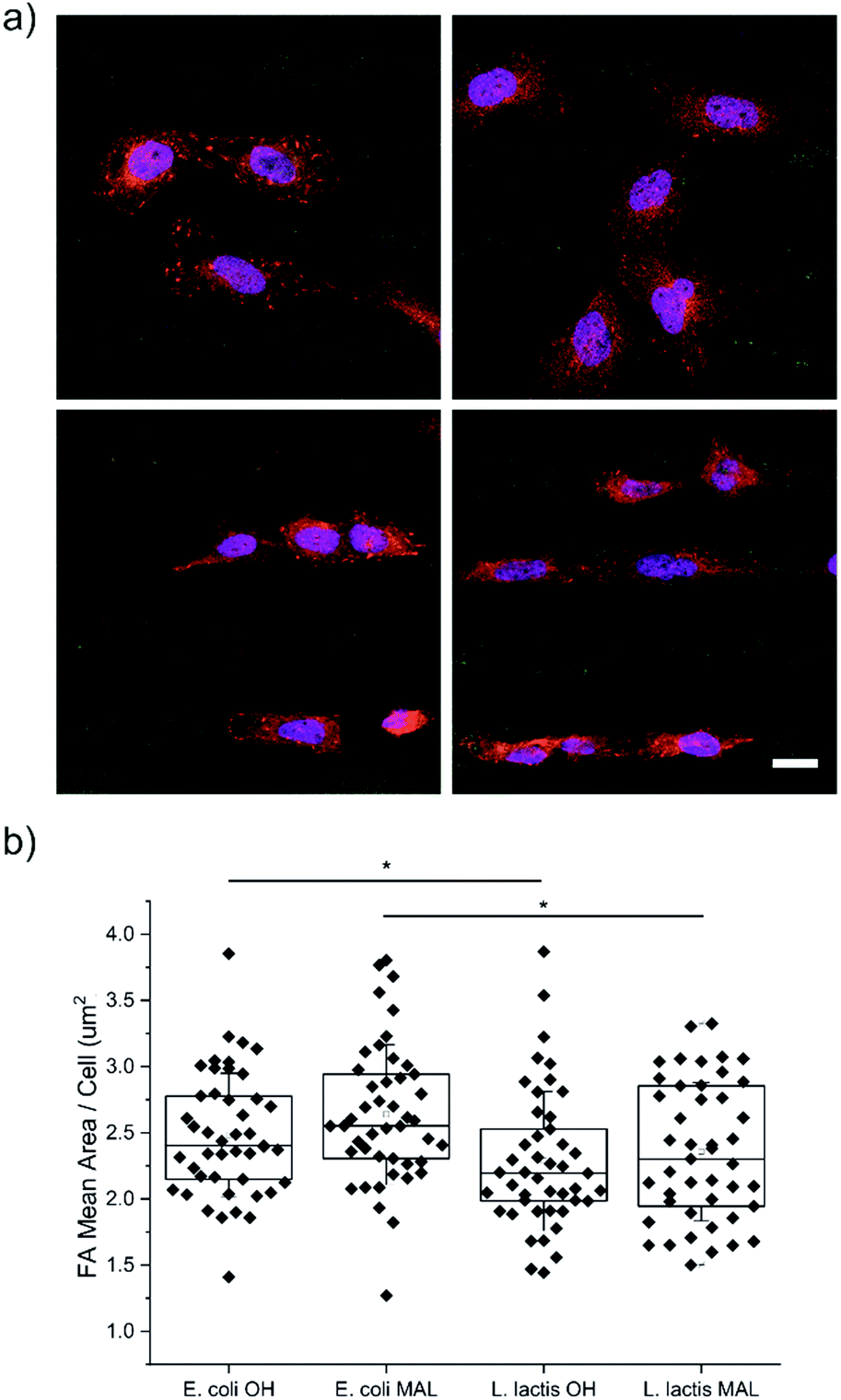 | ||
| Fig. 3 (a) Representative confocal fluorescence microscopy images of immunofluorescence staining against paxillin for FAs (red) and DAPI for the nuclei (blue) of U2-OS cells seeded on the (left) HO- and (right) maleimide-terminated SAMs with (top) E. coli and (bottom) L. lactis GFP pNP patterns (green). Scale bar = 20 μm. (b) FA mean area per cell in E. coli and L. lactis pNP patterned substrates. Nexperiments = 3; and Ncells = 45. Statistical analysis was performed using the Kruskal–Wallis ANOVA test. *p < 0.05. | ||
Cell adhesion quantification
Cell adhesion on surfaces can be studied by imaging the FAs, which are macromolecular complexes that connect cells with their ECM.49–52 Through these anchoring points, cells are able to transmit and receive mechanical forces and regulatory signals (Fig. 3a).53 Here, the FA areas were measured and their mean value per cell was determined (Fig. 3b), since their size is related to adhesion and migration processes.50 U2-OS cells cultured on surfaces with Coli-pNPs showed slightly larger FA mean areas per cell than those cultured with Lactis-pNPs. These results are significant for both HO- and maleimide-terminated SAMs. As discussed before, the role of the host organism in which the pNPs are produced can play an important role in the properties of the resulting material. In this case, Coli-pNP decorated surfaces show higher FA areas in comparison to surfaces decorated with Lactis-pNPs, even though these yielded a higher coverage and retention (Fig. 2). This increase could be justified by a higher roughness of the particulate material,54 which was observed on Coli-pNP decorated surfaces. In addition, larger FAs were observed when the pNPs, either produced in E. coli or L. lactis, were covalently bound to the surface, although no significant differences were obtained. In the following sections, we analyzed the potential relation between such higher FA areas and the morphology of the adhered cells.Cell elongation
Cell morphology was assessed on the four decorated surfaces by measuring their elongation through the AR-1 values, where AR is the aspect ratio (Fig. 4a). U2-OS cells seeded on Coli-pNP decorated surfaces with OH- and maleimide-terminated groups did not show significant differences in elongation. This could be due to the particular physicochemical properties of the Coli-pNPs, which might impair the covalent union between the maleimide-group and the cysteine groups from GFP. On the other hand, cell elongation was significantly higher on the Lactis-pNP decorated maleimide-terminated surface in comparison with the HO-terminated one. This difference is most likely due to the higher adhesion of pNPs to the maleimide-terminated surface, and the subsequent increase in pNP density and robustness of the pattern. Thus, the increased interaction of pNPs with the surface can be relevant for cells when following a pattern, since non-covalently bound pNPs are able to be displaced by cells. The formation of covalent bonds therefore enhances the robustness of the system and allows cells to adhere and elongate through the pattern without disrupting it.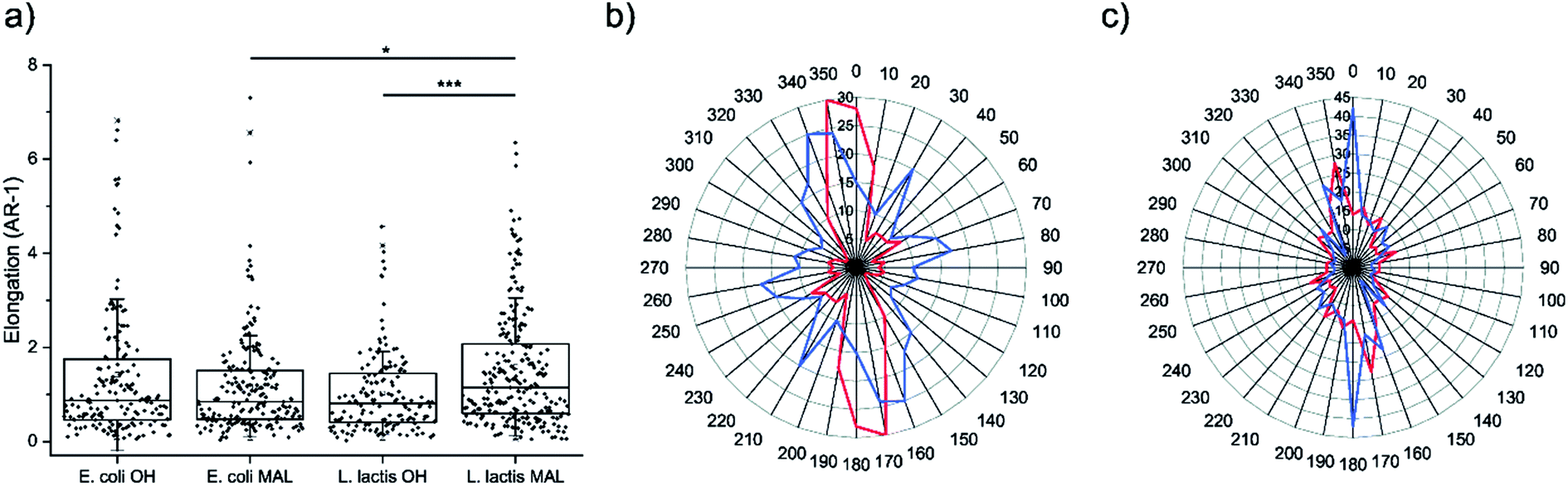 | ||
| Fig. 4 (a) Elongation of cells, measured as aspect ratio-1, cultured on HO- and maleimide-terminated surfaces micropatterned with E. coli and L. lactis produced pNPs. Nexperiments = 3; and Ncells ≥ 140. Statistical analysis was performed using the Kruskal–Wallis ANOVA test. *p < 0.05. ***p < 0.001. (b and c) Radial distribution plots of cell orientation versus the frequency of appearance of cells cultured on surfaces with striped micropatterns decorated with E. coli and L. lactis-produced pNPs, respectively, printed onto HO- (red line) and maleimide-terminated SAMs (blue line). The spindle-shaped plots show the strong orientation of cells along the pattern. Nexperiments = 3; and Ncells ≥ 140. | ||
Cell orientation
The orientation of the U2-OS cells in E. coli and L. lactis produced pNP striped micropatterns was assessed by measuring the angle between the long axis of cells and the axis of the striped pattern (Fig. 4b and c). This data correlates with the elongation data previously reported, and also provides information about how cells are able to follow the pattern when adhering to the surface. When evaluating the alignment of cells on surfaces decorated with Coli-pNPs, we could not appreciate a relevant difference between the surfaces that can establish covalent bonds and the others. However, on surfaces decorated with Lactis-pNPs, an alignment increase with the pattern was observed for the maleimide-terminated SAM in comparison with the HO-terminated one. In the latter, cells can probably displace the pNPs due to their low interaction with the surface, while the covalently bound pNPs form a denser and more robust pattern, providing more stable anchoring points for cells. Thus, we demonstrated the utility of the maleimide–thiol interfacial reaction for anchoring pNPs on surfaces, which is of high importance for longer cell culture experiments and applicability.Conclusions
Well-defined stripped micropatterns were prepared using novel IB-like pNPs by μCP. These pNPs were produced by E. coli and L. lactis and resulted in average sizes of 350 and 520 nm, respectively. Both pNPs were successfully attached to surfaces through covalent bonding, due to the interaction between the accessible thiol groups of exposed cysteine residues of pNPs and the maleimide groups of the surfaces. Patterns that featured covalent bonds were denser and allowed stronger cell adhesion and larger cell spreading than those where pNPs were only physisorbed to the HO-SAM surface, especially for Lactis-pNP patterns. Moreover, LAB produced pNPs present other advantages, such as high stability and activity, a cheap and scalable production process, and a lack of pyrogenic impurities.41 We can, therefore, conclude that pNPs produced in L. lactis represent a step forward in the fabrication of functional micropatterned surfaces with pNPs. In summary, the micropatterns presented here combined submicron topographic and biochemical cues, while providing a robust and endotoxin-free system for the study of cell adhesion and migration. The strong immobilization of functional pNPs could thus be useful for the design of new durable surfaces for the study of cell behavior in vitro.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors are grateful for the financial support received from MOTHER (MAT2016-80826-R) granted by the DGI (Spain), GenCat (SGR-918 and SGR-229) financed by DGR (Catalunya), the Spanish Ministry of Economy and Competitiveness (MINECO) through the “Severo Ochoa” Programme for Centres of Excellence in R&D (SEV-2015-0496), the COST Action CA15126 Between Atom and Cell, Fundació La Marató de TV3 (Nr. 201812), the European Social Fund, and EU to J. V., A. V., and I. R. (H2020-INFRAIA-2014-2015; NFFA-654360). J. G. is also grateful to MINECO for a “Ramón y Cajal” fellowship as well as to the Max Planck Society through the Max Planck Partner Group “Dynamic Biomimetics for Cancer Immunotherapy” in collaboration with the Max Planck Institute for Medical Research (Heidelberg, Germany). This study has been also supported by the Networking Research Center on Bioengineering, Biomaterials and Nanomedicine (CIBER-BBN), an initiative funded by the VI National R&D&I Plan, Iniciativa Ingenio 2010, Consolider Program, CIBER Actions and financed by the Instituto de Salud Carlos III with assistance from the European Regional Development Fund. The authors also acknowledge Micalis Institute, INRA, France that kindly provided us the strain clpP− htrA−-NZ9000 (patent Nr. EP1141337B1/US6994997B1). M. M. M. and O. C. G. received a PhD fellowship from MECD (FPU) and E. G. F. a post-doctoral fellowship from INIA (DOC-INIA, MINECO). A. V. received an ICREA ACADEMIA Award. This work has been developed under the biochemistry and biomedicine program of Universitat Autònoma de Barcelona (UAB). The characterization of the pNPs has been performed by the ICTS “NANBIOSIS”, more specifically by the Biomaterial Processing and Nanostructuring Unit (U6), Unit of the CIBER in Bioengineering, Biomaterials & Nanomedicine (CIBER-BBN) located at the Institute of Materials Science of Barcelona (ICMAB-CSIC).Notes and references
- B. Geiger, J. P. Spatz and A. D. Bershadsky, Nat. Rev. Mol. Cell Biol., 2009, 10, 21 CrossRef CAS PubMed.
- R. O. Hynes, Cell, 2002, 110, 673–687 CrossRef CAS PubMed.
- F. G. Giancotti and E. Ruoslahti, Science, 1999, 285, 1028–1032 CrossRef CAS PubMed.
- J. Seras-Franzoso, W. I. Tatkiewicz, E. Vazquez, E. Garcia-Fruitos, I. Ratera, J. Veciana and A. Villaverde, Nanomedicine, 2015, 10, 873–891 CrossRef CAS PubMed.
- D. Falconnet, G. Csucs, H. M. Grandin and M. Textor, Biomaterials, 2006, 27, 3044–3063 CrossRef CAS PubMed.
- E. Martinez, E. Engel, J. A. Planell and J. Samitier, Ann. Anat., 2009, 191, 126–135 CrossRef CAS PubMed.
- C. J. Bettinger, R. Langer and J. T. Borenstein, Angew. Chem., Int. Ed., 2009, 48, 5406–5415 CrossRef CAS PubMed.
- J. Guasch, B. Conings, S. Neubauer, F. Rechenmacher, K. Ende, C. G. Rolli, C. Kappel, V. Schaufler, A. Micoulet, H. Kessler, H.-G. Boyen, E. A. Cavalcanti-Adam and J. P. Spatz, Adv. Mater., 2015, 27, 3737–3747 CrossRef CAS PubMed.
- E. A. Cavalcanti-Adam, T. Volberg, A. Micoulet, H. Kessler, B. Geiger and J. P. Spatz, Biophys. J., 2007, 92, 2964–2974 CrossRef CAS PubMed.
- J. Guasch, C. A. Muth, J. Diemer, H. Riahinezhad and J. P. Spatz, Nano Lett., 2017, 17, 6110–6116 CrossRef CAS PubMed.
- J. Guasch, J. Diemer, H. Riahinezhad, S. Neubauer, H. Kessler and J. P. Spatz, Chem. Mater., 2016, 28, 1806–1815 CrossRef CAS.
- F. Rechenmacher, S. Neubauer, C. Mas-Moruno, P. M. Dorfner, J. Polleux, J. Guasch, B. Conings, H. G. Boyen, A. Bochen, T. R. Sobahi, R. Burgkart, J. P. Spatz, R. Fassler and H. Kessler, Chemistry, 2013, 19, 9218–9223 CrossRef CAS PubMed.
- C. Dirscherl and S. Springer, Eng. Life Sci., 2018, 18, 124–131 CrossRef CAS.
- A. C. von Philipsborn, S. Lang, A. Bernard, J. Loeschinger, C. David, D. Lehnert, M. Bastmeyer and F. Bonhoeffer, Nat. Protoc., 2006, 1, 1322–1328 CrossRef CAS PubMed.
- S. Zhang, L. Yan, M. Altman, M. Lassle, H. Nugent, F. Frankel, D. A. Lauffenburger, G. M. Whitesides and A. Rich, Biomaterials, 1999, 20, 1213–1220 CrossRef CAS PubMed.
- J.-T. Zhang, J. Nie, M. Mühlstädt, H. Gallagher, O. Pullig and K. D. Jandt, Adv. Funct. Mater., 2011, 21, 4079–4087 CrossRef CAS.
- S. A. Lange, V. Benes, D. P. Kern, J. K. Horber and A. Bernard, Anal. Chem., 2004, 76, 1641–1647 CrossRef CAS PubMed.
- S. Y. Yoo, W.-J. Chung, T. H. Kim, M. Le and S.-W. Lee, Soft Matter, 2011, 7, 363–368 RSC.
- A. R. Kyvik, C. Luque-Corredera, D. Pulido, M. Royo, J. Veciana, J. Guasch and I. Ratera, J. Phys. Chem. B, 2018, 122, 4481–4490 CrossRef CAS PubMed.
- S. Raghavan, R. A. Desai, Y. Kwon, M. Mrksich and C. S. Chen, Langmuir, 2010, 26, 17733–17738 CrossRef CAS PubMed.
- M. Bautista, A. Fernandez and F. Pinaud, Micromachines, 2019, 10, 810 CrossRef PubMed.
- P. Lanzerstorfer, U. Müller, K. Gordiyenko, J. Weghuber and C. M. Niemeyer, Biomolecules, 2020, 10, 540 CrossRef PubMed.
- Z. Xu, J. A. Orkwis, B. M. DeVine and G. M. Harris, J. Tissue Eng. Regener. Med., 2020, 14, 229–242 CrossRef CAS PubMed.
- E. Rodriguez-Carmona and A. Villaverde, Trends Microbiol., 2010, 18, 423–430 CrossRef CAS PubMed.
- E. Garcia-Fruitós, E. Vazquez, C. Diez-Gil, J. L. Corchero, J. Seras-Franzoso, I. Ratera, J. Veciana and A. Villaverde, Trends Biotechnol., 2012, 30, 65–70 CrossRef PubMed.
- E. Garcia-Fruitós, J. Seras-Franzoso, E. Vazquez and A. Villaverde, Nanotechnology, 2010, 21, 205101 CrossRef PubMed.
- W. I. Tatkiewicz, J. Seras-Franzoso, E. García-Fruitós, E. Vazquez, N. Ventosa, K. Peebo, I. Ratera, A. Villaverde and J. Veciana, ACS Nano, 2013, 7, 4774–4784 CrossRef CAS PubMed.
- L. Iversen, N. Cherouati, T. Berthing, D. Stamou and K. L. Martinez, Langmuir, 2008, 24, 6375–6381 CrossRef CAS PubMed.
- T. Cornish, D. W. Branch, B. C. Wheeler and J. T. Campanelli, Mol. Cell. Neurosci., 2002, 20, 140–153 CrossRef CAS PubMed.
- M. Scholl, C. Sprossler, M. Denyer, M. Krause, K. Nakajima, A. Maelicke, W. Knoll and A. Offenhausser, J. Neurosci. Methods, 2000, 104, 65–75 CrossRef CAS PubMed.
- B. T. Houseman, E. S. Gawalt and M. Mrksich, Langmuir, 2003, 19, 1522–1531 CrossRef CAS.
- F. Rusmini, Z. Zhong and J. Feijen, Biomacromolecules, 2007, 8, 1775–1789 CrossRef CAS PubMed.
- S. Mourtas, M. Kastellorizios, P. Klepetsanis, E. Farsari, E. Amanatides, D. Mataras, B. R. Pistillo, P. Favia, E. Sardella, R. d'Agostino and S. G. Antimisiaris, Colloids Surf., B, 2011, 84, 214–220 CrossRef CAS PubMed.
- G. Liu, Y. Zhang, M. Qi and F. Chen, Anal. Methods, 2015, 7, 5619–5626 RSC.
- M. Okan and M. Duman, Sens. Actuators, B, 2018, 256, 325–333 CrossRef CAS.
- S. J. Wakelin, I. Sabroe, C. D. Gregory, I. R. Poxton, J. L. Forsythe, O. J. Garden and S. E. Howie, Immunol. Lett., 2006, 106, 1–7 CrossRef CAS PubMed.
- F. Rueda, O. Cano-Garrido, U. Mamat, K. Wilke, J. Seras-Franzoso, E. Garcia-Fruitos and A. Villaverde, Appl. Microbiol. Biotechnol., 2014, 98, 9229–9238 CrossRef CAS PubMed.
- D. Petsch and F. B. Anspach, J. Biotechnol., 2000, 76, 97–119 CrossRef CAS PubMed.
- E. Garcia-Fruitós, Microb. Cell Fact., 2012, 11, 157 CrossRef PubMed.
- W. N. Konings, J. Kok, O. P. Kuipers and B. Poolman, Curr. Opin. Microbiol., 2000, 3, 276–282 CrossRef CAS PubMed.
- O. Cano-Garrido, A. Sanchez-Chardi, S. Pares, I. Giro, W. I. Tatkiewicz, N. Ferrer-Miralles, I. Ratera, A. Natalello, R. Cubarsi, J. Veciana, A. Bach, A. Villaverde, A. Aris and E. Garcia-Fruitos, Acta Biomater., 2016, 43, 230–239 CrossRef CAS PubMed.
- L. Gifre-Renom, O. Cano-Garrido, F. Fàbregas, R. Roca-Pinilla, J. Seras-Franzoso, N. Ferrer-Miralles, A. Villaverde, À. Bach, M. Devant and A. Arís, Sci. Rep., 2018, 8, 1–10 CrossRef CAS PubMed.
- L. Gifre-Renom, J. Seras-Franzoso, D. Rafael, F. Andrade, O. Cano-Garrido, F. Martinez-Trucharte, E. Ugarte-Berzal, E. Martens, L. Boon and A. Villaverde, Pharmaceutics, 2020, 12, 157 CrossRef PubMed.
- N. G. Cortes-Perez, I. Poquet, M. Oliveira, J. J. Gratadoux, S. M. Madsen, A. Miyoshi, G. Corthier, V. Azevedo, P. Langella and L. G. Bermudez-Humaran, Microbiology, 2006, 152, 2611–2618 CrossRef CAS PubMed.
- C. K. Riener, G. Kada and H. J. Gruber, Anal. Bioanal. Chem., 2002, 373, 266–276 CrossRef CAS PubMed.
- D. Chatzikleanthous, S. T. Schmidt, G. Buffi, I. Paciello, R. Cunliffe, F. Carboni, M. R. Romano, D. T. O’Hagan, U. D’Oro and S. Woods, J. Controlled Release, 2020, 323, 125–137 CrossRef CAS PubMed.
- C. P. Haycook, J. A. Balsamo, E. B. Glass, C. H. Williams, C. C. Hong, A. S. Major and T. D. Giorgio, Int. J. Nanomed., 2020, 15, 1215 CrossRef PubMed.
- J. Royes, O. Ilioaia, Q. Lubart, F. Angius, G. V. Dubacheva, M. Bally, B. Miroux and C. Tribet, Angew. Chem., Int. Ed., 2019, 58, 7395–7399 CrossRef CAS PubMed.
- H. Lodish, A. Berk and S. Lawrence Zipursky, Molecular Cell Biology, W. H. Freeman, New York, 4th edn, 2000 Search PubMed.
- D. H. Kim and D. Wirtz, FASEB J., 2013, 27, 1351–1361 CrossRef CAS PubMed.
- H. Truong and E. H. Danen, Cell Adhes. Migr., 2009, 3, 179–181 CrossRef PubMed.
- D. C. Worth, K. Hodivala-Dilke, S. D. Robinson, S. J. King, P. E. Morton, F. B. Gertler, M. J. Humphries and M. Parsons, J. Cell Biol., 2010, 189, 369–383 CrossRef CAS PubMed.
- K. Burridge, Annu. Rev. Cell Dev. Biol., 1988, 4, 487–525 CrossRef CAS PubMed.
- S. Oh, K. S. Brammer, Y. S. Li, D. Teng, A. J. Engler, S. Chien and S. Jin, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 2130–2135 CrossRef CAS PubMed.
- C. Diez-Gil, S. Krabbenborg, E. Garcia-Fruitos, E. Vazquez, E. Rodriguez-Carmona, I. Ratera, N. Ventosa, J. Seras-Franzoso, O. Cano-Garrido, N. Ferrer-Miralles, A. Villaverde and J. Veciana, Biomaterials, 2010, 31, 5805–5812 CrossRef CAS PubMed.
- J. Seras-Franzoso, C. Diez-Gil, E. Vazquez, E. Garcia-Fruitos, R. Cubarsi, I. Ratera, J. Veciana and A. Villaverde, Nanomedicine, 2012, 7, 79–93 CrossRef CAS PubMed.
- W. I. Tatkiewicz, J. Seras-Franzoso, E. Garcia-Fruitos, E. Vazquez, A. R. Kyvik, J. Guasch, A. Villaverde, J. Veciana and I. Ratera, ACS Appl. Mater. Interfaces, 2018, 10, 25779–25786 CrossRef CAS PubMed.
- W. Tatkiewicz, J. Seras-Franzoso, E. García-Fruitós, E. Vazquez, A. R. Kyvik, N. Ventosa, J. Guasch, A. Villaverde, J. Veciana and I. Ratera, ACS Biomater. Sci. Eng., 2019, 5(10), 5470–5480, DOI:10.1021/acsbiomaterials.9b01085.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0tb00702a |
| ‡ Present address: Department of Ruminant Production, Institut de Recerca i Tecnologia Agroalimentàries (IRTA), 08140 Caldes de Montbui, Spain. |
| This journal is © The Royal Society of Chemistry 2020 |