
Graphene oxide-coated porous titanium for pulp sealing: an antibacterial and dentino-inductive restorative material†
Abstract
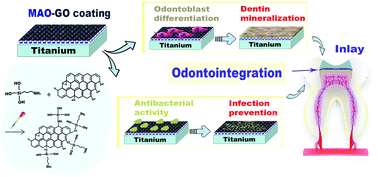
Pulp treatment techniques such as pulp capping, pulpotomy and pulp regeneration are all based on the principle of preserving vital pulp. However, specific dental restorative materials that can simultaneously protect pulp vitality and repair occlusal morphology have not been developed thus far. Traditional pulp capping materials cannot be used as dental restorative materials due to their long-term solubility and poor mechanical behavior. Titanium (Ti) is used extensively in dentistry and is regarded as a promising material for pulp sealing because of its favorable biocompatibility, processability and mechanical properties. Originally, we proposed the concept of “odontointegration”, which represents direct dentin-like mineralization contact between pulp and the surface of the pulp sealing material; herein, we report the fabrication of a novel antibacterial and dentino-inductive material via micro-arc oxidation (MAO), incorporating self-assembled graphene oxide (GO) for Ti surface modification. The hierarchical micro/nanoporous structure of the MAO coating provides a suitable microenvironment for odontogenic differentiation of human dental pulp stem cells, and GO loading contributes to antibacterial activity. Scanning electron microscopy (SEM), energy-dispersive X-ray spectroscopy and Raman spectroscopy were employed for structure and elemental analysis. In vitro studies, including cell adhesion, Live/Dead and CCK-8 assays, alkaline phosphatase activity and calcium deposition assay, real-time polymerase chain reaction, western blot analysis and immunofluorescence staining were used to examine cell adhesion, viability, proliferation, mineralization, and odontogenic differentiation ability. Antibacterial properties against Streptococcus mutans were analyzed by SEM, spread plate, Live/Dead and Alamar blue tests. The Ti–MAO–1.0 mg mL−1 GO group exhibited excellent cell adhesion, odontoblast differentiation, mineralization, and antibacterial ability, which are beneficial to odontointegration.
 Please wait while we load your content...
Please wait while we load your content...

