
Overcoming the biological barriers in the tumor microenvironment for improving drug delivery and efficacy
Yang
Zhou
a,
Xianchun
Chen
b,
Jun
Cao
 *cde and
Huile
Gao
*a
*cde and
Huile
Gao
*a
aKey Laboratory of Drug-Targeting and Drug Delivery System of the Education Ministry, Sichuan Engineering Laboratory for Plant-Sourced Drug and Sichuan Research Center for Drug Precision Industrial Technology, West China School of Pharmacy, Sichuan University, Chengdu, 610064, China. E-mail: gaohuile@scu.edu.cn; gaohuilescu@163.com
bCollege of Material Science and Engineering, Sichuan University, Chengdu, 610064, China
cNational Engineering Research Center for Biomaterials, Sichuan University, Chengdu, 610064, China. E-mail: caojun@scu.edu.cn
dDepartment of Biomedical Engineering, The University of Texas at Austin, Austin, TX, USA. E-mail: caojun@utexas.edu
eDivision of Molecular Pharmaceutics and Drug Delivery, College of Pharmacy, The University of Texas at Austin, Austin, TX, USA
First published on 1st April 2020
Abstract
The delivery of drugs to tumors by nanoparticles is a rapidly growing field. However, the complex tumor microenvironment (TME) barriers greatly hinder drug delivery to tumors. In this study, we first summarized the barriers in TME, including anomalous vasculature, rigid extracellular matrix, hypoxia, acidic pH, irregular enzyme level, altered metabolism pathway and immunosuppressive conditions. To overcome these barriers, many strategies have been developed, such as modulating TME, active targeting by ligand modification and biomimetic strategies, and TME-responsive drug delivery strategies to improve nanoparticle penetration, cellular uptake and drug release. Although extensive progress has been achieved, there are still many challenges, which are discussed in the last section. Overall, we carefully discuss the landscape of TME, development for improving drug delivery, and challenges that need to be further addressed.
 Yang Zhou | Yang Zhou received her BS Degree in pharmacy from West China School of Pharmacy in Sichuan University in 2018. Currently, she is pursuing her PhD degree under the supervision of Professor Huile Gao. Her current research mainly focuses on the development and biomedical applications of functional nanomaterials for cancer therapy. |
 Xianchun Chen | Xianchun Chen is an Associated Professor at Sichuan University. She obtained her PhD in Materials Science from Sichuan University in 2010 and then joined Prof. Guangfu Yin's group in the College of Material Science and Engineering, Sichuan University as a Lecturer. From 2014 to 2017, she was engaged in the management of scientific research projects in the Ministry of Science and Technology of China. Her research interests focus on the material design of nanocarriers for drug delivery. |
 Jun Cao | Jun Cao is an Associate Professor and a member of the National Engineering Research Center for Biomaterials and the Engineering Research Center in Biomaterials at Sichuan University. She received her PhD degree from the College of Polymer Science and Engineering at Sichuan University in 2013. Her current research interests focus on the design and synthesis of various smart polymers as drug delivery carriers for tumor diagnosis and therapy. |
 Huile Gao | Huile Gao is a Professor at Sichuan University. He obtained his PhD in Pharmaceutics from Fudan University in 2013 and then joined West China School of Pharmacy, Sichuan University as a lecturer. In 2017, Gao was promoted as a Professor. His research focuses on the rational design of drug delivery systems for central nervous system diseases and tumors. |
1. Introduction
Tumors are the second leading cause of death worldwide, threatening the lives of tens of millions of people.1,2 Although substantial drugs have been developed, their delivery is far from satisfactory. Regardless of oral administration or intravenous injection, drug delivery to tumors primarily depends on the chemical properties of the drugs, such as molecular weight, hydrophobicity and chemical units, which is plagued by enzymolysis and rapid clearance from circulation. To circumvent these problems, nanoparticles (herein referring to all particles in the nanometer scale, NPs), including organic (e.g., lipid-based, polymer-based and cell-derived NPs) and inorganic NPs (e.g., gold, silver, iron oxide and silica), have been developed to deliver drugs for cancer therapy, also termed “nanomedicine”.3–5 The basis for the design of current NPs is the enhanced permeability and retention (EPR) effect, where the hyperpermeable tumor vasculature allows the extravasation and passive diffusion of large particles coupled with impaired lymphatic drainage, reducing the elimination of NPs; thus, NPs are further retained and accumulate within tumors.6,7 Besides, NPs can enhance drug stability and extend the circulation half-life.Unfortunately, the clinical translation of nanomedicine still faces undesirable anti-tumor performances8 despite the achievement of some progress in lowering off-target side effects.9 As reported in a previous review, the median delivery efficiency of NPs into tumors is estimated to be merely 0.7% of an injected dose, completely diverging from the optimistic anticipation of nanomedicine.10 Accordingly, the golden standard in drug delivery, i.e., the EPR effect is somewhat oversimplified or exaggerated. In fact, for NPs, there is a long and bumpy journey to reach tumor cells. Besides abnormal vascular networks, other components in the tumor microenvironment (TME) play an essential role in deciding the in vivo fate of NPs. TME refers to all the physiological and biochemical compositions around tumor cells, which comprise various types of growth-supporting cells, fortified by a dense extracellular matrix (ECM) and irregular vascular networks.11 These cells consist of endothelial cells, pericytes, cancer-associated fibroblasts (CAFs) and immune-infiltrating cells, while ECM consists of collagens, fibronectin, integrins, elastin, and microfibrillar proteins. These compositions interweave with each other and provide a niche for tumor development, progression and metastasis.12 For NP-mediated anti-tumor therapy, some of the characteristics of TME, such as a high interstitial fluid pressure (IFP) and solid pressure, can impede drug delivery within the deep regions of tumors, while others, such as hypoxia and acidity, attenuate the antitumor effect and even cause drug resistance and metastasis.13 For example, photodynamic therapy (PDT) and radiotherapy, which are dependent on oxygen, are greatly restricted by hypoxia in tumor tissues. Thus, it is important to elucidate the biological barriers in TME and modulate these TME targets, which can help NPs overcome the barriers to enhance their delivery efficiency and therapeutic effectiveness.
In this perspective, we summarize the barriers in the TME, presenting the landscape of the TME. Subsequently, we review the strategies to overcome the barriers in the TME, including modulating the TME, active tumor targeting, and TEM-sensitive drug delivery, as shown in Fig. 1. Finally, the challenges to overcome the TME barriers are discussed.
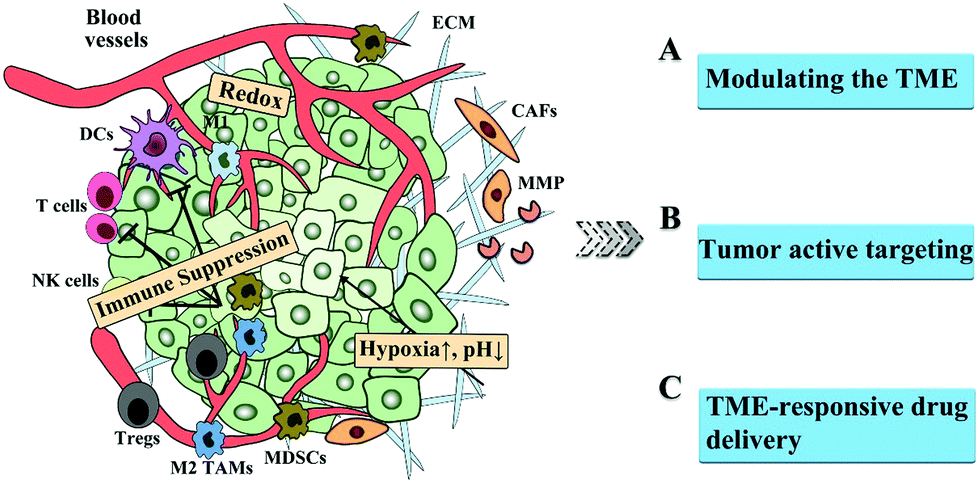 | ||
| Fig. 1 Schematic illustration of the barriers in the TME and the corresponding strategies for improved drug delivery. | ||
2. Barriers in the TME
Increasing evidence has indicated drug delivery using nanocarriers is, to a large extent, hindered by the tumor heterogeneity, including anomalous vasculature, rigid ECM, hypoxia, acidic pH, overproduced glutathione (GSH) and reactive oxygen species (ROS), and immune suppression, which are derived from cancer cells, and the crosstalk between them and the TME.14,15 Thus, the brief introduction of the dynamic tumor landscape will offer researchers better access to the developing direction of drug delivery in the future.2.1 TME composition
Strictly, a favourable vascular network is an essential prerequisite for the effective delivery of nanomedicine. However, this is not the case with tumors. The overgrowth of neoplastic cells demands dramatically more nutrients relative to normal cells. Consequently, they and the surrounding stromal cells secret angiogenic growth factors, such as vascular endothelial growth factor (VEGF), basic fibroblast growth factor and tumor necrosis factor (TNF), leading to substantial vessel growth.16 Notably, these newly-formed vessels are tortuous and leaky since aggressive angiogenesis is accompanied by a lack of paracellular junctions between endothelial cells and incomplete pericyte coverage.14,17,18 As discussed earlier, the “leaky” gaps and blockage of lymphatic return provide evidence for the EPR effect.19 However, this feature of the tumor vasculature is a double-edged sword. Excessive plasma and fluid can extravasate from leaky vessels to the interstitium, resulting in elevated IFP and viscosity,15 while poor lymphatic drainage aggravates the IFP, and thus causes blood reversal and further retards blood flow. Accordingly, abnormally high IFP becomes a daunting bottleneck limiting the penetration and accumulation of NPs. Similarly, it also provides the possibility of tumor metastasis or drug resistance. Additionally, the tortuous nature of vessels can result in geometric obstruction against blood flow. Thus, the Janus-faced nature of aberrant vascular networks should be considered carefully for drug delivery mediated by passive targeting.The tumor stroma is another roadblock, which comprises ECM, CAFs, mesenchymal stromal cells, osteoblasts, and chondrocytes.20 Unlike quiescent fibroblasts in normal tissues, CAFs are activated to enhance the proliferation, migration, and secretion of multiple cytokines, such as VEGF, TNF, interleukin-6 (IL-6), platelet-derived growth factor (PDGF) and transforming growth factor-beta (TGF-β), which support tumor growth and render tumor resistance towards traditional chemotherapeutics.21,22 More importantly, CAFs generate large amounts of ECM components, such as collagen and proteoglycans, which together offer a breeding ground for tumors. They also release vast enzymes, such as matrix metalloproteases (MMPs), disintegrins and plasmin, to degrade and remodel the ECM. Besides the intrinsic mesh-like physical resistance, the stiff stroma can elevate solid stress, increase IFP and isolate tumors from blood vessels, thus compromising the interstitial transport of nanotherapeutics.23 Also, the positively charged collagen and negatively charged hyaluronic acid (HA) can trap charged nanomedicines with high affinity.24,25
2.2 Alteration in metabolic and immune profiles
Basically, the interior of tumors, particularly their core, presents a hypoxic state, while the surroundings near the vessels are normoxic, which can be explained by the rapid proliferation of tumor cells and deficient oxygen supply from abnormal neovessels.26 Also, blood vessels exhibit an uneven distribution within tumors, which results in the local hypoxia of tumor regions.27 Hypoxia can induce the increased expression of hypoxia-induced factors (HIFs) and stabilize them, subsequently initiating the downstream signalling cascade, such as promoting epithelial–mesenchymal transition, metabolic reprogramming, angiogenesis and resistance to therapy.28 Metabolic adaption is a key step to satisfy the high energy demand and hypoxia. The change to anaerobic glycolysis alleviates the insufficient oxygen supply and increases lactate production, together with H+ ions. CO2 hydration in oxidative tumor areas via carbonic anhydrases (CAs) contributes to another part of the production of H+ ions. These excessive intracellular H+ ions can be exported via Na+/H+ exchangers (NHEs), monocarboxylate transporters (MCTs) and H+-ATPases, thus resulting in the acidosis of the TME, tumor proliferation and metastasis.29,30 Also, tumors increase the production of GSH to cope with the elevated ROS and maintain redox homeostasis. Thus, the above hallmarks can be exploited to develop responsive or TME-modulating therapeutic agents.In addition, it should be emphasized that another significant feature of the TME is the overall immunosuppressive microenvironment. Immune-infiltrating cells include immunomodulatory cells, such as TAMs, myeloid-derived suppressor cells (MDSCs), regulatory T cells (Tregs) and dendritic cells (DCs), as well as effector immune cells, such as T lymphocytes, B lymphocytes, and natural killer (NK) cells.31–33 These cells play a dominant and complex role in immune-escape mechanisms. Tumor cells themselves significantly reduce the expression of MHC and upregulate other immunosuppressive molecules, such as programmed death ligand-1 (PD-L1), immune-checkpoint molecules (CD47), and NK-cell ligands (factor associated suicide ligand (FASL)), and recruit anti-inflammatory immune cells.
TAMs represent the main component of myeloid cells, involving pro-inflammatory M1-like phenotype and anti-inflammatory M2-like phenotype, the latter of which is triggered particularly by the hypoxic TME. Within tumors, the predominant M2-like phenotype can facilitate angiogenesis, metastasis and decrease the immune response of T cells.34 Thus, depleting or switching TAMs can be a potent strategy to reverse immune suppression. Tregs and MDSCs are other partners implicated in the negative modulation of the immune system.35 Tregs produce TGF-β and IL-10 to directly suppress T cell function and release perforin and granzyme to destroy effector T cells, while MDSCs created a large amount of nitric oxide (NO), arginine (Arg)-1 and IL-10, which further decrease the T cell response and promote immunosuppression.36 The production of indoleamine 2,3 dioxygenase (IDO) can decrease the amino acids (e.g. tryptophan) and recruit Tregs to activate MDSCs, with the aim to suppress T cells. Besides, the immunosuppressive milieu, containing IL-10, TGF-β, PD-1 and prostaglandin E (PGE)-2, can reduce the antigen cross-presenting ability of DCs. Also, tumor cells downregulate NK cells to avoid the exposure of TNF-related apoptosis-inducing ligand (TRAIL).37 The major anti-tumor immune cells in the TME, CD8+ T cells, are unable to exert an inhibitory response against tumors due to the decreased trafficking into tumor sites (including down-regulation of CD62L on CD8+ T cells, and the adhesion molecules (ICAM-1 and VCAM-1) and IL-12) and a lack of activation elicited by above immunosuppressive TME.38 Thus, targeting immune cells or immune-related specific molecules can reshape the immune microenvironment and boost the efficacy of traditional treatments.
Thus, targeting angiogenesis, the ECM (e.g. degrading enzyme and CAFs) and endogenous stimulus-response (e.g. hypoxia, pH, GSH and ROS) can enhance the delivery of nanocarriers into tumors and keep the concentrations of drugs within effective therapeutic windows. Besides, modulating the hypoxic, acidic or immunosuppressive microenvironment is another strategy to overcome the dilemma in cancer therapy, and reduce tumor invasion and metastasis (Fig. 1).
3. Strategies to overcome the TME barriers
3.1 Strategies for modulating the TME
As mentioned before, the TME dominates the fate of nanocarriers within tumors, from impeding their delivery to resisting conventional therapies.11 In this section, particular attention is paid to feasible strategies to modulate the TME via the following aspects: remodeling the vascular networks, regulating stroma, manipulating hypoxia and pH, and reshaping the immune microenvironment, as summarized in Table 1.| Classification | Examples | Ref. |
|---|---|---|
| Remodelling the vascular networks | Vascular disruption: DMXAA, CA4P, radiation, ultrasound, and near infrared (NIR) light | 40–49 |
| Vascular normalization: Cediranib, Sunitinib, Bevacizumab, DC101, and NO | 50–53 | |
| Regulating stroma | Targeting the ECM: | 56–71 |
| ① inhibiting ECM biogenesis: prolyl-4-hydroxylase inhibitor, deferoxamine, and 4-methy-lumbelliferone | ||
| ② disrupting the ECM: PTT, ultrasound, collagenase, hyaluronidase, cyclopamine, relaxin and NO | ||
| ③ mimicking the ECM: laminin-mimicking peptide | ||
| Targeting CAFs: | 72–74 and 77–80 | |
| □ directly disrupting CAFs: FAP-targeting NPs, docetaxel-conjugate nanoparticles or delivering plasmids encoding TNF-related factor | ||
| □ reducing the activity of CAFs: losartan, quercetin, ellagic acid and dexamethasone | ||
| Manipulating hypoxia and pH | Modulating hypoxia: | 81–92 |
| □ improving blood flow: PTT | ||
| □ delivering oxygen: hemoglobin and perfluorocarbon | ||
| □ in situ oxygen production: MnO2, CaO2, catalase, and light-driven water splitting | ||
| □ decreasing oxygen consumption: atovaquone, metformin, tamoxifen, and NO | ||
| Modulating pH: | 30, 93 and 94 | |
| □ neutralizing acidity with buffers: sodium bicarbonate, imidazoles and lysine | ||
| □ interfering with pH-regulating enzyme: proton-pump inhibitors (e.g. omeprazole and esomeprazole), MCT inhibitors (e.g. AZD3965), and CAIX inhibitors (e.g. sulfonamide- and coumarin-related substances) | ||
| Reshaping immune microenvironment | Targeting immune cells: | 53 and 95–110 |
| □ TAMs: CSF-1, bisphosphonates (e.g. clodronate and zoledronic acid), TLR agonists, chloroquine, celecoxib, NO, and CO | ||
| □ Tregs: CTLA-4, Sunitinib | ||
| □ MDSCs: CDDO-Me, 6-thioguanine, and gemcitabine | ||
| Targeting immune molecules: | 111–114 | |
| □ IDO: NLG919 and indoximod | ||
| □ CXCL12: AMD 3100 or monoclonal antibody |
Vascular disruption was presented as an approach to destroy neovessels, leading to a dramatic decrease in blood supply and the necrosis of tumors. Several vascular disrupting agents such as 5,6-dimethylxanthenone-4-acetic acid (DMXAA) and combretastatin A4 phosphate (CA4P) have verified the practicability of this strategy. After administration, CA4, which is released from CA4P, inhibits tubulin polymerization and destabilizes the cytoskeleton, thus disrupting endothelial cells.40,41 Chen et al. first employed CA4-conjugated poly(L-glutamic acid)-graft-methoxy poly(ethylene glycol) nanodrugs (CA4-NPs) to disrupt vessels, subsequently increasing MMP-9 in tumor tissues. Then, they delivered an MMP9-activated doxorubicin prodrug (Fmoc-GPLGL-DOX), which enhanced the selective release by 3.7-fold.42 The sequential delivery of CA4-NPs and MMP9-DOX-NPs was a new paradigm for the next generation of tumor-selective enzyme-triggered prodrugs with reduced systemic toxicity. Other research combined this drug with photothermal therapy (PTT), hypoxia-sensitive drug (e.g. imiquimod and tirapazamine) and other agents.43–46 Besides, physical approaches such as radiation, ultrasound, and near infrared (NIR) light treatment can also trigger vascular disruption.47,48 Tang et al. applied αvβ3-targeting peptide c(RGDfE)-modified hollow copper sulfide NPs to load vinyl azide, which targeted tumor neovasculature and released nitrogen (N2) bubbles under NIR light-induced heating. These bubbles exploded and damaged the neovessels, inducing the ischemic necrosis of tumors, as indicated by photoacoustic imaging.49
Vascular normalization has emerged as an alternative to address the biological challenges in delivering NPs. Anti-angiogenic drugs, such as Cediranib, Sunitinib, Bevacizumab and DC101, have been shown to decrease vascular leakiness and improve vascular functionality.50–52 Recently, Chen and coworkers encapsulated [Fe(μ-SEt)2(NO)4] (NO donor, DNIC) into lipid-poly(lactic-co-glycolic acid) (PLGA) polymer NPs (NanoNO). After treatment with free DNIC, HUVECs obviously downregulated three pro-angiogenic genes (VEGFA, ANGPT2 and EGF) and increased the expression of two vessel maturation-related genes (S1PR1 and ANGPT1). In contrast with high-dosed NanoNO (1 mg kg−1), low-dosed NanoNO (0.1 mg kg−1) normalized tumor vessels by increasing pericyte coverage (increased pericyte marker NG2/vessel marker CD31) and functional vessel perfusion (lectin+/CD31+ area increased), thus enhancing the delivery and efficacy of other therapeutics, such as doxorubicin and recombinant TRAIL.53 More importantly, tumor vessel normalization is closely associated with immunostimulatory pathways, such as lymphocyte infiltration or activity, which provide new route to influence tumor progression.54 The above low-dosed NanoNO also further evidenced this theory since PD-L1 expression was hampered, anti-tumor M1-like TAMs increased and CD8+ T cell infiltration was enhanced in vivo. This study, for the first time, clearly indicated the difference between low- and high-dosed NO for vessel normalization and the versatility of NO therapy.
However, despite the progress in vascular remodelling, these two strategies have some inevitable drawbacks. For vascular disruption, the abnormal vasculature and impaired lymphatic drainage exacerbate the IFP and hypoxia, further reducing NP penetration and recruiting pro-inflammatory and pro-angiogenetic cells. Relatively, vascular normalization increases blood perfusion and mitigates the IFP to promote NP diffusion, but the reduced gaps between endothelial cells may block large-sized NPs.55
The ECM can be regulated via the following three routes, inhibiting ECM biogenesis, disrupting the ECM and mimicking the ECM. The synthesis of collagen can be interrupted by prolyl-4-hydroxylase inhibitor,56,57 deferoxamine and siRNA silencing heat shock protein 47,58 while 4-methylumbelliferone can block the synthesis of HA, another major component.59,60 Physical methods (e.g. PTT and ultrasound61,62), enzymes (e.g. collagenase and hyaluronidase (HAase)63–65) and chemical agents (e.g. cyclopamine, relaxin and NO66–69) can be applied to facilitate the degradation of the ECM and NP permeation into tumors. Cao et al. used NO to trigger MMP expression and disintegrate collagen, which rendered a 2.7-fold greater distribution of mesoporous silica NPs within tumors.67 However, it should be noted that the above two strategies may increase the chances that tumor cells will migrate and spread throughout the body. By contrast, the final means is based on the fact that cancer cells express ECM degrading enzyme to promote invasion and metastasis. Thus, strengthening the ECM, rather than disrupting it may address tumor metastasis.70 Considering this, Wang and coworkers constructed a laminin (essential component in ECM)-mimicking peptide (BP-KLVFFK-GGDGR-YIGSR) (Fig. 2A), where a Y-type RGD-YIGSR motif binding with tumor cells, KLVFF segments, formed the fibrous structure around tumors.71 This ECM-simulating strategy effectively hampered 82.3% and 50.0% of lung metastasis in breast and melanoma tumor models, respectively, as seen in Fig. 2B–D. This case provided new insight for the inhibition of tumor metastasis.
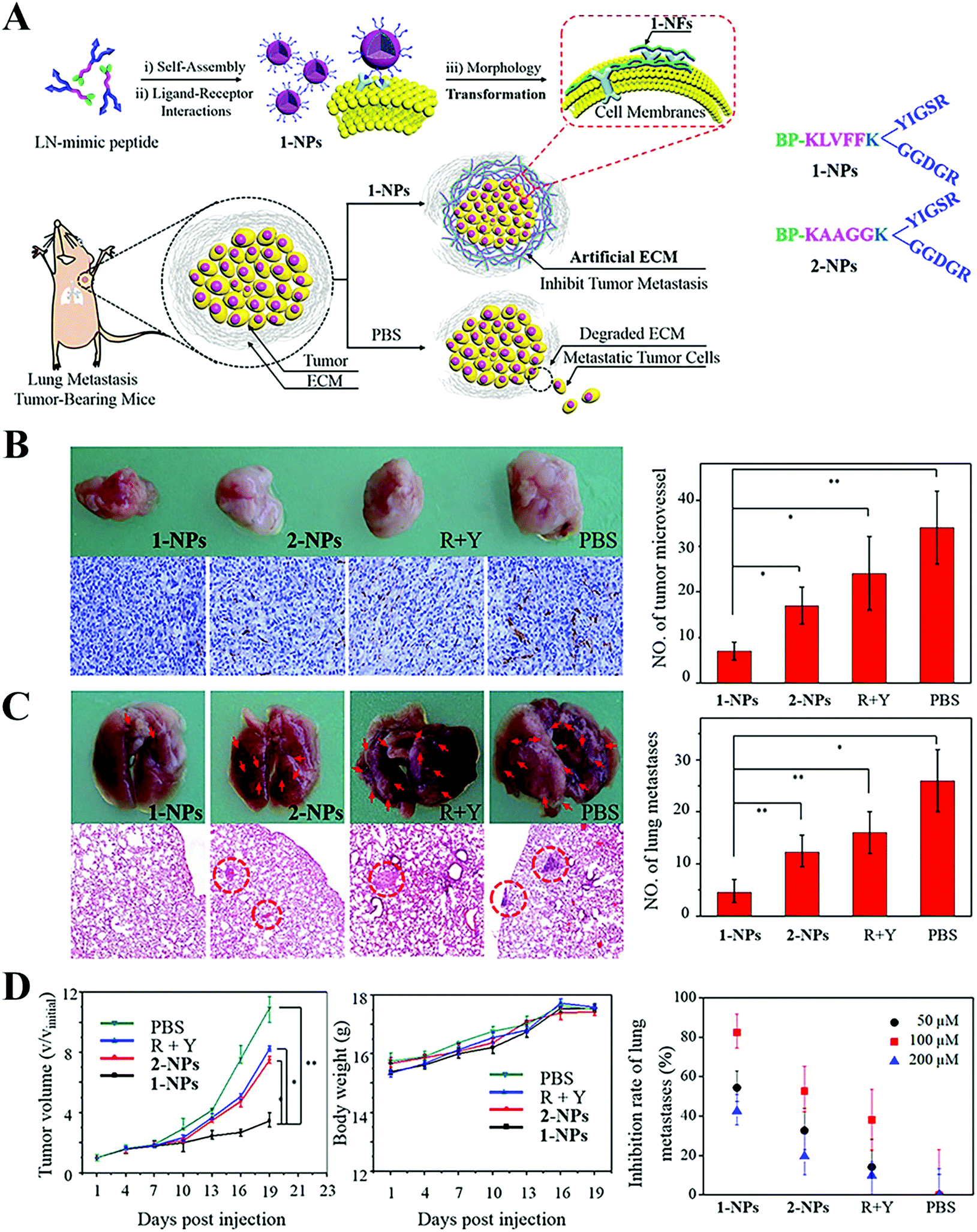 | ||
| Fig. 2 Transformable nanomaterials as an artificial extracellular matrix for inhibiting tumor invasion and metastasis. (A) Schematic Illustration. (B) Photos of original tumors (upper panel) and immuno-histochemical staining analyses for CD31 (lower panel) on day 19 after the first administration and the quantitative analysis of MVD (right panel). (C) Photos of the lungs (upper panel) and H&E stained lung slices (lower panel) and quantitative analysis of the lungs (left panel). (D) Tumor volume and body weight of the 1-NPs, 2-NPs, RGD + YIGSR and PBS treated mice and inhibition rate of lung metastases treated with different concentrations of 1-NPs. Data are presented as mean ± SD (n = 4). *p < 0.05 and **p < 0.01. Reproduced with permission from ref. 71. Copyright © 2017, American Chemical Society. | ||
The subsequent emphasis shifted to ECM origin-CAFs. CAFs are key mediators in the ECM, and contribute to limited drug delivery. Thus, directly disrupting CAFs may improve delivery, and fibroblast activation protein-targeting NPs, docetaxel-conjugate NPs and delivering plasmids encoding TNF-related factor into CAFs were all utilized to verify this hypothesis.72–74 It is worth noting that direct CAF depletion may promote tumor progression and metastasis, mainly because CAFs possess an anti-tumor phenotype to retard growth.75,76 Thus, perhaps reducing CAFs activity is a better alternative. There are several anti-inflammatory or anti-fibrotic drugs, such as losartan, quercetin, ellagic acid and dexamethasone.77–80 If CAFs accidentally internalize chemotherapeutics, this will induce them to secret damage response specific proteins, e.g., Wnt16, which become involved in drug resistance (e.g. cisplatin). For example, Huang et al. prepared lipid/calcium/phosphate NP-encapsulating quercetin, which greatly downregulated Wnt16 expression, blocked the growth of α-SMA+ CAFs and reduced collagen.78
Hypoxia is usually accompanied by an acidic microenvironment. As discussed earlier, the acidity also contributes to tumor progression and metastasis. The strategies modulating acidosis include neutralizing acidity with buffers and interfering with pH-regulating enzyme.30 Sodium bicarbonate, imidazoles and lysine were introduced to act as a buffer against acidity and reduce tumor aggressiveness. Proton-pump inhibitors (e.g. omeprazole and esomeprazole) can inhibit H+/K+-ATPase and enhance extracellular pH. The use of MCT inhibitors (e.g. AZD396593), which tackle lactate synthesis-derived H+ ions, is another attractive approach to manipulate pH. CAIX expression correlates with the poor prognosis of hypoxic tumors, and thus specific chemical inhibitors (e.g. sulfonamide- and coumarin-related substances) have been employed. Chen et al. presented the self-assembly of a CA inhibitor (benzenesulfonamide)-modified short peptide (N-pepABS), which formed fibrous structures around hypoxic tumor cells triggered by pH, concentrated CA inhibitors on the cell membrane and caused CA dysfunction.94 N-pepABS regulated the extracellular pH and inhibited cell migration, largely because N-pepABS not only suppresses CAIX enzymes, but its fibrous structure also restricts normal movements for cancer cells. They were also internalised into cancer cells via CAIX enzymes and further formed longer fibres in the lysosome, eventually blocking protective autophagy. This study exemplified the effect of regulating CAIX enzymes on acidic pH and hypoxia, while modulating them can reduce the resistance to traditional therapy.
Negative regulatory cells include TAMs, Tregs and MDSCs, which inhibit the anti-tumor immune response of T cells and NK cells. M2-like TAMs secret some immunosuppressive cytokines, such as IL-10, PGE-2 and TGF-β, and small amounts of proinflammatory cytokines (IL-6, TNF-α), resulting in evasion-mediated immune suppression.32 Colony-stimulating factor 1 receptor (CSF-1R) or bisphosphonates (e.g. clodronate and zoledronic acid) can directly inhibit the survival and proliferation of macrophages. However, the direct depletion of TAMs can kill M1-like macrophages, and thus researchers have modified several ligands to enhance the M2-targeting ability, such as M2pep and anti-CD204 immunotoxin.95 Reprogramming TAMs into an M1-like phenotype represents a relatively ideal method based on the excellent plasticity of macrophages. Metal NPs, such as gold, silver and iron oxide, can increase ROS within TAMs, thus activating M1-like macrophages.96,97 Several therapeutics also mediated this switching, such as TLR agonists,98 chemicals (chloroquine and celecoxib99,100), and gases (NO and CO53,101). For example, Yao et al. fabricated two look-like nanodrugs (GLnano), which were both established on antiangiogenic low molecular weight heparin (LMWH), and mixed together to administer to mice for melanoma treatment.102 One of the nanodrugs was doxorubicin (DOX)-loaded LMWH-chrysin (LCY) with DSPE-PEG-anisamide decoration (D-LCA nanodrugs) to actively kill tumor cells. The other was celecoxib-loaded LMWH-PLGLAG-containing peptide conjugation (C-Lpep), which could release celecoxib for M2-to-M1-like macrophage reeducation. This GLnano effectively inhibited tumor growth, and on day 10, the tumor size was only 1/2, 1/4, and 1/8 of that in the D-LCA, free DOX, and saline groups, respectively. For Tregs, the strategies can also be divided into three types, including depletion103,104 (using PDT or chemotherapy), function inhibition with anti-cytotoxic T-lymphocyte-associated protein 4 (CTLA-4) molecules and modulating Tregs by the signal transducer and activator of the transcription (STAT)3/5 pathway.105 Some studies revealed STAT3 of immune cells facilitate secretion of Th2 cytokines, thus suppressing the Th1 response and promoting the survival of Tregs.106 Sunitinib, a tyrosine kinase inhibitor, acts on this pathway and lowers the proportion of Tregs and MDSCs.107 In the case of MDSCs, derivatives of oleanolic acid (CDDO-Me),108 6-thioguanine109 and gemcitabine110 all were used to inhibit their function and development.
Besides targeting immune cells, soluble mediators such as IDO, MMP, TGF-β and CXCL12 play a vital role in promoting the immunosuppressive microenvironment. Thus, delivery of these mediator-related inhibitors is viewed as a viable approach to regulate the immune system. IDO can catalyze the degradation of tryptophan to kynurenine, and a low concentration of tryptophan impairs the survival and activity of cytotoxic T cells. IDO inhibitors such as NLG919 and indoximod with the use of nanocarriers exert an effective immune response against tumors, in combination with other treatments.111–113 Notably, IDO inhibitors alone cannot induce an intense immunostimulatory response. Wang et al. fabricated redox-responsible phospholipid–porphyrin conjugates to self-assemble into liposomes (IND@RAL), in which NLG-8189 was loaded in their lumen (Fig. 3A).111 After tumor cell internalization, IND@RAL responded to the high GSH levels in the tumor cells, then recovered the fluorescence signal and PDT activity (>100-fold). Strikingly, GSH-activated PDT induced immunogenic cell death at the primary tumor sites and recruited CD8+ T cells into the tumors. After combination with IDO inhibitor, the systemic antitumor immune response was further enhanced, as evidenced by the inhibition of metastatic sites (Fig. 3B). Tumor cells and mesenchymal cells secret chemokines (e.g. CXCL12) to activate and recruit immunosuppressive cells, such as Tregs, into the TME. Small-molecule inhibitors (AMD 3100) or monoclonal antibody can block this effect of CXCL12.114
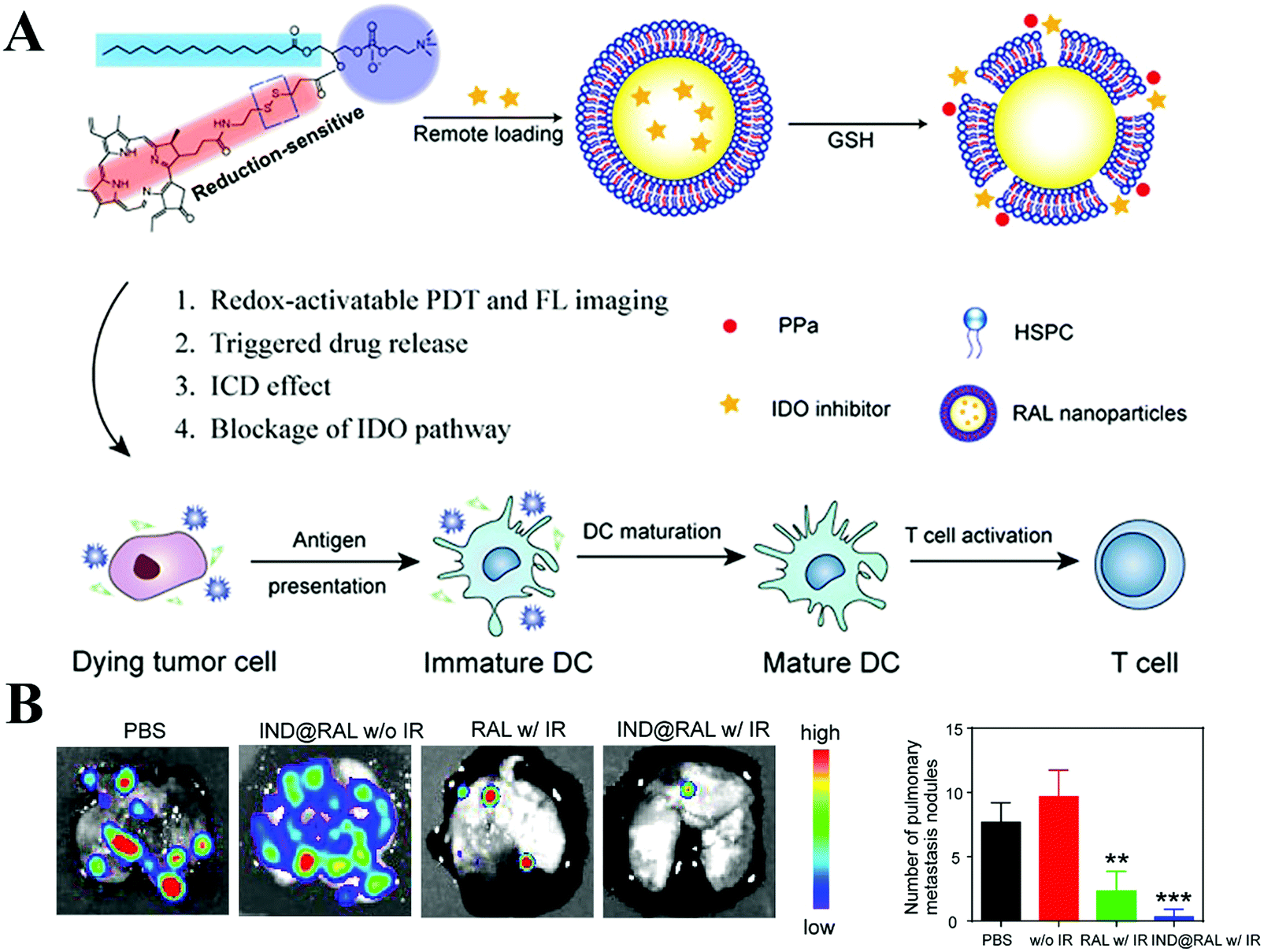 | ||
| Fig. 3 Porphyrin-based liposome loaded with IDO inhibitor for synergistic photoimmunotherapy. (A) Schematic Illustration. (B) Antimetastasis of each group imaged by BLI analysis (left panel) and the number of lung metastatic nodules of mice bearing 4T1-Luc tumors at the end of the antitumor study (right panel). Reproduced with permission from ref. 111. Copyright © 2019, American Chemical Society. | ||
3.2 Strategies for tumor active targeting
Besides TEM modulation, physical NPs feature, such as size, surface chemistry, shape and existence of targeting ligands, can also influence the tumor distribution and cellular uptake of NPs. In this section, we focus on how to enhance targeting capacity towards tumors or non-cancer cells in the TME for improved delivery and efficacy, including the modifying targeting ligand and biomimetic strategy, as depicted in Fig. 5.Initially, researchers attempted to use some natural ligands that could recognize proteins overexpressed on tumor cells or stromal cells. For example, transferrin is the most widely used ligand, which can specifically interact with transferrin receptor (TfR) overexpressed on tumor cells and endothelial cells.116 Also, oxaliplatin-loaded transferrin-modified liposomes are under clinical evaluation. Although high tumor selectivity and therapeutic activity were observed in the pre-clinical study, thrombocytopenia was found as the main toxicity in the Phase I study.117 Additionally, the most serious limitations of proteins as targeting ligands are their poor stability, large molecular weight, high immunogenicity, and high cost,118 which restrict the application and clinical translation of protein-decorated NPs.
To solve the above-mentioned problems, peptides with several or tens of amino acids in length were designed for tumor targeting.119 T7 peptide (HAIYPRH) is one such ligand that can specifically bind with TfR.120 However, the traditional L-form peptide still suffers from quick degradation by enzymes in the blood, while the retro-inverso D-form peptide was still integral after 8 h incubation.121 Additionally, D-form T7 peptide (D-T7) showed stronger binding affinity with TfR compared with L-formed T7.122 Thus, we conjugated D-T7 onto PEGylated bilirubin nanoparticles (BRNPs) for brain tumor targeting delivery.123 The results showed the brain tumor accumulation of D-T7-modified BRNPs (TBRNPs) was over 4-fold higher than that of BRNPs (Fig. 4A–C). According to the fluorescence distribution in the brain slices (Fig. 4D and E), the TBRNPs showed higher intensity in the brain tumor, and well co-localized with TfR, indicating the TfR-mediated active targeting of TBRNPs. Cediranib-loaded TBRNPs could better target neovessels and inhibit the tyrosine kinase activity of c-KIT and VEGFR1, 2 and 3 for anti-angiogenesis. As a result, cediranib and paclitaxel-loaded TBRNPs greatly prolonged the medium survival time of brain tumor-bearing mice from 16 days to 53 days, which was significantly longer than that for the unmodified BRNPs. Also, there are several other targeting ligands for tumors or other cells in the TME, such as RGD (targeting vascular receptor Integrin αvβ349), fibroblast activation protein antibody (targeting CAF),124 vitamin D ligand (targeting CAFs in pancreas),125 and mannose (targeting DCs126 or cancer cells127).
 | ||
| Fig. 4 Fluorescence distribution in vivo. (A) Fluorescence signal distribution of different nanoparticles with the extension of time in vivo. Bar represents radiant efficiency between 1935 and 6154. (B) Fluorescent imaging of organs ex vivo. Bar represents radiant efficiency between 8.26 × 107 and 3.11 × 109. (C) Semi-quantification of the fluorescence signals. (D and E) Fluorescence distribution of DiD@BRNPs and DiD@TBRNPs in glioma sections after intravenous injection at 24 h and 48 h, where green indicates DiD, blue indicates nucleus and red indicates TfR and CD34, respectively. N represents the normal brain, G represents the glioma, and bars represent 50 μm. Reproduced with permission from ref. 123. Copyright © 2019, American Chemical Society. | ||
Directly loading NPs or drugs into whole cells can also achieve active tumor targeting. Xue et al. used neutrophils to deliver paclitaxel-loaded liposomes (PTX-CL/NEs) for glioma recurrence treatment.134 After surgical removal of a glioma, local brain inflammation occurs accompanied with the release of inflammatory factors, such as TNFα, which can activate neutrophils to migrate to the inflammatory area.135 Compared with paclitaxel-loaded liposomes (PTX-CL), the blood brain barrier penetration ratio of PTX-CL/NEs increased from 1% to 38%, while the penetration into glioma spheroids by PTX-CL/NEs also increased significantly. Similarly, macrophages also show potential as carriers of drugs and NPs for tumor targeting delivery.136 Guo et al. loaded doxorubicin into M1 macrophages (M1-Dox) for the treatment of metastatic ovarian carcinoma.137 When M1-Dox interacted with the tumor cells, it formed tunnelling nanotubes to transport doxorubicin from the macrophages to tumor cells, providing an effective drug transportation pathway and leading to an effective antitumor effect.
Similarly, exosomes and vesicles can serve as drug carriers due to their long blood circulation time, immune escape and potential tumor targeting.138 Wang et al. loaded doxorubicin into macrophage-derived exosomes and then modified them with gold nanorods.139 After entering the tumor, the photothermal effect of the gold nanorods caused disruption of the exosomes and they released doxorubicin, boosting enhanced chemo-photothermal combinational therapy. Furthermore, some exosomes may directly kill tumor cells. A recent study showed engineered T cells expressing chimeric antigen receptor (CAR) could release exosomes, which also carried CAR on their surface.140 As a result, the cytotoxic molecule-expressed CAR-T cell exosomes could directly inhibit tumor growth. Additionally, the CAR exosomes did not express PD1, making them safer than CAR-T therapy.
Besides mimicking biological components in the body, the immune system can also be hitchhiked for tumor targeting delivery. Li et al. developed a type of cisplatin loaded nano-pathogenoid,141 which could in situ hitchhike circulating neutrophils. When the tumor was applied with PTT, PTT-induced inflammation recruited neutrophils together with the nano-pathogenoid. The inflammation factors in the tumor caused the release of the nano-pathogenoid from the neutrophils, which were then taken up by tumor cells. Finally, the cisplatin in the nano-pathogenoid led to apoptosis of the tumor cells. Thus, this hitchhiking strategy can effectively target tumors with a simple NP composition and preparation, which is promising for clinical translation.
3.3 Strategies for TME-responsive drug delivery
Compared to normal tissue, the unique characteristics of the TME, including low pH, hypoxia, and high levels of redox potential and enzymes, have guided the construction of stimuli-responsive NPs.142 Generally, the pre-designed functions of these NPs can be activated in response to the TME signals, as shown in Fig. 6, such as detachment of the PEG corona, surface charge conversion, ligand exposure, and on demand drug release, for enhanced deep tumor penetration, augmented drug release, and increased tumor cell uptake.143–146 Based on these strategies, the efficacy of TME-triggered cancer treatment has greatly improved.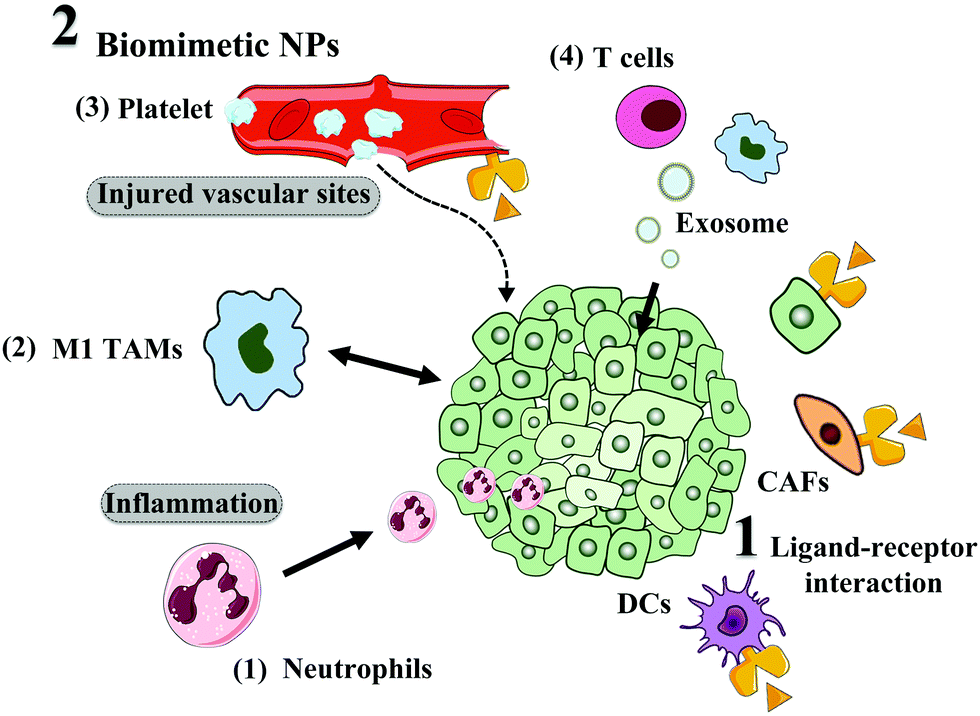 | ||
| Fig. 5 TME and tumor cell active targeting, including ligand modification and biomimetic strategies. | ||
 | ||
| Fig. 6 Size, charge, and ligand availability modifications to sequentially take advantage of favorable physicochemical properties during systemic circulation and tumor penetration. | ||
The first strategy has been established on the principle that TME cues will induce the detachment or degradation of the outer shell layer on NPs. In this regard, many studies have been devoted to design NPs with matrix metalloproteinase (MMP)-sensitive peptides or benzoic imine-conjugated PEG corona,155,156 which can respond to either a higher expression level of MMP or lower pH at the tumor site to form smaller NPs. These size-shrinkage characteristics result in improved penetration into the tumor site due to the easier diffusion throughout the tumor interstitial space. In addition, tethering small particles on the surface of other NPs or hiding them inside large particles through TME-sensitive linkers can also prompt the penetration of small NPs when the platform is exposed to the TME.157–160 Cui et al. fabricated P-DOX by conjugating doxorubicin (DOX) with poly(4-formylphenyl methacrylate-co-2-(diethylamino)ethyl methacrylate)-b-poly(oligo(ethylene glycol) methacrylate) (P(FPMA-co-DEA)-b-POEGMA) via imine linkage.161 The constructed pH sensitive NPs could dissociate into ultrasmall particles (∼10 nm), which are inherently favorable for deep tumor penetration.
Objectively, the deep penetration efficacy of any of the abovementioned methods will not be perfect due to the complexity of the TME. Hence, the combination of multiple strategies, including the modulation of NP properties (size shrinkage/surface charge conversion) and remodeling the TME, has become a promising approach to improve the penetration capacity.162,163 In addition, TME-triggered self-assembled NP can also be used to improve solid-tumor penetration. For instance, Wang et al.164 employed an in vivo self-assembly strategy and designed polymer–peptide conjugates (PPCs), which underwent an increase in acid-induced hydrophobicity under a narrow pH-response range. The results showed that at appropriate molecular concentrations (around the IC50 values of PPCs), in situ self-assembly in the TME enabled the drug to be delivered deeper into the tumor.
In addition, TME signals can target the sensitive bonds on NPs to recover hidden ligands, and thus achieve enhanced tumor internalization. Generally, targeting ligands should be shielded during the blood circulation to circumvent their non-specific interaction with surrounding healthy tissue. One effective approach to achieve this purpose utilizes enzyme- and pH-sensitive PEG coatings. The overexpressed enzymes in the TME can cleave bonds (e.g. MMP-2 and MMP-9) to deshield PEG and expose the targeting ligands to promote efficient cellular uptake. In their pioneering research article, Ji et al. constructed enzyme-sensitive GEM nanovectors through the conjugation of matrix metalloproteinase-9 (MMP-9) detachable PEG (Fig. 7A).169 When the nanovectors accumulate at the tumor site via the EPR effect, the PEG corona can be detached through overexpressed MMP-9 to expose RGD for facilitating cellular internalization, as seen in Fig. 7B.
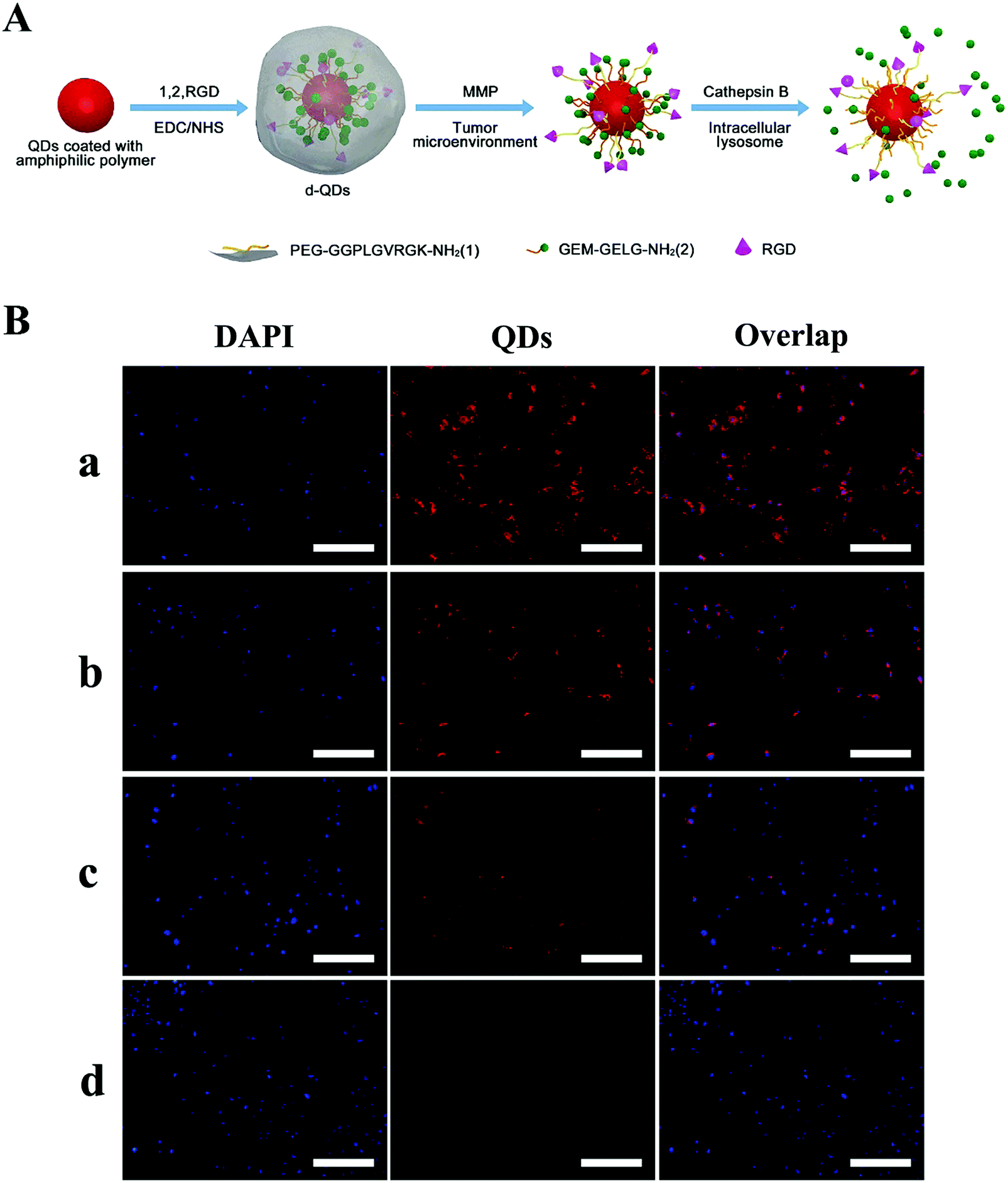 | ||
| Fig. 7 Dual enzymatic reaction-assisted GEM nanovectors achieving multistage tumor cell targeting and efficient drug release. (A) Schematic illustration of the preparation of the nanovectors and their enzyme-sensitive behavior. (B) Fluorescence microscopy images of BxPC-3 cells incubated with d-QDs in the (a) absence and (b) presence of the MMP inhibitor, (c) cb-QDs (cathepsin B-sensitive QDs without MMP-9 responsiveness, with an uncleavable PEG shell), and (d) d-QDs (nr) (dual-enzyme-sensitive QDs without RGD targeting ability) with a concentration of 200 nM QDs for 4 h. The scale bar is 200 μm. Reproduced with permission from ref. 169. Copyright © 2017, American Chemical Society. | ||
Besides, cleaving various TME-sensitive linkers to degrade NPs is another strategy for the efficient release of loaded therapeutics. Recently, hypoxia-mediated drug release has aroused attention, in which hypoxia-responsive molecules such as nitroimidazole (NI) are incorporated into NPs. When exposed to the tumor hypoxic environment, hydrophobic NI segments are converted into hydrophilic 2-aminoimidazole, thereby facilitating the release of the drug encapsulated in NPs.171 Furthermore, azobenzene (AZO), as a hypoxia-sensitive linker, can also be used to conjugate carboxymethyl dextran (CMD) with black hole quencher 3 (BHQ3) to load DOX. When these particles enter the hypoxic tumor site, the release rate of DOX remarkably increases through the cleavage of the azo bond in BHQ3.172 In addition, redox-sensitive linkages, including disulfide (S–S) and diselenide (Se–Se) linkers, are integrated into the backbone or side chains to attain redox-sensitive drug release.173
Notably, the acidic pH in the TME can induce the collapse of some inorganic NPs, such as manganese dioxide (MnO2), calcium carbonate (CaCO3) and calcium phosphates (Ca3(PO4)2) by facilitating the release of loaded cargo.174 Additionally, the utilization of TME signals to trigger the generation of gas (e.g. CO2 and H2S) demonstrates great potential for accelerated drug release.175
4. Challenges of overcoming TME barriers
Based on the distinguished TME, many types of TME-modulating strategies and NPs have been developed to overcome the biological barriers and improve drug delivery efficiency. However, although impressive progress has been made, there are still many issues remaining to be elucidated and improved.4.1 Cross talk of the complex TME
The complex properties of the TME construct a network in which the alternation in a specific character will influence many other characters. As described above, angiogenesis is an important character of tumors, resulting in leaky vasculature and EPR effect, where the latter is the most widely accepted rationale for designing nanoscale drug delivery systems.176 However, the leaky vasculature also leads to extravasation of blood components, such as proteins, resulting in high IFP, and finally inhibits the passive diffusion of drugs and NPs. Therefore, many researchers have developed tumor vessel normalization strategies by anti-angiogenesis to reduce IFP and improve the penetration of NPs.11 Although these studies have reported enhanced drug delivery and antitumor effect, anti-angiogenesis greatly reduces blood perfusion of tumors, which aggravates the deficiency of oxygen in tumors, and unfortunately, hypoxia is considered as a contributor to tumor resistance towards chemotherapy.Similarly, inhibition of the tumor matrix synthesis can reduce the density of the tumor matrix, which can improve tumor penetration. For example, oral administration of losartan can apparently reduce the synthesis of collagen via recognition of the angiotensin II type I receptor, which can downregulate tumor transforming growth factor-β1.177 Thus, the combination of oral losartan with NPs can improve the tumor targeting delivery of NPs.178 However, the loose tumor matrix also facilitates the extravasation of tumor cells, which will elevate metastasis potential. Unfortunately, most studies do not pay much attention to this concern.
4.2 Active targeting and cell uptake
Ligand modification is the most widely used strategy to overcome tumor biological barriers, improve tumor active targeting and facilitate cell uptake.115 Although the targeting capacity of ligands has been widely accepted, and some ligand-modified nanomedicines are under clinical evaluation, the efficacy of ligand modification is challenged by the formation of a protein corona.179 Upon interaction with biological fluid, proteins in the fluid can adsorb on NPs and form a protein layer, called the protein corona. Several studies indicate that the formation of a protein corona may diminish or even eliminate the specific interaction of a ligand with its receptors, while some studies indicate the specific interaction is still reserved in in vivo conditions.180,181 However, regardless if the targeting is diminished or eliminated, the protein corona indeed influences the active targeting and cellular uptake of NPs. Therefore, it is useful to develop strategies that can overcome the influence of the protein corona.Surface de-shielding may be an applicable strategy to overcome the influence of the protein corona. A PEG layer on the surface of NPs can reduce the adsorption of protein and improve the blood circulation time, and thus PEGylation is widely used in the construction of drug delivery systems.182 However, the PEG layer also inhibits the interaction between NPs and cells. Therefore, PEG should be conjugated onto NPs through a cleavable linker.183 When the NPs arrive at the tumor, specific stimuli, such as acidic condition and overexpressed enzyme, will trigger the detachment of PEG from the NPs and the protein corona will be detached correspondingly, resulting in the exposure of the inner ligand on the NPs and specific targeting to tumor cells.184
4.3 Specificity and efficiency of stimulus response
Responsive drug delivery systems are designed in response to the stimuli in tumors to improve drug delivery, such as acidic condition, GSH, ROS and overexpressed enzymes. Thus, it is important to ensure the response occurs in the right location, the tumor. Although the conditions in the tumor are different compared with that of other tissues, they indeed exist in normal tissues. For example, the concentration of GSH in tumor cells, especially in the cytoplasm, may be as high as 1–10 mM, which can be used for designing GSH-sensitive drug delivery systems.185 However, GSH is distributed throughout the body. The concentration of GSH in the blood is about 2–20 μM, which is the first place that NPs enter after intravenous injection. Due to the long time that NPs circulate in the blood, it is important to prohibit premature drug release in response to GSH. Thus, to circumvent this problem, dual-responsive drug delivery systems have been designed, where the effective response requires the existence of two stimuli.186Besides the responsive specificity, responsive efficiency is another important issue. Although NPs display an enhanced retention effect compared with free drugs, their retention time in tumors is still limited, with a half-life of several hours. Therefore, NPs should respond to stimuli as quickly and efficiently as possible before they are cleared from the tumor. Although the degrading reaction of specific substrates by enzymes is very rapid, the full response of NPs is time-consuming. For example, gelatin NPs incubated with MMP-2 were reduced only to half of their initial size in about 8 h.187 Unfortunately, more than half of the NPs will be pumped back into the blood within 8 h. This time-consuming procedure may explain the limited benefit of enzyme-sensitive drug delivery system.
5. Conclusion
The knowledge about the barriers in the TME for drug delivery have been greatly extended. In the present review, we summarized the knowledge about the barriers, and then discussed the strategies to overcome these barriers and improve drug delivery. Although promising development has been made in this area, there are still some issues that need to be given great attention. In addition to the challenges discussed in last section, the properties of NPs are also very important. There are numerous studies that show the surface moieties, charge, size, shape, softness and even the inner core of NPs can influence their behaviour. This means there is no general rule that all NPs obey. Thus, case-by-case evaluation should be performed before obtaining a good drug delivery system. The safety of drug delivery systems is another critical concern. Although the strategies in this review showed the potential of improving drug delivery to tumors, most NPs, at least over 90%, will go to normal tissues. Thus, biocompatible and biodegradable materials should be used in the construction of drug delivery systems. Recently, some carrier-free NPs have been developed with tumor targeting capacity. Due to the termination of the use of toxic materials, carrier-free NPs hold great promise in clinical application.In short, great improvement has been made in TME-targeting drug delivery. A better understanding about the TME will further improve the drug delivery efficiency and antitumor outcome.
Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
The work was supported by National Natural Science Foundation of China (81961138009), Research Funds of Sichuan Science and Technology Department (19YYJC2250), 111 Project (B18035) and the Fundamental of Research Funds for the Central Universities.References
- F. Bray, J. Ferlay, I. Soerjomataram, R. L. Siegel, L. A. Torre and A. Jemal, Ca-Cancer J. Clin., 2018, 68, 394–424 CrossRef PubMed.
- World Health Organization. Cancer, https://www.who.int/health-topics/cancer.
- M. Björnmalm, K. J. Thurecht, M. Michael, A. M. Scott and F. Caruso, ACS Nano, 2017, 11, 9594–9613 CrossRef PubMed.
- D. Peer, J. M. Karp, S. Hong, O. C. Farokhzad, R. Margalit and R. Langer, Nat. Nanotechnol., 2007, 2, 751–760 CrossRef CAS PubMed.
- R. van der Meel, E. Sulheim, Y. Shi, F. Kiessling, W. J. M. Mulder and T. Lammers, Nat. Nanotechnol., 2019, 14, 1007–1017 CrossRef CAS PubMed.
- J. W. Nichols and Y. H. Bae, J. Controlled Release, 2014, 190, 451–464 CrossRef CAS PubMed.
- Y. S. Youn and Y. H. Bae, Adv. Drug Delivery Rev., 2018, 130, 3–11 CrossRef CAS PubMed.
- J. Shi, P. W. Kantoff, R. Wooster and O. C. Farokhzad, Nat. Rev. Cancer, 2017, 17, 20–37 CrossRef CAS PubMed.
- U. Bulbake, S. Doppalapudi, N. Kommineni and W. Khan, Pharmaceutics, 2017, 9(2), 12 CrossRef PubMed.
- S. Wilhelm, A. J. Tavares, Q. Dai, S. Ohta, J. Audet, H. F. Dvorak and W. C. W. Chan, Nat. Rev. Mater., 2016, 1, 16014 CrossRef CAS.
- S. Yang and H. Gao, Pharmacol. Res., 2017, 126, 97–108 CrossRef CAS PubMed.
- S. C. Gupta, J. H. Kim, S. Prasad and B. B. Aggarwal, Cancer Metastasis Rev., 2010, 29, 405–434 CrossRef CAS PubMed.
- H. Gao, Curr. Drug Metab., 2016, 17, 731–736 CrossRef CAS PubMed.
- M. Overchuk and G. Zheng, Biomaterials, 2018, 156, 217–237 CrossRef CAS PubMed.
- M. R. Junttila and F. J. de Sauvage, Nature, 2013, 501, 346–354 CrossRef CAS PubMed.
- R. K. Jain, Nat. Med., 2003, 9, 685–693 CrossRef CAS PubMed.
- S. Azzi, J. K. Hebda and J. Gavard, Front. Oncol., 2013, 3, 211 Search PubMed.
- K. D. Barlow, A. M. Sanders, S. Soker, S. Ergun and L. J. Metheny-Barlow, Cancer Microenviron., 2013, 6, 1–17 CrossRef PubMed.
- J. Fang, H. Nakamura and H. Maeda, Adv. Drug Delivery Rev., 2011, 63, 136–151 CrossRef CAS PubMed.
- K. C. Valkenburg, A. E. de Groot and K. J. Pienta, Nat. Rev. Clin. Oncol., 2018, 15, 366–381 CrossRef PubMed.
- E. G. Neilson, N. A. Bhowmick and H. L. Moses, Nature, 2004, 432, 332–337 CrossRef PubMed.
- R. Kalluri, Nat. Rev. Cancer, 2016, 16, 582–598 CrossRef CAS PubMed.
- X. Han, Y. Xu, M. Geranpayehvaghei, G. J. Anderson, Y. Li and G. Nie, Biomaterials, 2020, 232, 119745 CrossRef PubMed.
- O. Lieleg, R. M. Baumgartel and A. R. Bausch, Biophys. J., 2009, 97, 1569–1577 CrossRef CAS PubMed.
- T. Stylianopoulos, M. Poh, N. Insin, M. G. Bawendi, D. Fukumura, L. L. Munn and R. K. Jain, Biophys. J., 2010, 99, 1342–1349 CrossRef CAS PubMed.
- G. Qiu, M. Jin, J. Dai, W. Sun, J. Feng and W. Jin, Trends Pharmacol. Sci., 2017, 38, 669–686 CrossRef CAS PubMed.
- C. Roma-Rodrigues, R. Mendes, P. Baptista and A. Fernandes, Int. J. Mol. Sci., 2019, 20, 840 CrossRef CAS PubMed.
- L. Schito and G. L. Semenza, Trends Cancer, 2016, 2, 758 CrossRef PubMed.
- G. Jiménez-Valerio and O. Casanovas, Trends Cancer, 2017, 3, 10–18 CrossRef PubMed.
- C. Corbet and O. Feron, Nat. Rev. Cancer, 2017, 17, 577–593 CrossRef CAS PubMed.
- S. Musetti and L. Huang, ACS Nano, 2018, 12, 11740–11755 CrossRef CAS PubMed.
- H. Phuengkham, L. Ren, I. W. Shin and Y. T. Lim, Adv. Mater., 2019, 31, e1803322 CrossRef PubMed.
- J. Nam, S. Son, K. S. Park, W. Zou, L. D. Shea and J. J. Moon, Nat. Rev. Mater., 2019, 4, 398–414 CrossRef.
- B. Zhang, Y. Hu and Z. Pang, Front. Pharmacol., 2017, 8, 952 CrossRef PubMed.
- D. H. Munn and V. Bronte, Curr. Opin. Immunol., 2016, 39, 1–6 CrossRef CAS PubMed.
- J. E. Talmadge and D. I. Gabrilovich, Nat. Rev. Cancer, 2013, 13, 739–752 CrossRef CAS PubMed.
- I. Waldhauer and A. Steinle, Oncogene, 2008, 27, 5932–5943 CrossRef CAS PubMed.
- M. Buoncervello, L. Gabriele and E. Toschi, Int. J. Mol. Sci., 2019, 20, 4320 CrossRef CAS PubMed.
- T. Ojha, V. Pathak, Y. Shi, W. E. Hennink, C. T. W. Moonen, G. Storm, F. Kiessling and T. Lammers, Adv. Drug Delivery Rev., 2017, 119, 44–60 CrossRef CAS PubMed.
- D. W. Siemann, Cancer Treat. Rev., 2010, 37, 63–74 CrossRef PubMed.
- W. Song, Z. Tang, D. Zhang, H. Yu and X. Chen, Small, 2015, 11, 3755–3761 CrossRef CAS PubMed.
- J. Jiang, N. Shen, T. Ci, Z. Tang, Z. Gu, G. Li and X. Chen, Adv. Mater., 2019, 31, 1904278 CrossRef CAS PubMed.
- Y. Liang, Y. Hao, Y. Wu, Z. Zhou, J. Li, X. Sun and Y. Liu, ACS Appl. Mater. Interfaces, 2019, 11, 21381–21390 CrossRef CAS PubMed.
- N. Shen, J. Wu, C. Yang, H. Yu, S. Yang, T. Li, J. Chen, Z. Tang and X. Chen, Nano Lett., 2019, 19, 8021–8031 CrossRef CAS PubMed.
- S. Yang, Z. Tang, C. Hu, D. Zhang, N. Shen, H. Yu and X. Chen, Adv. Mater., 2019, 31, 1805955 CrossRef PubMed.
- H. Qin, H. Yu, J. Sheng, D. Zhang, N. Shen, L. Liu, Z. Tang and X. Chen, Adv. Sci., 2019, 6, 1900327 CrossRef PubMed.
- S. Kunjachan, A. Detappe, R. Kumar, T. Ireland, L. Cameron, D. E. Biancur, V. Motto-Ros, L. Sancey, S. Sridhar and G. M. Makrigiorgos, et al. , Nano Lett., 2015, 15, 7488–7496 CrossRef CAS PubMed.
- M. Guan, Y. Zhou, S. Liu, D. Chen, J. Ge, R. Deng, X. Li, T. Yu, H. Xu and D. Sun, et al. , Biomaterials, 2019, 213, 119218 CrossRef CAS PubMed.
- W. Gao, S. Li, Z. Liu, Y. Sun, W. Cao, L. Tong, G. Cui and B. Tang, Biomaterials, 2017, 139, 1–11 CrossRef CAS PubMed.
- V. P. Chauhan, T. Stylianopoulos, J. D. Martin, Z. Popović, O. Chen, W. S. Kamoun, M. G. Bawendi, D. Fukumura and R. K. Jain, Nat. Nanotechnol., 2012, 7, 383–388 CrossRef CAS PubMed.
- T. Stylianopoulos and R. K. Jain, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 18632–18637 CrossRef CAS PubMed.
- R. S. Heist, D. G. Duda, D. V. Sahani, M. Ancukiewicz, P. Fidias, L. V. Sequist, J. S. Temel, A. T. Shaw, N. A. Pennell and J. W. Neal, et al. , Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 1547–1552 CrossRef CAS PubMed.
- Y. Sung, P. Jin, L. Chu, F. Hsu, M. Wang, C. Chang, S. Chiou, J. T. Qiu, D. Gao and C. Lin, et al. , Nat. Nanotechnol., 2019, 14, 1160–1169 CrossRef CAS PubMed.
- L. Tian, A. Goldstein, H. Wang, L. E. Dobrolecki, X. Zhang, N. Putluri, A. Sreekumar, M. A. M. William and X. H. F. Zhang, Nature, 2017, 544, 250–254 CrossRef CAS PubMed.
- W. Yu, R. Liu, Y. Zhou and H. Gao, ACS Cent. Sci., 2020, 6, 100–116 CrossRef CAS PubMed.
- L. Schuster, O. Seifert, S. Vollmer, R. E. Kontermann, B. Schlosshauer and H. Hartmann, Mol. Pharmaceutics, 2015, 12, 3146–3157 CrossRef CAS PubMed.
- G. Xiong, L. Deng, J. Zhu, P. G. Rychahou and R. Xu, BMC Cancer, 2014, 14, 1 CrossRef PubMed.
- X. Han, Y. Li, Y. Xu, X. Zhao, Y. Zhang, X. Yang, Y. Wang, R. Zhao, G. J. Anderson and Y. Zhao, et al. , Nat. Commun., 2018, 9, 3318–3390 CrossRef PubMed.
- A. G. Kohli, S. Kivimäe, M. R. Tiffany and F. C. Szoka, J. Controlled Release, 2014, 191, 105–114 CrossRef CAS PubMed.
- A. Kultti, S. Pasonen-Seppänen, M. Jauhiainen, K. J. Rilla, R. Kärnä, E. Pyöriä, R. H. Tammi and M. I. Tammi, Exp. Cell Res., 2009, 315, 1914–1923 CrossRef CAS PubMed.
- V. Raeesi and W. C. W. Chan, Nanoscale, 2016, 8, 12524–12530 RSC.
- S. Lee, H. Han, H. Koo, J. H. Na, H. Y. Yoon, K. E. Lee, H. Lee, H. Kim, I. C. Kwon and K. Kim, J. Controlled Release, 2017, 263, 68–78 CrossRef CAS PubMed.
- A. Zinger, L. Koren, O. Adir, M. Poley, M. Alyan, Z. Yaari, N. Noor, N. Krinsky, A. Simon and H. Gibori, et al. , ACS Nano, 2019, 13, 11008–11021 CrossRef CAS PubMed.
- H. Gong, Y. Chao, J. Xiang, X. Han, G. Song, L. Feng, J. Liu, G. Yang, Q. Chen and Z. Liu, Nano Lett., 2016, 16, 2512–2521 CrossRef CAS PubMed.
- H. Zhou, Z. Fan, J. Deng, P. K. Lemons, D. C. Arhontoulis, W. B. Bowne and H. Cheng, Nano Lett., 2016, 16, 3268–3277 CrossRef CAS PubMed.
- B. Zhang, T. Jiang, S. Shen, X. She, Y. Tuo, Y. Hu, Z. Pang and X. Jiang, Biomaterials, 2016, 103, 12–21 CrossRef CAS PubMed.
- X. Dong, H. Liu, H. Feng, S. Yang, X. Liu, X. Lai, Q. Lu, J. F. Lovell, H. Chen and C. Fang, Nano Lett., 2019, 19, 997–1008 CrossRef PubMed.
- D. F. Mardhian, G. Storm, R. Bansal and J. Prakash, J. Controlled Release, 2018, 290, 1–10 CrossRef CAS PubMed.
- L. Qin and H. Gao, Asian J. Pharm. Sci., 2019, 14, 380–390 CrossRef PubMed.
- Z. Guo, K. Hu, J. Sun, T. Zhang, Q. Zhang, L. Song, X. Zhang and N. Gu, ACS Appl. Mater. Interfaces, 2014, 6, 10963–10968 CrossRef CAS PubMed.
- X. Hu, P. He, G. Qi, Y. Gao, Y. Lin, C. Yang, P. Yang, H. Hao, L. Wang and H. Wang, ACS Nano, 2017, 11, 4086–4096 CrossRef CAS PubMed.
- T. Ji, Y. Ding, Y. Zhao, J. Wang, H. Qin, X. Liu, J. Lang, R. Zhao, Y. Zhang and J. Shi, et al. , Adv. Mater., 2015, 27, 1865–1873 CrossRef CAS PubMed.
- M. J. Ernsting, B. Hoang, I. Lohse, E. Undzys, P. Cao, T. Do, B. Gill, M. Pintilie, D. Hedley and S. Li, J. Controlled Release, 2015, 206, 122–130 CrossRef CAS PubMed.
- L. Miao, Q. Liu, C. M. Lin, C. Luo, Y. Wang, L. Liu, W. Yin, S. Hu, W. Y. Kim and L. Huang, Cancer Res., 2017, 77, 719–731 CrossRef CAS PubMed.
- G. Ishii, A. Ochiai and S. Neri, Adv. Drug Delivery Rev., 2016, 99, 186–196 CrossRef CAS PubMed.
- X. Chen and E. Song, Nat. Rev. Drug Discovery, 2019, 18, 99–115 CrossRef CAS PubMed.
- Y. Zhao, J. Cao, A. Melamed, M. Worley, A. Gockley, D. Jones, H. T. Nia, Y. Zhang, T. Stylianopoulos and A. S. Kumar, et al. , Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 2210–2219 CrossRef PubMed.
- K. Hu, L. Miao, T. J. Goodwin, J. Li, Q. Liu and L. Huang, ACS Nano, 2017, 11, 4916–4925 CrossRef CAS PubMed.
- Y. Wei, Y. Wang, D. Xia, S. Guo, F. Wang, X. Zhang and Y. Gan, ACS Appl. Mater. Interfaces, 2017, 9, 25138–25151 CrossRef CAS PubMed.
- J. D. Martin, M. Panagi, C. Wang, T. T. Khan, M. R. Martin, C. Voutouri, K. Toh, P. Papageorgis, F. Mpekris and C. Polydorou, et al. , ACS Nano, 2019, 13, 6396–6408 CrossRef CAS PubMed.
- J. Liu, Q. Chen, L. Feng and Z. Liu, Nano Today, 2018, 21, 55–73 CrossRef CAS.
- G. Song, C. Liang, H. Gong, M. Li, X. Zheng, L. Cheng, K. Yang, X. Jiang and Z. Liu, Adv. Mater., 2015, 27, 6110–6117 CrossRef CAS PubMed.
- W. L. Liu, T. Liu, M. Z. Zou, W. Y. Yu, C. X. Li, Z. Y. He, M. K. Zhang, M. D. Liu, Z. H. Li and J. Feng, et al. , Adv. Mater., 2018, 30, e1802006 CrossRef PubMed.
- M. Gao, C. Liang, X. Song, Q. Chen, Q. Jin, C. Wang and Z. Liu, Adv. Mater., 2017, 29, 1701429 CrossRef PubMed.
- W. Zhu, Z. Dong, T. Fu, J. Liu, Q. Chen, Y. Li, R. Zhu, L. Xu and Z. Liu, Adv. Funct. Mater., 2016, 26, 5490–5498 CrossRef CAS.
- L. H. Liu, Y. H. Zhang, W. X. Qiu, L. Zhang, F. Gao, B. Li, L. Xu, J. X. Fan, Z. H. Li and X. Z. Zhang, Small, 2017, 13, 1701621 CrossRef PubMed.
- P. Liu, X. Xie, X. Shi, Y. Peng, J. Ding and W. Zhou, ACS Appl. Mater. Interfaces, 2019, 11, 48261–48270 CrossRef CAS PubMed.
- D. Zheng, B. Li, C. Li, J. Fan, Q. Lei, C. Li, Z. Xu and X. Zhang, ACS Nano, 2016, 10, 8715–8722 CrossRef CAS PubMed.
- Y. Fan, T. Zhou, P. Cui, Y. He, X. Chang, L. Xing and H. Jiang, Adv. Funct. Mater., 2019, 29, 1806708 CrossRef.
- X. Song, L. Feng, C. Liang, M. Gao, G. Song and Z. Liu, Nano Res., 2017, 10, 1200–1212 CrossRef CAS.
- Z. Yang, Q. Chen, J. Chen, Z. Dong, R. Zhang, J. Liu and Z. Liu, Small, 2018, 14, e1803262 CrossRef PubMed.
- W. Yu, T. Liu, M. Zhang, Z. Wang, J. Ye, C. X. Li, W. Liu, R. Li, J. Feng and X. Z. Zhang, ACS Nano, 2019, 13, 1784–1794 CAS.
- B. M. Bola, A. L. Chadwick, F. Michopoulos, K. G. Blount, B. A. Telfer, K. J. Williams, P. D. Smith, S. E. Critchlow and I. J. Stratford, Mol. Cancer Ther., 2014, 13, 2805–2816 CrossRef CAS PubMed.
- J. Li, K. Shi, Z. F. Sabet, W. Fu, H. Zhou, S. Xu, T. Liu, M. You, M. Cao and M. Xu, et al. , Sci. Adv., 2019, 5, x937 Search PubMed.
- M. Ovais, M. Guo and C. Chen, Adv. Mater., 2019, 31, 1808303 CrossRef PubMed.
- S. Zanganeh, G. Hutter, R. Spitler, O. Lenkov, M. Mahmoudi, A. Shaw, J. S. Pajarinen, H. Nejadnik, S. Goodman and M. Moseley, et al. , Nat. Nanotechnol., 2016, 11, 986–994 CrossRef CAS PubMed.
- V. Mulens-Arias, J. M. Rojas, S. Pérez-Yagüe, M. P. Morales and D. F. Barber, Biomaterials, 2015, 52, 494–506 CrossRef CAS PubMed.
- H. Shime, M. Matsumoto, H. Oshiumi, S. Tanaka, A. Nakane, Y. Iwakura, H. Tahara, N. Inoue and T. Seya, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 2066–2071 CrossRef CAS PubMed.
- C. Xu, S. Yang, Z. Jiang, J. Zhou and J. Yao, Nano Lett., 2020, 20, 372–383 CrossRef CAS PubMed.
- D. Chen, J. Xie, R. Fiskesund, W. Dong, X. Liang, J. Lv, X. Jin, J. Liu, S. Mo and T. Zhang, et al. , Nat. Commun., 2018, 9, 873 CrossRef PubMed.
- Z. Nemeth, E. Csizmadia, L. Vikstrom, M. Li, K. Bisht, A. Feizi, S. Otterbein, B. Zuckerbraun, D. B. Costa and P. P. Pandolfi, et al. , Oncotarget, 2016, 7, 23919 CrossRef PubMed.
- C. Xu, S. Yang, Z. Jiang, J. Zhou and J. Yao, Nano Lett., 2019, 20, 372–383 CrossRef PubMed.
- K. Sato, N. Sato, B. Xu, Y. Nakamura, T. Nagaya, P. L. Choyke, Y. Hasegawa and H. Kobayashi, Sci. Transl. Med., 2016, 8, 110r–352r Search PubMed.
- Q. Song, Y. Yin, L. Shang, T. Wu, D. Zhang, M. Kong, Y. Zhao, Y. He, S. Tan and Y. Guo, et al. , Nano Lett., 2017, 17, 6366–6375 CrossRef CAS PubMed.
- M. Saeed, J. Gao, Y. Shi, T. Lammers and H. Yu, Theranostics, 2019, 9, 7981–8000 CrossRef PubMed.
- H. Yu, H. Lee, A. Herrmann, R. Buettner and R. Jove, Nat. Rev. Cancer, 2014, 14, 736–746 CrossRef CAS PubMed.
- M. Huo, Y. Zhao, A. B. Satterlee, Y. Wang, Y. Xu and L. Huang, J. Controlled Release, 2017, 245, 81–94 CrossRef CAS PubMed.
- Y. Zhao, M. Huo, Z. Xu, Y. Wang and L. Huang, Biomaterials, 2015, 68, 54–66 CrossRef CAS PubMed.
- L. Jeanbart, I. C. Kourtis, A. J. van der Vlies, M. A. Swartz and J. A. Hubbell, Cancer Immunol. Immunother., 2015, 64, 1033–1046 CrossRef CAS PubMed.
- M. S. Sasso, G. Lollo, M. Pitorre, S. Solito, L. Pinton, S. Valpione, G. Bastiat, S. Mandruzzato, V. Bronte and I. Marigo, et al. , Biomaterials, 2016, 96, 47–62 CrossRef CAS PubMed.
- D. Liu, B. Chen, Y. Mo, Z. Wang, T. Qi, Q. Zhang and Y. Wang, Nano Lett., 2019, 19, 6964–6976 CrossRef CAS PubMed.
- X. Han, K. Cheng, Y. Xu, Y. Wang, H. Min, Y. Zhang, X. Zhao, R. Zhao, G. J. Anderson and L. Ren, et al. , J. Am. Chem. Soc., 2020, 142, 2490–2496 CrossRef CAS PubMed.
- H. Huang, C. Jiang, S. Shen, A. Liu, Y. Gan, Q. Tong, S. Chen, Z. Gao, J. Du and J. Cao, et al. , Nano Lett., 2019, 19, 5356–5365 CrossRef CAS PubMed.
- L. Miao, J. Li, Q. Liu, R. Feng, M. Das, C. M. Lin, T. J. Goodwin, O. Dorosheva, R. Liu and L. Huang, ACS Nano, 2017, 11, 8690–8706 CrossRef CAS PubMed.
- C. Li, J. Wang, G. Wei, Y. Huang, H. Yu, Y. Gan, Y. Wang, L. Mei, H. Chen and H. Hu, et al. , Acta Pharm. Sin. B, 2019, 9, 1145–1162 CrossRef PubMed.
- Z. M. Qian, Pharmacol. Rev., 2002, 54, 561–587 CrossRef CAS PubMed.
- M. Merino, S. Zalba and M. J. Garrido, J. Controlled Release, 2018, 275, 162–176 CrossRef CAS PubMed.
- H. Gao, Acta Pharm. Sin. B, 2016, 6, 268–286 CrossRef PubMed.
- F. Araste, K. Abnous, M. Hashemi, S. M. Taghdisi, M. Ramezani and M. Alibolandi, J. Controlled Release, 2018, 292, 141–162 CrossRef CAS PubMed.
- J. H. Lee, J. A. Engler, J. F. Collawn and B. A. Moore, Eur. J. Biochem., 2001, 268, 2004–2012 CrossRef CAS PubMed.
- X. Wei, C. Zhan, X. Chen, J. Hou, C. Xie and W. Lu, Mol. Pharmaceutics, 2014, 11, 3261–3268 CrossRef CAS PubMed.
- H. Zhang, T. Wu, W. Yu, S. Ruan, Q. He and H. Gao, ACS Appl. Mater. Interfaces, 2018, 10, 9094–9103 CrossRef CAS PubMed.
- M. Yu, D. Su, Y. Yang, L. Qin, C. Hu, R. Liu, Y. Zhou, C. Yang, X. Yang and G. Wang, et al. , ACS Appl. Mater. Interfaces, 2018, 11, 176–186 CrossRef PubMed.
- E. K. Duperret, A. Trautz, D. Ammons, A. Perales-Puchalt, M. C. Wise, J. Yan, C. Reed and D. B. Weiner, Clin. Cancer Res., 2018, 24, 1190–1201 CrossRef CAS PubMed.
- M. H. Sherman, R. T. Yu, D. D. Engle, N. Ding, A. R. Atkins, H. Tiriac, E. A. Collisson, F. Connor, T. Van Dyke and S. Kozlov, et al. , Cell, 2014, 159, 80–93 CrossRef CAS PubMed.
- R. Yang, J. Xu, L. Xu, X. Sun, Q. Chen, Y. Zhao, R. Peng and Z. Liu, ACS Nano, 2018, 12, 5121–5129 CrossRef CAS PubMed.
- S. Ruan, L. Qin, W. Xiao, C. Hu, Y. Zhou, R. Wang, X. Sun, W. Yu, Q. He and H. Gao, Adv. Funct. Mater., 2018, 28, 1802227 CrossRef.
- R. Li, Y. He, S. Zhang, J. Qin and J. Wang, Acta Pharm. Sin. B, 2018, 8, 14–22 CrossRef PubMed.
- Y. Lu, Q. Hu, C. Jiang and Z. Gu, Curr. Opin. Biotechnol., 2019, 58, 81–91 CrossRef CAS PubMed.
- M. Zhang, J. Ye, Y. Xia, Z. Wang, C. Li, X. Wang, W. Yu, W. Song, J. Feng and X. Zhang, ACS Nano, 2019, 13, 14230–14240 CrossRef CAS PubMed.
- B. Nieswandt and S. P. Watson, Blood, 2003, 102, 449–461 CrossRef CAS PubMed.
- M. Feng, J. Y. Chen, R. Weissman-Tsukamoto, J. Volkmer, P. Y. Ho, K. M. McKenna, S. Cheshier, M. Zhang, N. Guo and P. Gip, et al. , Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 2145–2150 CrossRef CAS PubMed.
- R. Liu, Y. An, W. Jia, Y. Wang, Y. Wu, Y. Zhen, J. Cao and H. Gao, J. Controlled Release, 2020, 321, 589–601 CrossRef CAS PubMed.
- J. Xue, Z. Zhao, L. Zhang, L. Xue, S. Shen, Y. Wen, Z. Wei, L. Wang, L. Kong and H. Sun, et al. , Nat. Nanotechnol., 2017, 12, 692–700 CrossRef CAS PubMed.
- E. Kolaczkowska and P. Kubes, Nat. Rev. Immunol., 2013, 13, 159–175 CrossRef CAS PubMed.
- Y. Huang, X. Gao and J. Chen, Acta Pharm. Sin. B, 2018, 8, 4–13 CrossRef PubMed.
- L. Guo, Y. Zhang, Z. Yang, H. Peng, R. Wei, C. Wang and M. Feng, ACS Nano, 2019, 13, 1078 CAS.
- D. Ha, N. Yang and V. Nadithe, Acta Pharm. Sin. B, 2016, 6, 287–296 CrossRef PubMed.
- J. Wang, Y. Dong, Y. Li, W. Li, K. Cheng, Y. Qian, G. Xu, X. Zhang, L. Hu and P. Chen, et al. , Adv. Funct. Mater., 2018, 28, 1707360 CrossRef.
- W. Fu, C. Lei, S. Liu, Y. Cui, C. Wang, K. Qian, T. Li, Y. Shen, X. Fan and F. Lin, et al. , Nat. Commun., 2019, 10, 4355 CrossRef PubMed.
- M. Li, S. Li, H. Zhou, X. Tang, Y. Wu, W. Jiang, Z. Tian, X. Zhou, X. Yang and Y. Wang, Nat. Commun., 2020, 11, 1126 CrossRef CAS PubMed.
- J. Z. Du, H. J. Li and J. Wang, Acc. Chem. Res., 2018, 51, 2848–2856 CrossRef CAS PubMed.
- Y. R. Zhang, R. Lin, H. J. Li, W. L. He, J. Z. Du and J. Wang, Wiley Interdiscip. Rev.: Nanomed. Nanobiotechnol., 2019, 11, e1519 Search PubMed.
- B. Chen, W. Dai, B. He, H. Zhang, X. Wang, Y. Wang and Q. Zhang, Theranostics, 2017, 7, 538–558 CrossRef CAS PubMed.
- W. Gao, Y. Hu, L. Xu, M. Liu, H. Wu and B. He, Chin. Chem. Lett., 2018, 29, 1795–1798 CrossRef CAS.
- C. Chen, Y. Li, X. Yu, Q. Jiang, X. Xu, Q. Yang and Z. Qian, Chin. Chem. Lett., 2018, 29, 1609–1612 CrossRef CAS.
- J. Ding, J. Chen, L. Gao, Z. Jiang, Y. Zhang, M. Li, Q. Xiao, S. S. Lee and X. Chen, Nano Today, 2019, 29, 100800 CrossRef CAS.
- X. Feng, H. Dixon, H. Glen Ravenhill, S. Karaosmanoglu, Q. Li, L. Yan and X. Chen, Adv. Ther., 2019, 2, 1900093 CrossRef.
- W. Yu, M. Shevtsov, X. Chen and H. Gao, Chin. Chem. Lett., 2020 DOI:10.1016/j.cclet.2020.02.036.
- Z. Zhang, H. Wang, T. Tan, J. Li, Z. Wang and Y. Li, Adv. Funct. Mater., 2018, 28, 1801840 CrossRef.
- C. Hu, X. Cun, S. Ruan, R. Liu, W. Xiao, X. Yang, Y. Yang, C. Yang and H. Gao, Biomaterials, 2018, 168, 64–75 CrossRef CAS PubMed.
- J. Chen, J. Ding, Y. Wang, J. Cheng, S. Ji, X. Zhuang and X. Chen, Adv. Mater., 2017, 29 CAS.
- R. Kumari, D. Sunil and R. S. Ningthoujam, J. Controlled Release, 2020, 319, 135–156 CrossRef CAS PubMed.
- Y. Niu, J. Zhu, Y. Li, H. Shi, Y. Gong, R. Li, Q. Huo, T. Ma and Y. Liu, J. Controlled Release, 2018, 277, 35–47 CrossRef CAS PubMed.
- X. Zhang, L. An, Q. Tian, J. Lin and S. Yang, J. Mater. Chem. B, 2020 10.1039/d0tb00030b.
- Y. Li, X. Xu, X. Zhang, Y. Li, Z. Zhang and Z. Gu, ACS Nano, 2017, 11, 416–429 CrossRef CAS PubMed.
- Q. Lei, S. Wang, J. Hu, Y. Lin, C. Zhu, L. Rong and X. Zhang, ACS Nano, 2017, 11, 7201–7214 CrossRef CAS PubMed.
- S. Ruan, X. Cao, X. Cun, G. Hu, Y. Zhou, Y. Zhang, L. Lu, Q. He and H. Gao, Biomaterials, 2015, 60, 100–110 CrossRef CAS PubMed.
- Y. L. Su, T. W. Yu, W. H. Chiang, H. C. Chiu, C. H. Chang, C. S. Chiang and S. H. Hu, Adv. Funct. Mater., 2017, 27, 1700056 CrossRef.
- R. Liu, C. Hu, Y. Yang, J. Zhang and H. Gao, Acta Pharm. Sin. B, 2019, 9, 410–420 CrossRef PubMed.
- C. Song, Y. Li, T. Li, Y. Yang, Z. Huang, J. M. de la Fuente, J. Ni and D. Cui, Adv. Funct. Mater., 2020, 1906309 CrossRef CAS.
- R. Liu, W. Xiao, C. Hu, R. Xie and H. Gao, J. Controlled Release, 2018, 28, 127–139 CrossRef PubMed.
- W. Xiao, S. Ruan, W. Yu, R. Wang, C. Hu, R. Liu and H. Gao, Mol. Pharmaceutics, 2017, 14, 3489–3498 CrossRef CAS PubMed.
- Y. Cong, L. Ji, Y. J. Gao, F. H. Liu, D. B. Cheng, Z. Hu, Z. Y. Qiao and H. Wang, Angew. Chem., Int. Ed., 2019, 58, 4632–4637 CrossRef CAS PubMed.
- Y. Li, H. Y. Yang, T. Thambi, J. Park and D. S. Lee, Biomaterials, 2019, 217, 119299 CrossRef CAS PubMed.
- X. Chen, L. Liu and C. Jiang, Acta Pharm. Sin. B, 2016, 6, 261–267 CrossRef PubMed.
- M. Kanamala, W. R. Wilson, M. Yang, B. D. Palmer and Z. Wu, Biomaterials, 2016, 85, 152–167 CrossRef CAS PubMed.
- J. Cao, X. Xie, A. Lu, B. He, Y. Chen, Z. Gu and X. Luo, Biomaterials, 2014, 35, 4517–4524 CrossRef CAS PubMed.
- H. Han, D. Valdeperez, Q. Jin, B. Yang, Z. Li, Y. Wu, B. Pelaz, W. J. Parak and J. Ji, ACS Nano, 2017, 11, 1281–1291 CrossRef CAS PubMed.
- S. Li, W. Wu, K. Xiu, F. Xu, Z. Li and J. Li, J. Biomed. Nanotechnol., 2014, 10, 1480–1489 CrossRef CAS PubMed.
- T. Thambi, V. G. Deepagan, H. Y. Yoon, H. S. Han, S. Kim, S. Son, D. Jo, C. Ahn, Y. D. Suh and K. Kim, et al. , Biomaterials, 2014, 35, 1735–1743 CrossRef CAS PubMed.
- S. Son, N. V. Rao, H. Ko, S. Shin, J. Jeon, H. S. Han, V. Q. Nguyen, T. Thambi, Y. D. Suh and J. H. Park, Int. J. Biol. Macromol., 2018, 110, 399–405 CrossRef CAS PubMed.
- J. Yan, H. Zhang, F. Cheng, Y. He, T. Su, X. Zhang, M. Zhang, Y. Zhu, C. Li and J. Cao, et al. , Int. J. Nanomed., 2018, 13, 8247–8268 CrossRef CAS PubMed.
- Z. Dong, L. Feng, W. Zhu, X. Sun, M. Gao, H. Zhao, Y. Chao and Z. Liu, Biomaterials, 2016, 110, 60–70 CrossRef CAS PubMed.
- K. H. Min, H. S. Min, H. J. Lee, D. J. Park, J. Y. Yhee, K. Kim, I. C. Kwon, S. Y. Jeong, O. F. Silvestre and X. Chen, et al. , ACS Nano, 2015, 9, 134–145 CrossRef CAS PubMed.
- Q. Sun, Z. Zhou, N. Qiu and Y. Shen, Adv. Mater., 2017, 29, 1606628 CrossRef PubMed.
- H. C. Dietz, J. Clin. Invest., 2010, 120, 403–406 CrossRef CAS PubMed.
- X. Cun, S. Ruan, J. Chen, L. Zhang, J. Li, Q. He and H. Gao, Acta Biomater., 2016, 31, 186–196 CrossRef CAS PubMed.
- W. Xiao and H. Gao, Int. J. Pharm., 2018, 552, 328–339 CrossRef CAS PubMed.
- A. Salvati, A. S. Pitek, M. P. Monopoli, K. Prapainop, F. B. Bombelli, D. R. Hristov, P. M. Kelly, C. Åberg, E. Mahon and K. A. Dawson, Nat. Nanotechnol., 2013, 8, 137–143 CrossRef CAS PubMed.
- W. Xiao, J. Xiong, S. Zhang, Y. Xiong, H. Zhang and H. Gao, Int. J. Pharm., 2018, 538, 105–111 CrossRef CAS PubMed.
- A. Kolate, D. Baradia, S. Patil, I. Vhora, G. Kore and A. Misra, J. Controlled Release, 2014, 192, 67–81 CrossRef CAS PubMed.
- Y. Fang, J. Xue, S. Gao, A. Lu, D. Yang, H. Jiang, Y. He and K. Shi, Drug Delivery, 2017, 24, 22–32 CrossRef CAS PubMed.
- V. Juang, C. H. Chang, C. S. Wang, H. E. Wang and Y. L. Lo, Small, 2019, 15, 1903296 CrossRef CAS PubMed.
- R. Cheng, F. Feng, F. Meng, C. Deng, J. Feijen and Z. Zhong, J. Controlled Release, 2011, 152, 2–12 CrossRef CAS PubMed.
- Z. Fang, S. Pan, P. Gao, H. Sheng, L. Li, L. Shi, Y. Zhang and X. Cai, Int. J. Pharm., 2019, 575, 118841 CrossRef PubMed.
- S. Ruan, X. Cao, X. Cun, G. Hu, Y. Zhou, Y. Zhang, L. Lu, Q. He and H. Gao, Biomaterials, 2015, 60, 100–110 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2020 |