
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
14N–1H HMQC solid-state NMR as a powerful tool to study amorphous formulations – an exemplary study of paclitaxel loaded polymer micelles†
Marvin
Grüne
a,
Robert
Luxenhofer
 b,
Dinu
Iuga
c,
Steven P.
Brown
c and
Ann-Christin
Pöppler
*a
b,
Dinu
Iuga
c,
Steven P.
Brown
c and
Ann-Christin
Pöppler
*a
aInstitute of Organic Chemistry, University of Würzburg, Am Hubland, 97074 Würzburg, Germany. E-mail: ann-christin.poeppler@uni-wuerzburg.de
bLehrstuhl für Chemische Technologie der Materialsynthese, University of Würzburg, Röntgenring 11, 97070 Würzburg, Germany
cDepartment of Physics, University of Warwick, Coventry, CV4 7AL, UK
First published on 29th May 2020
Abstract
Amorphous drug–polymer formulations are complex materials and often challenging to characterize, even more so if the small molecule component itself is increasingly complex. In this work, we present 14N–1H HMQC magic-angle spinning (MAS) NMR experiments in the solid state as a promising tool to study amorphous formulations. Poly(2-oxazoline) based polymer micelles loaded with different amounts of the cancer drug paclitaxel serve to highlight the possibilities offered by these experiments: while the dense core of these polymeric micelles prevents NMR spectroscopic analysis in solution and the very similar 15N chemical shifts hamper a solid-state NMR characterization based on this nucleus, 14N is a very versatile alternative. 14N–1H HMQC experiments yield well-separated signals, which are spread over a large ppm range, and provide information on the symmetry of the nitrogen environment and probe 14N–1H through-space proximities. In this way, the overall complexity can be narrowed down to specific N-containing environments. The results from the experiments presented here represent a valuable puzzle piece, which helps to improve the structural understanding of drug–polymer formulations. It can be straightforwardly combined with complementary NMR spectroscopic experiments and other analytical techniques.
Introduction
Paclitaxel (PTX, Scheme 1) is an effective anti-cancer drug for a wide range of tumours, but it exhibits a very low aqueous solubility of 0.4 μg mL−1.1,2 Therefore, a variety of different formulations for PTX has been developed.3 This includes the protein-based nanoparticle Abraxane®, the polymer-conjugate OpaxioTM, Genexol-PM® and NK105.4,5 The latter two contain PTX in polymer micelles formed of polyethylene glycol–polylactic acid (Genexol-PM®) and polyethylene glycol–poly(amino acid) (NK105) copolymers. In general, the development of formulations and their clinical trials require very long times with highly uncertain outcomes. The above mentioned formulations have undergone multiple phase II/III clinical trials over the past decades, e.g. a recently published phase III clinical trial of NK105,6 and only Abraxane® has been successfully marketed with FDA approval since 2007. Furthermore, they are all characterized by a relatively low PTX loading not exceeding 25 wt%. In this context, Luxenhofer et al. reported promising preclinical data for polymer micelles comprising a poly(2-oxazoline) (POx) based triblock copolymer loaded with up to 50 wt% of PTX.7,8 Compared to the clinically approved PTX formulations, their formulations showed higher maximum tolerated doses, elevated drug exposure to tumour tissues and prolonged survival of mice bearing A2780 human ovarian tumours. Schulz et al.,9 Jaksch et al.10 and Sochor et al.11 studied their micellar morphology for different drug loadings (PTX and curcumin) using dynamic light scattering, atomic force microscopy, (cryogenic) transmission electron microscopy, and small-angle neutron scattering (SANS). They found that the pure polymer self-assembles into wormlike and spherical micelles in aqueous solution, while the incorporation of PTX led to the exclusive formation of spherical particles. Interestingly, SANS data revealed small, PTX rich domains partly submerged within the micellar core. Apart from such morphological aspects and capability to design improved drug delivery systems, it is very important to also understand the complex structural arrangements in such amorphous drug–polymer formulations on the molecular level. NMR probes the local environment of molecules and is thus an excellent tool to report on various possible interactions. This doesn’t necessarily require crystalline or well-ordered structures, and also gives excellent results for disordered as well as amorphous samples.12–14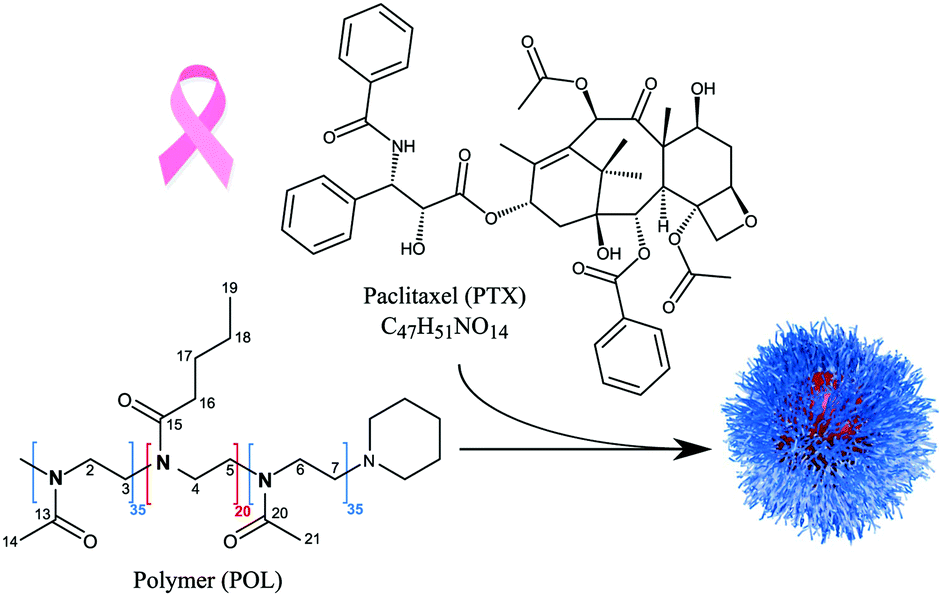 | ||
| Scheme 1 Structural formula of the components used in this study: the amphiphilic block copolymer poly(2-methyl-2-oxazoline)-block-poly(2-n-butyl-2-oxazoline)-block-poly(2-methyl-2-oxazoline) (POL) encapsulates paclitaxel (PTX) by self-assembly into polymeric micelles (schematic drawing on the right). | ||
In particular, solid-state NMR has been established as a powerful tool for structure elucidation in pharmaceutical contexts such as identification of polymorphs or investigation of amorphous solid dispersions.15–17 The sometimes strongly reduced mobility of encapsulated drugs within drug formulations can hinder the characterization by NMR in solution due to strong broadening of the signals, which makes solid-state NMR the corresponding method of choice. Callari et al. as well as Pöppler et al. recently showcased how solid-state NMR at moderate to fast Magic Angle Spinning (MAS) helps to obtain loading dependent structural insights into micellar formulations with an assumed core–shell structure.18,19 Both groups found that increasing the loading of two different types of polymeric micelles did not just affect the micellar core but interestingly also affected the surrounding shell, which served as a basis to explain their physicochemical and biological properties such as reduced cellular uptake and inferior dissolution rates. Hirsh et al. have recently also shown how 1H MAS measurements enable the rapid characterization of general pharmaceutical dosage forms.20 The enormous progress made with respect to hardware development, e.g. MAS frequencies of up to 130 kHz being available now,21 also enables high-resolution detection of protons in the solid state with longer coherence lifetimes.22–24 The latter are essential prerequisites for sophisticated homonuclear as well as heteronuclear 2D experiments, which are exciting tools to learn more about the intermolecular proximities in a large range of materials.
However, for the solid-state NMR spectroscopic characterization of PTX and its formulations as discussed above, a series of complexities arises: PTX alone with its 51 protons and thus a variety of strongly overlapping chemical environments already poses a challenge. In addition, crystalline PTX contains two individual molecules in the asymmetric unit (Z′ = 2),25 which leads to the doubling of the expected resonances. Consequentially, to date, only 13C solid-state NMR data with assignment based on comparison with structurally related fragments and calculations has been published,26,27 while 1H solid-state NMR data of PTX is still missing. Taking the polymer component and potential drug–polymer interactions into account further complicates the spectral evaluation and thus requires narrowing down the search space, e.g. by focusing on specific functionalities. For example, spectral simplification can be achieved by making use of NMR active heteronuclei.
The frequent involvement of nitrogen atoms in hydrogen bonding in pharmaceuticals makes this nucleus a promising starting point for a detailed investigation of PTX containing formulations through 2D nitrogen-proton correlation experiments, especially since PTX has only one nitrogen environment per molecule. There are two NMR-active nitrogen isotopes with low magnetogyric ratios: 15N (I = 1/2) possesses a low natural abundance of only 0.37%, whereas 14N has a natural abundance of 99.6% but exhibits quadrupole broadening due to its nuclear spin quantum number I = 1. The substantial disadvantage of the first-order quadrupole interaction, causing a broadening in the range of several MHz, can be avoided by setting the indirect 14N spectral width of the 2D correlation equal to the spinning frequency. Tatton et al. successfully applied this in the form of a 14N–1H heteronuclear multiple-quantum correlation (HMQC) experiment to study crystalline model compounds,28 hydrogen bonding in co-crystals and attempting the first transfer to amorphous solid dispersions.29 So far, the majority of 14N–1H HMQC experiments have been performed for highly ordered, crystalline compounds.28,30–39 In contrast, disordered systems with significant differential dynamics are more challenging systems and there are thus very seldom reports on employing the 14N–1H HMQC experiment for their characterization. However, the characterization of disordered and amorphous materials could substantially benefit from the additional 14N second-order isotropic quadrupolar shifts (see the ESI† for detailed equations) as opposed to the 15N isotropic chemical shifts alone. The additional 14N second-order isotropic quadrupolar shifts will result in the spreading of the nitrogen signals over a larger ppm range (hundreds of ppm) and can thus disperse very similar or even overlapping 15N NMR signals. This could prove to be crucial for the analysis of amorphous drug formulations, either when the nitrogen in both, the drug and polymer, is a part of the same functional moiety (e.g. amides in the previously mentioned POx/PTX formulations) or when the distribution of similar local environments as observed in amorphous forms makes the differentiation between peaks challenging. This is underlined by the fact that around 84% of unique small-molecule drugs contain a nitrogen atom.40 As a result, there is a large, yet largely unexplored potential of this experiment for the analysis and increased understanding of such materials. In solid-state NMR, there are two different ways to create 14N–1H HMQCs, either heteronuclear through-bond 14N–1H J-couplings and residual second-order quadrupole–dipolar couplings or by 14N–1H through-space dipolar couplings.33,34,41 By varying the recoupling time in the latter case, it is then possible to obtain different through-space proximities with longer recoupling times probing longer range intra- and intermolecular N⋯H distances.28 A practical guideline with a focus on experimental procedures and parameters can be found in the recent literature.42 Combining all aspects, 14N–1H HMQC experiments represent a very promising tool for the investigation of the complex drug–polymer assemblies.
In the present work, we investigate the suitability and potential of 14N–1H solid-state NMR experiments for the analysis of amorphous polymer–PTX formulations, for which structural information is challenging to obtain due to the lack of long-range order. Therefore, differently loaded paclitaxel formulations based on the amphiphilic triblock copolymer poly(2-methyl-2-oxazoline)-block-poly(2-n-butyl-2-oxazoline)-block-poly(2-methyl-2-oxazoline) (pMeOx-b-pBuOx-b-pMeOx = A-pBuOx-A = POL) (Scheme 1) serve as the set of samples.8 Complemented by samples of the individual components, the following points will be addressed: (i) general feasibility of the experimental setup for the individual components, (ii) exploring the potential of 14N–1H HMQC experiments for the analysis of their amorphous micellar formulations, (iii) extracting information about 14N–1H proximities for intermolecular through-space contacts and (iv) their interpretation with respect to (loading dependent) structural features as a complementary source of information to existing data obtained by techniques such as SANS.
Experimental section
Materials
Paclitaxel in its crystalline form was purchased from LC Laboratories and used without further purification. The ABA triblock polymer poly(2-methyl-2-oxazoline)-block-poly(2-n-butyl-2-oxazoline)-block-poly(2-methyl-2-oxazoline) (POL) was synthesized according to previously published protocols.43 Subsequent preparation of the differently loaded PTX–POL formulations also followed the literature known procedures.8,9 A short description can be found in the ESI.† Resulting drug loadings were determined using HPLC analysis. The corresponding micellar formulation denoted as POL-2-PTX, POL-4-PTX and POL-9-PTX contain 17 wt% (10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2), 29 wt% (10:4) and 47 wt% (10:9) PTX.
:2), 29 wt% (10:4) and 47 wt% (10:9) PTX.
NMR
14N–1H HMQC experiments were performed using a Bruker Avance III spectrometer at a 1H Larmor frequency of 850 MHz. A Bruker 1.3 mm triple resonance probe operating in the double resonance mode at a MAS frequency of 60 kHz was used. A pulse sequence diagram is shown in Fig. S1 (ESI†). Heteronuclear dipolar couplings were reintroduced by applying rotary resonance recoupling (R3)44 under the n = 2 condition, as proposed by Gan et al.,33 using x,−x phase inversion.45 Each recoupling block had a length of 16.67 μs. The durations of the 1H pulses/14N pulses were 1.6 μs/3.2 μs for PTX and POL-9-PTX, 1.55 μs/3.1 μs for POL-2-PTX, POL-4-PTX, and 1.55 μs/3.2 μs for POL. A four-step nested phase cycle was used to select changes in the coherence order Δp = ±1 (on the first 1H pulse, two steps) and Δp = ±1 (on the final 14N pulse, two steps). Correct calibration of the magic angle is crucial for this experiment. All NMR data including the compiled pulse sequences for the results presented in this paper can be found in the Warwick Research Archive Portal (WRAP). Magic angle and pulse calibrations were performed with the dipeptide β-AspAla. 14N chemical shifts were referenced to saturated NH4Cl at −352.9 ppm, corresponding to a primary reference of CH3NO2 at 0 ppm. 15N experiments recorded at 9.4 T were referenced to unlabelled α-glycine at −342.0 ppm and those at 20 T were referenced to labelled L-histidine, which has an NH3+ resonance at −333.1 ppm. Both ways of 15N referencing correspond to a CH3NO2 reference at 0 ppm. The 13C NMR data were measured with a Bruker Avance III HD 600 MHz spectrometer and a 3.2 mm double-channel probe. The duration of the 1H pulses was 2.5 μs in the 13C CP MAS under ramped cross-polarization conditions optimized using a ramp from 90 to 100 with 100 increments and the α-glycine sample. SPINAL-6446 heteronuclear decoupling was applied during an acquisition period of 23 ms at a 1H rf nutation frequency of 100 kHz, with an optimized pulse length of 4.9–5.1 μs. The 15N data of POL-9-PTX were recorded with a Bruker Avance III spectrometer at a 1H Larmor frequency of 850 MHz and a duration of the 1H pulses of 2.5 μs under the optimized cross-polarization conditions using histidine. The 15N data of crystalline PTX and POL were recorded using a Bruker Avance Neo 400 MHz spectrometer. In both cases, 4 mm rotors with a sample volume of 80 μL were used. The duration of the 1H pulses was 2.5 μs under the optimized cross-polarization conditions using α-glycine. KBr was used for magic angle calibration. 13C chemical shifts were referenced to the methylene carbon of adamantane at 38.48 ppm.47Results
As the starting point for the spectral assignment and further investigation of the three differently loaded formulations of the poorly water-soluble cancer-drug paclitaxel containing 17 wt% (POL-2-PTX), 29 wt% (POL-4-PTX) and 47 wt% (POL-9-PTX) PTX, NMR experiments in solution were performed (Sections SI 4–SI 6, ESI†). For the assignment of PTX, data from NMR measurements in CDCl3 were used. However, in aqueous polymer micellar formulations, only extremely broad, unresolved PTX signals can be observed in the 1H NMR spectra (Section SI 6 with Fig. S4, ESI†), underlining the necessity to focus on solid-state NMR for the analysis of such formulations. Consequently, a set of different solid-state NMR spectra at moderate to fast MAS spinning frequencies were recorded for the formulations and as-received PTX, amorphous PTX and the neat polymer.Both the 13C CP MAS NMR spectra recorded at 24 kHz (Fig. 1a) as well as powder X-ray diffraction data (Fig. S5, ESI†) confirm that the formulations are amorphous materials in agreement with a single glass transition temperature published in a previous work by one of the authors.9 While crystalline paclitaxel (black spectrum) shows two sets of carbon resonances due to its two independent molecules in the asymmetric unit, amorphous PTX and an exemplary formulation (POL-9-PTX, highest PTX content) (grey and blue spectrum) show broad signals as expected for materials lacking long-range order and containing a distribution of environments and thus chemical shifts. Consequently, several moieties of PTX appear as joint signals complicating a straightforward extraction of spectral changes upon incorporation into the micelles in a similar way as recently shown for related polymer micelles loaded with curcumin as the model compound.19 Moreover, additional complications arise from the overlap between PTX and POL resonances as can be seen from a comparison with the 13C CP MAS spectrum of the neat polymer (purple spectrum). Note, that for better comparability, the neat polymer was heated above its glass transition temperature Tg (Tg(POL) = 56 °C)48 prior to the measurement to account for similar conditions during the preparation of the formulations. Consequently, the analysis of the carbon spectra and extraction of reliable structural information is difficult and requires complementary tools such as quantum chemical calculations, which will be explored in detail in a separate work.
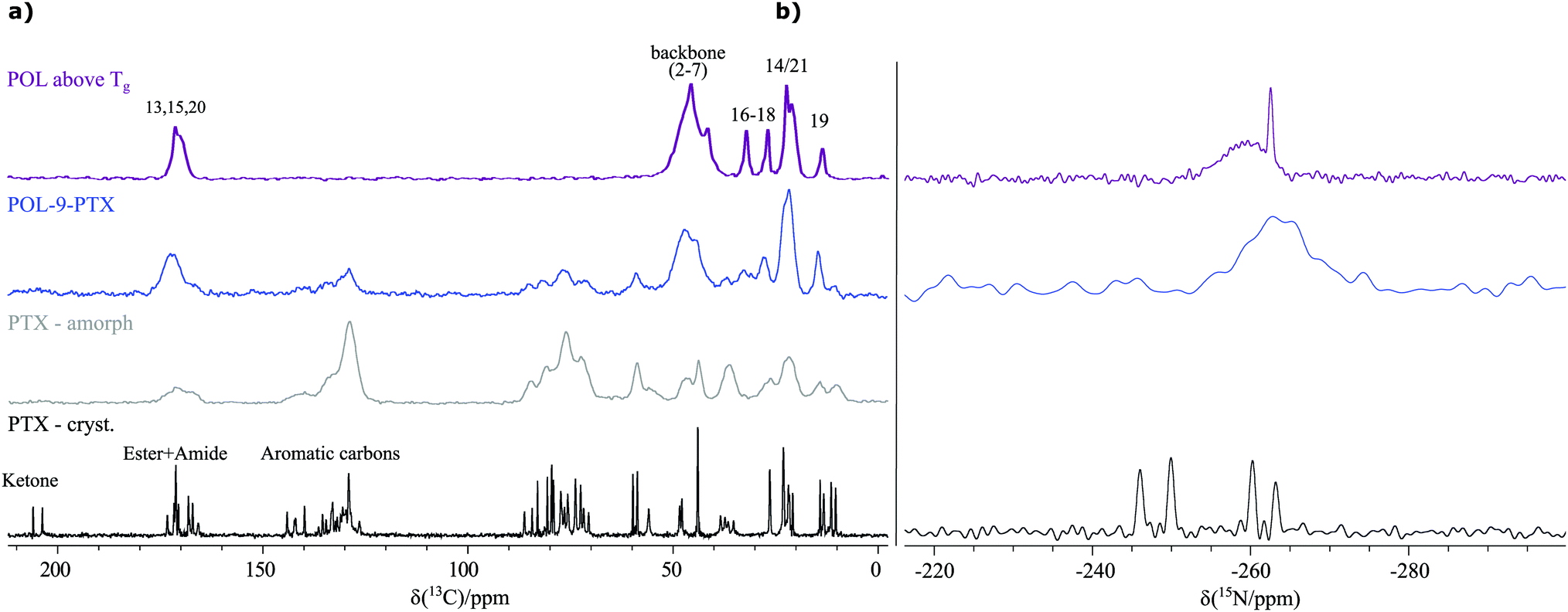 | ||
| Fig. 1 (a) 13C CP MAS NMR spectra of the pure polymer (purple), an exemplary PTX formulation (blue), amorphous (grey) and crystalline PTX (black). Partial peak assignment is indicated. All spectra were recorded at 14.1 T and 24 kHz MAS with a contact time of 2.5 ms and the following parameters (co-added transients/recycle delay): 1756/2.5 s (purple), 2048/2.0 s (blue), 23532/2.5 s (grey) and 1756/2.5s (black). (b) Corresponding 15N CP MAS NMR spectra recorded at 9.4 T (PTX, POL) and 20 T (POL-9-PTX) using a contact time of 1 ms for POL and 2 ms for PTX. 82796 (PTX), 16991 (POL) and 25600 (POL-9-PTX) co-added transients were measured with a recycle delay of 5.0 s (PTX, POL) and 3.0 s (POL-9-PTX) resulting in an overall experimental time of 4 d 19 h, 24 h and 21 h, respectively. Please note that the 15N NMR spectrum of crystalline PTX is the only dataset, which was recorded on a new batch of PTX containing two differently hydrated PTX phases and thus showing an additional set of signals. As PTX exists in its amorphous state in the formulations and is dissolved for preparation, the initial degree of incorporated water in the respective phase is not relevant for the analysis of the formulations. | ||
Ideally, for a thorough characterization of the formulations we would like to use the direct information from 1H NMR data at fast MAS due to the high sensitivity of the 1H nucleus to intermolecular interactions, through space proximities and subtle changes in the packing arrangement. The 1H MAS NMR spectra recorded at 60 kHz are shown as external projection in the corresponding 14N–1H HMQC spectra in Fig. 2 and 3. As can be seen, the spectra are dominated by severe signal overlaps despite being recorded in a high magnetic field (20 T). Consequently, spectral simplification is required and the presence of only one secondary amide moiety per PTX enables this through N–H correlation experiments. Additionally, the tertiary amide functional groups in the polymer could also serve as another source of structural information. Therefore, 15N CP MAS spectra were recorded and it is very interesting to note that the secondary amides from PTX (Z′ = 2, crystal structure in SI 10, ESI†) and the tertiary amide of the polymer show resonance in the same spectral region (Fig. 1b). For the POL-9-PTX formulation, only a very broad resonance can be observed, again in a similar spectral region and thus showing the need for a dispersion of the signals as expected for the quadrupolar 14N nucleus.
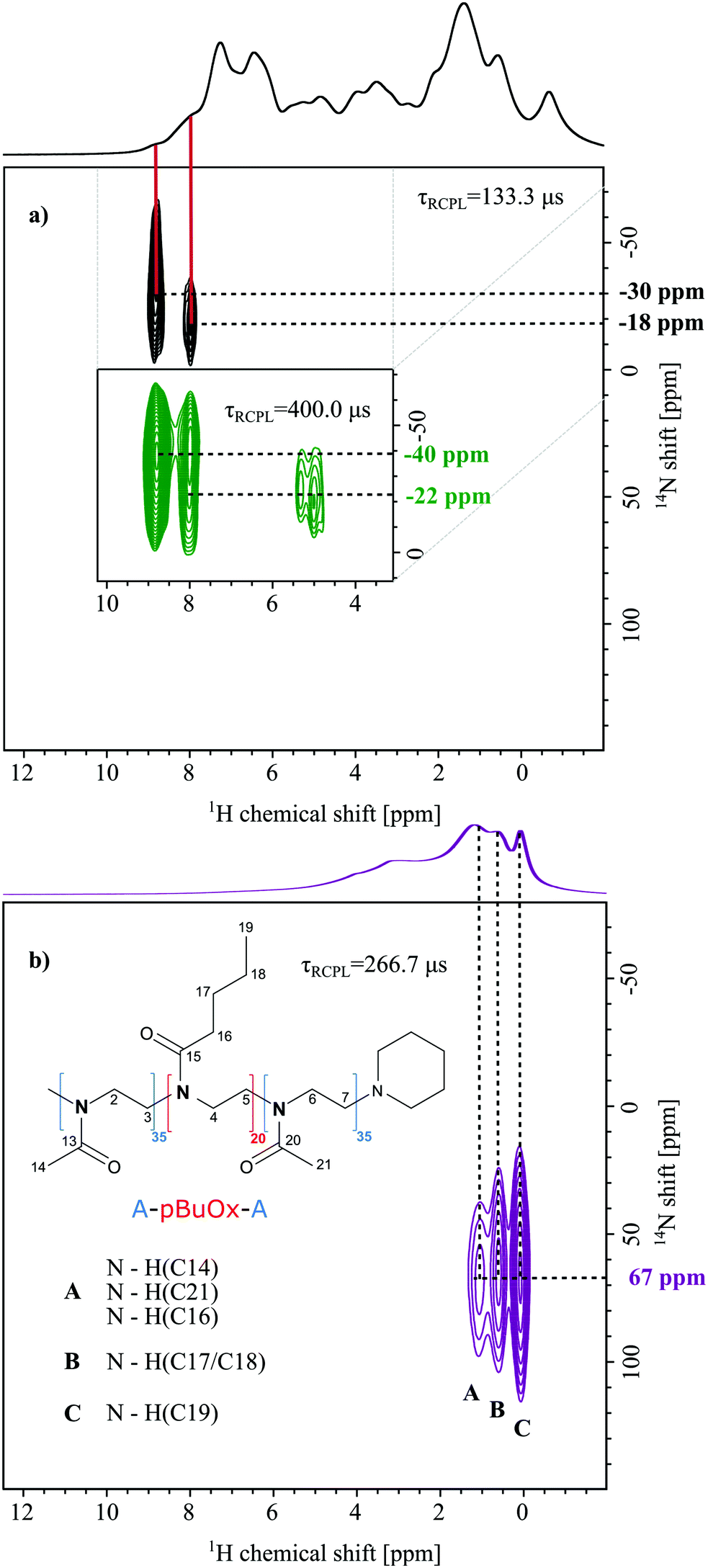 | ||
| Fig. 2 (a) 14N–1H HMQC spectra (20 T, 60 kHz) of crystalline PTX recorded with recoupling times of τRCPL = 133.3 μs, acquired with 34 t1 FIDs (black) and τRCPL = 400.0 μs, acquired with 98 t1 FIDs (green, as an inset), each with 128 co-added transients for a recycle delay of 1.5 s (experimental time: 1 h 50 min (black) and 5 h 18 min (green)). The base contour level is at 30% (black), 34% (green) and 22% of the maximum peak height. The red bars point out the corresponding peaks in the 1D 1H one-pulse MAS NMR spectrum. (b) The 14N–1H HMQC spectrum of POL recorded with a recoupling time τRCPL = 266.7 μs, 16 t1 FIDs, each with 2048 co-added transients for a recycle delay of 1.3 s (experimental time 1 h 30 min), and corresponding assignment of the N⋯H cross-peaks. The skyline projections correspond to 1H one-pulse MAS NMR spectra of each sample. | ||
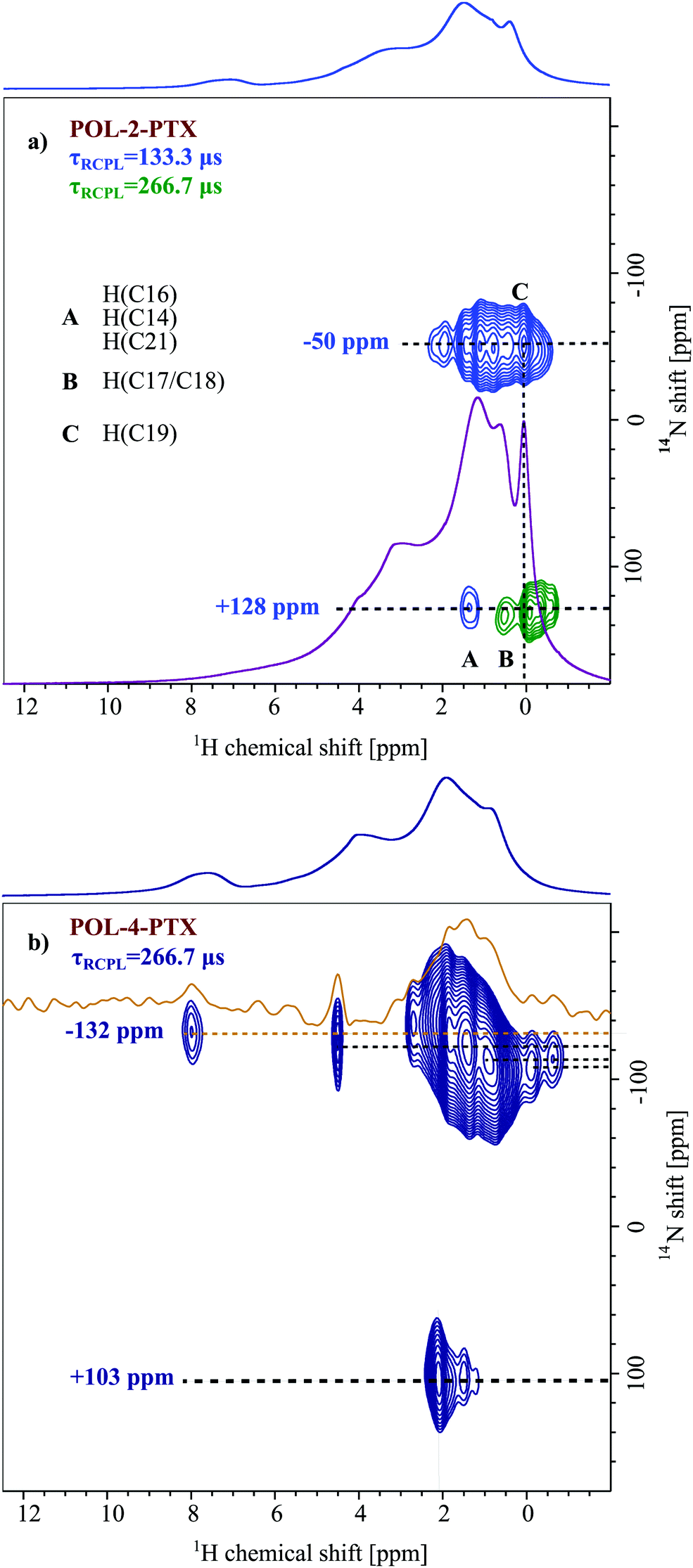 | ||
| Fig. 3 14N–1H HMQC spectra (20 T, 60 kHz) of (a) the POL-2-PTX formulation recorded with recoupling times τRCPL = 133.3 μs (light blue) and τRCPL = 266.7 μs (green), both acquired with 18 t1 FIDs, each with 1024 co-added transients for a recycle delay of 1.3 s (experimental time: 6 h 46 min), including tentative assignment of N⋯H proximities. The base contour level is at 34% of the maximum peak height. The internal overlaid 1H spectrum (purple) is that for POL. (b) The POL-4-PTX formulation recorded with a recoupling time τRCPL = 266.7 μs (dark blue), and 12 t1 FIDs each acquired with 1200 co-added transients for a recycle delay of 1.3 s (experimental time: 5 h 17 min). The internal overlaid spectrum (orange) is the extracted slice at a 14N shift of −132 ppm. The base contour level is at 22% of the maximum peak height. The skyline projections represent the 1H one-pulse MAS NMR spectra of each sample. | ||
14N–1H HMQC MAS NMR experiments are particularly powerful in this context, require only a small amount of sample and have been applied to study a variety of crystalline compounds.33,35 Therefore, in the first step, 14N–1H HMQC spectra of the pure compound were recorded to identify their nitrogen shifts and ensure sufficient separation of the PTX and polymer nitrogen environments.
Pure compounds
Fig. 2 shows 14N–1H HMQC spectra of crystalline PTX as well as of the pure polymer. For PTX, correlations with two different dipolar recoupling times were measured and are illustrated in black (133.3 μs) and green (400.0 μs). Similar recoupling times in the 14N–1H HMQC experiments were first tested for the dipeptide β-AspAla showing the expected appearance of longer range through space proximities upon increase of the recoupling time (see Fig. S6 (ESI†) and Tatton et al.28). For PTX, using short recoupling times and in agreement with the X-ray diffraction data, two cross peaks at 8.8/−30 ppm (NA) and 8.0/−18 ppm (NB) were observed for the NH groups of the two individual molecules of PTX in the asymmetric unit. Consequently, one can deduce that NA is involved in stronger hydrogen bonding than NB. This agrees with a longer direct NA⋯H distance, e.g. stronger involvement in hydrogen bonding, as observed in the crystal structure reported by Vella-Zarb et al for PTX anhydrate (Table 1, CSD code: RIGLEA).25| N⋯H proximity | Distance/Å |
|---|---|
| a Intermolecular proximities in italics. Mobile aromatic units are shown in bold. N and H atoms of interest are underlined. | |
![[N with combining low line]](https://www.rsc.org/images/entities/char_004e_0332.gif) A A![[H with combining low line]](https://www.rsc.org/images/entities/char_0048_0332.gif) |
0.979 |
| AH–CA32 |
1.642 |
| AH–CA3′ |
2.031 |
| AH–(CA2′–OH) |
2.601 |
![[N with combining low line]](https://www.rsc.org/images/entities/b_char_004e_0332.gif) A
H–C
A38
A
H–C
A38
![[H with combining low line]](https://www.rsc.org/images/entities/b_char_0048_0332.gif)
|
2.887 |
| B |
0.846 |
| BH–CB3′ |
1.915 |
| BH–(CB2′H–O) |
2.486 |
![[N with combining low line]](https://www.rsc.org/images/entities/i_char_004e_0332.gif) B
H–C
A31
B
H–C
A31
![[H with combining low line]](https://www.rsc.org/images/entities/i_char_0048_0332.gif) 3
3
|
2.787 |
|
B
H–C
B32
|
2.965 |
With a three times higher recoupling time, two changes of the previous peaks for shorter recoupling time are noticeable: the intensity of the peak corresponding to nitrogen NB increases compared to NA, the peak seems elongated in the 14N dimension and two additional signals at around 5 ppm of proton chemical shift are observed. Small changes in the 14N shift (NA: −30 to −40 ppm and NB: −18 to −22 ppm) could be caused by a temperature increase in the sample caused by the longer irradiation through the recoupling blocks. As the recoupling time for the dipolar couplings increases, it is possible to sample longer N⋯H distances through space. Consequently, the relative signal increase of NB can be explained by proximities of NA and NB to aromatic protons, whose chemical shifts are overlapping with those of the proton attached to NB in agreement with an increased 1H intensity at 8.0 ppm. To confirm this, intra- and intermolecular close 14N⋯1H distances of up to 3 Å were extracted from PTX crystallographic data (Table 1, CSD code: RIGLEA). The threshold was chosen based on the maximum distances previously observed for comparable recoupling times.28 Indeed, the shortest contact for N1 is found for the ortho-CH of the adjacent aromatic ring denoted as carbon 32 in the ESI.† In turn, the shortest NH distances for the second molecule were observed for the adjacent aliphatic CH and OH units (C3′H and C2′OH), which could explain the cross peaks observed at 5.0 and 5.3 ppm in the 1H dimension. However, the distances in Table 1 can only serve as indication and should be viewed carefully as these distances originate from PXRD data at high temperature (360 K). Furthermore, it is still not fully understood why, for some small molecule compounds, fewer contacts than expected are observed.42 In the 14N–1H HMQC MAS NMR spectrum of the neat polymer (Fig. 2b) recorded at an intermediate recoupling time of 266.7 μs (no direct NH), three cross peaks at a 14N shift of 67 ppm and distinct 1H chemical shifts are observed. Peak A results from the through space proximity of the nitrogen with the methyl groups C14 and C21 as well as the methylene group C16. Peak B indicates proximity to the CH2 unit of the hydrophobic butyl sidechain (C17/18) and peak C is related to N⋯H proximities including the methyl protons of the butyl sidechain (C19). Please note, that due to micelle formation, these cross-peaks are most likely of intermolecular nature. Interestingly, no cross peaks between N and the polymer backbone CH2 groups could be observed. For a more detailed discussion on the signal assignment and appearance of specific cross-peaks, the reader is referred to the SI 4 (ESI†). Overall, the comparison of the two individual components shows that their nitrogen environments can be clearly distinguished in the 14N–1H HMQC experiment, which is not the case if 15N chemical shifts are observed (see Fig. 2b and Table 1). Additionally, distinct correlation peaks are obtained in the 2D NMR spectra significantly reducing the spectral complexity in the 1H dimension. This is an essential prerequisite for the following analysis of the formulations. While revealing valuable intra- and intermolecular N⋯H proximities, the HMQC experiments also feature considerably shorter experimental times (1 h 30 min up to 7 h) than the 1D 15N experiments (min 1 d), while also using significantly lower samples volumes (80 μL in 4 mm rotors vs. 1.5 μL in 1.3 mm rotors).
Formulations
Fig. 3 depicts 14N–1H HMQC MAS NMR spectra of two differently loaded formulations (a) POL-2-PTX and (b) POL-4-PTX, both with their 1H MAS NMR spectra as external projections. In both spectra, two different 14N shifts, one at positive and one at negative ppm values, can be observed with the cross-peaks at negative values being more intense than the corresponding signals at positive 14N shifts. For the formulation with the lowest PTX loading (POL-2-PTX), all cross-peaks appear in the aliphatic 1H chemical shift region below 2 ppm, the extracted 14N shifts are −50 and +128 ppm. With increasing PTX loading of the formulations, both nitrogen shifts decrease to −132 and +103 ppm. Taking a closer look at the characteristic peaks for the medium loaded formulation, POL-4-PTX, cross-peaks are again observed in the aliphatic 1H region and additional cross peaks at a proton chemical shift of 4.5 and 8.0 ppm can be clearly distinguished alongside weaker signals at 5.6 and 6.4 ppm (orange slice). The high 1H chemical shift values are indicative of aromatic or hydrogen bonding environments. Interestingly, for this sample and in contrast to POL-2-PTX, multiple, defined 14N shifts between −135 and −110 ppm are observed.To complete the set of differently loaded formulations and subsequently extract trends, which might reveal information on changes of the local environment in the formulations, 14N–1H HMQC data were also recorded for POL-9-PTX, a formulation with almost 50 wt% PTX loading (Fig. 4). Due to hardware problems (untraceable spikes appearing in the FIDs) and despite multiple measurement attempts at different times (August 2019 and November 2019), unfortunately no artefact free dataset could be recorded. Therefore, the 14N–1H HMQC spectrum for POL-9-PTX was generated by addition of the individual FIDs of several datasets with different recoupling times ranging from 133.3 to 533.3 μs. The averaging of different datasets comes at the cost of resolution, particularly in the 1H dimension. Consequently, only the 14N shifts and relative signal intensities are discussed in the course of this work. The values −273 and +86 ppm are consistent with the observed decrease in shift upon increasing the PTX loading.
 | ||
| Fig. 4 14N–1H HMQC (20 T, 60 kHz) spectrum of the POL-9-PTX formulation. The base contour level is at 34% of the maximum peak height. The skyline projection shows the corresponding 1H one-pulse MAS NMR spectrum. Due to problems with the probe, the recorded datasets for this sample contained artefacts. Therefore, multiple datasets with different recoupling times varying from τRCPL = 133.3 μs to τRCPL = 533.3 μs (recycle delay of 1.0 s) were added to obtain the spectrum. Consequently, only the 14N shift values were used for comparison. | ||
All extracted values for both the individual components and the formulations are summarized in Table 1, which also includes selected 15N chemical shifts extracted from the spectra shown in Fig. 1b. This first examination of the 14N–1H HMQC spectra of the three formulations is very promising as two different nitrogen environments can be distinguished for each sample, which follow a clear trend of decreasing 14N shift with increasing loading. An analogous trend could not have been observed based on 15N solid-state NMR data as indicated by the similarity of the extracted chemical shifts for the polymer and the formulation with highest PTX loading.
Discussion
In the next step, the observed trends and key N⋯H proximities have to be analyzed and transformed into chemical knowledge to improve our understanding of the studied formulations on the molecular level. To do so, 14N quadrupolar shifts δQiso (for detailed explanation see SI 1, ESI†) can be estimated based on the hypothesis that the 14N isotropic chemical shift are approximately identical to the 15N isotropic chemical shifts. Consequently, the quadrupolar shift values can be determined by subtraction giving the values indicated in Table 2. If available, the corresponding experimental 15N chemical shifts were used. For all formulations, a 15N value of −263 ppm was used. For as-received and amorphous PTX, relatively large 14N quadrupolar shifts of around 200 ppm are observed, which is indicative of the more asymmetric environment of the 14N within a secondary amide. The data for the pure polymer, which only contains tertiary amides, reveals even larger quadrupolar shifts of above 300 ppm. Interestingly, the resulting quadrupolar shifts of all three formulations remain similarly sized for the 14N signal observed at positive ppm values, which is further comparable in size to the neat polymer, while the quadrupolar shift of the 14N signal at negative ppm values is decreasing with increasing PTX loading. This indicates a more symmetric N-environment at higher PTX loadings with the very low experimentally determined quadrupolar shift for POL-9-PTX verging on that of an almost tetrahedral arrangement. How can this major change in symmetry be explained?| δ 14N neg. ppm | δ 14N pos. ppm | δ 15N/ppm | δQiso/ppm | |
|---|---|---|---|---|
| a Calculated as δ(14N)–δ(15N) using an average 15N chemical shift of −255 ppm. | ||||
| Crystalline PTX | −18/−30 | — | −246/−260 | 228/230 |
| Amorphous PTX | −55/−70 | — | 200a/185a | |
| Pure POL | — | +67 | −258/−263 | 325/330 |
| POL-2-PTX | −50 | +128 | 205/383 | |
| POL-4-PTX | −132 | +103 | 123/358 | |
| POL-9-PTX | −273 | +86 | −263 | −10/349 |
To approach this question, it is first necessary to discuss the assignment of the two 14N shifts observed for the formulations. At a first glance, the 2D NMR data of individual components, PTX and POL, showing positive as well as negative 14N shifts in a similar region to the two shifts found for the formulations, suggest an analogous assignment for these two environments. However, for the lowest PTX loading (POL-2-PTX), only 2.2% of all nitrogen atoms in the sample belong to PTX, while the majority of nitrogen atoms originate from the polymer. Furthermore, no intramolecular NH contact like the one found in pure PTX is observed in the formulation with the lowest loading. For POL-4-PTX and POL-9-PTX, 4.5% and 10.1% of the nitrogen atoms in the sample are found in PTX molecules. With this in mind, we hypothesize that both 14N signals arise from polymeric environments, the more intense signal at negative ppm values being assigned to amides in close proximity to PTX molecules acting as hydrogen bond donors and the signal at positive ppm resulting from the remaining amide fragments of the triblock copolymer. This agrees with the signal at negative ppm gaining intensity with respect to the other 14N environment upon increasing the PTX loading. The tertiary amides can act as hydrogen bond acceptors and in doing so the nitrogen environment gets more symmetric, which results in a smaller quadrupolar shift. For example, this has been observed for small di- and oligopeptides, where nitrogen atoms with three bonds and thus a less tetrahedral environment due to the lone pair showed high second order quadrupolar shifts, while in the case of zwitterionic RNH3+ environments (proton transfer being the extreme case of hydrogen bonding interaction) only a very small shift was observed.28,36 Finally, the formulation with medium loading gives further insights into the drug–polymer assembly. While the amount of PTX in POL-2-PTX is very low and the 14N–1H HMQC is dominated by polymer–polymer contacts, distinct NH contacts with protons resonating above 4 ppm can be observed for the 14N at negative ppm values in the 14N–1H HMQC spectrum of POL-4-PTX. All these cross-peaks can only arise from intermolecular drug–polymer contacts as the polymer itself does not show any proton chemical shifts above 3.9 ppm. Specifically, cross-peaks at 8.0, 6.4, 5.6 and 4.5 ppm were observed. In comparison with the NMR spectroscopy data of pure PTX, the cross-peak above 8 ppm can be assigned to a tertiary amide⋯HN proximity, while the remaining cross-peaks could result from interactions between the amide and different OH environments within PTX. Additionally, the spectrum of POL-4-PTX contains diverse 14N shifts. As this feature is neither observed for the neat polymer nor for POL-2-PTX, where only one distinct 14N shift is observed, it must be attributed to the increasing amounts of PTX present in the nanoparticles. Taking a closer look at the known structures of PTX with increasing number of water molecules could help to shed light on this: in the three structures published by Vella-Zarb et al.,25 the NH group and all three OH-groups act as hydrogen bond donors to adjacent PTX and water molecules suggesting that this would also be the case for PTX–polymer interactions. The polymer amide nitrogen atoms serve as the hydrogen bond acceptor. From the previous work on similar polymers with PTX and curcumin,11,19,49 we know that, with increasing loading, the poorly water-soluble guest not only interacts with the more hydrophobic pBuOx polymer block, but also with the hydrophilic pMeOx units. The critical drug loading, where the corona become significantly involved appears to be between 20–30 wt%. This would explain, why we don’t observe this for the lowest loading. Furthermore, steric hindrance between the different PTX interaction sites and pBuOx vs. pMeOx would result in a differing deviation from ideal hydrogen bond geometries with respect to bond distances and bond angles, while, overall, the hydrogen bonded amide nitrogen atom would be found in a more symmetric, closer to tetrahedral environment as compared to the planar, sp2 hybridized initial environment (Fig. 5).
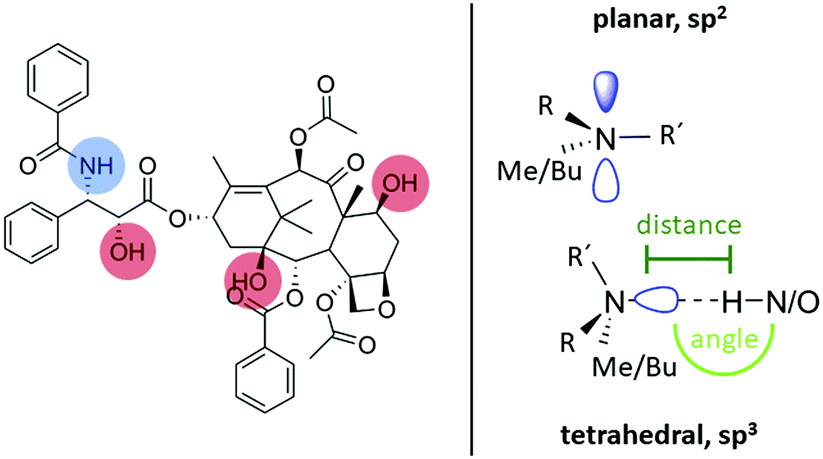 | ||
| Fig. 5 Summary of the hydrogen bond donating moieties in PTX alongside nitrogen atom geometries and environments for initial tertiary amides and hydrogen bonded functionalities. | ||
This underlines that 14N–1H HMQC NMR experiments are not only spreading out the nitrogen NMR signals but are also a promising tool to study the local symmetry and detect structure determining intermolecular interactions in complex and amorphous drug–polymer formulations. To further improve the structural understanding, these finding now need to be complemented by additional experimental and detailed theoretical data.
Summary and conclusion
In this work, we investigated amorphous polymer formulations of the anti-cancer drug paclitaxel. For both the polymer and the drug, 15N chemical shifts in a very similar spectral area were observed, which results in significant signal overlap, especially if amorphous samples are involved. Consequently, for this set of samples, it is very challenging to distinguish small changes in the spectra, extract trends and information on the interactions of specific moieties, e.g. upon increasing the drug loading of the formulations. Therefore, 14N–1H HMQC NMR experiments were investigated as a valuable tool to disperse signals due to the additional quadrupolar shift when observing 14N instead of 15N. We could show that such experiments can be a rich source of information for the characterization of molecular interactions in amorphous polymer–drug nanoformulations, which is otherwise difficult to obtain. On the one hand, the 14N signals were dispersed so that two distinct 14N environments could be extracted for each formulation and the complexity in the 1H dimension could be reduced through this correlation experiment. Thus, it was possible to extract trends for different drug loadings, distinguish cross peaks and extract quadrupolar parameters, which altogether enabled us to learn about the local amide environment, its symmetry and potential interacting motifs between the drug and polymer in such complex amorphous mixtures. Overall, the 14N–1H HMQC NMR experiments have great potential for the analysis of disordered and amorphous drug-delivery systems, which can be transferred and should be explored for other systems. Here, the 14N–1H experiments could also be insightful for nanocrystalline materials such as those found in Pluronic F127 stabilized PTX nanocrystals.50It is of importance to stress for the broader applicability of the 14N–1H HMQC experiment that while fast MAS (≥60 kHz) is essential for these experiments due to the narrowing of the 1H signal and most importantly increased 1H coherence lifetimes,28 the experiments are not limited to high magnetic fields. With the second order quadrupolar interaction being inversely proportional to the strength of the magnetic field, narrower lines are obtained at higher fields. However, a larger dispersion of the signals is counterbalancing this at lower magnetic field strengths. In fact, many examples for the application of this and other 14N based experiments in the literature show their feasibility at 400–600 MHz.30,34,36,51
Furthermore, in a next step we intend to combine these experiments with additional data and complementary calculations to learn more about these PTX formulations and ultimately derive realistic structural models on a molecular level. In this context, molecular dynamics simulations could provide important insights due to the possibility to reproduce distributions of environments encountered for amorphous drug–polymer formulations.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Dr Michael Lübtow for providing the samples and Dr Matthias Grüne for helpful discussion and proofreading of the manuscript. Dr Rüdiger Bertermann kindly measured the 15N data of the individual components. We further thank Dominik Heuler for his support with the PXRD measurements. This work was supported by the Newton International Fellowship Alumni Follow on Funding of the Royal Society (AL\180018). The UK 850 MHz solid-state NMR Facility used in this research was funded by the EPSRC and the BBSRC (contract reference PR140003), and the University of Warwick including part funding from the Birmingham Science City Advanced Materials Projects 1 and 2 supported by Advantage West Midlands (AWM) and the European Regional Development Fund (ERDF). The experimental data for this study are provided as a supporting dataset from WRAP, the Warwick Research Archive Portal at https://wrap.warwick.ac.uk/136941.Notes and references
- P. Ma and R. J. Mumper, J. Nanomed. Nanotechnol., 2013, 4, 1000164 CrossRef PubMed.
- T.-H. Wang, H.-S. Wang and Y.-K. Soong, Cancer, 2000, 88, 2619–2628 CrossRef CAS PubMed.
- A. M. Sofias, M. Dunne, G. Storm and C. Allen, Adv. Drug Delivery Rev., 2017, 122, 20–30 CrossRef CAS PubMed.
- Y. S. Youn and Y. H. Bae, Adv. Drug Delivery Rev., 2018, 130, 3–11 CrossRef CAS PubMed.
- Z. He, A. Schulz, X. Wan, J. Seitz, H. Bludau and D. Y. Alakhova, et al. , J. Controlled Release, 2015, 208, 67–75 CrossRef CAS PubMed.
- Y. Fujiwara, H. Mukai, T. Saeki, J. Ro, Y.-C. Lin and S. E. Nagai, et al. , Br. J. Cancer, 2019, 120, 475 CrossRef CAS PubMed.
- Z. He, X. Wan, A. Schulz, H. Bludau, M. A. Dobrovolskaia and S. T. Stern, et al. , Biomaterials, 2016, 101, 296–309 CrossRef CAS PubMed.
- R. Luxenhofer, A. Schulz, C. Roques, S. Li, T. K. Bronich and E. V. Batrakova, et al. , Biomaterials, 2010, 31, 4972–4979 CrossRef CAS PubMed.
- A. Schulz, S. Jaksch, R. Schubel, E. Wegener, Z. Di and Y. Han, et al. , ACS Nano, 2014, 8, 2686–2696 CrossRef CAS PubMed.
- S. Jaksch, A. Schulz, Z. Di, R. Luxenhofer, R. Jordan and C. M. Papadakis, Macromol. Chem. Phys., 2016, 217, 1448–1456 CrossRef CAS.
- B. Sochor, Ö. Düdükcü, M. M. Lübtow, B. Schummer, S. Jaksch and R. Luxenhofer, Langmuir, 2020, 36, 3494–3503 CrossRef CAS PubMed.
- R. F. Moran, D. M. Dawson and S. E. Ashbrook, Int. Rev. Phys. Chem., 2017, 36, 39–115 Search PubMed.
- S. E. Ashbrook and P. Hodgkinson, J. Chem. Phys., 2018, 149, 040901 CrossRef PubMed.
- P. Florian and F. Fayon, Disordered Solids. Modern Methods in Solid-state NMR: A Practitioner's Guide, The Royal Society of Chemistry, 2018, ch. 12, pp. 356–90 Search PubMed.
- R. K. Harris, J. Pharm. Pharmacol., 2007, 59, 225–239 CrossRef CAS PubMed.
- H. G. Brittain. Polymorphism in Pharmaceutical Solids, CRC Press, 2nd edn, 2009 Search PubMed.
- S. Baghel, H. Cathcart and N. J. O'Reilly, J. Pharm. Sci., 2016, 105, 2527–2544 CrossRef CAS PubMed.
- M. Callari, P. L. De Souza, A. Rawal and M. H. Stenzel, Angew. Chem., Int. Ed., 2017, 56, 8441–8445 CrossRef CAS PubMed.
- A. C. Pöppler, M. M. Lübtow, J. Schlauersbach, J. Wiest, L. Meinel and R. Luxenhofer, Angew. Chem., Int. Ed., 2019, 58, 18540–18546 CrossRef PubMed.
- D. A. Hirsh, A. V. Wijesekara, S. L. Carnahan, I. Hung, J. W. Lubach and K. Nagapudi, et al. , Mol. Pharmaceutics, 2019, 16, 3121–3132 CrossRef CAS PubMed.
- S. K. Vasa, P. Rovó and R. Linser, Acc. Chem. Res., 2018, 51, 1386–1395 CrossRef CAS PubMed.
- S. P. Brown, Solid State Nucl. Magn. Reson., 2012, 41, 1–27 CrossRef CAS PubMed.
- R. Zhang, K. H. Mroue and A. Ramamoorthy, Acc. Chem. Res., 2017, 50, 1105–1113 CrossRef CAS PubMed.
- Y. Nishiyama, Solid State Nucl. Magn. Reson., 2016, 78, 24–36 CrossRef CAS PubMed.
- L. Vella-Zarb, U. Baisch and R. E. Dinnebier, J. Pharm. Sci., 2013, 102, 674–683 CrossRef CAS PubMed.
- J. K. Harper, D. H. Barich, E. M. Heider, D. M. Grant, R. R. Franke and J. H. Johnson, et al. , Cryst. Growth Des., 2005, 5, 1737–1742 CrossRef CAS.
- E. M. Heider, J. K. Harper and D. M. Grant, Phys. Chem. Chem. Phys., 2007, 9, 6083–6097 RSC.
- A. S. Tatton, J. P. Bradley, D. Iuga and S. P. Brown, Z. Phys. Chem., 2012, 226, 1187–1204 CrossRef CAS.
- A. S. Tatton, T. N. Pham, F. G. Vogt, D. Iuga, A. J. Edwards and S. P. Brown, Mol. Pharmaceutics, 2013, 10, 999–1007 CrossRef CAS PubMed.
- E. K. Corlett, H. Blade, L. P. Hughes, P. J. Sidebottom, D. Walker and R. I. Walton, et al. , Solid State Nucl. Magn. Reson., 2020 DOI:10.1016/j.ssnmr.2020.101662.
- A. G. M. Rankin, J. Trébosc, P. Paluch, O. Lafon and J.-P. Amoureux, J. Magn. Reson., 2019, 303, 28–41 CrossRef CAS PubMed.
- G. N. M. Reddy, D. S. Cook, D. Iuga, R. I. Walton, A. Marsh and S. P. Brown, Solid State Nucl. Magn. Reson., 2015, 65, 41–48 CrossRef CAS PubMed.
- Z. Gan, J. P. Amoureux and J. Trebosc, Chem. Phys. Lett., 2007, 435, 163–169 CrossRef CAS.
- S. Cavadini, Prog. Nucl. Magn. Reson. Spectrosc., 2010, 56, 46–77 CrossRef CAS PubMed.
- K. Maruyoshi, D. Iuga, O. N. Antzutkin, A. Alhalaweh, S. P. Velaga and S. P. Brown, Chem. Commun., 2012, 48, 10844–10846 RSC.
- Y. l. Hong, T. Asakura and Y. Nishiyama, ChemPhysChem, 2018, 19, 1841–1845 CrossRef CAS PubMed.
- A. S. Tatton, T. N. Pham, F. G. Vogt, D. Iuga, A. J. Edwards and S. P. Brown, CrystEngComm, 2012, 14, 2654–2659 RSC.
- A. S. Tatton, H. Blade, S. P. Brown, P. Hodgkinson, L. P. Hughes and S. O. N. Lill, et al. , Cryst. Growth Des., 2018, 18, 3339–3351 CrossRef CAS.
- D. Bernasconi, S. Bordignon, F. Rossi, E. Priola, C. Nervi and R. Gobetto, et al. , Cryst. Growth Des., 2020, 20, 906–915 CrossRef CAS.
- E. Vitaku, D. T. Smith and J. T. Njardarson, J. Med. Chem., 2014, 57, 10257–10274 CrossRef CAS PubMed.
- S. Cavadini, A. Abraham and G. Bodenhausen, Chem. Phys. Lett., 2007, 445, 1–5 CrossRef CAS.
- S. P. Brown, High-resolution 1H 2D Magic-angle Spinning Techniques for Organic Solids. Modern Methods in Solid-state NMR, A Practitioner's Guide: The Royal Society of Chemistry, 2018, ch. 2, pp. 39–74 Search PubMed.
- M. M. Lübtow, L. Hahn, M. S. Haider and R. Luxenhofer, J. Am. Chem. Soc., 2017, 139, 10980–10983 CrossRef PubMed.
- T. Oas, R. Griffin and M. Levitt, J. Chem. Phys., 1988, 89, 692–695 CrossRef CAS.
- P. Costa, J. Gross, M. Hong and R. Griffin, Chem. Phys. Lett., 1997, 280, 95–103 CrossRef CAS.
- B. Fung, A. Khitrin and K. Ermolaev, J. Magn. Reson., 2000, 142, 97–101 CrossRef CAS PubMed.
- C. R. Morcombe and K. W. Zilm, J. Magn. Reson., 2003, 162, 479–486 CrossRef CAS.
- M. M. Lübtow, M. S. Haider, M. Kirsch, S. Klisch and R. Luxenhofer, Biomacromolecules, 2019, 20, 3041–3056 CrossRef PubMed.
- M. S. Haider, M. M. Lübtow, S. Endres, V. Aseyev, A.-C. Pöppler and R. Luxenhofer, ACS Appl. Mater. Interfaces, 2020, 12, 24531–24543 CrossRef CAS PubMed.
- J. Deng, L. Huang and F. Liu, Int. J. Pharm., 2010, 390, 242–249 CrossRef CAS PubMed.
- D. Carnevale, X. Ji and G. Bodenhausen, J. Chem. Phys., 2017, 147, 184201 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0tb00614a |
| This journal is © The Royal Society of Chemistry 2020 |