
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe rise of bio-inspired polymer compartments responding to pathology-related signals
Luisa
Zartner
,
Moritz S.
Muthwill
 ,
Ionel Adrian
Dinu
,
Cora-Ann
Schoenenberger
and
Cornelia G.
Palivan
*
,
Ionel Adrian
Dinu
,
Cora-Ann
Schoenenberger
and
Cornelia G.
Palivan
*
Chemistry Department, University of Basel, Mattenstr. 24a, BPR1096, Basel, Switzerland. E-mail: cornelia.palivan@unibas.ch
First published on 1st May 2020
Abstract
Self-organized nano- and microscale polymer compartments such as polymersomes, giant unilamellar vesicles (GUVs), polyion complex vesicles (PICsomes) and layer-by-layer (LbL) capsules have increasing potential in many sensing applications. Besides modifying the physicochemical properties of the corresponding polymer building blocks, the versatility of these compartments can be markedly expanded by biomolecules that endow the nanomaterials with specific molecular and cellular functions. In this review, we focus on polymer-based compartments that preserve their structure, and highlight the key role they play in the field of medical diagnostics: first, the self-assembling abilities that result in preferred architectures are presented for a broad range of polymers. In the following, we describe different strategies for sensing disease-related signals (pH-change, reductive conditions, and presence of ions or biomolecules) by polymer compartments that exhibit stimuli-responsiveness. In particular, we distinguish between the stimulus-sensitivity contributed by the polymer itself or by additional compounds embedded in the compartments in different sensing systems. We then address necessary properties of sensing polymeric compartments, such as the enhancement of their stability and biocompatibility, or the targeting ability, that open up new perspectives for diagnostic applications.
Introduction
There is a growing demand for new approaches that allow for the easy and sensitive detection of pathological conditions. Thus, engineering novel polymer-based materials that are able to sense in a fast and specific manner different parameters associated with different states of diseases is a compelling research topic. Moreover, the precise detection and control of fluctuations in analyte bioactivity or concentration will lead to a better understanding of self-regulation processes in living organisms. Predominant environmental changes that can trigger a specific response from a polymer assembly include the variation of pH or ionic strength, the presence of reactive oxygen species, redox agents and other bio- or chemical molecules.9–15 Externally applied stimuli, such as light, temperature, ultrasound and electrical or magnetic fields can also trigger a response in polymer-based assemblies,12,17–20 but we did not include them in this overview.To act as biosensors, polymer systems should minimally consist of two main components: (i) a receptor molecule or moiety that functions as a recognition element by specifically interacting with the stimulus and (ii) a transducer that converts the analyte–receptor interaction into a measurable signal. For example, a specific pathological condition can trigger a detectable color transition or a change in fluorescence in a polymer assembly specifically designed to respond to such changes by the incorporation of dye molecules.3,22–25
Among the novel responsive materials that are being developed as biosensors, polymer systems play a major role because they offer multiple advantages including a large diversity of chemical features, variable molecular weight and composition of the repeating units, changeable functionalities and a variety of architectures. In this review, we present spherical polymer compartments with dimensions from the nano to the microscale and how they are rendered responsive to changes in their environment. First, we briefly introduce the synthesis strategies of polymer building blocks that assemble compartments with a spherical architecture: vesicles, polyion complex vesicles (PICsomes) and layer-by-layer (LbL) capsules. The main driving forces that promote the self-organization of individual polymer chains into such compartments are: (i) hydrophobic interactions that induce the segregation of polymer blocks during the self-assembly of amphiphilic block copolymers (vesicles) and (ii) electrostatic interactions between oppositely charged polymer blocks and/or homopolymers (PICsomes and LbL capsules).10,27–35 There are a number of excellent reviews that summarize the recent advances in the development of polymeric nanovesicles and other assemblies for biomedical applications.13,36–43 These reviews predominantly discuss systems that respond to one or multiple stimuli by disassembling the polymeric membrane, leading to the release of the dye or a drug cargo. Moreover, many of the stimuli that these systems respond to are externally applied.
Here, we want to provide an overview over the current progress in the development of polymeric structures that stay intact for extended periods of time and thus, have the potential to function as sensors under pathological conditions. We focus on polymeric compartments because they allow the encapsulation of hydrophilic cargoes, the insertion of hydrophobic molecules, or even the simultaneous encapsulation and insertion thereof.2,10,28,36,45–48 Moreover, the efficacy of polymer compartments can be increased by localizing them via targeting moieties to specific sites in the body.15,29,45,49
Specifically, we present how polymer biosensors can be tailored to change their properties in response to the presence of stimuli in a bioinspired manner based on the behavior of natural organelles inside cells or cells that respond to specific intracellular or intercellular signals while preserving their integrity. Stimuli-responsiveness is a key property both for drug delivery systems where physical or chemical changes of the polymer induce the release ‘on demand’ of the encapsulated cargo, and for sensors that detect changes associated with pathological conditions. While in the case of drug delivery systems, the majority of compartments are designed to rupture and release their cargo,8,24,37,50–56 in sensors, the compartment integrity should be preserved in the presence of a stimulus, preferably for a long period of time.
We review different approaches to design compartments for sensing purposes: either by combining “reporter” molecules with compartments made of intrinsically stimuli-responsive polymers, or by the incorporation/insertion of sensitive biomolecules into non-responsive compartments for converting the stimulus into a detectable signal. The second type of “sensing” compartment depends on the insertion/attachment of a stimuli-responsive biomolecule or on the encapsulation of active molecules able to perform an in situ reaction inside the compartment. This type of compartment is known as nanoreactors or catalytic compartments that represent simple bio-inspired models for cells or organelles. They have served as confined spaces for model reactions, as biosensors, or for the design of functional compartments with higher complexity, such as artificial organelles and cell mimics.2,16,21,57–70 In this review, we include catalytic compartments with demonstrated biosensing abilities and systems with other functions, which also have potential for biosensing.
There are numerous systems proposed as carriers for contrast agents, dyes or other imaging molecules/nano-objects that specifically improve the imaging of pathological regions. Here, the carrier containing the “imaging” component is usually stable, without response to specific stimuli that are associated with the respective pathological conditions. However, we provide a complementary overview of bio-inspired compartments at the nano- and micro-scale whose response to the presence of stimuli together with a rapid signal will open new avenues for the efficient biosensing of various pathological conditions.
Polymer building blocks for the assembly of compartments
The building blocks of compartments are polymers and copolymers that are based on a variety of hydrophobic and hydrophilic monomers, and are synthesized by various routes.71–77 Depending on their chemical nature, the resulting homo- and copolymers serve to build either compartments with intrinsic stimuli-responsive properties (Table 1) or compartments with stable, non-responsive architectures that can be combined with stimuli-responsive molecules (Table 2). Conventional free radical polymerization (free RP) can be employed in the polymerization of vinyl monomers under mild conditions. However, polymers synthesized via this technique have limited use in the preparation of spherical compartments as biosensors (only for LbL capsules) since with this method there is no control over the molecular weight, polydispersity, chemical composition, chain architecture or end-group functionality. To obtain homo-polymers and copolymers with well-defined properties and narrow polydispersity, several synthetic strategies have been developed, such as: (a) living ionic polymerization, (b) controlled/quasi-‘living’ radical polymerization (CRP), and (c) ring-opening polymerization (ROP). As all these synthetic pathways have been comprehensively reviewed,71–75,78–83 we only present a short description thereof and point at their advantages and limitations.| Polymer building block | Abbreviation | Structure | Method of synthesis | pKa values | Ref. |
|---|---|---|---|---|---|
| Hydrophilic | |||||
| Poly(acrylic acid) | PAA |
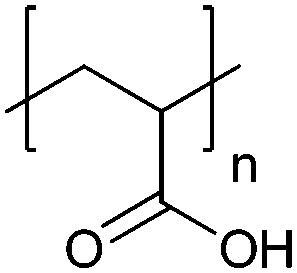
|
Free RP, CRP | ∼4.5 | 102 and 103 |
| Poly(styrene sulfonate) | PSS |
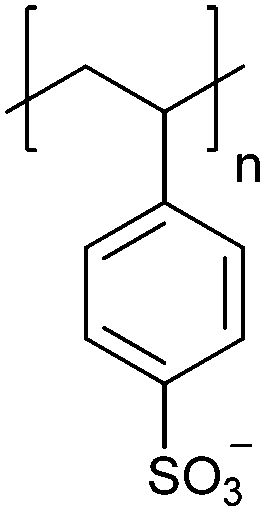
|
CRP | ∼1.6 | 104–106 |
| Poly(aspartic acid) | PAsp |
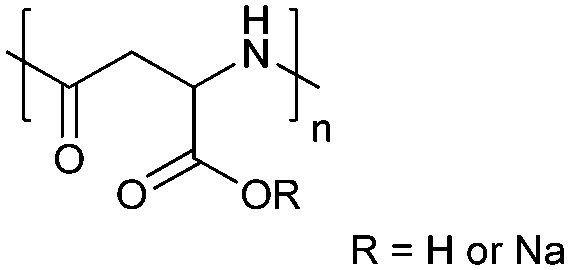
|
ROP | ∼3.7 | 52, 60 and 107 |
| Poly(allylamine hydrochloride) | PAH |
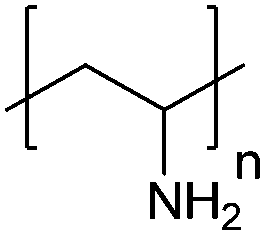
|
Free RP | ∼8.5 | 106, 108 and 109 |
| Poly[2-(dimethylamino)ethyl methacrylate] | PDMAEMA |
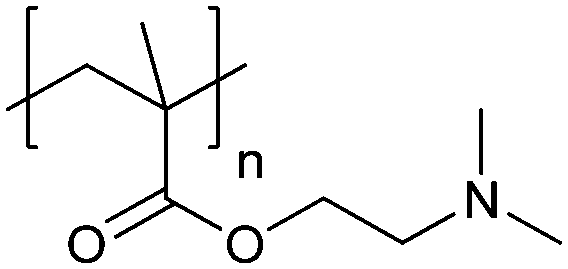
|
CRP | ∼7.5 | 65 and 110–112 |
| Poly(diallyldimethylammonium chloride) | PDADMAC |
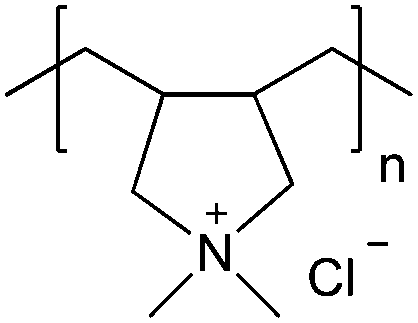
|
Free RP | Not attainable | 108 and 113 |
| Poly(ethylenimine) | PEI |
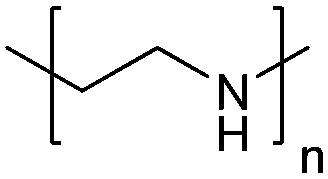
|
ROP | ∼9.7 | 114 and 115 |
| Hydrophobic or hydrophilic, depending on the stimulus | |||||
| Poly[2-(diethylamino)ethyl methacrylate] | PDEAEMA |
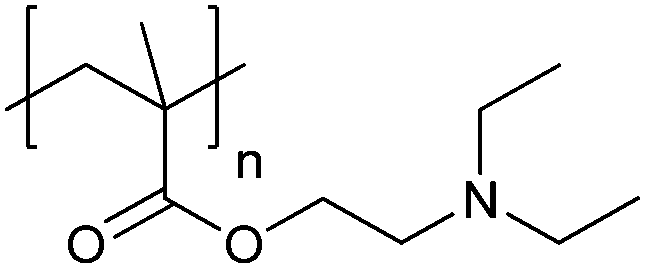
|
CRP | ∼7.3 | 116–118 |
| Poly[2-(diisopropylamino)ethyl methacrylate] | PDPA |
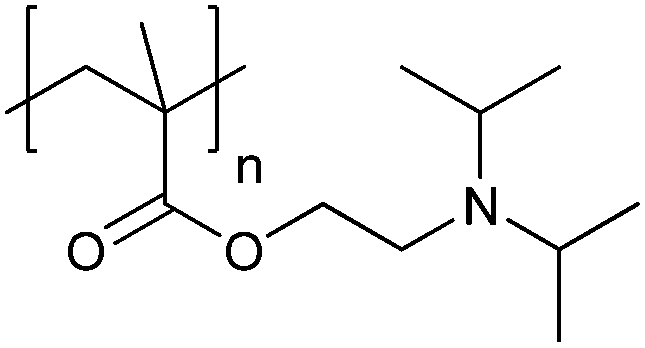
|
CRP | ∼6.3 | 105 |
| Poly(N-isopropylacrylamide) | PNIPAM |
|
Free RP, CRP | Thermo-responsive | 53 |
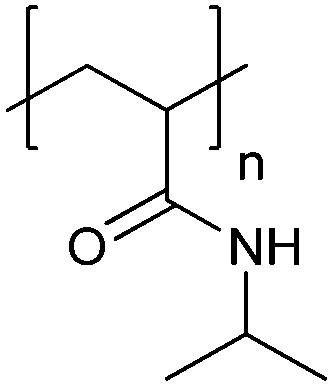
| Poly[(N-amidino)dodecyl acrylamide] | PAD |
|
CRP | ∼5.4 | 114 |
| Polymer building block | Abbreviation | Structure | Method of synthesis | Ref. |
|---|---|---|---|---|
| Hydrophilic | ||||
| Poly(ethylene glycol)/poly(ethylene oxide) | PEG/PEO |
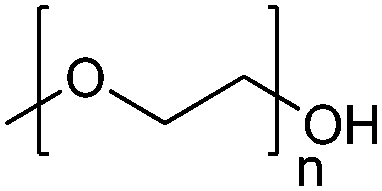
|
ROP | 7, 60, 107, 114 and 117 |
| Poly(2-methyl-2-oxazoline) | PMOXA |
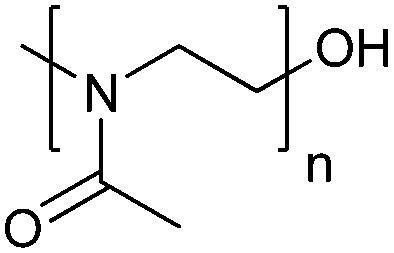
|
ROP | 1, 3, 59 and 119–123 |
| Poly(2-hydroxypropyl methacrylate) | PHPMA |
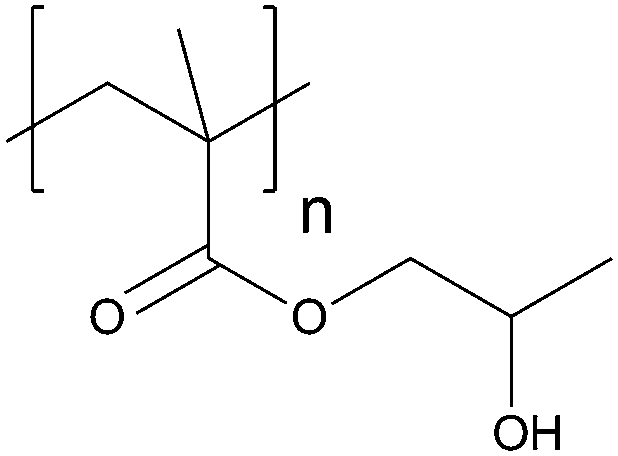
|
CRP | 7 and 124 |
| Hydrophobic | ||||
| Poly(ε-caprolactone) | PCL |
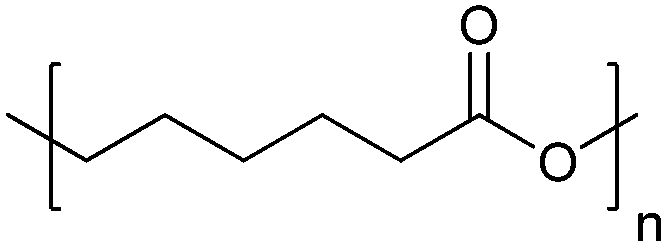
|
ROP | 103 and 117 |
| Poly(butyl methacrylate) | PBMA |
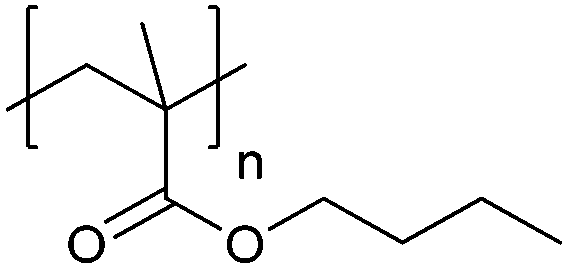
|
CRP | 112 |
| Polydimethylsiloxane | PDMS |
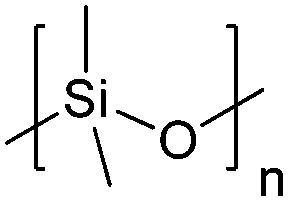
|
ROP | 1, 3, 59 and 119–123 |
| Polystyrene | PS |
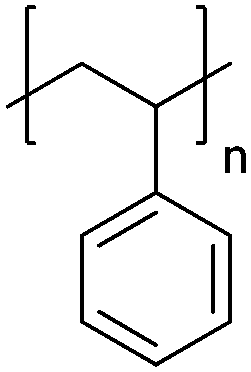
|
CRP; anionic | 125 |
Living ionic polymerization (LIP), either anionic or cationic, is frequently used for the synthesis of polymers with desired molecular weights, molecular weight distribution, polymer chain composition, and tailored end-chain moieties that can be added due to the absence of irreversible chain transfer and chain termination processes.71–73 In LIP, all polymer chains are spontaneously initiated, and the highly reactive propagating chains (anions or cations) are growing simultaneously in the presence of their ionic counterparts (counterions), such as to preserve the system's overall electroneutrality. However, the ends of growing ionic chains must retain their reactivity over the whole reaction process for further extension of the polymer chain or functionalization with the desired terminal groups. In this respect, it is necessary to ‘control’ the reactivity of the ionic species by carefully optimizing the reaction conditions (adequate selection of monomers, initiators, temperature, solvent, and terminating agents) and avoiding the presence of impurities. To overcome these limitations, controlled ionic polymerizations have been recently performed under ambient conditions in unconventional reaction media, which endow the growing chains with high stability and excellent chain-end fidelity.84,85 Various non-responsive polymers, such as polystyrene and polydienes or poly[alkyl (meth)acrylates], have been obtained by LIP (Table 2). In addition, styrene derivatives substituted with various functional groups (OH, NH2, SH, alkyne, SiOH, aldehyde, keto, and COOH) or other vinyl monomers carrying N atoms (2- or 4-vinyl pyridine) can also undergo successful ionic polymerizations after the appropriate protection of the functional groups. Finally, the removal of ‘masking’ groups after polymerization allows for the regeneration of the original functionality.74,86
‘Controlled’/quasi-‘living’ radical polymerization (CRP), also known as reversible deactivation radical polymerization (RDRP), is one of the most investigated strategies for the synthesis of (co)polymers. CRP includes various synthesis techniques, such as atom transfer radical polymerization (ATRP), reversible addition–fragmentation chain transfer polymerization (RAFT), and nitroxide-mediated radical polymerization (NMP).78,79,87–91 These methods have been used in the synthesis of stimuli-responsive polymers based on acrylates and acrylamides, such as poly(acrylic acid) (PAA), poly(styrene sulfonate) (PSS), poly[2-(dimethylamino)ethyl methacrylate] (PDMAEMA) and poly[2-(diisopropylamino)ethyl methacrylate] (PDPA) (Table 1), or non-responsive polymers, such as polystyrene (PS) and poly(butyl methacrylate) (PBMA). CRP synthetic pathways have various advantages compared to LIP including low-cost, conditions that promote the chain growth more favorably compared to ionic processes, low dispersity, high chain-end fidelity and a lower impact of impurities.92,93 Moreover, the growing chains are in a “dormant” state, and therefore, can be reactivated and functionalized or can react with other types of monomers to obtain block copolymers. The limitations of CRP are the low oxygen tolerance, the increased number of “dead chains” or the decrease of end chain functionality, especially when polymers with high molecular weight are being synthesized.82,94
Starting from cyclic monomers, ring-opening polymerization (ROP) techniques allow the synthesis of polymers with well-defined molecular weight and chemical composition.95–99 For example, polyesters have been synthesized from cyclic esters (lactones), polysiloxanes from cyclosiloxanes, and polyamides from cyclic amides (lactams) (Tables 1 and 2). However, the ROP mechanism and the polydispersity of the resulting polymer are strongly influenced by the type and concentration of the monomer and the catalyst which calls for extensive optimization procedures. An additional drawback of these methods is the use of heavy metal compounds as catalysts. In particular, when polymers are used in bio-applications, ROP is problematic because complete removal of the catalyst is challenging. This drawback of anionic ROP reactions was recently overcome by metal-free ROPs, where the activation of monomers is performed in the presence of organocatalysts, such as N-heterocyclic carbenes, N-heterocyclic olefins or phosphazene bases.100,101
Stimuli with relevance for sensing pathological conditions
Gradients and fluctuations of pH are the most explored triggers of biosensors since they are associated with a broad variety of diseases, for example cancer or Alzheimer's disease.126 A slightly acidic extracellular microenvironment (pH 6.5–7.2) and a slightly basic intracellular pH are typical for many solid tumors, while intracellular compartments, such as endosomes and lysosomes have pH values of 5.0–6.0 and 4.5–5.0, respectively.127,128 The blood pH of a healthy person is considered to be between 7.35 and 7.45. Alteration to a pH below 7.35 and above 7.45 is called acidosis and alkalosis, respectively, and can indicate pathological metabolic conditions as seen in diabetes or chronic renal disease.129 Another trigger of medical relevance is changes in CO2 levels. For example, elevated CO2 levels in the blood occur in a medical condition called hypercapnic acidosis which is related to other respiratory diseases including cystic fibrosis and chronic obstructive pulmonary disease (COPD).130The cellular redox state reflects the redox environment of a cell and defines its potential to maintain the redox balance and eliminate reactive oxygen species (ROS).131 More precisely, the redox state can be derived from the actual cellular reduction potential related to one specific redox pair. The most important cellular redox pairs are 2GSH/GSSG (glutathione), NAD(P)+/NAD(P)H (nicotinamide adenine dinucleotide (phosphate)) and TrxSS/Trx(SH)2 (thioredoxin),132 of which glutathione is the main redox buffer of the cell.133 It plays a role in a plethora of functions, including the protection of cells from oxidative stress, regulation of cellular signaling, and apoptosis.134,135 Oxidative stress resulting from elevated ROS of endogenous and exogenous sources is well known to damage biomacromolecules, and is associated with a variety of pathological conditions, in particular so-called “free radical diseases” such as cancer, neurodegeneration, atherosclerosis or inflammation.131 Besides ROS, also reactive nitrogen species (RNS) and reactive sulfur species (RSS) can cause oxidative stress.37
Other pathological conditions are associated with an imbalance in ion concentrations or the presence of various biomolecules such as uric acid,136 lactate,137 glucose,138 sugar alcohols,139 urea140 and cholesterol,141 which can be metabolites, substrates in enzymatic catalysis, mediators in many cellular processes, or can be part of signaling pathways.
This diversity of inter- and intracellular changes serves as the basis for specifically designing sensing systems that allow for a rapid and efficient detection of such changes. The diversity and extent of these, as well as the simultaneous presence of multiple changes, lead to a complex scenario of requirements for which sophisticated systems need to be developed.
Polymersomes as compartments for developing biosensors
Polymer vesicles in the nanometer range, usually called polymersomes, are generated by the self-assembly of amphiphilic block-copolymers with a specific hydrophilic-to-hydrophobic ratio in aqueous dilute solutions.142 Polymersome formation can be achieved by different methods such as the “solvent switch” method, the “solvent-free” method and polymerization-induced self-assembly (PISA).The “solvent switch” method for polymersome assembly is based on dissolving the copolymer in an organic solvent, followed by its dropwise addition into an aqueous solution. The high dilution induces the self-assembly into nanostructures and subsequently, the organic solvent is removed by evaporation or dialysis.143
In the “solvent-free” method, the copolymer solution is first dried to an anhydrous copolymer film. Slowly hydrating the film with an aqueous buffer induces the self-assembly process which results in the formation of nano-assemblies (polymersomes, micelles, and nanotubes). Additional shaking or stirring can fasten the process provided the assemblies are not perturbed.143
PISA is an assembly procedure allowing for polymersome formation in parallel to polymerization. More precisely, a hydrophilic macroinitiator block in aqueous solution is coupled with a second, water-soluble monomer. The latter, which initially is hydrophilic, is polymerized and becomes more hydrophobic with ongoing polymerization. With increasing block length and depending on the hydrophilic-to-hydrophobic block ratio, the resulting copolymers assemble in situ into supramolecular structures. The reaction can be stopped when the copolymer reaches a block ratio that will induce the assembly of a desired structure, e.g. polymersomes. PISA has become an effective strategy to produce polymeric assemblies with various morphologies at higher polymer concentrations than by conventional methods (<1%).27,144–146
The location of the “reporting” cargoes (molecules/nano-objects) as key functional elements is an important factor in obtaining efficient biosensor systems. When aiming at the encapsulation of reporting cargoes by polymersomes, the preparation methods need to be carefully chosen to avoid degradation of these cargoes. For example, the film rehydration method, due to its mild conditions, is suitable for the encapsulation of sensitive hydrophilic molecules (enzymes,5,147,148 proteins,149 DNA,150 or dyes151) and the entrapment or insertion of hydrophobic molecules (drugs152 and membrane proteins153), while solvent switch methods can only be used for molecules that are not sensitive to organic solvents (e.g. solvent-stable enzymes).154 Similarly, PISA also allows for the encapsulation of cargo molecules in aqueous solution under mild conditions, as reported in several examples for functional proteins and enzymes,7,155 nanoparticles,156–158 and for hydrophilic and hydrophobic dyes and drugs.159,160 A prerequisite for the accessibility of reporting cargoes inside polymersomes is that polymer membranes are permeable to the stimulus, either intrinsically or induced by the corresponding stimuli, or by the insertion of biopores. Chemical approaches to obtain permeable membranes are based on: (i) selection of copolymers forming porous membranes for example poly(styrene)-block-poly[isocyanoalanine(2-thiophene-3-yl-ethyl)amide] (PS-PIAT),147,161,162 (ii) chemical modification of the membrane with a hydroxyalkylphenone163 or by exposure to visible light,164 and (iii) selection of polymers forming membranes that change their permeability in a stimuli-responsive manner, e.g. in response to pH changes.165,166 However, these approaches result in membrane permeability without or with very limited selectivity as the pore size determines the molecular cut-off of constituents that can pass through. At the same time, in the case of permeabilizing polymersome membranes for macromolecules or nanoparticles, the softness rather than the size of this aimed cargo is a major factor influencing the pH-induced permeability of polymersome membranes towards these macromolecules or nanoparticles.167 Alternatively, the membrane is permeabilized by the insertion of membrane proteins or biopores that, depending on their specific bio-functionality, achieve non-selective or selective permeability.168 This strategy is bio-inspired from the cell membrane that exchanges molecules with the extracellular environment by a variety of membrane proteins and biopores. Membranes based on poly(2-methyl-oxazoline)-block-poly(dimethylsiloxane)-block-poly(2-methyl-oxazoline) (PMOXA-b-PDMS-b-PMOXA) copolymers, reported as being impermeable to molecules/ions other than oxygen and superoxide radical anions (O2˙−),169 have been permeabilized by insertion of channel porins such as OmpF,170,171 Aquaporin Z172, aquaglyceroporin Glpf119 or biopores such as Melittin.153
Polymersomes sensing pH changes
pH responsiveness is promoted (i) by polymer membranes with an intrinsic pH sensitivity and (ii) by pH-sensitive pores inserted in non-responsive membranes.When appropriately selected, copolymers can form polymersomes with “breathing” membranes that reversibly swell at specific pH values and shrink at others. This effect has been reported for copolymers containing protonatable tertiary amine groups. For example, a change from pH 5 to pH 10 induced reversible swelling in polymersomes consisting of copolymers with a PDEAEMA (poly[2-(diethylamino)ethyl methacrylate]) block.118 Polymersomes which breath upon pH changes are interesting candidates for sensing when they are loaded with reporting molecules (e.g. sensitive dyes) that emit a pH-dependent signal. For instance, polymersomes assembled from poly(ethylene oxide)45-block-polystyrene130-block-poly[2-(diethylamino)ethyl methacrylate]120 (PEO45-b-PS130-b-PDEAEMA120), which showed reversible breathing between pH 10.4 and 3.4, were used to encapsulate a pH sensitive pyrene-derived dye.125 Under acidic conditions, the pH-dependent breathing led to a dramatic increase in membrane permeability towards protons, which shifted the absorption maximum of the encapsulated dye to a second peak at a smaller wavelength and thus allowed for proton concentration determination by calculating an excitation intensity ratio measured at two wavelengths.
Combination of the chromophore from the green fluorescent protein with an amphiphilic, intrinsically pH-sensitive block copolymer allowed for the development of pH-sensing polymersomes bearing the bio-inspired sensing moiety covalently integrated in the membrane (Fig. 1).4 The chromophore (IE) was inserted between a hydrophilic polyethylene glycol (PEG) and a protonatable PDEAEMA block (PEG-IE-b-PDEAEMA). Protonation and deprotonation of the PDEAEMA block were achieved by CO2 and N2, respectively (Fig. 1A). Although the study did not include size data reflecting the reversible swelling and shrinking of the polymersomes, it revealed a reversible change of fluorescence intensity from pH 6.8 to pH 5 (Fig. 1B). When the PDEAEMA block is deprotonated and hydrophobic, the chromophore is protected from water and showed high fluorescence intensity, whereas when PDEAEMA is protonated and more hydrophilic, exposure of the chromophore to water reduced the fluorescence intensity.
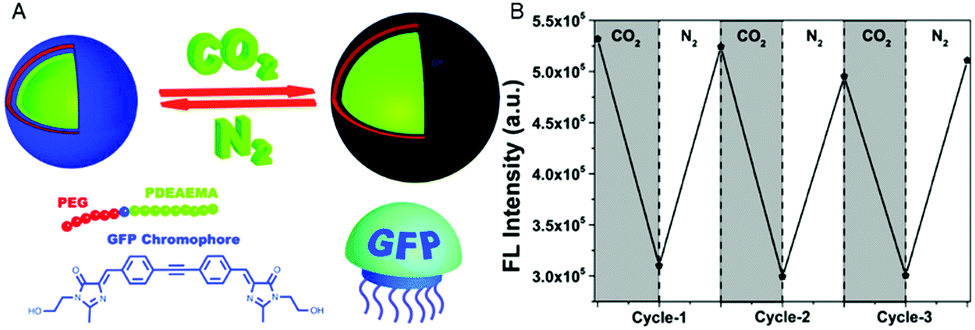 | ||
| Fig. 1 pH-Sensing polymersomes with the GFP chromophore. (A) The chromophore (IE) of the green fluorescent protein is inserted between two polymer blocks, yielding PEG-IE-b-PDEAEMA polymersomes with pH-responsive breathing character, induced by CO2 and N2 introduction. (B) Reversible change of fluorescence intensity of IE-containing polymersome sensors upon pH decrease from 6.8 to 5 (after CO2 treatment) and vice versa (after N2 treatment). Adapted with permission from ref. 4. Published by the PCCP Owner Societies. | ||
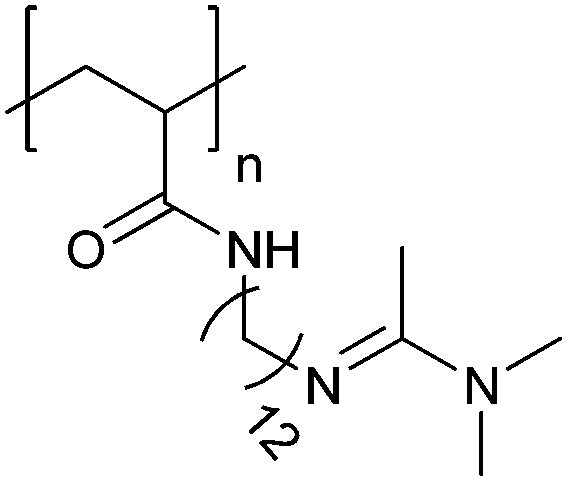
Similarly, reversible permeability of the membrane was reported for polymersomes assembled from poly(ethylene glycol)-b-poly[(N-amidino)dodecyl acrylamide] (PEG-b-PAD) diblock copolymers which resulted from the protonation and deprotonation of the polyamidine block in the presence of CO2 and N2 in solution.114 Depending on the time of CO2 incubation, PEG-b-PAD vesicles expanded in diameter and pores were formed with a tuneable diameter as visualized by a size-dependent release of dye-labeled poly(ethylene imine) nanoparticles (PEI-NPs) that were encapsulated inside the PEG-b-PAD polymersomes. Size-specific release of PEI-NPs indicated an increase in pore size from ≤4 nm to more than 10 nm upon the stepwise transformation from a hydrophobic, entangled polyamidine block to a hydrophilic, stretched polyamidinium block. The increased pore size allowed an enzyme, trypsin, to diffuse into the polymersome cavity where it catalyzed the hydrolysis of the encapsulated oxygenated myoglobin, thereby inducing a measurable photometric change. Permeabilization that enables even larger biomolecules to pass the membrane was observed with polymersomes assembled from a hyperbranched poly(ε-caprolactone)-b-(poly[2-(diethylamino)-ethyl methacrylate]-b-poly(ethylene glycol)) (PCL-b-PDEAEMA-b-PEG) triblock copolymer.117 With increasing CO2 exposure, the vesicles first swelled and after 15 min, sudden release of fluorescent rhodamine occurred. Eventually, macropores of several 100 nm in diameter were revealed by transmission electron microscopy. These examples show that enhanced pore formation can be corroborated by sensing moieties such as large enzymes which produce a fluorescence signal or the release of labelled nanoparticles. Further studies are needed to precisely understand how different pH values gradually affect permeabilization/pore formation of these polymersomes.
A similar concept resulting in pH-responsive polymersomes is to specifically induce the membrane to become porous only at a certain pH. A copolymer of acrylic acid and distearin acrylate poly(AA-co-DSA) formed polymersomes which showed reversible opening (pH 8) and closing (pH 5) due to the deprotonation of PAA under basic conditions.102 Ensuing pores allowed the influx of calcein which could then be detected in the compartment interior via confocal laser scanning microscopy (CLSM). Thus, PAA turned the polymersomes into a pH-sensor with an output signal that was detectable by fluorimetry.
A different means of achieving pH responsiveness is reflected in so-called “schizophrenic vesicles” that are based on zwitterionic diblock copolymers or polyampholytes, whose blocks are interchangeable in terms of hydrophilicity. More specifically, a change in pH induces the change of the hydrophilic block to a hydrophobic block and vice versa, predominantly through changes in conformation and charge. There are many polymers that exhibit this behaviour. Yet, only a few examples applicable in sensing exist because the most important part, the sensing moiety, is lacking. Not so in PDEAEMA-b-PAP (poly(N-acryloyl-L-phenylalanine)) functionalized at the PDEAEMA block with a fluorescent pyrene group as a pH sensing entity.116 This polymer formed “flower-like” polymersomes with switchable coronas and thus schizophrenic behaviour upon a pH change from 2 to 12. At pH 2, PDEAEMA is positively charged and hydrophilic, and forms the corona, while PAP is uncharged and hydrophobic. At high pH, PAP is negatively charged and hydrophilic and PDEAEMA is uncharged and hydrophobic, and the corona is inverse.
Besides the above-mentioned strategies that are based on pH responsiveness inherent to the polymer, a completely different strategy to endow polymersomes with pH responsiveness is to integrate biomolecules with specific functions. For example, by inserting gramicidin (gA), a small pore forming polypeptide, into the membrane of PMOXA-b-PDMS-b-PMOXA polymersomes, the pH-sensitive dye encapsulated in the cavity was able to respond to pH changes in the polymersomes’ environment.173 The bio-inspired strategy has been further extended in various ways. For instance, pH-sensitivity was achieved by inserting chemically or genetically modified channel porins in the polymersome membrane where they function as pH-triggered “gates”.58,120 In particular, the outer membrane protein F (OmpF) from E. coli, which allows diffusion of molecules up to 600 Da, has been modified by a pH-responsive molecular cap that was chemically attached to the channel.58 When the pH in the polymersome environment was lowered from 7.4 to 5.5, the OmpF pore was unplugged. A genetic modification of OmpF, which enabled the stable conjugation of a pH-responsive peptide cap, turned OmpF into a “biovalve” that reversibly allowed molecular flow when the pH increased from 6 to 7.4.120 In both cases of polymersomes equipped with modified OmpF channels, the sensor capacity was established by encapsulating horseradish peroxidase (HRP), thereby transforming the polymersomes into catalytic nanocompartments: in response to external pH changes, the OmpF pores opened and the substrate (Amplex UltraRed) gained access to the encapsulated HRP, which catalyzed its conversion to a fluorescent product (Resorufin) in situ.
Polymersomes sensing changes in the reductive conditions of the surrounding
Polymersomes that are inherently sensitive to the surrounding reduction/oxidation conditions have been reported to function as drug delivery systems, but their architecture disintegrated to release the cargo.50,174–176 Nevertheless, intact polymersomes exist that are turned into reduction-sensitive systems by biomolecules. A recent example of reduction-responsive polymersomes with preserved integrity has been obtained by inserting genetically engineered OmpF in the membrane of PMOXA6–PDMS44–PMOXA6-based polymersomes.3 The genetic modification allowed the conjugation of a fluorescent molecular cap to OmpF via disulphide bonds that are cleaved in a reducing environment (Fig. 2A). As a result of the cleavage, the pores open to a substrate, which is then converted to a fluorescent product by the HRP confined in the polymersome cavity (Fig. 2B). This biosensor served as an artificial organelle that was able to sense reductive conditions in vivo.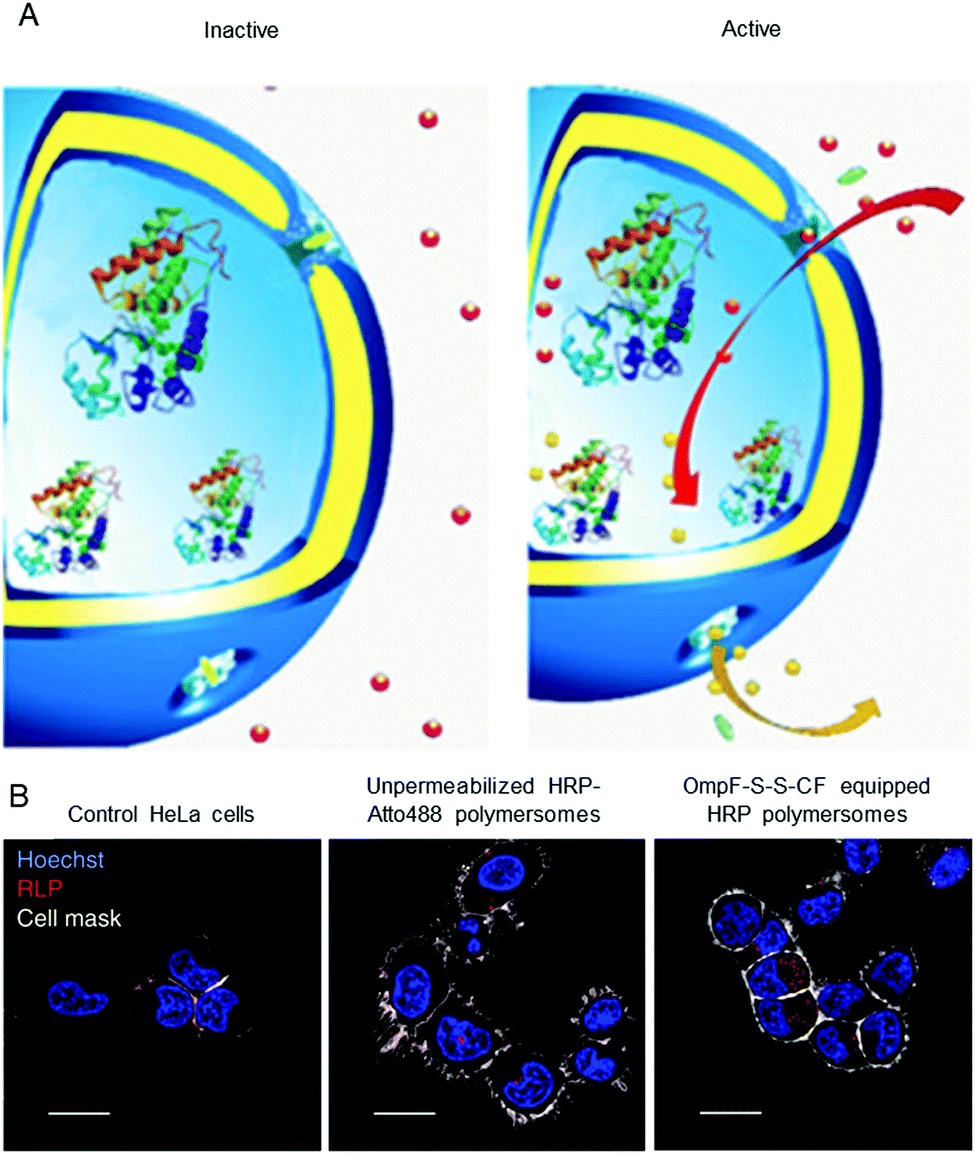 | ||
| Fig. 2 Catalytic nanocompartments as sensors for reduction, based on incorporation of reduction-sensitive membrane protein OmpF. Two reduction-sensitive cysteine residues of genetically engineered OmpF were chemically conjugated with a molecular cap. (A) Scheme of biosensor with modified OmpF as a membrane gate and encapsulated horseradish peroxidase acting as a catalytic moiety. In the inactive state (left), the gates stay closed and are impermeable for the substrate. Upon activation by reduction stimulus (right), the gates open and the enzyme produces a fluorescent product, representing the sensing moiety. (B) Polymersome biosensors are taken up and activated in HeLa cells. Comparison between CLSM images of untreated cells, cells treated with unpermeabilised catalytic compartments and catalytic compartments permeabilised with reduction-sensitive OmpF (OmpF-SS-CF) (from left to right). Blue: nucleus (Hoechst 33342 nuclear stain); grey: plasma membrane (CellMask Deep Red Plasma membrane stain); red: resorufin-like product (RLP) from activated catalytic biosensors (scale bar: 20 μm). Adapted with permission from ref. 3. Copyright 2018® Springer Nature. | ||
Ion-sensing polymersomes
Ion-sensing polymersomes that maintain their structural integrity have similarly been obtained by intrinsically permeable membranes and by the insertion of biomolecules in non-responsive polymersomes. For example, the membranes of polymersomes based on PMOXA-b-PDMS-b-PMOXA copolymers are permeable to different oxygen species.Accordingly, superoxide anions diffused through the membrane and were enzymatically converted to hydrogen peroxide and oxygen by the superoxide dismutase (SOD) encapsulated in the polymersome cavity.169 By co-encapsulation of a second enzyme, lactoperoxidase (LPO) or catalase, an in situ cascade reaction occurred, which further degraded H2O2 to water and oxygen.177 Such detoxifying polymersomes internalized by HeLa cells remained active as indicated by an increased fluorescence of a reporter product. Thus, catalytic compartments with a confined SOD–LPO cascade reaction represent a straightforward way to simultaneously detect and degrade superoxide radicals and associated H2O2, both well known to be involved in oxidative stress.
The specific ion sensitivity of PMOXA-b-PDMS-b-PMOXA polymersomes was also achieved by insertion of biopores such as the gramicidin A (gA) into the membranes. Polymersomes permeabilized with gA allowed for the diffusion of Na+ and K+, which was detected by a change in the fluorescence intensity of the encapsulated reporting molecules.173 The responsiveness of such bio-equipped polymersomes can be adjusted by selecting specific biomolecules for insertion. For example, PMOXA-b-PDMS-b-PMOXA polymersomes have been rendered Ca2+-sensitive by the incorporation of ionomycin, a specific ionophore for calcium ions (Fig. 3A).16 The sensor detected the presence of Ca2+ ions in a range corresponding to the intra- and extracellular concentrations via an encapsulated Ca2+-sensitive dye (Fig. 3B). Integration of such ion channels and transporters may pave the way for the development of advanced cellular ion sensors.
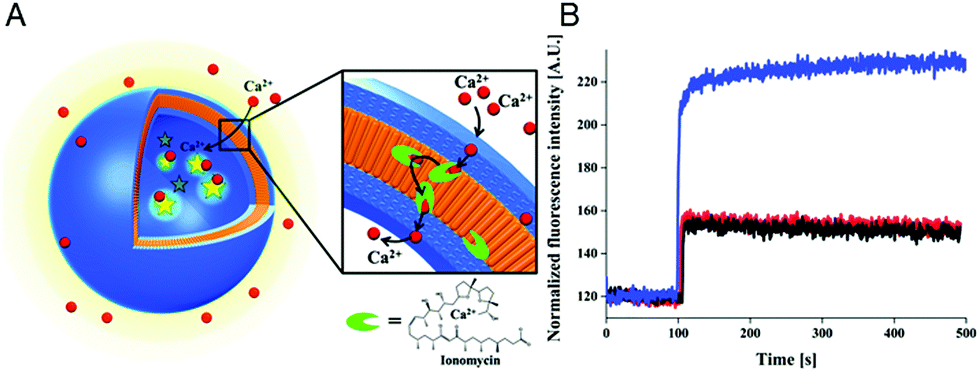 | ||
| Fig. 3 Scheme of polymersome sensors for Ca2+. (A) The Ca2+ transporter ionomycin is inserted into polymersome membranes, allowing for the sensing of Ca2+ ions by the encapsulated Ca2+-sensitive fluorescent dye Asante Calcium Green (ACG). (B) Ca2+ influx through PMOXA6-b-PDMS44-b-PMOXA6 triblock copolymer membrane. The graph compares the fluorescence intensities of ACG encapsulated in unpermeabilised polymersomes (black curve), in unpermeabilised polymersomes in the presence of EtOH (red curve), which was used for ionomycin solubilisation and insertion into the membrane, and in ionomycin-permeabilised polymersomes in the presence of 830 mM CaCl2 (blue curve). Adapted with permission from ref. 16. Published by the PCCP Owner Societies. | ||
Biomolecule-sensitive catalytic polymersomes
The concept of producing catalytic nanocompartments by incorporating enzymes in polymersomes has been reported to induce responsiveness to specific surrounding biomolecules which are substrates for the enzymes encapsulated inside. Due to its potential in cancer therapy,24,51 the glucose-sensitive enzyme glucose oxidase (GOx) lends itself to encapsulation into catalytic polymersomes. H2O2, which is quantitatively produced by the catalytic activity of GOx, can participate in a second reaction catalysed by a peroxidase (such as horseradish peroxidase, HRP) that is able to oxidize specific substrates easily detectable by fluorimetry or UV/Vis spectroscopy. The GOx–HRP enzyme cascade was integrated into glucose-sensing catalytic polymersomes assembled from poly(ethylene glycol)-b-poly(2-hydroxypropyl methacrylate) (PEG-b-PHPMA) which were formed by visible light-mediated PISA (photo-PISA).7 These polymersomes possessed intrinsic permeability for glucose and H2O2. Glucose was indirectly sensed via the formation of a chromophoric product (Fig. 4). The same reaction has been exploited by GOx catalytic polymersomes assembled from PMOXA-b-PDMS-b-PMOXA which were rendered permeable by the pore-forming peptide Melittin.153 These catalytic nanocompartments (CNCs) produced H2O2 which was used to convert nonfluorescent Amplex Red to the fluorescent resorufin in a reaction catalyzed by horseradish peroxidase (HRP).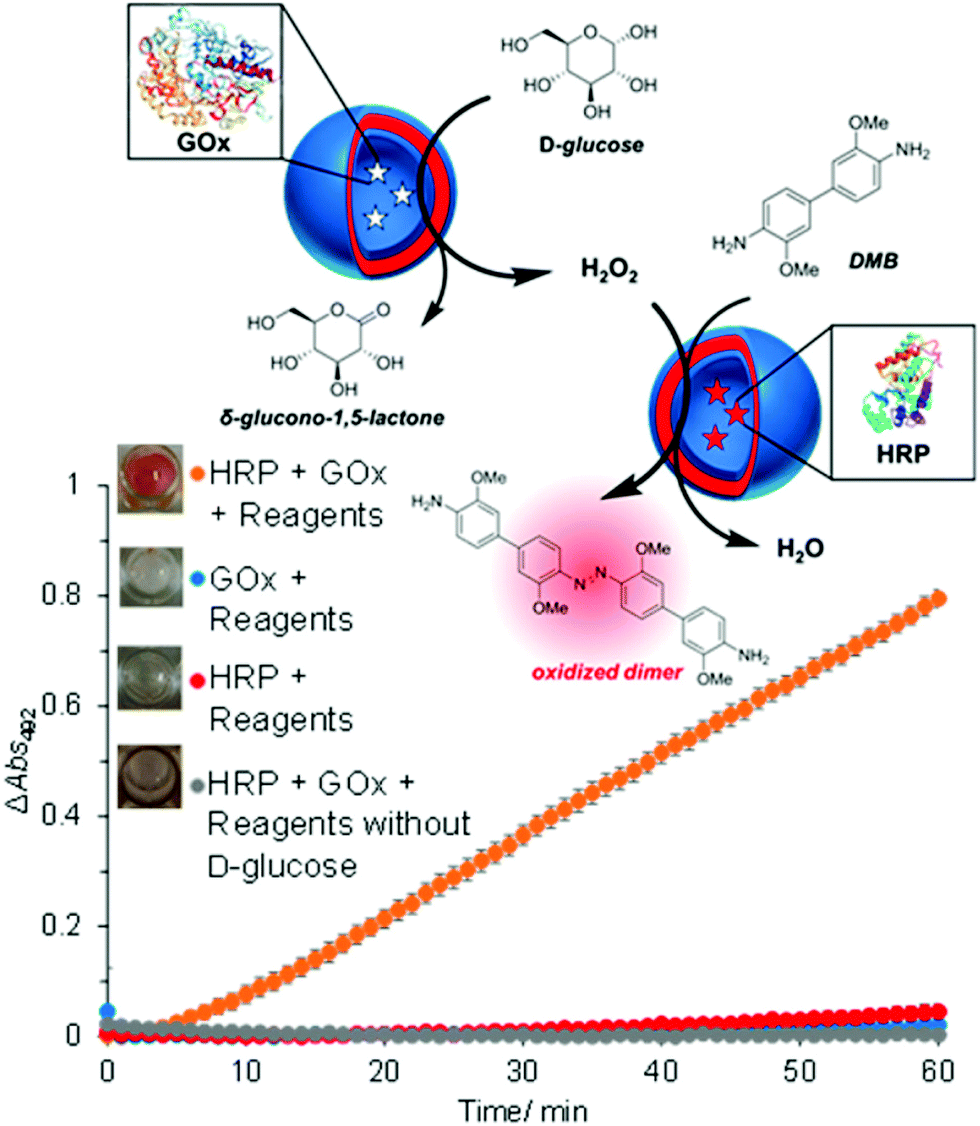 | ||
| Fig. 4 Glucose-sensing catalytic polymersomes working in tandem, generating a chromophoric product. Upper scheme shows polymersomes assembled from PEG-b-PHPMA, which possess intrinsic permeability. Enzymes GOx and HRP were encapsulated into separate, communicating compartments, where the GOx polymersomes represent the sensing moiety and the HRP polymersomes represent the signal-generating moiety. The graph shows the product formation of the cascade reaction between HRP and GOx-loaded vesicles with negative controls. Error bars show the standard deviation from four repeats. Insets: End-point photographs of wells after 1.5 h. Adapted with permission from ref. 7. Published by the American Chemical Society. | ||
Another biomolecule that has been efficiently detected by catalytic nanocompartments is uric acid, known to be associated with the occurrence of gout. By encapsulation of uricase into CNCs assembled from PMOXA6–PDMS44–PMOXA6 equipped with OmpF, uric acid was successfully detected by a second enzyme present in serum acting as a catalyst.178 Taking the development of sensing devices one step further, catalytic nanocompartments containing uricase or lactoperoxidase that were equipped with a biopore for permeabilization of their membrane, were immobilized in tandem on a surface.5 Specifically, CNCs were immobilized via streptavidin–biotin interactions on polymer brushes synthesized on a silicon surface (Fig. 5). This sensor system successfully sensed uric acid present in the environment of the surface. Such active surfaces have large potential for the development of biosensors on a chip.
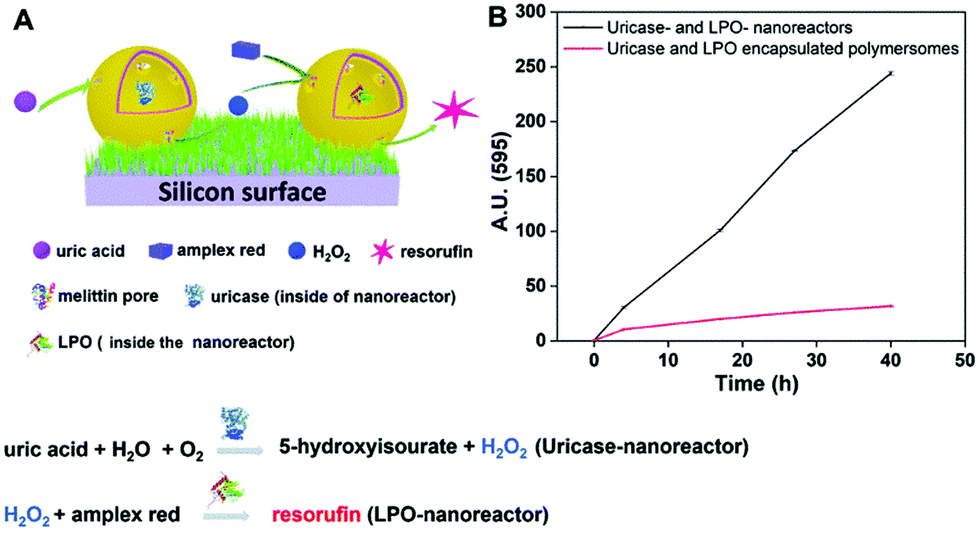 | ||
| Fig. 5 Uric acid-sensing nanoreactors immobilized on a surface. (A) Schematic diagram of the cascade reaction on uricase and LPO-nanoreactor immobilized surface. (B) Fluorescence intensity of resorufin in PBS solution produced by uricase and LPO nanoreactor immobilized soft surface (black curve) and uricase and LPO encapsulated polymersome (without melittin as control) immobilized soft surface (red curve). Adapted with permission from ref. 5. Published by The Royal Society of Chemistry. | ||
A biosensor for sugar alcohols that has similar potential to be used on a chip was developed by the encapsulation of the enzyme ribitol dehydrogenase (RDH) into polymersomes which were selectively permeable for sugar alcohols.119 The selective permeability was accomplished by insertion and reconstitution of the aquaglyceroporin Glpf from E. coli, which conducts water and linear polyalcohols across the inner bacterial membrane, into the polymersome membrane.
Polymersomes with dual sensitivity
Extending the responsiveness to multiple stimuli evidently renders the systems more efficient in the detection of certain pathological conditions. Furthermore, it offers the possibility of inducing a specific response by the external application of a second stimulus. However, there are only very few examples of the dual-responsiveness of polymersomes simultaneously preserving their integrity during the sensing period of time. The combination of pH-sensitive “breathing” polymersomes with the biological principle of feedback-regulated systems was used to create catalytic nanocompartments able to sense the presence of an acidic “fuel” (Fig. 6A and B).21 Addition of “fuel” (urea acidified with HCl) changed the catalytic compartments from an “OFF” to an “ON” state. In the ON state, hydrolysis of the fuel by urease produced basic ammonia. The increased pH turned the catalytic compartments to the “OFF” state again. Based on the co-encapsulation of HRP and the turnover of a chromogenic substrate, the urease activity could be followed over time-programmed several cycles (Fig. 6C). A complementary approach has been obtained by clustering polymersomes together to use them as distinct but colocalized nanocompartments: one type containing the enzyme human Dopa decarboxylase (DDC) and the second type loaded with fluorescent probes for the detection, respectively (Fig. 6D and E). The catalytic compartment containing the active DDC triggers the cellular expression of a secreted reporter enzyme via production of dopamine, while the second type of polymersomes allows tractability via dye-loading as the imaging component.26 Such bio-inspired systems have high potential for the development of dual-responsive sensors with the advantage, to allow for detoxification or conversion of the biomolecules as well.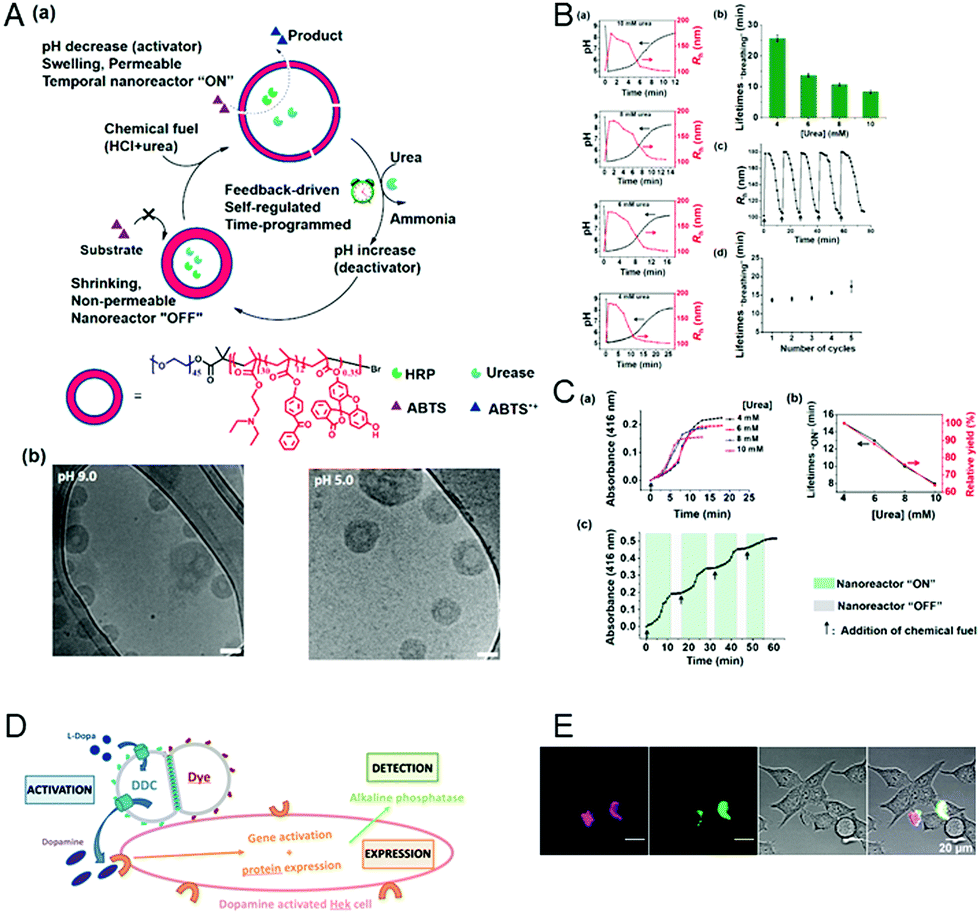 | ||
| Fig. 6 (A) (a) Schematic overview of feedback-induced temporal control of polymersome nanoreactors. (b) Cryo-TEM images of polymersomes at pH 9.0 (left) and pH 5.0 (right). All scale bars are 100 nm. (B) (a) Influence of the urea concentration on the size changes of the polymersome nanoreactor solution. Urea concentrations from top to bottom: 10, 8, 6, and 4 mM. (b) Average transient periods of polymersome “breathing” as a function of the urea concentration. (c) Reversible hydrodynamic radius (Rh) changes of the polymersomes in time following repeated additions of chemical fuel (6 mM urea). (d) Average transient periods of “breathing” within five cycles (6 mM urea in all cases). Concentration of urease in the polymersomes is 30 U mL−1.” (C) UV absorbance at 416 nm of the oxidation of ABTS by nanoreactors upon the addition of different concentrations of urea. (b) Lifetimes (black line) of the nanoreactor “ON” state and relative yield (red line) as a function of urea concentration. (c) Reversible nanoreactor “ON–OFF” modulation in time following repeated additions of 6.0 mM urea. Experimental conditions: urease, 30 U mL−1; HRP, 10 U mL−1; ABTS, 8 mM; H2O2, 5 mM. Adapted with permission from ref. 21. Published by the American Chemical Society. (D) Schematic representation of segregated clustered nanocompartments containing DDC and imaging compound.26 (E) CLSM pictures from four different locations of dye-loaded nanocompartment clusters attached to the surface of HEKREWARD cells44 after 24 h of incubation, with separated DY-633 and Atto-488 channels, transmission channel and merged images (scale bar = 20 μm). (D and E) Unpublished data. | ||
Synthetic giant unilamellar vesicles (GUVs) as potential compartments for biosensors
Giant unilamellar vesicles (GUVs) are usually based on a chemical composition similar to that of polymersomes but they differ in size. GUVs typically have diameters in the micrometer range (10–100 μm) and thus, represent an elegant tool to prepare cell-sized compartments that can sense changes in their environment.68,69,179–182 There are different methods to prepare polymer GUVs, including “solvent switch” and “solvent-free” approaches,46,183 which have been described above for the generation of polymersomes, others specific for GUV production, such as electroformation and gel-assisted hydration of a dry polymer film.46,122,184,185 Unfortunately, each of the solvent-free techniques also has its own drawbacks. Overall, they are less controllable than the “solvent switch” methods, generating polymer GUVs with a broader size distribution. Moreover, the encapsulation of reporting molecules is more challenging, especially with the electroformation method, which involves the application of an electric field.183 The advantage of polymer GUVs is that they allow for the direct visualization of the interactions and reactions occurring inside their cavity by established microscopic techniques. Thus, they are suitable for investigating in real time pathological changes at the cellular level. However, because of the instability of their membrane, GUVs are prone to burst and release their content, which excludes them from more advanced applications.GUVs reporting changes in pH and the presence of ions
To induce selective permeability for ions and molecules in GUVs or to sustain reactions inside their cavity while preserving the architecture is a challenging goal. Membrane insertion of ionophores or biopores has been achieved only in few cases. For example, ionophores including Lasalocid A, Alamethicin, N,N-dicyclohexyl-N′,N′-dioctadecyl-3-oxapentane-1,5-diamide and calcimycin inserted into the membranes of GUVs based on PMOXA–PDMS–PMOXA block copolymers made them permeable to calcium ions,186,187 while gramicidin rendered GUV sensitive to pH and monovalent ions.59 The stability of giant compartments and biopore functionality were assessed by encapsulating specific dyes that respond to the presence of the corresponding ions as a straightforward means of detection.GUVs sensing changes in the reductive conditions of the surrounding
An elegant approach to sense the reductive conditions of the GUV environment was achieved by forming GUVs from a mixture of amphiphilic block copolymers PMOXA-b-PDMS-b-PMOXA and PDMS-b-heparin.1 The resulting GUV allowed the redox reagent dithiothreitol (DTT) to access the cavity where it disassembled encapsulated nanoparticles loaded with reporter dyes. By encapsulating different nanoparticles, each loaded with a specific biomolecule, inside the GUVs, DTT was able to induce signaling cascades that mimic cell signaling. As a first step inside the GUV, DTT triggered the disassembly of nanoparticles loaded with gA. The released gA inserted in the GUV membrane and rendered it sensitive to the presence of ions which then function as a second stimulus (Fig. 7A). In addition, enzymes confined in the GUV's cavity converted the corresponding substrates and provided biopores or channel porins that were inserted in the GUV membrane let them enter the cavity as in the case of polymersomes. Compared to polymersomes, insertion of biopores/membrane proteins in the GUV membrane is by far more challenging because of its instability. To present, only one example of PMOXA-b-PDMS-b-PMOXA based GUVs that contained a functional HRP was reported.2 When OmpF was inserted in the GUV's membrane, H2O2 and AmplexRed could enter the cavity and react with the encapsulated HRP to form fluorescent resorufin as a “reporter” for H2O2 (Fig. 7B). | ||
| Fig. 7 (A) Schematic representation (top) of gramicidin (gA)-mediated sensing of sodium ions. Upon DTT addition, the encapsulated gA is released from nanoparticles and inserts into the GUV membrane boundary. This allows the flux of sodium ions into the GUV cavity where they activate the sodium sensitive dye ANG2. Bright field image (left), fluorescence image (middle), and histogram along the diagonal of the fluorescence image (right) in the presence (middle) and absence (bottom) of DTT.1 (B) Schematic representation of a synthetic giant unilamellar vesicle (GUV) (top left) with encapsulated enzyme (horseradish peroxidase – HRP) and selective permeability to substrates (Amplex UltraRed – AR) induced by reconstitution of outer membrane protein F (OmpF). Top right, flow cytometry analysis of PMOXA7–PDMS49–PMOXA7 GUVs with (red) and without (blue) OmpF membrane insertion. Bottom, CLSM fluorescence micrographs of an individual HRP-containing GUV with OmpF membrane insertion, recorded over time after the addition of H2O2 and Amplex UltraRed. Adapted from ref. 2 with permission of John Wiley & Sons. | ||
PICsomes as compartments for the development of biosensors
PICsomes are special types of polymersomes, which are based on a polyion complex (PIC).188 PICsomes are formed by self-assembling mixtures of anionic and cationic block copolymers selected so that the resulting assembly is neutral. The mixture for PIC systems is usually based on poly(ethylene glycol) PEG block “aniomer” (negatively charged polymer) and a second “homocatiomer” (positively charged polymer) neutralizing each other's charge.189 Due to the immiscibility of the two components and the large steric hindrance of PEG, a rather small volume of PEG is considered to preferentially form hollow spheres. When long PEG chains were mixed with the homocatiomer chains, micelles were formed due to the large volume of PEG-units.107An advantage of PICsomes is their intrinsic membrane permeability which allows ions and small compounds (e.g. O2 and Na2S2O4) to pass through, while molecules with larger molecular weight (enzymes and proteins) remain inside and are protected from pathological attack.190 In addition, PICsomes are only formed in aqueous media, which makes them particularly suitable for combining with sensitive biomolecules such as enzymes.107 In aqueous solutions with a physiological salt concentration (150 mM NaCl), micron-sized vesicles were obtained, which are called “the first generation PICsomes” (1st-G PICsomes). In the absence of NaCl, submicron-sized PICsomes were formed presenting the second generation of PICsomes that were named “Nano-PICsomes”.107 Nano-PICsomes are well suited for medical applications because they showed longevity (a mean residence time of over 40 h) and a prolonged blood circulation if they were smaller than 200 nm.191,192 As PICsomes are formed by aniomers and catiomers, depending on their pKa values, these polymers facilitate specific charge changes within the PICsome membrane that induce structural fluctuations (tightness of the membrane and size of the PICsome). Because such structural modifications can be coupled to a fast and easily measurable signal, e.g. fluorescence, PICsomes lend themselves to designing biosensors.
Intrinsically pH sensitive PICsomes
PICsomes have already been developed and exploited for the use as diagnostic nanocarriers with sensitivity towards changes in pH. 1st-G PICsomes based on PEG–poly[(5-aminopentyl)-α,β-aspartamide] (PEG–P(Asp-AP)) as a homo-catiomer and PEG–poly(α,β-aspartic acid) (PEG–PAsp) as an aniomer were able to sense slight, acid-based pH changes by reversibly changing their morphology.107 This kind of PICsomes maintained their structure at pH 7.4, while they fragmented into smaller particles below a pH of 3.8. When the pH changed again to neutral, the PICsomes were reformed. During this regeneration process, it was possible to encapsulate macromolecules. In addition, 1st-G PICsomes showed permeability changes in response to different pH values. This was observed by measuring the fluorescence of dextran conjugates (Mn = 70![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000) that were able to get through the PICsome membrane at pH 5.8 but not at pH 6.2 to 7.4.193 Such pH-sensitive properties of the membrane components make PICsomes a promising tool for cancer diagnosis since tumor cells exhibit an increased glucose metabolism that results in a higher H+-concentration outside the cell.54
000) that were able to get through the PICsome membrane at pH 5.8 but not at pH 6.2 to 7.4.193 Such pH-sensitive properties of the membrane components make PICsomes a promising tool for cancer diagnosis since tumor cells exhibit an increased glucose metabolism that results in a higher H+-concentration outside the cell.54
Layer-by-layer (LbL) capsules as compartments for the development of biosensors
LbL capsules are hollow microcapsules prepared by the assembly of a polyelectrolyte multilayer (PEM) film onto colloidal particles that serve as a spherical template. Layers of positively and negatively charged polyelectrolytes are alternatively adsorbed onto the template (Fig. 8). In addition, various charged nano-objects (e.g. magnetic nanoparticles or quantum dots, QDs) can be incorporated within the multilayers in order to achieve different functionalities of the capsules.8 After the assembly process, the CaCO3 or silica based core194 is removed by dissolving under mild pH conditions (Fig. 8).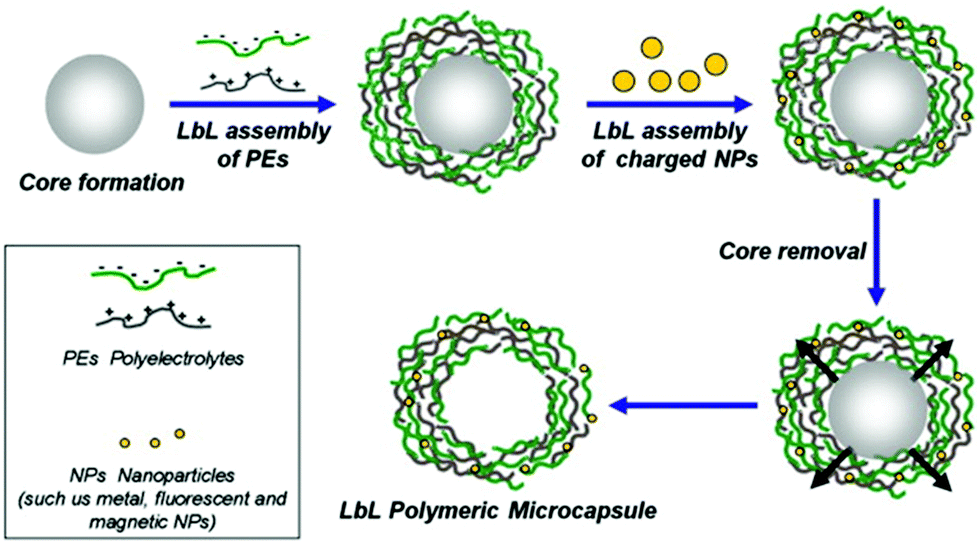 | ||
| Fig. 8 Schematic representation of LbL assembly. PEM capsules are produced by LbL adsorption of oppositely charged polyelectrolytes (green and gray) onto spherical charged spheres, followed by dissolution of sacrificial templates. During LbL coating, various charged elements can be incorporated within the multilayers to add functionality to the capsules. Adapted from ref. 8 with permission of Elsevier B.V. | ||
LbL microcapsules have attracted interest for diagnostic applications because they enable the incorporation of sensitive fluorophores and enzymes in the same capsule for multiplexed detection of different pathological signals. LbL capsules containing “reporting” molecules/nano-assemblies have been used for sensing (i) pH, (ii) ions and (iii) biomolecules (Fig. 9). The functionality of pH- or ion-sensitive capsules was based on “reporting molecules” that functioned either as ratiometric ion-sensitive indicators or non-ratiometric indicator dyes with a reference dye inside the cavity (Type 1). A more complex scenario allowing detection of biomolecules has been achieved by co-loading their cavity with enzymes and dye indicators (Type II), while multiple detection of ions has been performed by incorporating QD-based barcodes in the shell of Type I LbL (Type III).8
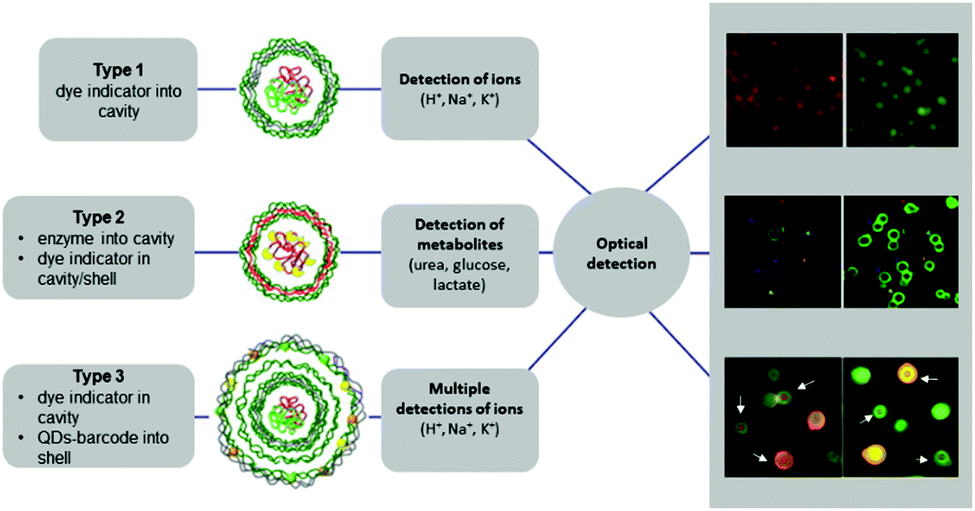 | ||
| Fig. 9 Different types of LbL-based sensors. Type 1 (typically for ion detection): ion-sensitive indicators reside in the cavity. Type 2 (typically for metabolite detection): metabolite-degrading enzymes and indicator dyes are present inside the cavity, or indicator dyes are incorporated in the outer shell. Type 3 (for multiplex ion detection): Type 1 with QD-based barcode placed in the outermost shell. Modified from ref. 8 with permission of Elsevier B.V. | ||
pH and ion sensitive LbL capsules
LbL capsules based on anionic and cationic polymer layers were able to reversibly respond to changes in pH or ionic strength because the polymer chains became uncharged if proton or ion concentrations increased. This reduction of charge caused an electrostatic repulsion between the different layers. As a result, the capsules swelled194 with a concomitant increase in membrane permeability.195 By decreasing the ion concentration to the starting conditions, the capsules shrunk again.For sensing pH-changes in particular, LbL microcapsules based on sodium polystyrene sulfonate (PSS) as an anionic and poly(allylamine hydrochloride) (PAH) as a cationic layer were modified with pH-responsive dextran-conjugates that emit a strong fluorescence signal lasting for several days.106,196 The resulting capsules were able to exhibit different colorimetric changes depending on alkaline, neutral and acidic pH values. To obtain pH-sensitivity, capsules were prepared that contain microparticles loaded with seminaphthorhodafluor (SNARF)–dextran conjugates.
Using the same PEM but an adapted preparation method, other LbL sensing systems based on incorporated fluorophores were developed that allowed for the optical ratiometric sensing of Na+, K+ or Cl−. Besides H+-sensor capsules based on fluorescein isothiocyanate (FITC) or SNARF, Na+-sensors based on sodium-binding benzofuran isophthalate (SBFI), K+-sensors based on PBFI and Cl−-sensors based on the halide-sensitive fluorescence indicator amino-MQAE were reported.32 All the different capsules were equipped with a reference dye (AF594) and with the respective ion-sensitive fluorophore. In the case of the cation sensors (H+, Na+ and K+), the response of the fluorophore reflected the ability of the capsules to sense low, medium and high concentrations of these ions.
The simultaneous detection of multiple ions is a main goal in diagnostics but is impeded by the emission cross-talk between different reporter fluorophores. To sidestep this limitation, porous microcapsules which carry a reference dye (Dy647) and one of 3 different ion-sensitive (FITC, SBFI, and PBFI) probes in their inner cavity were each marked with a unique fluorescent quantum dot (QD) barcode in their outermost wall to identify individual sensors.6 In mixtures of H+-sensor, Na+-sensor and K+-sensor capsules, the fluorescence emitted by the QDs was used for sensing the type of ion (orange: H+, green: Na+ and yellow: K+), while the ratio of the measured intensities of the ion-sensitive fluorophore and reference dye reflected the corresponding ion-concentration (Fig. 10). Thus, based on ion-sensitive QD barcode identification, combined H+-sensor, Na+-sensor and K+-sensor capsules could be used for the multiplexed analysis of proton, sodium, and potassium ions in solution. The engineering of QD barcodes to capsule walls represents a promising strategy for optical multianalyte measurements.
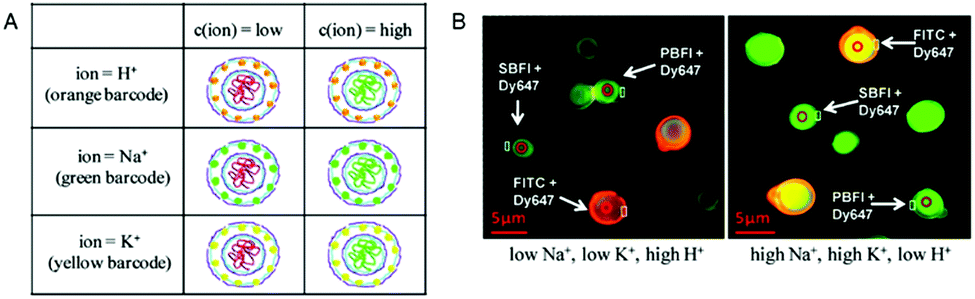 | ||
| Fig. 10 Multiplexed measurements of ions with QD barcoded polyelectrolyte sensor capsules. (A) Three different types of capsules loaded with Dy647-modified dextran and with either FITC-, SBFI-, or PBFI-modified dextran. These capsules were labeled with a QD barcode incorporated in their outer wall. (B) Fluorescence image of a mixture of the three different types of capsules in two solutions with different ion concentrations. By changing from a low Na+- and K+-concentration (5 mM) at pH 5 (left panel) to a high Na+- and K+-concentration (140 mM) at pH 9 (right panel), the ISBFI/IDy647 ratio of Na+-responsive capsules increased. Adapted and modified from ref. 6. Copyright® 2011 American Chemical Society. | ||
Oxygen and metabolite-sensitive LbL capsules
Although LbL capsules based on PSS as an anionic layer and PAH as a cationic layer were initially used for sensing pH changes or different ions, the same LbL capsules have been intensively explored for their ability to sense different molecules, such as oxygen (O2),197,198 or metabolites, for example urea,109 lactate,108 cholesterol199 and glucose.200–203 LbL capsules allowed the ratiometric detection of oxygen by entrapment of a ruthenium-based oxygen fluorophore (Ru(bpy)) together with fluorescein serving as reference.197 With increasing oxygen concentration, the fluorescence emitted by Ru(bpy) decreased while the fluorescein signal remained constant. All metabolite-sensitive capsules used one of the following two strategies for sensing: (i) the capsules contained an O2-quenchable fluorophore in order to detect a decreasing O2-concentration caused by the respective metabolite and (ii) LbL capsules served as catalytic compartments that produce a fluorescent product in the presence of a certain metabolite.(i) Besides the ratiometric detection of oxygen, different O2-quenchable fluorophores have been used for sensing different types of metabolites in coupled reactions. LbL capsules sensing cholesterol were based on cholesterol oxidase (ChOx) immobilized in alginate–silica cores together with platin(II)-octaethylporphine (PtOEP) as an extraordinary O2-quenchable luminescent dye.199 The resulting sensing mechanism for cholesterol involves the ChOx-catalyzed production of Δ-cholestenone and H2O2. This reaction results in a decreased O2-concentration causing the characteristic luminescence of PtOEP in a highly sensitive and selective manner. Glucose-detecting capsules present a very attractive platform for the clinical monitoring of the blood glucose in patients with diabetes. The sensing concept is based on incorporated glucose oxidase (GOx) for the selective catalyzed reaction of glucose and O2 to gluconic acid and H2O2. GOx-loaded alginate hydrogel microspheres served as the core and as the immobilization matrix for the enzyme. The O2-quenchable ruthenium fluorophore Rh(dpp) was distributed throughout this alginate-core along with GOx to observe a decreased O2-concentration in the presence of glucose.200
(ii) A complementary strategy to obtain metabolite-responsive LbL capsules involved the development of catalytic LbL capsules with metabolite-degrading enzymes inside. Enzyme catalyzed reactions were used to convert the respective metabolite into a product that reacts with sensitive dyes to cause a colorimetric response. The selective permeability of the capsule shells allowed the metabolite to pass through freely, while the enzymes inside remained protected. For example, urea-sensing LbL capsules are based on the co-encapsulation of urease as metabolite-sensitive enzyme and a SNARF-dextran conjugate as a pH-sensitive fluorophore.109 The urease-catalyzed hydrolysis of urea produces carbon dioxide and ammonia. The latter results in an increased pH that is reflected in the change in fluorescence emission of the pH-sensitive indicator SNARF. This system was suggested to sense rather high natural urea concentrations usually found in blood or urine. Lactate-biosensors based on LbL assemblies follow a similar principle.109
The negatively charged polyelectrolyte PSS was labelled with dihydrorhodamine 123 as a peroxide-sensitive fluorophore prior to PEM assembly. Lactate oxidase and peroxidase were incorporated into the capsules by loading the CaCO3 core with both enzymes. The enzyme catalyzed detection of lactate includes two consecutive steps: lactate oxidase converts the lactate to pyruvate and H2O2 which is then used by the peroxidase to catalyze the reaction of the nonfluorescent ROS indicator dihydrorhodamine 123 to fluorescent rhodamine 123. Another type of glucose-sensing LbL capsule used a coupled reaction involving co-encapsulated HRP to monitor the enzymatic activity of GOx.201 In this bienzymatic catalytic system, H2O2 produced by the GOx activity was used in a following HRP-catalyzed reaction to convert Amplex Red to fluorescent resorufin. By measuring the fluorescence, these LbL capsules enabled real-time monitoring of the glucose consumption and showed high potential for technologies sensing minimal amounts glucose.
Other LbL capsules based on alginate microspheres were investigated for their potential use as implantable fluorescent biosensors, so called “smart tattoos”.202,203 Such implants were intended for injections directly into the dermis in order to measure local changes in blood glucose levels. The optical glucose-sensors worked on the principle of fluorescence resonance energy transfer (FRET) between GOx and an FITC dextran-conjugate that took place in the absence of glucose. Due to the high binding affinity of GOx towards glucose, glucose was able to displace FITC as a donor dye resulting in a measurable decrease of energy transfer.
Another bio-inspired approach for sensing intracellular hydrogen peroxide (H2O2) has been introduced by using LbL capsules based on a combination of PSS and poly(diallyldimethylammonium chloride) (PDADMAC).113 Inside these capsules, H2O2-sensitive bovine serum albumin-capped gold nanoclusters (BSA-AuNCs) were co-encapsulated with insensitive FluoSpheres as reference fluorescent nanoparticles, allowing for the ratiometric detection of peroxide.
Perspectives of bio-inspired compartments as efficient sensors
As illustrated by the examples above, considerable efforts have been made for the development of bio-inspired compartments that rapidly produce a measurable signal reflecting the changes related to a pathological condition. Although this field is in its early stage of research and development the potential of polymer nanocompartments is immense. Yet, there is a complex scenario of factors that need to be significantly improved to optimise such nano- and micro-sized sensing systems and make them viable as novel efficient medical solutions.First, there are few polymer systems with intrinsic responsiveness to bio-relevant stimuli other than pH changes or the presence of specific ions, and also preserve their integrity during the detection process. In this respect, there are different options to expand the chemistry of polymersome membranes such as to achieve responsiveness to other stimuli. For example, polymers containing phenylboronic acid were reported to possess glucose-responsiveness,204,205 and thus, represent a viable option for making glucose-sensitive polymersomes. Furthermore, self-assembly of diacetylene monomers (DAs) into spherical structures followed by polymerization leads to polydiacetylene vesicles (PDAs) that exhibit a unique chromatic transition from red to blue induced by several stimuli, including pH, temperature, mechanical stress or the presence of various chemical compounds.206–210 Considering this interesting property of PDA assemblies, various colorimetric sensors have been developed for detecting pathogens and their toxins, DNA,209 histamine,211 and urease,212 and for the discrimination of lysine enantiomers.213 This variability underlines the high potential in developing PDAs as components of polymer multicompartments, for example inside of other spherical assemblies or attached to their surface, that will be able to signal the presence of different environmental stimuli.
In the case of PICsomes or LbL capsules, their responsiveness can be extended to stimuli other than pH by coupling self-assembling natural polymers, lipids, proteins and enzymes to hollow polyelectrolyte structures. For example, LbL capsules based on hemoglobin and glucose oxidase were reported as glucose-sensitive multilayers that show enhanced permeability when consuming glucose.214 In addition to distinct glucose-responsiveness, these LbL capsules showed higher biocompatibility due to their environmentally friendly self-assembled components. To this day, this system is regarded as a carrier for insulin delivery and release, but we consider such LbL capsules also as an option towards developing non-toxic glucose-biosensors. Forming sensing systems based on biodegradable, natural polymers will overcome the actual issues of toxicity, which is described more precisely in this section.
It is equally important to optimise the methods for the generation of compartments with intrinsic stimuli-responsiveness such as to improve the resulting architecture, loading efficiency, and controlled range of responsiveness. For example, more controllable PICsome formation is one of the gaps remaining to be closed in the development of PICsome-based sensors. Molecular factors such as (i) the degree of cross-linking, (ii) molecular weight and (iii) the degree of branching of the polymer backbone showed a high impact on PICsomes’ shape and size.52 These different influencers of PIC formation should be exploited in order to achieve more controllable PIC systems as the next generation of diagnostic sensors. The increase of cargo loading efficiency in the biosensors is another important point that needs to be addressed, as is decreasing the amount of polymer necessary for these systems. In this respect, microfluidic approaches are expected to significantly increase the loading efficiency, as demonstrated for lipid-based GUVs215,216 or non-responsive polymers.217
As architectural integrity of the bio-inspired compartments is a prerequisite for the detection process, a key factor is their stability in biological environments. Polymersomes can be rendered more stable by cross-linking approaches, but only if the resulting assemblies retain a very low cytotoxicity.53,55,110,115,218
The structure of PICsomes was stabilized in different smart ways that should be adopted for future PICsome-based sensors. The contributions of the molecular backbone play a very important role in enhancing the stability of PICsomes and in expanding the preparation conditions.219 The introduction of guanidinium groups to the side chains of polycations, for example, led to a very narrow PICsome distribution and stability in the blood-stream.220 The reason is that guanidinium groups facilitated intermolecular hydrogen bonding in addition to electrostatic interactions within the backbone. A corresponding approach might be applied to establish sensing PICsomes in order to improve their stability without the need for chemical cross-linking. Omitting the chemical cross-linking may also result in lower PICsome toxicity which would be a definite advantage for diagnostic applications. Regarding the specific stabilization of PIC membranes, PIC systems formed by a charge-neutral diblock copolymer and a reversible metal-coordination polymer have considerable potential because the resulting PICsomes were very robust but permeable enough to allow rapid water exchange.35
For further improvement of the stability, sensors with a stable silica core and additional silica NPs in the outer shell have been fabricated by the LbL method.221 The resulting porous microspheres have been proven to be promising candidates for ratiometric acidic pH sensing. In the future, such structures could be optimized by using a diversity of dyes to allow multianalyte detection.
A crucial factor when the bio-inspired compartments are used in vivo is their toxicity. Therefore, while several of the examples presented above have potential for sensing changes in their environment, not all have been tested in terms of their cytotoxicity. For example, biogenic LbL capsules have been developed to enhance biocompatibility and biodegradability. Compared to the former capsules based on PSS and PAH, biogenic capsules are biomimetic systems containing polyelectrolyte structures with incorporated biological materials (lipids, proteins, enzymes, natural polymers, and human serum albumin)222 or consist entirely of self-assembled bio-macromolecules such as hemoglobin which are inherently less toxic.214 Bio-inspired capsules like traditional LbL assemblies afford complete encapsulation of guest molecules in their inner compartment while blocking various external molecules due to their adjustable permeability and versatile mechanical properties. Therefore, when loaded with the corresponding reporting molecules/nanoobjects, bio-inspired capsules have great potential as hybrid platforms that sense environmental signals in biomedical applications.223
Catalytic nano/micro-compartments, where non-sensitive compartments are rendered sensitive by the combination with biomolecules and reporters producing a specific readout, represent a robust tool for the further development of biosensors.3,7,21,58,119,120,153,169,178,224 Their advantage over intrinsically stimuli-responsive compartments is not only improved stability, but a larger diversity and better control over the types of stimuli that can be detected in the surrounding environment. Such catalytic compartments can be further refined by: (i) the co-encapsulation of enzymes that function in tandem provided their activities do not interfere,177 (ii) the combination of individual, communicating compartments178 and (iii) engineering multicompartment architectures where small internal compartments loaded with biomolecules act as sensors while the GUV's boundary protects and concentrates them on the inside.1,225 In this context, mimicking the subcellular compartmentalization of cells and controlling the responsiveness and/or communication inside of an artificial polymer-based architecture are two major goals for the development of new bio-inspired materials with potential application as sensors in biomedicine. The large variety of biomolecules, their specificity and precise functionality support the extension of the sensing ability of such bio-inspired compartments and provide ideal conditions for multiplexed sensing approaches. However, in order to advance as viable diagnostic solutions, catalytic compartments have to be still optimized both in terms of the compartment properties (stability, biocompatibility, and biodegradability) and the encapsulated enzymes (encapsulation efficiency). Besides, inducing stimuli-responsiveness of the compartments by insertion of membrane proteins remains a significant challenge because of the differences between cell or lipid-based membranes and synthetic membranes (flexibility and thickness). First examples of scrutinizing the factors that play a role in the successful insertion of membrane proteins in synthetic membranes23,105,226 indicate that the conditions for compartment assembly have to be carefully optimized to preserve the functionality of the membrane proteins in a synthetic environment.
Another important aspect to be taken into account for the development of efficient biosensors is their localization to relevant pathologic regions such as to allow rapid detection of the associated changes. In this respect, the combination of bio-inspired sensing compartments with targeting approaches is a necessary step for improving these systems. Ligand–receptor and antibody–antigen interactions lead to specific accumulation of surface-functionalized polymer compartments at the target site where they either (i) are taken up by the cells, (ii) identify the presence of certain biomolecules, and/or (iii) attach to the cell membrane and signal the changes to and from the cell. For example, attachment of cyclic arginylglycylaspartic acid (cRGD) peptides onto PICsomes supported their accumulation predominantly in the neovascular system, and the tumor imaging by encapsulated contrast agents in vivo was improved maintaining a signal for more than 6 h after administration.49
The prevailing view in the field is that the design of polymer assemblies with proven in vivo functionality and persistent biosensing properties for the diagnosis of pathological changes remains a challenging task. Overcoming these challenges provides incentive for many research groups to systematically explore approaches that can cope with the complexity of the real-time biosensing.
Conclusions
We here present an overview of bio-inspired polymer compartments whose response to the presence of stimuli together with rapid signal generation allow an effective, immediate sensing of changes associated with different pathologies. The supramolecular hollow architectures are versatile polymer compartments that can exhibit a distinct response to various stimuli. They represent a fundamental for nontoxic biosensors that in future will be valuable in diagnostic fields. When the polymers were assembled into nano- and micro-sized compartments, they have shown great potential in the selective detection of different disease-related signals, both in vitro and in vivo, via triggered reactions that convert the signals into specific read-outs such as fluorescence. With a properly tuned design and corresponding surface modifications, these assemblies have been proven to be promising stimuli-responsive platforms. Additionally, several approaches have been already developed to improve their stability, biocompatibility, biodegradability, and non-toxicity, in order to put in vivo applications into effect.Due to the vast possibilities of combining various polymer building blocks and the ability to integrate biomolecules or bio-mimicking molecules, polymer vesicles represent a promising tool for biomedical applications as sensors. Moreover, self-assembled 3D architectures could be chemically designed through their polymer components to respond to a large variety of stimuli, and thus, provide new opportunities for broadening the knowledge about disease-causing factors and pathological conditions. Many efforts have been made so far to improve the existing biosensing systems. However, there is still a pressing need for developing new responsive materials. Novel components include both polymer building blocks and biomolecules. Moreover, there is an increasing demand from the scientific community and funding agencies to develop in vitro surrogates for important tissues and organs. This clear request is motivated by the complexity and high-cost of using animal models. Certain aspects of pathogenesis are also too fleeting to be recorded in animal models and, therefore, in vitro analysis is needed. Our vision is that biosensors will be integrated within cell-culture systems and will be used to dissect the signaling underlying tissue injury or regeneration. In general, we foresee that the use of biosensors for cell analysis will expand exponentially during the next several years and provide novel functionalities in the fields of biology and medicine. In conclusion, our perspective briefly highlighted different types of hollow polymer compartments that are promising candidates in biosensing. We pointed at new opportunities for the combination of polymer compartments with a plethora of active biomolecules to create bio-inspired detection platforms. In addition, we provided outlooks and forecasts on various pathways to be followed when designing novel stimuli-responsive polymer-based compartments as diagnostic tools with improved sensing properties.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge the financial support provided by the Swiss National Science Foundation, the Swiss Nanoscience Institute, the University of Basel and the National Centre of Competence in Research – Molecular Systems Engineering (NCCR-MSE).Notes and references
- S. Thamboo, A. Najer, A. Belluati, C. Planta, D. Wu, I. Craciun, W. Meier and C. G. Palivan, Adv. Funct. Mater., 2019, 29, 1904267 CrossRef.
- M. Garni, T. Einfalt, R. Goers, C. G. Palivan and W. Meier, ACS Synth. Biol., 2018, 7, 2116–2125 CrossRef CAS PubMed.
- T. Einfalt, D. Witzigmann, C. Edlinger, S. Sieber, R. Goers, A. Najer, M. Spulber, O. Onaca-Fischer, J. Huwyler and C. G. Palivan, Nat. Commun., 2018, 9, 1127 CrossRef CAS PubMed.
- L. Xu, N. Ren, J. Pang, H. Deng, X. Zhu, M. Sun and D. Yan, Polym. Chem., 2017, 8, 6283–6288 RSC.
- C. E. Housecroft, D. Wu, S. Rigo, S. Di Leone, A. Belluati, E. C. Constable and C. G. Palivan, Nanoscale, 2019, 12, 1551–1562 Search PubMed.
- L. L. del Mercato, A. Z. Abbasi, M. Ochs and W. J. Parak, ACS Nano, 2011, 5, 9668–9674 CrossRef CAS PubMed.
- L. D. Blackman, S. Varlas, M. C. Arno, A. Fayter, M. I. Gibson and R. K. O’Reilly, ACS Macro Lett., 2017, 6, 1263–1267 CrossRef CAS PubMed.
- L. L. del Mercato, M. M. Ferraro, F. Baldassarre, S. Mancarella, V. Greco, R. Rinaldi and S. Leporatti, Adv. Colloid Interface Sci., 2014, 207, 139–154 CrossRef CAS PubMed.
- J. Hu and S. Liu, Macromolecules, 2010, 43, 8315–8330 CrossRef CAS.
- X. Hu, Y. Zhang, Z. Xie, X. Jing, A. Bellotti and Z. Gu, Biomacromolecules, 2017, 18, 649–673 CrossRef CAS PubMed.
- Y. Gao, M. Wei, X. Li, W. Xu, A. Ahiabu, J. Perdiz, Z. Liu and M. J. Serpe, Macromol. Res., 2017, 25, 513–527 CrossRef CAS.
- J. M. Korde and B. Kandasubramanian, Ind. Eng. Chem. Res., 2019, 58, 9709–9757 CrossRef CAS.
- L. Hu, Q. Zhang, X. Li and M. J. Serpe, Mater. Horiz., 2019, 6, 1774–1793 RSC.
- M. Wei, Y. Gao, X. Li and M. J. Serpe, Polym. Chem., 2017, 8, 127–143 RSC.
- A. M. Pethe and K. S. Yadav, Artif. Cells, Nanomed., Biotechnol., 2019, 47, 395–405 CrossRef CAS PubMed.
- M. Lomora, F. Itel, I. A. Dinu and C. G. Palivan, Phys. Chem. Chem. Phys., 2015, 17, 15538–15546 RSC.
- F. Doberenz, K. Zeng, C. Willems, K. Zhang and T. Groth, J. Mater. Chem. B, 2020, 8, 607–628 RSC.
- Y. Zhou, R. Chen, H. Yang, C. Bao, J. Fan, C. Wang, Q. Lin and L. Zhu, J. Mater. Chem. B, 2020, 8, 727–735 RSC.
- A. Abdollahi, H. Roghani-Mamaqani and B. Razavi, Prog. Polym. Sci., 2019, 98, 101149 CrossRef CAS.
- A. P. Blum, J. K. Kammeyer, A. M. Rush, C. E. Callmann, M. E. Hahn and N. C. Gianneschi, J. Am. Chem. Soc., 2015, 137, 2140–2154 CrossRef CAS PubMed.
- H. Che, S. Cao and J. C. M. Van Hest, J. Am. Chem. Soc., 2018, 140, 5356–5359 CrossRef CAS PubMed.
- S. H. C. Askes, N. Bossert, J. Bussmann, V. S. Talens, M. S. Meijer, R. E. Kieltyka, A. Kros, S. Bonnet and D. Heinrich, Biomaterials, 2018, 168, 54–63 CrossRef CAS PubMed.
- R. Goers, J. Thoma, N. Ritzmann, A. Di Silvestro, C. Alter, G. Gunkel-Grabole, D. Fotiadis, D. J. Müller and W. Meier, Commun. Chem., 2018, 1, 35 CrossRef.
- W. Ke, J. Li, F. Mohammed, Y. Wang, K. Tou, X. Liu, P. Wen, H. Kinoh, Y. Anraku, H. Chen, K. Kataoka and Z. Ge, ACS Nano, 2019, 13, 2357–2369 CAS.
- M. E. Idrissi, C. E. Meyer, L. Zartner and W. Meier, J. Nanobiotechnol., 2018, 16, 63 CrossRef PubMed.
- C. E. Meyer, J. Liu, I. Craciun, D. Wu, H. Wang, M. Xie, M. Fussenegger and C. G. Palivan, Small, 2020, 1906492, DOI:10.1002/smll.201906492.
- Y. Zhu, B. Yang, S. Chen and J. Du, Prog. Polym. Sci., 2017, 64, 1–22 CrossRef CAS.
- A. Larrañaga, M. Lomora, J. R. Sarasua, C. G. Palivan and A. Pandit, Prog. Mater. Sci., 2017, 90, 325–357 CrossRef.
- S. T. Poschenrieder, S. K. Schiebel and K. Castiglione, Eng. Life Sci., 2017, 17, 58–70 CrossRef CAS.
- N. Aibani, T. N. Khan and B. Callan, Int. J. Pharm. X, 2020, 2, 100040 Search PubMed.
- A. H. Groschel and A. H. Muller, Nanoscale, 2015, 7, 11841–11876 RSC.
- L. L. del Mercato, A. Z. Abbasi and W. J. Parak, Small, 2011, 7, 351–363 CrossRef CAS PubMed.
- K. C. Bentz and D. A. Savin, Polym. Chem., 2018, 9, 2059–2081 RSC.
- M. G. Bah, H. M. Bilal and J. Wang, Soft Matter, 2020, 16, 570–590 RSC.
- W. Zhou, J. Wang, P. Ding, X. Guo, M. A. Cohen Stuart and J. Wang, Angew. Chem., Int. Ed., 2019, 58, 8494–8498 CrossRef CAS PubMed.
- C. G. Palivan, R. Goers, A. Najer, X. Zhang, A. Car and W. Meier, Chem. Soc. Rev., 2016, 45, 377–411 RSC.
- Z. Deng, J. Hu and S. Liu, Macromol. Rapid Commun., 2017, 38, 1600685 CrossRef PubMed.
- X. Hu, Y. Zhang, Z. Xie, X. Jing, A. Bellotti and Z. Gu, Biomacromolecules, 2017, 18, 649–673 CrossRef CAS PubMed.
- M. E. Idrissi, C. E. Meyer, L. Zartner and W. Meier, J. Nanobiotechnol., 2018, 16, 63 CrossRef PubMed.
- J. Leong, J. Y. Teo, V. K. Aakalu, Y. Y. Yang and H. Kong, Adv. Healthcare Mater., 2018, 7, 1701276 CrossRef PubMed.
- C. Lu and M. W. Urban, Prog. Polym. Sci., 2018, 78, 24–46 CrossRef CAS.
- B. Iyisan and K. Landfester, Macromol. Rapid Commun., 2019, 40, 1800577 CrossRef PubMed.
- O. Rifaie-Graham, E. A. Apebende and N. Bruns, Chimia, 2019, 73, 21–24 CrossRef CAS PubMed.
- K. Rössger, G. Charpin-El Hamri and M. Fussenegger, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 18150 CrossRef PubMed.
- M. Mohammadi, M. Ramezani, K. Abnous and M. Alibolandi, Int. J. Pharm., 2017, 519, 287–303 CrossRef CAS PubMed.
- E. Rideau, F. R. Wurm and K. Landfester, Polym. Chem., 2018, 9, 5385–5394 RSC.
- S. Yorulmaz Avsar, M. Kyropoulou, S. Di Leone, C. A. Schoenenberger, W. P. Meier and C. G. Palivan, Front. Chem., 2018, 6, 645 CrossRef PubMed.
- L. Rodriguez-Arco, A. Poma, L. Ruiz-Perez, E. Scarpa, K. Ngamkham and G. Battaglia, Biomaterials, 2019, 192, 26–50 CrossRef CAS PubMed.
- W. Kawamura, Y. Miura, D. Kokuryo, K. Toh, N. Yamada, T. Nomoto, Y. Matsumoto, D. Sueyoshi, X. Liu, I. Aoki, M. R. Kano, N. Nishiyama, T. Saga, A. Kishimura and K. Kataoka, Sci. Technol. Adv. Mater., 2015, 16, 035004 CrossRef PubMed.
- Z. Sun, G. Liu, J. Hu and S. Liu, Biomacromolecules, 2018, 19, 2071–2081 CrossRef CAS PubMed.
- J. Li, Y. Li, Y. Wang, W. Ke, W. Chen, W. Wang and Z. Ge, Nano Lett., 2017, 17, 6983–6990 CrossRef CAS PubMed.
- O. F. Mutaf, Y. Anraku, A. Kishimura and K. Kataoka, Polymer, 2017, 133, 1–7 CrossRef CAS.
- F. Du, S. Bobbala, S. Yi and E. A. Scott, J. Controlled Release, 2018, 282, 90–100 CrossRef CAS PubMed.
- L. Liang, J. Fu and L. Qiu, ACS Appl. Mater. Interfaces, 2018, 10, 5213–5226 CrossRef CAS PubMed.
- H. Gumz, S. Boye, B. Iyisan, V. Kronert, P. Formanek, B. Voit, A. Lederer and D. Appelhans, Adv. Sci., 2019, 6, 1801299 CrossRef PubMed.
- X. Liu, X. Wang, B. Voit and D. Appelhans, Chemistry, 2019, 25, 13694–13700 CrossRef CAS PubMed.
- S. Chuanoi, Y. Anraku, M. Hori, A. Kishimura and K. Kataoka, Biomacromolecules, 2014, 15, 2389–2397 CrossRef CAS PubMed.
- T. Einfalt, R. Goers, I. A. Dinu, A. Najer, M. Spulber, O. Onaca-Fischer and C. G. Palivan, Nano Lett., 2015, 15, 7596–7603 CrossRef CAS PubMed.
- M. Lomora, M. Garni, F. Itel, P. Tanner, M. Spulber and C. G. Palivan, Biomaterials, 2015, 53, 406–414 CrossRef CAS PubMed.
- Y. Anraku, A. Kishimura, M. Kamiya, S. Tanaka, T. Nomoto, K. Toh, Y. Matsumoto, S. Fukushima, D. Sueyoshi, M. R. Kano, Y. Urano, N. Nishiyama and K. Kataoka, Angew. Chem., Int. Ed., 2016, 55, 560–565 CrossRef CAS PubMed.
- P. Baumann, M. Spulber, O. Fischer, A. Car and W. Meier, Small, 2017, 13, 1603943 CrossRef PubMed.
- M. Godoy-Gallardo, M. J. York-Duran and L. Hosta-Rigau, Adv. Healthcare Mater., 2018, 7, 1700917 CrossRef PubMed.
- L. Klermund and K. Castiglione, Bioprocess Biosyst. Eng., 2018, 41, 1233–1246 CrossRef CAS PubMed.
- T. S. Schwarzer, L. Klermund, G. Wang and K. Castiglione, Nanotechnology, 2018, 29, 44LT01 CrossRef PubMed.
- Y. Zhang, N. Gal, F. Itel, I. N. Westensee, E. Brodszkij, D. Mayer, S. Stenger, M. Castellote-Borrell, T. Boesen, S. R. Tabaei, F. Hook and B. Stadler, Nanoscale, 2019, 11, 11530–11541 RSC.
- A. Najer, D. Wu, A. Bieri, F. Brand, C. G. Palivan, H.-P. Beck and W. Meier, ACS Nano, 2014, 8, 12560–12571 CrossRef CAS PubMed.
- T. Osaki and S. Takeuchi, Anal. Chem., 2017, 89, 216–231 CrossRef CAS PubMed.
- A. Peyret, E. Ibarboure, J. F. Le Meins and S. Lecommandoux, Adv. Sci., 2018, 5, 1700453 CrossRef PubMed.
- E. Rideau, R. Dimova, P. Schwille, F. R. Wurm and K. Landfester, Chem. Soc. Rev., 2018, 47, 8572–8610 RSC.
- T. Einfalt, M. Garni, D. Witzigmann, S. Sieber, N. Baltisberger, J. Huwyler, W. Meier and C. G. Palivan, Adv. Sci., 2020, 7, 1901923 CrossRef CAS PubMed.
- K. Hong, W. Wang and J. W. Mays, Reference Module in Materials Science and Materials Engineering, 2016 DOI:10.1016/b978-0-12-803581-8.03738-3.
- S. G. Roy, S. Banerjee and P. De, Reference Module in Materials Science and Materials Engineering, 2016 DOI:10.1016/b978-0-12-803581-8.01357-6.
- S. Kanaoka and S. Aoshima, Polymer Science: A Comprehensive Reference, 2012, pp. 527–558 DOI:10.1016/b978-0-444-53349-4.00075-3.
- A. Hirao, R. Goseki and T. Ishizone, Macromolecules, 2014, 47, 1883–1905 CrossRef CAS.
- M. Hong, J. Chen and E. Y. Chen, Chem. Rev., 2018, 118, 10551–10616 CrossRef CAS PubMed.
- W. N. Ottou, H. Sardon, D. Mecerreyes, J. Vignolle and D. Taton, Prog. Polym. Sci., 2016, 56, 64–115 CrossRef CAS.
- M. Kamigaito, K. Satoh and M. Uchiyama, J. Polym. Sci., Part A: Polym. Chem., 2018, 57, 243–254 CrossRef.
- A. Kermagoret and D. Gigmes, Tetrahedron, 2016, 72, 7672–7685 CrossRef CAS.
- G. Moad, Polym. Chem., 2017, 8, 177–219 RSC.
- K. Matyjaszewski, Adv. Mater., 2018, 30, e1706441 CrossRef PubMed.
- X. Pan, M. Fantin, F. Yuan and K. Matyjaszewski, Chem. Soc. Rev., 2018, 47, 5457–5490 RSC.
- D. J. Walsh, M. G. Hyatt, S. A. Miller and D. Guironnet, ACS Catal., 2019, 9, 11153–11188 CrossRef CAS.
- S. L. Baker, B. Kaupbayeva, S. Lathwal, S. R. Das, A. J. Russell and K. Matyjaszewski, Biomacromolecules, 2019, 20, 4272–4298 CrossRef CAS PubMed.
- A. Sanchez-Condado, G. A. Carriedo, A. Presa Soto, M. J. Rodriguez-Alvarez, J. Garcia-Alvarez and E. Hevia, ChemSusChem, 2019, 12, 3134–3143 CrossRef CAS PubMed.
- V. Kottisch, J. O'Leary, Q. Michaudel, E. E. Stache, T. H. Lambert and B. P. Fors, J. Am. Chem. Soc., 2019, 141, 10605–10609 CrossRef CAS PubMed.
- M.-J. Kim, Y.-G. Yu, N.-G. Kang, B.-G. Kang and J.-S. Lee, Macromol. Chem. Phys., 2017, 218, 1600445 CrossRef.
- K. Matyjaszewski and N. V. Tsarevsky, J. Am. Chem. Soc., 2014, 136, 6513–6533 CrossRef CAS PubMed.
- J. Rieger, Macromol. Rapid Commun., 2015, 36, 1458–1471 CrossRef CAS PubMed.
- C. Boyer, N. A. Corrigan, K. Jung, D. Nguyen, T. K. Nguyen, N. N. Adnan, S. Oliver, S. Shanmugam and J. Yeow, Chem. Rev., 2016, 116, 1803–1949 CrossRef CAS PubMed.
- J. C. Theriot, B. G. McCarthy, C. H. Lim and G. M. Miyake, Macromol. Rapid Commun., 2017, 38, 1700040 CrossRef PubMed.
- T. G. Ribelli, F. Lorandi, M. Fantin and K. Matyjaszewski, Macromol. Rapid Commun., 2019, 40, e1800616 CrossRef PubMed.
- M. H. Stenzel and C. Barner-Kowollik, Mater. Horiz., 2016, 3, 471–477 RSC.
- D. J. Lunn, E. H. Discekici, J. Read de Alaniz, W. R. Gutekunst and C. J. Hawker, J. Polym. Sci., Part A: Polym. Chem., 2017, 55, 2903–2914 CrossRef CAS.
- J. Yeow, R. Chapman, A. J. Gormley and C. Boyer, Chem. Soc. Rev., 2018, 47, 4357–4387 RSC.
- K. Kempe, Macromol. Chem. Phys., 2017, 218, 1700021 CrossRef.
- S. Hu, J. Zhao, G. Zhang and H. Schlaad, Prog. Polym. Sci., 2017, 74, 34–77 CrossRef CAS.
- Y. Xia and J. Zhao, Polymer, 2018, 143, 343–361 CrossRef CAS.
- H. Li, R. M. Shakaroun, S. M. Guillaume and J. F. Carpentier, Chemistry, 2020, 26, 128–138 CrossRef CAS PubMed.
- D. Bandelli, J. Alex, C. Weber and U. S. Schubert, Macromol. Rapid Commun., 2020, 41, e1900560 CrossRef PubMed.
- J. Herzberger, K. Niederer, H. Pohlit, J. Seiwert, M. Worm, F. R. Wurm and H. Frey, Chem. Rev., 2016, 116, 2170–2243 CrossRef CAS PubMed.
- S. Liu, C. Ren, N. Zhao, Y. Shen and Z. Li, Macromol. Rapid Commun., 2018, 39, e1800485 CrossRef PubMed.
- H.-C. Chiu, Y.-W. Lin, Y.-F. Huang, C.-K. Chuang and C.-S. Chern, Angew. Chem., Int. Ed., 2008, 47, 1875–1878 CrossRef CAS PubMed.
- J. Qin, Q. Liu, J. Zhang, J. Chen, S. Chen, Y. Zhao and J. Du, ACS Appl. Mater. Interfaces, 2015, 7, 14043–14052 CrossRef CAS PubMed.
- R. Enomoto, M. Khimani, P. Bahadur and S.-I. Yusa, J. Taiwan Inst. Chem. Eng., 2014, 45, 3117–3123 CrossRef CAS.
- J. Gaitzsch, S. Hirschi, S. Freimann, D. Fotiadis and W. Meier, Nano Lett., 2019, 19, 2503–2508 CrossRef CAS PubMed.
- O. Kreft, A. M. Javier, G. B. Sukhorukov and W. J. Parak, J. Mater. Chem., 2007, 17, 4471–4476 RSC.
- A. Kishimura, Polym. J., 2013, 45, 892–897 CrossRef CAS.
- L. I. Kazakova, L. I. Shabarchina, S. Anastasova, A. M. Pavlov, P. Vadgama, A. G. Skirtach and G. B. Sukhorukov, Anal. Bioanal. Chem., 2013, 405, 1559–1568 CrossRef CAS PubMed.
- L. I. Kazakova, L. I. Shabarchina and G. B. Sukhorukov, Phys. Chem. Chem. Phys., 2011, 13, 11110–11117 RSC.
- W. Yang, G. Zhu, S. Wang, G. Yu, Z. Yang, L. Lin, Z. Zhou, Y. Liu, Y. Dai, F. Zhang, Z. Shen, Y. Liu, Z. He, J. Lau, G. Niu, D. O. Kiesewetter, S. Hu and X. Chen, ACS Nano, 2019, 13, 3083–3094 CrossRef CAS PubMed.
- H. Gumz, T. H. Lai, B. Voit and D. Appelhans, Polym. Chem., 2017, 8, 2904–2908 RSC.
- X. Liu, D. Appelhans and B. Voit, J. Am. Chem. Soc., 2018, 140, 16106–16114 CrossRef CAS PubMed.
- A. Biswas, S. Banerjee, E. V. Gart, A. T. Nagaraja and M. J. McShane, ACS Omega, 2017, 2, 2499–2506 CrossRef CAS PubMed.
- Q. Yan, J. Wang, Y. Yin and J. Yuan, Angew. Chem., Int. Ed., 2013, 52, 5070–5073 CrossRef CAS PubMed.
- X. Xu, W. Yang, Q. Liang, Y. Shi, W. Zhang, X. Wang, F. Meng, Z. Zhong and L. Yin, Nano Res., 2019, 12, 659–667 CrossRef CAS.
- J. Du and R. K. O'Reilly, Macromol. Chem. Phys., 2010, 211, 1530–1537 CrossRef CAS.
- A. Feng, J. Liang, J. Ji, J. Dou, S. Wang and J. Yuan, Sci. Rep., 2016, 6, 23624 CrossRef CAS PubMed.
- B. Iyisan, J. Kluge, P. Formanek, B. Voit and D. Appelhans, Chem. Mater., 2016, 28, 1513–1525 CrossRef CAS.
- X. Zhang, M. Lomora, T. Einfalt, W. Meier, N. Klein, D. Schneider and C. G. Palivan, Biomaterials, 2016, 89, 79–88 CrossRef CAS PubMed.
- C. Edlinger, T. Einfalt, M. Spulber, A. Car, W. Meier and C. G. Palivan, Nano Lett., 2017, 17, 5790–5798 CrossRef CAS PubMed.
- S. L. Loo, W. Siti, M. Thiyagarajan, J. Torres, R. Wang and X. Hu, Langmuir, 2017, 33, 12336–12343 CrossRef CAS PubMed.
- L. Martin, P. Gurnani, J. Zhang, M. Hartlieb, N. R. Cameron, A. M. Eissa and S. Perrier, Biomacromolecules, 2019, 20, 1297–1307 CrossRef CAS PubMed.
- D. Wu, S. Rigo, S. Di Leone, A. Belluati, E. C. Constable, C. E. Housecroft and C. G. Palivan, Nanoscale, 2020, 12, 1551–1562 RSC.
- S. Varlas, J. C. Foster, P. G. Georgiou, R. Keogh, J. T. Husband, D. S. Williams and R. K. O'Reilly, Nanoscale, 2019, 11, 12643–12654 RSC.
- S. Yu, T. Azzam, I. Rouiller and A. Eisenberg, J. Am. Chem. Soc., 2009, 131, 10557–10566 CrossRef CAS PubMed.
- V. I. Martynov, A. A. Pakhomov, I. E. Deyev and A. G. Petrenko, Biochim. Biophys. Acta, Gen. Subj., 2018, 1862, 2924–2939 CrossRef CAS PubMed.
- D. Schmaljohann, Adv. Drug Delivery Rev., 2006, 58, 1655–1670 CrossRef CAS PubMed.
- P. Swietach, R. D. Vaughan-Jones, A. L. Harris and A. Hulikova, Philos. Trans. R. Soc., B, 2014, 369, 20130099 CrossRef PubMed.
- J. Pizzorno, Integr. Med. (Encinitas), 2015, 14, 8–12 Search PubMed.
- I. T. Helenius, T. Krupinski, D. W. Turnbull, Y. Gruenbaum, N. Silverman, E. A. Johnson, P. H. S. Sporn, J. I. Sznajder and G. J. Beitel, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 18710–18715 CrossRef CAS PubMed.
- E. Georgieva, D. Ivanova, Z. Zhelev, R. Bakalova, M. Gulubova and I. Aoki, Anticancer Res., 2017, 37, 5373–5381 CAS.
- F. Q. Schafer and G. R. Buettner, Free Radicals Biol. Med., 2001, 30, 1191–1212 CrossRef CAS PubMed.
- H. F. Gilbert, Adv. Enzymol. Relat. Areas Mol. Biol., 1993, 63, 69 Search PubMed.
- Y. Hatori, Y. Yan, K. Schmidt, E. Furukawa, N. M. Hasan, N. Yang, C.-N. Liu, S. Sockanathan and S. Lutsenko, Nat. Commun., 2016, 7, 10640 CrossRef CAS PubMed.
- C. J. Dunnill, K. Ibraheem, A. Mohamed, J. Southgate and N. T. Georgopoulos, Oncogene, 2016, 36, 2515–2528 CrossRef PubMed.
- F. Perez-Ruiz, N. Dalbeth and T. Bardin, Adv. Ther., 2015, 32, 31–41 CrossRef CAS PubMed.
- S. Kushimoto, S. Akaishi, T. Sato, R. Nomura, M. Fujita, D. Kudo, Y. Kawazoe, Y. Yoshida and N. Miyagawa, Acute Med. Surg., 2016, 3, 293–297 CrossRef PubMed.
- M. Stumvoll, B. J. Goldstein and T. W. Van-Haeften, Lancet, 2005, 365, 1333–1346 CrossRef CAS.
- K. K. Mäkinen, Int. J. Dent., 2016, 2016, 1–16 CrossRef PubMed.
- R. Vanholder, T. Gryp and G. Glorieux, Nephrol., Dial., Transplant., 2018, 33, 4–12 CrossRef CAS PubMed.
- C.-J. Lin, C.-K. Lai, M.-C. Kao, L.-T. Wu, U. G. Lo, L.-C. Lin, Y.-A. Chen, H. Lin, J.-T. Hsieh, C.-H. Lai and C.-D. Lin, Biomedicine, 2015, 5, 7 CrossRef PubMed.
- D. E. Discher, Science, 2002, 297, 967–973 CrossRef CAS PubMed.
- J. Cui, M. P. Van Koeverden, M. Müllner, K. Kempe and F. Caruso, Adv. Colloid Interface Sci., 2014, 207, 14–31 CrossRef CAS PubMed.
- J. Gaitzsch, X. Huang and B. Voit, Chem. Rev., 2016, 116, 1053–1093 CrossRef CAS PubMed.
- J.-L. Six and K. Ferji, Polym. Chem., 2019, 10, 45–53 RSC.
- N. J. W. Penfold, J. Yeow, C. Boyer and S. P. Armes, ACS Macro Lett., 2019, 8, 1029–1054 CrossRef CAS.
- S. M. Kuiper, M. Nallani, D. M. Vriezema, J. J. L. M. Cornelissen, J. C. M. Van Hest, R. J. M. Nolte and A. E. Rowan, Org. Biomol. Chem., 2008, 6, 4315 RSC.
- M. Spulber, P. Baumann, S. S. Saxer, U. Pieles, W. Meier and N. Bruns, Biomacromolecules, 2014, 15, 1469–1475 CrossRef CAS PubMed.
- G. Liu, S. Ma, S. Li, R. Cheng, F. Meng, H. Liu and Z. Zhong, Biomaterials, 2010, 31, 7575–7585 CrossRef CAS PubMed.
- H. Lomas, I. Canton, S. Macneil, J. Du, S. P. Armes, A. J. Ryan, A. L. Lewis and G. Battaglia, Adv. Mater., 2007, 19, 4238–4243 CrossRef CAS.
- H. O. Kim, J. W. Lim, J. Choi, H. Lee, H. Y. Son, J. Kim, G. Park, H. Chun, D. Song, Y. M. Huh and S. Haam, J. Mater. Chem. B, 2017, 5, 9571–9578 RSC.
- H. E. Colley, V. Hearnden, M. Avila-Olias, D. Cecchin, I. Canton, J. Madsen, S. Macneil, N. Warren, K. Hu, J. A. McKeating, S. P. Armes, C. Murdoch, M. H. Thornhill and G. Battaglia, Mol. Pharmaceutics, 2014, 11, 1176–1188 CrossRef CAS PubMed.
- A. Belluati, V. Mikhalevich, S. Yorulmaz Avsar, D. Daubian, I. Craciun, M. Chami, W. P. Meier and C. G. Palivan, Biomacromolecules, 2019, 21, 701–715 CrossRef PubMed.
- S. F. van Dongen, M. Nallani, J. J. Cornelissen, R. J. Nolte and J. C. van Hest, Chemistry, 2009, 15, 1107–1114 CrossRef CAS PubMed.
- L. D. Blackman, S. Varlas, M. C. Arno, Z. H. Houston, N. L. Fletcher, K. J. Thurecht, M. Hasan, M. I. Gibson and R. K. O’Reilly, ACS Cent. Sci., 2018, 4, 718–723 CrossRef CAS PubMed.
- R. Deng, M. J. Derry, C. J. Mable, Y. Ning and S. P. Armes, J. Am. Chem. Soc., 2017, 139, 7616–7623 CrossRef CAS PubMed.
- J. Tan, H. Sun, M. Yu, B. S. Sumerlin and L. Zhang, ACS Macro Lett., 2015, 4, 1249–1253 CrossRef CAS.
- J. Tan, D. Liu, X. Zhang, C. Huang, J. He, Q. Xu, X. Li and L. Zhang, RSC Adv., 2017, 7, 23114–23121 RSC.
- X.-F. Xu, C.-Y. Pan, W.-J. Zhang and C.-Y. Hong, Macromolecules, 2019, 52, 1965–1975 CrossRef CAS.
- B. Karagoz, C. Boyer and T. P. Davis, Macromol. Rapid Commun., 2014, 35, 417–421 CrossRef CAS PubMed.
- I. Louzao and J. C. M. Van Hest, Biomacromolecules, 2013, 14, 2364–2372 CrossRef CAS.
- D. M. Vriezema, P. M. L. Garcia, N. Sancho Oltra, N. S. Hatzakis, S. M. Kuiper, R. J. M. Nolte, A. E. Rowan and J. C. M. Van Hest, Angew. Chem., Int. Ed., 2007, 119, 7522–7526 CrossRef.
- M. Spulber, A. Najer, K. Winkelbach, O. Glaied, M. Waser, U. Pieles, W. Meier and N. Bruns, J. Am. Chem. Soc., 2013, 135, 9204–9212 CrossRef CAS.
- O. Rifaie-Graham, S. Ulrich, N. F. B. Galensowske, S. Balog, M. Chami, D. Rentsch, J. R. Hemmer, J. Read de Alaniz, L. F. Boesel and N. Bruns, J. Am. Chem. Soc., 2018, 140, 8027–8036 CrossRef CAS.
- J. Gaitzsch, D. Appelhans, L. Wang, G. Battaglia and B. Voit, Angew. Chem., Int. Ed., 2012, 51, 4448–4451 CrossRef CAS PubMed.
- D. Gräfe, J. Gaitzsch, D. Appelhans and B. Voit, Nanoscale, 2014, 6, 10752–10761 RSC.
- H. Gumz, S. Boye, B. Iyisan, V. Krönert, P. Formanek, B. Voit, A. Lederer and D. Appelhans, Adv. Sci., 2019, 6, 1801299 CrossRef PubMed.
- M. Garni, T. Einfalt, M. Lomora, A. Car, W. Meier and C. G. Palivan, Chimia, 2016, 70, 424–427 CrossRef CAS.
- F. Axthelm, O. Casse, W. H. Koppenol, T. Nauser, W. Meier and C. G. Palivan, J. Phys. Chem. B, 2008, 112, 8211–8217 CrossRef CAS PubMed.
- C. Nardin, S. Thoeni, J. Widmer, M. Winterhalter and W. Meier, Chem. Commun., 2000, 1433–1434, 10.1039/b004280n.
- W. Meier, C. Nardin and M. Winterhalter, Angew. Chem., Int. Ed., 2000, 39, 4599–4602 CrossRef CAS.
- M. Kumar, M. Grzelakowski, J. Zilles, M. Clark and W. Meier, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 20719–20724 CrossRef CAS PubMed.
- M. Lomora, M. Garni, F. Itel, P. Tanner, M. Spulber and C. G. Palivan, Biomaterials, 2015, 53, 406–414 CrossRef CAS PubMed.
- J. Zhang, L. Wu, F. Meng, Z. Wang, C. Deng, H. Liu and Z. Zhong, Langmuir, 2012, 28, 2056–2065 CrossRef CAS.
- A. Zhu, K. Miao, Y. Deng, H. Ke, H. He, T. Yang, M. Guo, Y. Li, Z. Guo, Y. Wang, X. Yang, Y. Zhao and H. Chen, ACS Nano, 2015, 9, 7874–7885 CrossRef CAS PubMed.
- L. Ding, W. Gu, Y. Zhang, S. Yue, H. Sun, J. J. L. M. Cornelissen and Z. Zhong, Biomacromolecules, 2019, 20, 3855–3863 CrossRef CAS PubMed.
- P. Tanner, V. Balasubramanian and C. G. Palivan, Nano Lett., 2013, 13, 2875–2883 CrossRef CAS PubMed.
- A. Belluati, I. Craciun, J. Liu and C. G. Palivan, Biomacromolecules, 2018, 19, 4023–4033 CrossRef CAS PubMed.
- K. Kamiya, R. Kawano, T. Osaki, K. Akiyoshi and S. Takeuchi, Nat. Chem., 2016, 8, 881–889 CrossRef CAS PubMed.
- K. Kamiya and S. Takeuchi, J. Mater. Chem. B, 2017, 5, 5911–5923 RSC.
- M. Weiss, J. P. Frohnmayer, L. T. Benk, B. Haller, J. W. Janiesch, T. Heitkamp, M. Borsch, R. B. Lira, R. Dimova, R. Lipowsky, E. Bodenschatz, J. C. Baret, T. Vidakovic-Koch, K. Sundmacher, I. Platzman and J. P. Spatz, Nat. Mater., 2018, 17, 89–96 CrossRef CAS.
- Y. Ai, R. Xie, J. Xiong and Q. Liang, Small, 2019, e1903940, DOI:10.1002/smll.201903940.
- E. Rideau, F. R. Wurm and K. Landfester, Adv. Biosyst., 2019, 3, 1800324 CrossRef.
- A. Kubilis, A. Abdulkarim, A. M. Eissa and N. R. Cameron, Sci. Rep., 2016, 6, 32414 CrossRef CAS.
- A. C. Greene, I. M. Henderson, A. Gomez, W. F. Paxton, V. VanDelinder and G. D. Bachand, PLoS One, 2016, 11, e0158729 CrossRef PubMed.
- M. Sauer, T. Haefele, A. Graff, C. Nardin and W. Meier, Chem. Commun., 2001, 2452–2453, 10.1039/b107833j.
- A. Picker, H. Nuss, P. Guenoun and C. Chevallard, Langmuir, 2011, 27, 3213–3218 CrossRef CAS.
- A. Koide, A. Kishimura, K. Osada, W.-D. Jang, Y. Yamasaki and K. Kataoka, J. Am. Chem. Soc., 2006, 128, 5988–5989 CrossRef CAS.
- Y. Anraku, A. Kishimura, M. Oba, Y. Yamasaki and K. Kataoka, J. Am. Chem. Soc., 2010, 132, 1631–1636 CrossRef CAS PubMed.
- A. Kishimura, A. Koide, K. Osada, Y. Yamasaki and K. Kataoka, Angew. Chem., Int. Ed., 2007, 46, 6085–6088 CrossRef CAS.
- Y. Anraku, A. Kishimura, A. Kobayashi, M. Oba and K. Kataoka, Chem. Commun., 2011, 47, 6054–6056 RSC.
- N. Bertrand and J.-C. Leroux, J. Controlled Release, 2012, 161, 152–163 CrossRef CAS.
- A. Kishimura, S. Liamsuwan, H. Matsuda, W.-F. Dong, K. Osada, Y. Yamasaki and K. Kataoka, Soft Matter, 2009, 5, 529–532 RSC.
- T. Mauser, C. Déjugnat and G. B. Sukhorukov, Macromol. Rapid Commun., 2004, 25, 1781–1785 CrossRef CAS.
- A. A. Antipov and G. B. Sukhorukov, Adv. Colloid Interface Sci., 2004, 111, 49–61 CrossRef CAS PubMed.
- P. Rivera Gil, M. Nazarenus, S. Ashraf and W. J. Parak, Small, 2012, 8, 943–948 CrossRef CAS PubMed.
- M. J. Mcshane, J. Q. Brown, K. B. Guice and Y. M. Lvov, J. Nanosci. Nanotechnol., 2002, 2, 2–3 CrossRef PubMed.
- B. B. Collier, S. Singh and M. McShane, Analyst, 2011, 136, 962–967 RSC.
- J. Prasad, A. Joshi, R. D. Jayant and R. Srivastava, Biotechnol. Bioeng., 2011, 108, 2011–2021 CrossRef CAS PubMed.
- J. Q. Brown, R. Srivastava and M. J. McShane, Biosens. Bioelectron., 2005, 21, 212–216 CrossRef CAS.
- E. W. Stein, D. V. Volodkin, M. J. McShane and G. B. Sukhorukov, Biomacromolecules, 2006, 7, 710–719 CrossRef CAS.
- A. Chaudhary, M. J. McShane and R. Srivastava, Analyst, 2010, 135, 2620–2628 RSC.
- R. Srivastava, R. D. Jayant, A. Chaudhary and M. J. McShane, J. Diabetes Sci. Technol., 2011, 5, 76–85 CrossRef.
- Q. Wang, G. Li, W. Xiao, H. Qi and G. Li, Sens. Actuators, B, 2006, 119, 695–700 CrossRef CAS.
- K. Lacina, P. Skladal and T. D. James, Chem. Cent. J., 2014, 8, 60 CrossRef PubMed.
- X. Chen, G. Zhou, X. Peng and J. Yoon, Chem. Soc. Rev., 2012, 41, 4610–4630 RSC.
- S. Lee, J. Y. Kim, X. Chen and J. Yoon, Chem. Commun., 2016, 52, 9178–9196 RSC.
- J. Huo, Q. Deng, T. Fan, G. He, X. Hu, X. Hong, H. Chen, S. Luo, Z. Wang and D. Chen, Polym. Chem., 2017, 8, 7438–7445 RSC.
- E. Lebègue, C. Farre, C. Jose, J. Saulnier, F. Lagarde, Y. Chevalier, C. Chaix and N. Jaffrezic-Renault, Sensors, 2018, 18, 599 CrossRef PubMed.
- F. Fang, F. Meng and L. Luo, Mater. Chem. Front., 2020, 4, 1089–1104 RSC.
- Q. Li, S. Ren, Y. Peng, Y. Lv, W. Wang, Z. Wang and Z. Gao, Anal. Chem., 2020, 92, 1611–1617 CrossRef CAS PubMed.
- F. Jannah and J.-M. Kim, Dyes Pigm., 2019, 169, 15–21 CrossRef CAS.
- G. Yang, J. Ji, S. Zhang, G. Li and B. Li, Anal. Methods, 2020, 12, 673–678 RSC.
- W. Qi, X. Yan, L. Duan, Y. Cui, Y. Yang and J. Li, Biomacromolecules, 2009, 10, 1212–1216 CrossRef CAS PubMed.
- S. Joshi, M. T. Hussain, C. B. Roces, G. Anderluzzi, E. Kastner, S. Salmaso, D. J. Kirby and Y. Perrie, Int. J. Pharm., 2016, 514, 160–168 CrossRef CAS PubMed.
- W. S. Lin and N. Malmstadt, Eur. Biophys. J., 2019, 48, 549–558 CrossRef CAS PubMed.
- L. J. C. Albuquerque, V. Sincari, A. Ja Ger, R. Konefa, J. I. Pa Nek, P. C. Ernoch, E. Pavlova, P. S. T. Pa Nek, F. C. Giacomelli and E. Z. Ja Ger, Langmuir, 2019, 35, 8363–8372 CAS.
- G. Wang, A. Hoornweg, H. T. Wolterbeek, L. E. Franken, E. Mendes and A. G. Denkova, J. Phys. Chem. B, 2015, 119, 4300–4308 CrossRef CAS PubMed.
- K. Naoyama, T. Mori, Y. Katayama and A. Kishimura, Macromol. Rapid Commun., 2016, 37, 1087–1093 CrossRef CAS PubMed.
- M. Hori, H. Cabral, K. Toh, A. Kishimura and K. Kataoka, Biomacromolecules, 2018, 19, 4113–4121 CrossRef CAS PubMed.
- I. Acquah, J. Roh and D. J. Ahn, Macromol. Res., 2017, 25, 950–955 CrossRef CAS.
- Y. Wang, J. Zhou, X. Guo, Q. Hu, C. Qin, H. Liu, M. Dong and Y. Chen, Mater. Sci. Eng., C, 2017, 81, 13–19 CrossRef CAS PubMed.
- M. Xuan, J. Zhao, J. Shao, C. Du, W. Cui, L. Duan, W. Qi and J. Li, J. Colloid Interface Sci., 2017, 487, 107–117 CrossRef CAS PubMed.
- S. M. Kuiper, M. Nallani, D. M. Vriezema, J. J. Cornelissen, J. C. van Hest, R. J. Nolte and A. E. Rowan, Org. Biomol. Chem., 2008, 6, 4315–4318 RSC.
- S. Thamboo, A. Najer, A. Belluati, C. von Planta, D. L. Wu, I. Craciun, W. Meier and C. C. Palivan, Adv. Funct. Mater., 2019, 29, 1904267 CrossRef.
- A. Belluati, V. Mikhalevich, S. Yorulmaz Avsar, D. Daubian, I. Craciun, M. Chami, W. P. Meier and C. G. Palivan, Biomacromolecules, 2020, 21, 701–715 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2020 |