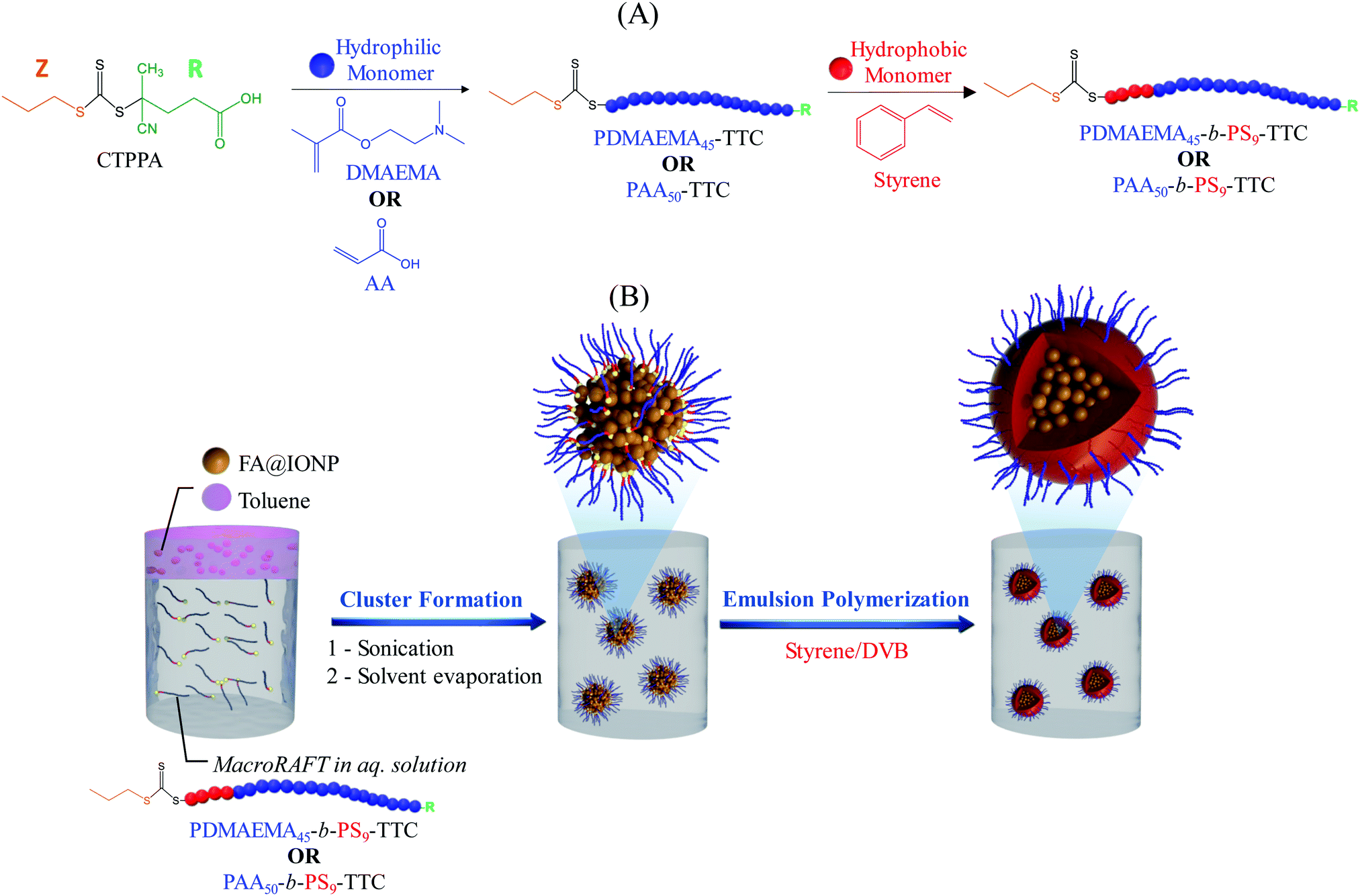
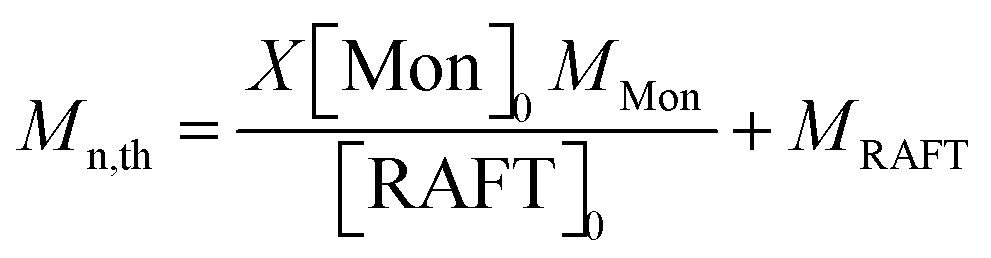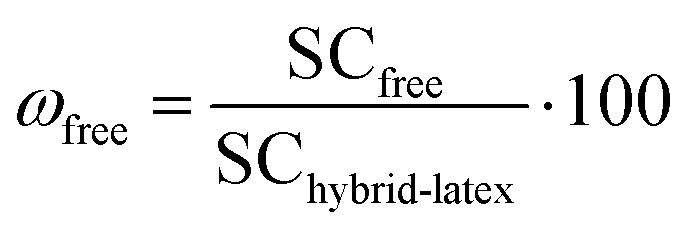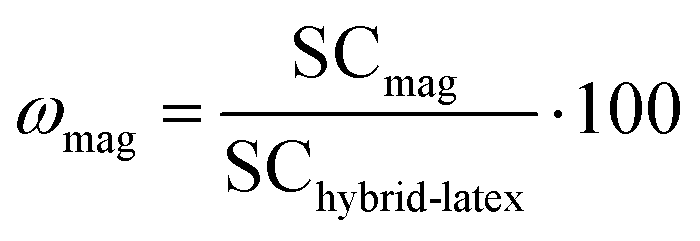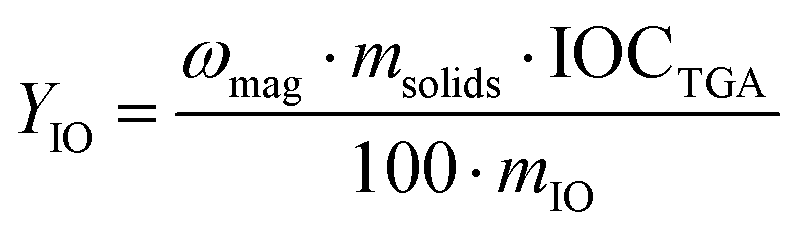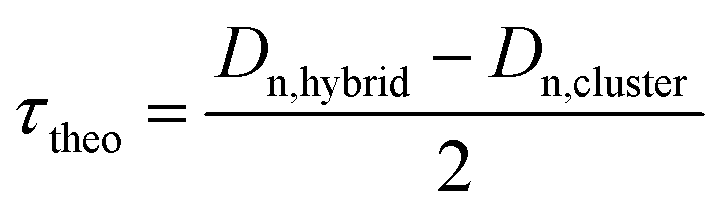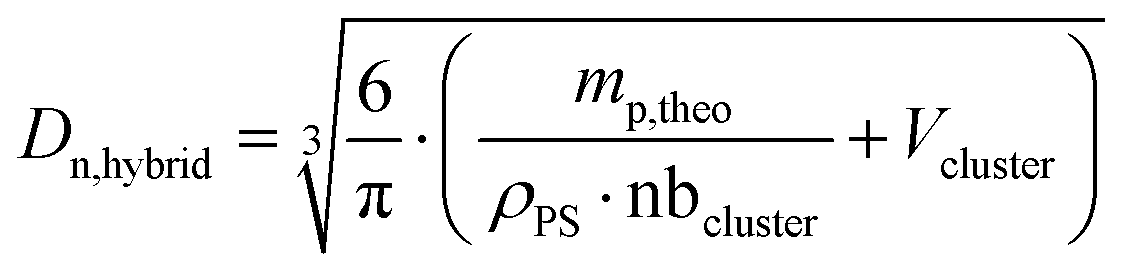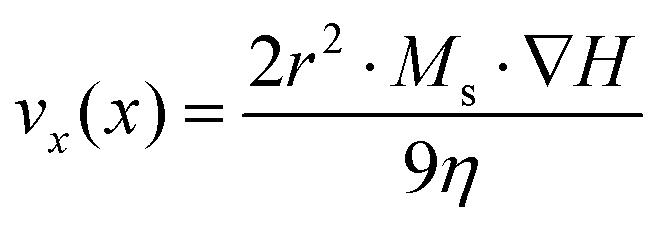Polymer-encapsulation of iron oxide clusters using macroRAFT block copolymers as stabilizers: tuning of the particle morphology and surface functionalization†
Received
11th February 2020
, Accepted 10th April 2020
First published on 14th April 2020
Abstract
We report the successful synthesis of superparamagnetic latex particles with a high fraction of magnetic materials and a fast magnetic response. Commercial fatty acid-modified iron oxide (IO) nanoparticles were first assembled into spherical clusters through an emulsification/solvent evaporation method. The resulting particles were stabilized with poly(2-dimethylaminoethyl methacrylate)-b-polystyrene (PDMAEMA-b-PS) amphiphilic block copolymers obtained by RAFT, and used as seeds in the emulsion copolymerization of styrene and divinylbenzene (DVB), used as cross-linking agent. The latter revealed to be key in preserving the integrity of the clusters during the emulsion polymerization reaction, and a minimum amount (i.e. 10 wt%) was necessary to obtain stable latexes composed of a core of densely packed IO nanoparticles surrounded by a thin polymer shell. DVB also had a strong influence on the particle morphology as the core–shell morphology of the composite particles could be tuned with either a smooth polymer shell or a raspberry-like surface by adjusting the DVB-to-monomer weight ratio and the feeding conditions. The amphiphilic macroRAFT not only provides colloidal stability to the magnetic latexes, but also offers a versatile platform for the design of composite particles with tailored surface properties by an appropriate choice of the hydrophilic block. Our strategy was thus successfully extended to poly(acrylic acid)-b-polystyrene (PAA-b-PS) copolymers, leading to PAA-stabilized composite particles. Both kinds of IO-encapsulated particles showed superparamagnetic properties (magnetizations at saturation of 35 and 31 emu g−1 for PDMAEMA and PAA systems, respectively), and could thus find interesting applications as magnetic carriers in the biological field due to their thermo- (for PDMAEMA) and pH- (for PDMAEMA and PAA) responsive properties.
1. Introduction
Superparamagnetic iron oxide nanoparticles (SPIONs) have been successfully used as magnetic carriers in applications that require the isolation of specific targets from a complex liquid medium. For instance, magnetic separation can be successfully used in the isolation and/or enrichment of proteins,1,2 peptides,3,4 viruses,5,6 bacteria7–11 and DNA/RNA amino acids.1,12–14 Nonetheless, the use of bare SPIONs is restricted as the hydrophilic surface of the nanoparticles can promote irreversible interactions with living organisms or tissues. In addition, their small size (generally lower than typically 20 nm) can lead to slow magnetic separation under a magnetic field, leading to time-consuming procedures. A way to overcome both issues is based on the design of composite particles with multi-encapsulated SPIONs protected by a shell that can be either inorganic, such as silica,15 titania3 or gold,16 or organic, like polymers.17–19 Hence, magnetic carriers with high iron oxide content (IOC) can be obtained resulting in a fast response upon exposure to an external magnetic field. In addition to a high IOC and a protective shell, the composite magnetic particles must also present superparamagnetic properties. The particles become magnetized on applying the magnetic field up to their saturation magnetization, but display negligible or no remanent magnetization when the external magnetic field is removed. This property is size-dependent and generally arises when the diameter of the iron oxide (IO) nanoparticles (magnetite and/or maghemite), is lower than typically 15 nm.20 This superparamagnetic behavior is essential for magnetic carriers (composite particles or SPIONs) used in applications involving magnetic separation, as a remanent magnetization (Mr > 0) would lead to particle aggregation due to interparticle magnetic forces, hindering their redispersion.21 Last but not least, an important characteristic of magnetic carriers is their surface functionalization, which, obviously, is a key parameter to afford suitable interactions with biological targets of interest.
Emulsion polymerization is a process widely investigated for the synthesis of IO/polymer particles,17 which can fulfill most of the aforementioned criteria, since particles with relatively large sizes (diameters >100 nm) are obtained with a relatively good control of particle surface functionalization depending on the stabilizer utilized.17,18,22 In addition, by tuning the compatibility between the inorganic and organic phases, magnetic particles exhibiting encapsulated morphology can be obtained. However, most of the examples described in the literature report composite particles with low magnetization at saturation due to low IO contents, resulting in a very slow magnetic separation.23–25 Alternatively, the formation of IO clusters prior to their encapsulation has demonstrated to be a successful strategy to obtain particles with high IOCs endowing them with a fast magnetic response.26–28 In that case, the IO clusters are encapsulated instead of the IONPs themselves. Clusters are indeed aggregates of SPIONs that, despite their relatively big particle size (50–300 nm), still display superparamagnetic properties.28–31 The relatively big cluster size, compared to the IONPs, results in a very fast response to a magnetic field, making them excellent candidates as magnetic carriers. IO clusters can be prepared via different methods such as solvothermal synthesis,29 polyelectrolyte/iron oxide self-assembly,32 solvent displacement30 and emulsification/solvent evaporation.28,31 As mentioned above, a protective shell is however required for their use as carriers, and polymers can efficiently play this role.27,28 Successful examples of IO cluster encapsulation via seeded emulsion polymerization have been reported in the literature resulting in the formation of hybrid particles with high IOCs (up to 80 wt%26).27,28 The resulting hybrid particles can display various morphologies such as multi-encapsulated or core–shell, with either a smooth or a rough polymer shell, forming in the latter case so-called raspberry-like particles.33–35 This variation in the morphology is generally attributed to the difference in the interfacial tension between the hydrophilic IO-core and the hydrophobic polymeric shell. This feature can be exploited in the field of magnetic separation to modulate the surface area of the magnetic carrier, and thus their interaction with the biological targets. However, most of the strategies relying on cluster formation result in composite particles with limited control of their surface properties. Conventional surfactants (e.g. sodium dodecyl sulfate (SDS)) can be replaced by a non-ionic poly(ethylene glycol)-based stabilizer or by an amphiphilic copolymer incorporating carboxylic acid units.34 Functional co-monomers can also be used to introduce carboxyl,33,35 hydroxyl,33 and epoxy33 groups onto the composite particles. However, such examples remain limited.
In the context of inorganic–organic hybrid materials, a strategy based on reversible addition–fragmentation chain transfer (RAFT) polymerization has recently emerged as an effective method for the preparation of composite particles with controlled morphologies and well-defined surface functionalization.36–38 The process, coined RAFT-assisted encapsulating emulsion polymerization (REEP), relies on the use of water-soluble amphipathic macroRAFT agents containing functional monomer units with lateral groups able to promote their adsorption at the inorganic surface, and an active chain end that can be reactivated for the polymerization of hydrophobic monomers in a subsequent emulsion polymerization step. The adsorbed macroRAFT agents not only encourage the polymerization to take place at the inorganic surface but also contribute to the stabilization of the resulting composite particles. This approach was first reported by the group of Hawkett for the encapsulation of titanium dioxide pigments39 and gibbsite clay platelets,40 and subsequently used by our group with success for the encapsulation of a large variety of inorganic particles such as cerium dioxide,41 silica,42 layered double hydroxides43–45 and montmorillonite clay platelets.46 REEP was also recently successfully used for the preparation of magnetic latex particles (MLPs) using poly(acrylic acid) (PAA)-based macroRAFT agents.47,48 In the work of Li et al.,47 the resulting MLPs displayed relatively large particle sizes (diameter ≈ 100 nm) and high IOCs (28%) leading to a fast and efficient magnetic separation: i.e., 99 wt% of the particles were collected after 1 min of exposure to an external magnetic field.47 Nguyen and coworkers48 were able to synthesize not only spherical magnetic particles but also magnetic rods by applying a magnetic field during the macroRAFT adsorption. Besides, Rhodamine was attached to the surface of the spheres, and the obtained dye-conjugated MLPs displayed no cytotoxicity and were successfully used for cell labeling. Although the REEP method can afford composite particles with well-defined surface functionalities, it has a limited level of customizability, as the hydrophilic monomer must be selected to promote good interaction with the inorganic nanomaterial, which restricts the range of polymers that can potentially be incorporated at the composite particle surface.
In a previous work, we demonstrated how we can exploit RAFT polymerization to design MLPs with a well-defined surface functionalization and a high IOC.49 Our reported strategy (Scheme 1) was based on the initial preparation of a poly(2-dimethylaminoethyl methacrylate)-b-polystyrene (PDMAEMA-b-PS) amphiphilic block copolymer via a two-step RAFT solution polymerization. An aqueous dispersion of IO clusters was next prepared via emulsification/solvent evaporation using the block copolymer as a stabilizer. The clusters were subsequently used as seeds in the emulsion copolymerization of styrene/divinylbenzene (DVB) resulting in polymer-encapsulated IO composite particles. One particular advantage of this process is that the surface functionality of the resulting magnetic particles can be tuned by changing the composition of the hydrophilic part of the amphiphilic macroRAFT agent. Building on this previous work, the influence of the cross-linking agent and polymer content are fully investigated in the present paper. The versatility and robustness of this MLP surface functionalization approach is also demonstrated through the preparation and use of a second amphiphilic macroRAFT block copolymer, namely poly(acrylic acid)-b-polystyrene (PAA-b-PS). Finally, the magnetic properties of both types of MLPs are evaluated.
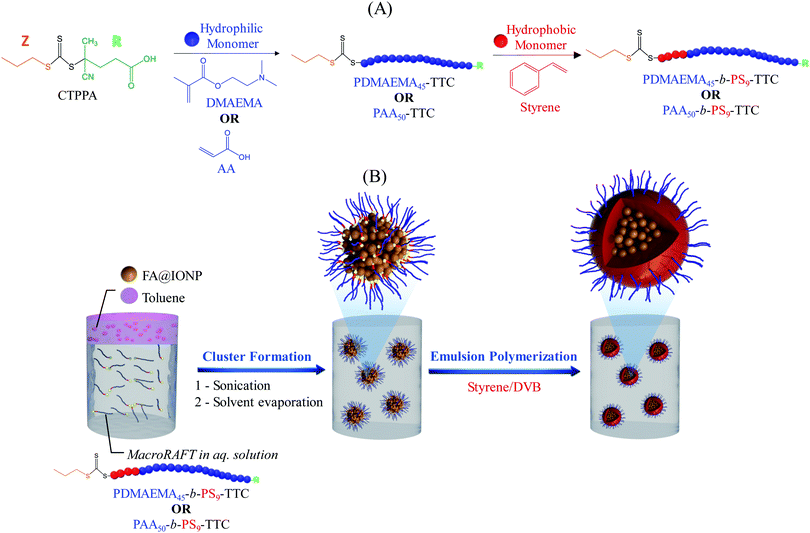 |
| | Scheme 1 Schematic representation of (A) synthesis of amphiphilic block copolymer (PDMAEMA45-b-PS9-TTC or PAA50-b-PS9-TTC), and (B) IO cluster formation via emulsification/solvent evaporation and synthesis of magnetic latex particles via emulsion polymerization using the macroRAFT-stabilized clusters as seeds. | |
2. Materials and methods
2.1 Materials
The RAFT agent, 4-cyano-4-thiothiopropylsulfanylpentanoic acid (CTPPA), was synthesized according to a protocol previously reported (1H NMR in Fig. S1, ESI†).50 The fatty acid-modified iron oxide nanoparticles (hereafter denoted as FA@IONPs, commercial name: EMG1200) were purchased from Ferrotec©. The initiators, 4,4′-azobis(cyanopentanoic acid) (ACPA, ≥98%, Sigma-Aldrich), 2,2′-azobis[2-(2-imidazolin-2-yl)propane]dihydrochloride (VA-044, 99%, Wako) and ammonium persulfate (APS, 98%, Acros Organics) were used without further purification. The solution of the methylation agent tri(methylsilyl)diazomethane (2 M in diethyl ether, Sigma-Aldrich), sodium hydroxide (NaOH 0.1 M, standard, Acros Organics), hydrochloric acid (HCl 0.1 M, standard, Acros Organics), styrene (S, 99%, Acros Organics) and toluene (>99%, Biosolve) were all used as received. Sodium nitrate (NaNO3, 99%), 1,3,5-trioxane (>99%), 1,4-dioxane (puriss p.a., >99.5%), dimethyl sulfoxide-d6 (DMSO-d6, 99%), chloroform-d (CDCl3, 99%), acrylic acid (AA, anhydrous, 99%), 2-dimethylaminoethyl methacrylate (DMAEMA, 99%) and petroleum ether were all purchased from Sigma-Aldrich and used without further purification. The cross-linking agent, divinylbenzene (DVB, 80%, Sigma-Aldrich), was composed of a mixture of four isomers (57.0 vol% m-DVB, 26.7 vol% p-DVB, 9.3 vol% m-ethyl vinylbenzene (EVB) and 7.0 vol% p-EVB as quantified by gas chromatography), and used as received. Tetrahydrofuran (THF, HPLC, stabilized/butylated hydroxytoluene, Sigma Aldrich) and dimethyl sulfoxide (DMSO, HPLC, Biosolve) were used for SEC analyses.
2.2 Methods
Synthesis of amphiphilic macroRAFT copolymers.
Amphiphilic block copolymers were synthesized in a two-step RAFT solution polymerization (Scheme 1A). Hydrophilic macroRAFT agents (PDMAEMA and PAA, MR2 and MR5, Table S1 and Fig. S2–S5, ESI†) were first synthesized, all of which carrying a reactivatable trithiocarbonate (TTC) end group. The CTPPA RAFT agent, 1,3,5-trioxane, the monomer (DMAEMA or AA), ACPA and 1,4-dioxane were introduced in a round-bottom glass flask according to recipes shown in Table S1 (ESI†), and the mixture was purged with nitrogen for 30 min. The flask was then immersed in an oil bath at 90 °C for DMAEMA and 80 °C for AA, and the polymerization stopped at a moderate conversion, i.e. around 50%, to guarantee high chain-end functionality and avoid irreversible termination reactions often observed at high conversions. A kinetic study was performed for both systems in a preliminary step (MR1 and MR4, Table S1, ESI†) to establish the time required to reach 50% conversion (105 and 72 min for PDMAEMA and PAA, respectively, Fig. S2 and S3, ESI†). The synthesized hydrophilic macroRAFTs were purified by precipitation, at least 3 times, in a large volume of petroleum ether, and characterized by size exclusion chromatography (SEC). The amphiphilic macroRAFTs were next synthesized via macroRAFT chain extension reactions using styrene as a hydrophobic monomer (Scheme 1A). Styrene polymerizations (MR3 and MR6, Table S1, ESI†) were conducted in 1,4-dioxane at 80 °C in the presence of the previously synthesized hydrophilic PDMAEMA-TTC (MR2) and PAA-TTC (MR5) macroRAFT agents.
The theoretical molar masses (Mn,th) of the hydrophilic and amphiphilic macroRAFTs were determined using eqn (1):
| | 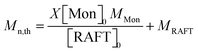 | (1) |
where
X is the monomer conversion determined by
1H NMR;
MMon, [Mon]
0,
MRAFT and [RAFT]
0 are the molar masses and the initial concentrations of monomer and of the RAFT agent (
i.e. CTPPA for MR1, MR2, MR4 and MR5, and hydrophilic macroRAFT agent for MR3 and MR6, Table S1, ESI
†), respectively. The degree of polymerization of the polystyrene block in the amphiphilic macroRAFT copolymers was determined from the relative integration of the aromatic peak from polystyrene and the characteristic peaks of PDMAEMA or PAA (Fig. S4 and S5, ESI
†).
Preparation of IO clusters.
The strategy developed for the formation of the IO clusters is based on the emulsification/solvent evaporation technique reported by Paquet et al.28,31 Initially, the commercial fatty acid-modified IO nanoparticles (EMG1200 from Ferrotec©) were dispersed in toluene at a concentration of 100 g L−1 leading to the formation of an organic ferrofluid. In parallel, aqueous solutions of amphiphilic macroRAFTs were prepared at pH 4 and pH 10 (for PDMAEMA-b-PS (MR3) and PAA-b-PS (MR6), respectively, see Table S1, ESI†). Then, 24.0 g of the toluene-based ferrofluid was introduced into 95.0 g of the aqueous solution of PDMAEMA- or PAA-based amphiphilic macroRAFT agents, and the mixture was sonicated using an ultrasonic processor (maximum output power of 750 W) with a standard probe (12 mm) during 240 s with an output power set at 150 W (amplitude = 20%). A small aliquot was collected and characterized by dynamic light scattering (DLS) to determine the droplet diameter. Toluene was then extracted by rotary evaporation under vacuum at a maximum temperature of 45 °C to prevent premature degradation of the RAFT agent. Water was added intermittently to the round-bottom flask to maintain a constant volume. The obtained PDMAEMA@clusters and PAA@clusters were purified by magnetic washing, and characterized by DLS and transmission electron microscopy (TEM). The results are summarized in Table S2 and Fig. S6 (ESI†).
Seeded emulsion polymerization.
Surfactant-free emulsion polymerization of styrene (with or without DVB) was carried out using IO clusters as seeds in semi-continuous experiments. In a typical experiment (Exp. 4, Table 1), 15 g of the cluster dispersion with a solid content (SC) of approximately 2% were introduced into a 50 mL double-jacketed round-bottom flask equipped with a condenser, a nitrogen inlet, a syringe pump and a mechanical stirring system (Fig. S7, ESI†). Then, VA-044 (7.6 mg, 1.7 10−3 M) and a minimum amount of monomer mixture (0.06 g, sty![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :DVB 80:20 wt%) were introduced in the reactor. The dispersion was deoxygenated with nitrogen for 30 min. After this interval, water at 60 °C was recirculated through the double jacket marking the beginning of the reaction. The monomer mixture (1.5 g, sty:DVB 80:20 wt%) was fed into the reactor; experimental details of the feeding profile are presented in Table S3 (ESI†). The polymerization reaction was conducted during 6 h and samples were periodically withdrawn to follow the evolution of monomer conversion as a function of time, by gravimetric analysis. The experimental details of all the polymerizations are gathered in Tables 1, 2 and Table S3 (ESI†).
:DVB 80:20 wt%) were introduced in the reactor. The dispersion was deoxygenated with nitrogen for 30 min. After this interval, water at 60 °C was recirculated through the double jacket marking the beginning of the reaction. The monomer mixture (1.5 g, sty:DVB 80:20 wt%) was fed into the reactor; experimental details of the feeding profile are presented in Table S3 (ESI†). The polymerization reaction was conducted during 6 h and samples were periodically withdrawn to follow the evolution of monomer conversion as a function of time, by gravimetric analysis. The experimental details of all the polymerizations are gathered in Tables 1, 2 and Table S3 (ESI†).
Table 1 Experimental conditions and results for seeded semi-batch emulsion polymerization of styrene in the presence of DVB (except Exp. 1) using PDMAEMA@clusters as seedsa
| Exp. |
SCtheob (wt%) |
PCtheob (wt%) |
DVBc (wt%) |
IOCtheod (wt%) |
X
(%)/t (h) |
Z
av
(nm)/PdI |
D
n (nm)/Dw/Dng |
ω
free/ωmagh (wt%) |
IOCTGAi (wt%) |
Y
IO
(wt%) |
Coag.k (wt%) |
|
Temperature = 60 °C; PDMAEMA@cluster dispersion: 15 g; VA-044: 0.5 wt% of overall monomer mass.
Theoretical solid content (SCtheo) and polymer content (PCtheo) considering 100% monomer conversion (see Table S3, ESI for feeding conditions).
DVB content based on overall monomer mass.
IOC based on overall monomer mass.
Monomer conversion/reaction time.
Determined by DLS.
Determined by statistical analysis of 90–360 particles from the TEM images (histograms presented in ESI).
Determined using eqn (2) and (3) after 10 min of magnetic separation.
Determined by TGA.
Yield of iron oxide incorporation in the magnetic fraction calculated using eqn (4).
Coagulum content (wt%) based on the total mass of latex determined after filtration on a 160 mesh grid.*Unstable latex. |
| 1 |
13.2 |
11.3 |
0 |
12 |
81/6 |
589/0.35 |
— |
15/85 |
14 |
97 |
— |
| 2* |
13.4 |
11.6 |
5 |
11 |
16/6 |
Unstable |
— |
— |
— |
— |
1.96 |
| 3 |
14.3 |
12.5 |
10 |
10 |
26/7 |
462/0.24 |
246/1.06 |
9/91 |
14 |
52 |
— |
| 4 |
14.0 |
12.1 |
20 |
11 |
34/6 |
596/0.21 |
197/1.18 |
11/89 |
44 |
89 |
0.02 |
| 5 |
4.5 |
2.5 |
12 |
56 |
66/7 |
228/0.09 |
— |
— |
— |
— |
0.01 |
| 6 |
4.6 |
2.6 |
23 |
53 |
63/7 |
290/0.17 |
— |
6/94 |
46 |
95 |
0.02 |
| 7 |
4.6 |
2.5 |
38 |
53 |
55/7 |
290/0.20 |
142/1.28 |
6/94 |
54 |
82 |
0.21 |
Table 2 Experimental conditions and results for seeded semi-batch emulsion polymerization of styrene in the presence of DVB using PAA@clusters as seedsa
| Exp. |
Initiator/T (°C) |
pH0b |
X
(%)/t (h) |
Z
av
(nm)/PdI |
D
n (nm)/Dw/Dne |
ω
free/ωmagf (wt%) |
IOCTGAg (wt%) |
Y
IO
(wt%) |
Coag.i (wt%) |
|
PAA@clusters dispersion: 15 g; PCth ≈ 2.5%; SCtheo ≈ 4.5%; IOCtheo = 55% (based on overall monomer mass); [initiator] = 1.7 mM; DVB ≈ 38% based on total monomer (see Table S3, ESI for feeding conditions).
Initial pH of the polymerization medium.
Monomer conversion/reaction time.
Determined by DLS.
Determined by statistical analysis of around 350 particles from the TEM images (the histogram is presented in Fig. S18, ESI).
Determined using eqn (2) and (3) after 10 min of magnetic separation.
Determined by TGA.
Yield of iron oxide incorporation in the magnetic fraction calculated using eqn (4).
Coagulum content (wt%) based on the total mass of latex determined after filtration on a 160 mesh grid.*Unstable latexes. |
| 8* |
VA-044/60 |
7.5 |
* |
Unstable |
— |
— |
— |
— |
1.31* |
| 9* |
ACPA/70 |
7.5 |
* |
Unstable |
— |
— |
— |
— |
0.46* |
| 10 |
ACPA/70 |
9.6 |
41/22 |
203/0.11 |
— |
— |
— |
— |
0.01 |
| 11 |
ACPA/80 |
9.5 |
47/22 |
650/0.43 |
129/1.21 |
3/97 |
59 |
100 |
0.16 |
| 12* |
APS/75 |
7.4 |
* |
Unstable |
— |
— |
— |
— |
0.87* |
| 13 |
APS-NaHCO3/75 |
7.3 |
43/21 |
266/0.30 |
— |
— |
— |
— |
0.13 |
Magnetic separation.
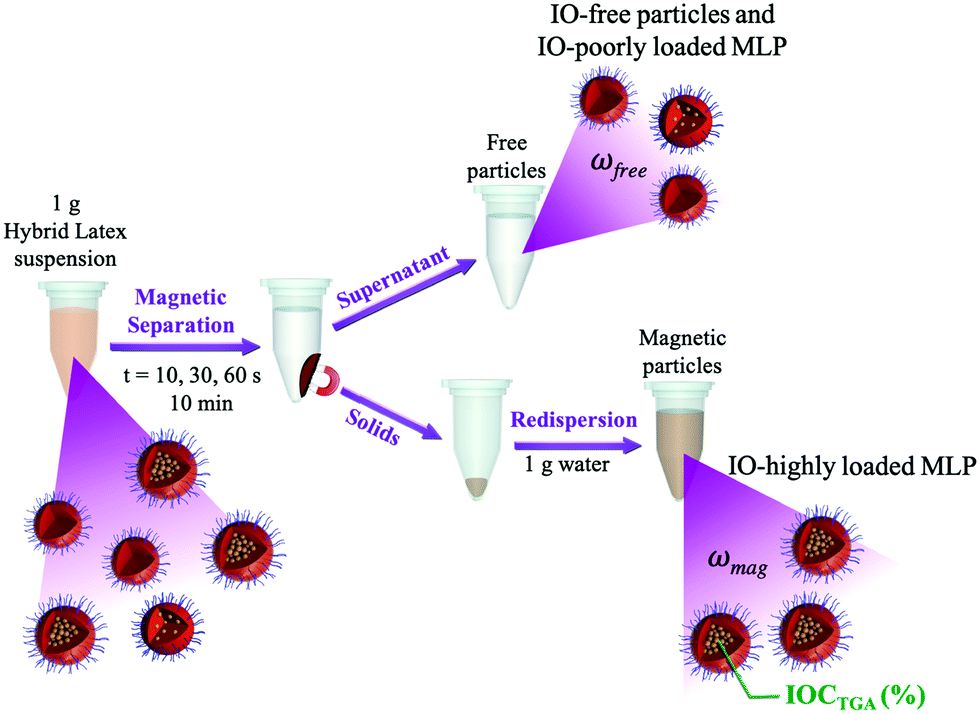
The resulting hybrid latexes were characterized in order to determine the fraction of magnetic latex particles and of “free” latex particles (i.e., latex particles that are not attracted by the magnet), as schematically shown in Fig. 1. Firstly, the hybrid latexes were exposed to a magnetic field using a magnet (Dynamag – 2 from ThermoFisher Scientific) during different times: 10, 30, 60 or 600 s, to separate the magnetic fraction from the non-magnetic latex fraction (which contains latex particles devoid of IO or composite particles with a very low amount of IONPs). The supernatant was collected, and the solid content was determined gravimetrically (SCfree). The magnetic fraction was redispersed in water and its solid content was again determined by gravimetry (SCmag). The fractions of MLPs (ωmag, wt%) and of free latex particles (ωfree, wt%) were then calculated using eqn (2) and (3), where SChybrid-latex is the experimental solid content of the hybrid latexes before magnetic separation. The magnetic fraction (ωmag) was then dried and characterized by thermogravimetric analysis in order to determine the iron oxide content (IOCTGA, wt%).| |  | (2) |
| |  | (3) |
The yield of iron oxide incorporation in the magnetic fraction, YIO (%), was then determined as follows:| | 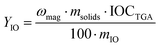 | (4) |
where mIO and msolids are, respectively, the mass of iron oxide and of total solid in the latex suspension.
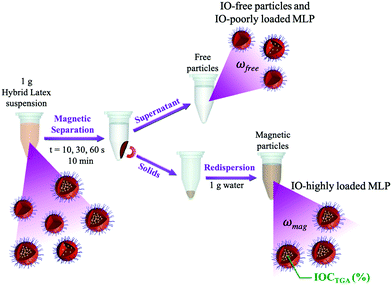 |
| | Fig. 1 Scheme of magnetic separation of IO/polymer composite latex particles. | |
2.3 Characterization techniques
Nuclear magnetic resonance (NMR).
Samples taken during the RAFT solution polymerization (synthesis of macroRAFT) were characterized by 1H liquid NMR spectroscopy (300 MHz Bruker) in DMSO-d6 at room temperature to determine the individual monomer conversion from the relative integration of the vinylic protons and the protons of 1,3,5-trioxane.
Size exclusion chromatography (SEC).
Before analyses of the homopolymers or block copolymers composed of AA units, the carboxylic acid groups were methylated in a THF/H2O (90/10) mixture using tri(methylsilyl)diazomethane as a methylation agent to prevent interactions between the polar groups and the stationary phase.51,52 All the samples were filtered through a 0.45 μm pore size membrane and analyzed at 4 mg mL−1. SEC analysis in dimethylformamide (DMF-SEC/LiBr, 0.01 mol L−1) was performed at 70 °C with a flow rate of 1.0 mL min−1 using an Eco-SEC semimicro SEC system from Tosoh. The separation was carried out using two PSS GRAM columns (7 μm, 300 × 7.5 mm). The setup was equipped with a dual flow refractive index (RI) detector and a UV detector. SEC measurements in THF (THF-SEC) were carried out at 40 °C with a flow rate of 1 mL min−1. The separation was carried out on three columns from PSS Instruments [PSS SDV analytical (8 × 300 mm)]. The device (Viscotek TDA305) was equipped with an RI detector (λ = 670 nm). The average molar masses (number-average molar mass Mn and weight-average molar mass Mw) and the molar mass distributions (Đ = Mw/Mn) were derived from the RI signal using a calibration curve based on poly(methyl methacrylate) (PMMA) or PS standards from Polymer Laboratories. For SEC-THF analysis of the homopolymers, the Mark–Houwink–Sakurada (MHWS) parameters were also used to calculate Mn, Mw and Đ using the following parameters: K = 3.81 × 10−4 and α = 0.63 (THF; 30 °C)53 for poly(methyl acrylate) (PMA) and K = 4.98 × 10−5 and α = 0.73 (THF; 30 °C)22 for PDMAEMA.
Dynamic light scattering (DLS).
The intensity-weighted mean diameters (Zav) were measured by dynamic light scattering (DLS) using a Zetasizer NanoZS instrument from Malvern. The data were collected at 173° scattering angle using the fully automatic mode of the Zetasizer system and fitted with the cumulant analysis. The broadness of the distribution was given by a dimensionless number called PdI (the higher this value, the broader was the size distribution).
Electron microscopy.
The morphology and particle size of the hybrid latexes were evaluated by transmission electron microscopy (TEM). The hybrid latex suspensions were diluted 20 times in water, and then deposited on a formvar–carbon 200-mesh copper grid. The observations were made using a Philips CM120 microscope operating at an accelerating voltage of 120 kV (Centre Technologique des Microstructures (CTμ), platform of the Université Claude Bernard Lyon 1, Villeurbanne, France). The number and weight average particle diameters (Dn and Dw, respectively), and the polydispersity index (Dw/Dn) were calculated using Dn = ∑niDi/∑ni and Dw = ∑niDi4/∑niDi3, where ni is the number of particles (1100–1300 particles) with diameter Di. The theoretical polymer shell thickness (τtheo) of the hybrid particles was calculated as follows:| | 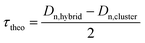 | (5) |
where Dn,cluster is the number-average diameter of the clusters determined by TEM (Fig. S6, ESI†) and Dn,hybrid is the theoretical diameter of the hybrid latex particles calculated with the following equation:| | 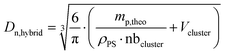 | (6) |
where Vcluster and nbcluster are, respectively, the number-average volume of one cluster and the number of clusters in the sample (determined from Dn,cluster), ρPS is the polystyrene density (1.05 g cm−3) and mp,theo is the theoretical mass of the polymer. The experimental shell thickness was determined directly from the TEM micrographs, with manual measurements on 100–250 particles. The morphology of the hybrid particles was also evaluated via scanning electron microscopy (SEM). The hybrid latex suspensions were diluted 20 times in water, and then deposited on a formvar–carbon 200-mesh copper grid, dried and covered by a thin layer of copper (sputtering Cu with Baltec MED020 – 10 nm). The observations were made using a FEI QUANTA 250 FEG scanning electron microscope at an acceleration voltage of 10 kV (CTμ).
Thermogravimetric analysis (TGA).
TGA was performed using a TGA/DSC1 STARe system from Mettler Toledo. In a typical experiment, the sample (5–15 mg) was heated under a nitrogen or air atmosphere from room temperature to 900 °C with a heating rate of 10 °C min−1.
Superconducting quantum interference device (SQUID) analysis.
Magnetic measurements were carried out using a SQUID MPMS-XL5 magnetometer (Quantum Design) equipped with an integrated helium liquefier.
Brunauer–Emmett–Teller (BET) surface area analysis.
Nitrogen adsorption–desorption isotherms were recorded using a Micromeritics ASAP 2000 device. Prior to analysis, the samples were purged under vacuum at 120 °C overnight to remove surface water. The specific surface area was determined using the BET method.
3. Results and discussion
3.1 Synthesis of PDMAEMA-stabilized magnetic latex particles (PDMAEMA@MLPs)
In our previous work, we demonstrated that the use of a cross-linker (namely, DVB) led to the encapsulation of IO clusters in a well-defined core–shell morphology.49 In fact, for the system prepared in the absence of cross-linker in the monomer charge (100% styrene), the spherical clusters disassembled generating MLPs with a multi-encapsulated instead of a core–shell morphology. In the present work, seeded emulsion polymerization of styrene was thus first investigated using increasing amounts of DVB, while maintaining a fixed polymer content (PCtheo = 12 wt%) and a fixed IO content (IOCtheo = 10–11 wt%, based on overall monomer mass) (Exp. 2–4, Table 1). For the sake of comparison, a reference experiment was also performed in the absence of DVB (Exp. 1, Table 1). The PDMAEMA@clusters were prepared as reported previously,49 and exhibited a spherical shape with a number average diameter of 71 nm and a relatively large particle size distribution (Dw/Dn = 1.58, Table S2, ESI†). The suspension was purified by magnetic washing to remove non-adsorbed macroRAFTs, and used as seeds in emulsion polymerization of styrene using DVB as a cross-linking agent (Table 1). In addition, to avoid the excess of monomer in the reaction medium during the initial stages of the reaction, which could promote swelling of the clusters and affect their integrity, all polymerizations were carried out in semi-batch conditions as reported in Table S3 (ESI†). Samples were taken at regular intervals and analyzed by gravimetry to follow the polymerization kinetics. As shown in Fig. 2A, all the experiments performed in the presence of DVB (Exp. 2–4 in Table 1) exhibited low polymerization rates and limited conversions. This can be explained by the thermodynamic balance involved in the stabilization of swollen cross-linked particles in emulsion polymerization. The Gibbs free energy balance involved in this equilibrium can be expressed as follows:54| | | ΔḠ* = ΔḠm + ΔḠt + ΔḠel | (7) |
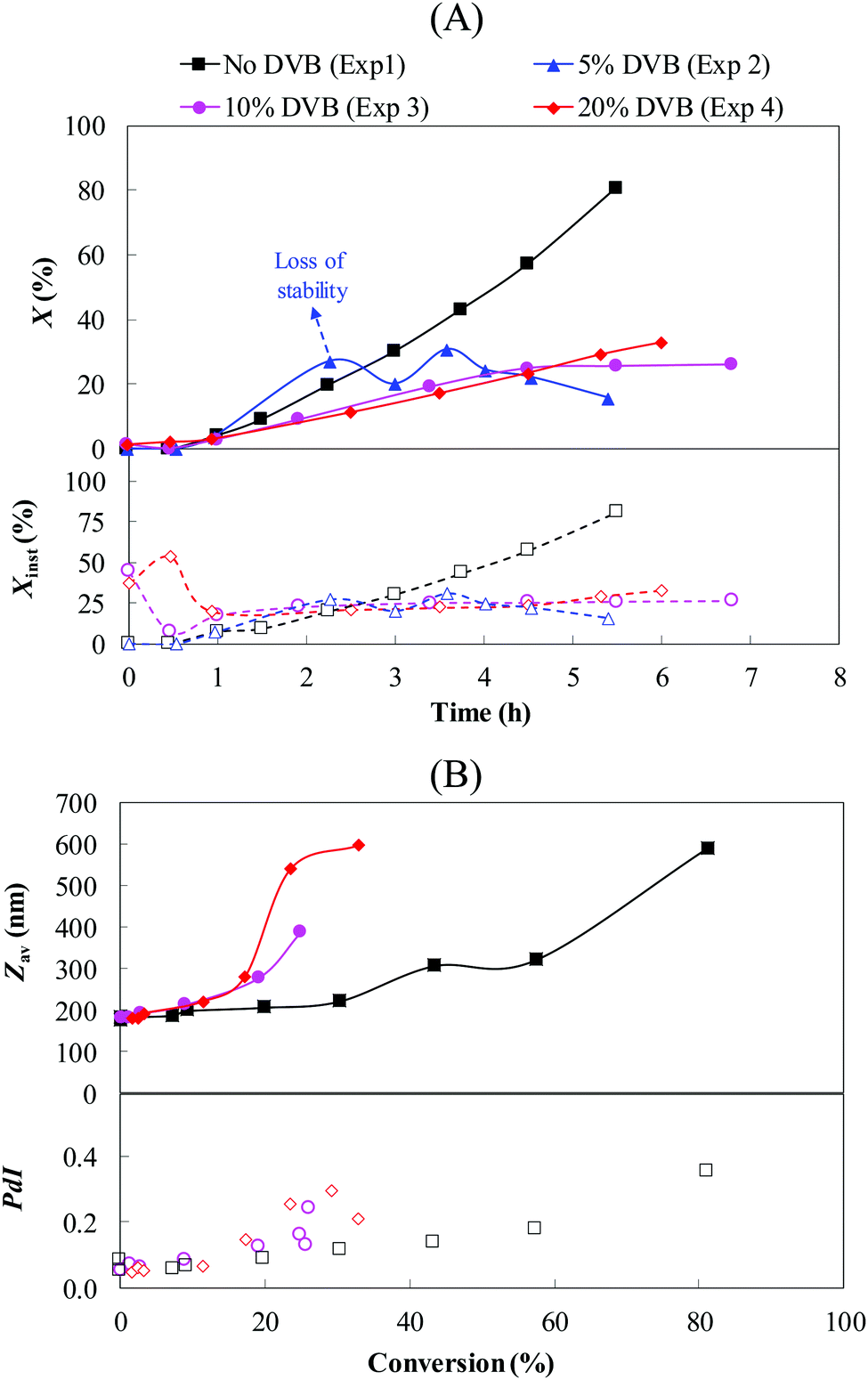
where ΔḠ* is the molar free energy, ΔḠm is the contribution of monomer–polymer mixing forces, ΔḠt is the particle-water interfacial tension force and ΔḠel the polymer network elastic-retractile force. In the absence of DVB (Exp. 1, Fig. 2A), only the two first parameters of eqn (7) must be considered.55 Given that the monomer is generally a good solvent for the polymer, the ΔḠ* value remains negative over the polymerization keeping the system energetically favorable towards polymer-monomer mixing. In the presence of DVB (Exp. 2–4), the term ΔḠel is added to the energy balance to take into account the changes in the polymer network configuration.56 This elastic force is directly affected by the cross-linking degree, and the positive increment of ΔḠel leads to a less energetically favorable solvation of the polymer particles. This induces a gradual decrease of the monomer concentration into the polymer particles resulting in lower polymerization rates (Fig. 2A). Indeed, at high cross-linker content the particles become solid, which means that the monomer cannot diffuse inside the particles and the polymerization is thus quenched, which limits the final conversion. Errede et al.57 reported that for a styrene/DVB system, the critical cross-linking degree is attained for 20 mol% of DVB in the polymer chains. In our case, taking into account the high reactivity of the first double bond of DVB58 and the fact that the second double bond should have approximately the same reactivity as styrene,59,60 the first chains generated are likely rapidly reaching this critical value generating the plateau of conversion.
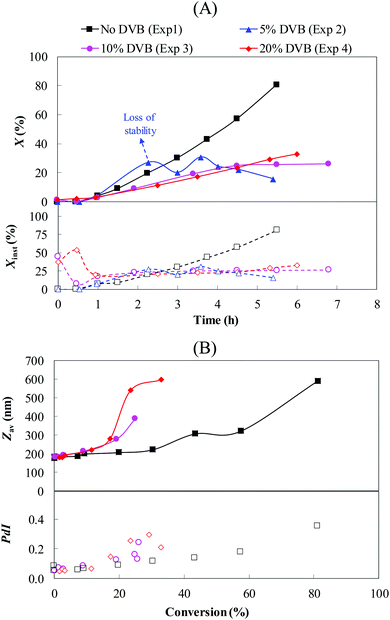 |
| | Fig. 2 Effect of DVB content on (A) the instantaneous (Xinst, empty symbols) and global conversions (X, filled symbols) with time and (B) the particle size (Zav, filled symbols) and size dispersity (PdI, empty symbols) with conversion, for the seeded semi-batch emulsion polymerization of styrene using PDMAEMA@clusters as seeds. The theoretical polymer and IO contents were fixed at around 12 and 11 wt%, respectively (Exp. 1–4, Table 1). | |
The experiments performed with 10 and 20 wt% DVB led to stable latexes (Table 1), and so did the experiment carried out without DVB (Table 1). The good colloidal stability of our system is illustrated by the relatively low coagulum content observed in these experiments (Table 1), generally lower than 0.2%, except for Exp. 2 (5 wt% DVB), which exhibited poor colloidal stability with a coagulum content of 1.96% (14.5 wt% of the theoretical solids in the system). The evolution of particle size with conversion is shown in Fig. 2B. It can be clearly seen that the size gradually increased with increasing conversion suggesting that the polymer chains were effectively captured by the IO clusters, which contributed to increase their size. This is accompanied by a sharp increase of particle size for conversions higher than 60% and 20% for Exp. 1 and Exp. 3–4, respectively, which could be associated with a lack of colloidal stability. However, these results must be considered with care, as it is well known that the intensity-weighted mean diameter (Zav) measured by DLS is strongly affected by the presence of big particles (>500 nm). The relatively big particles detected in the systems containing DVB, associated with the increase of the PdI values with conversion, could be the result of some bridging between the particles, i.e. inter-particle cross-linking reactions. Partial burial of the PDMAEMA hydrophilic segment (stabilizer) into the particles could also lead to colloidal stability issues, and thus to particle sizes higher than expected. Even if the system does not contain a large number of these relatively big particles, the latter can lead to a substantial increase of the average particle size.
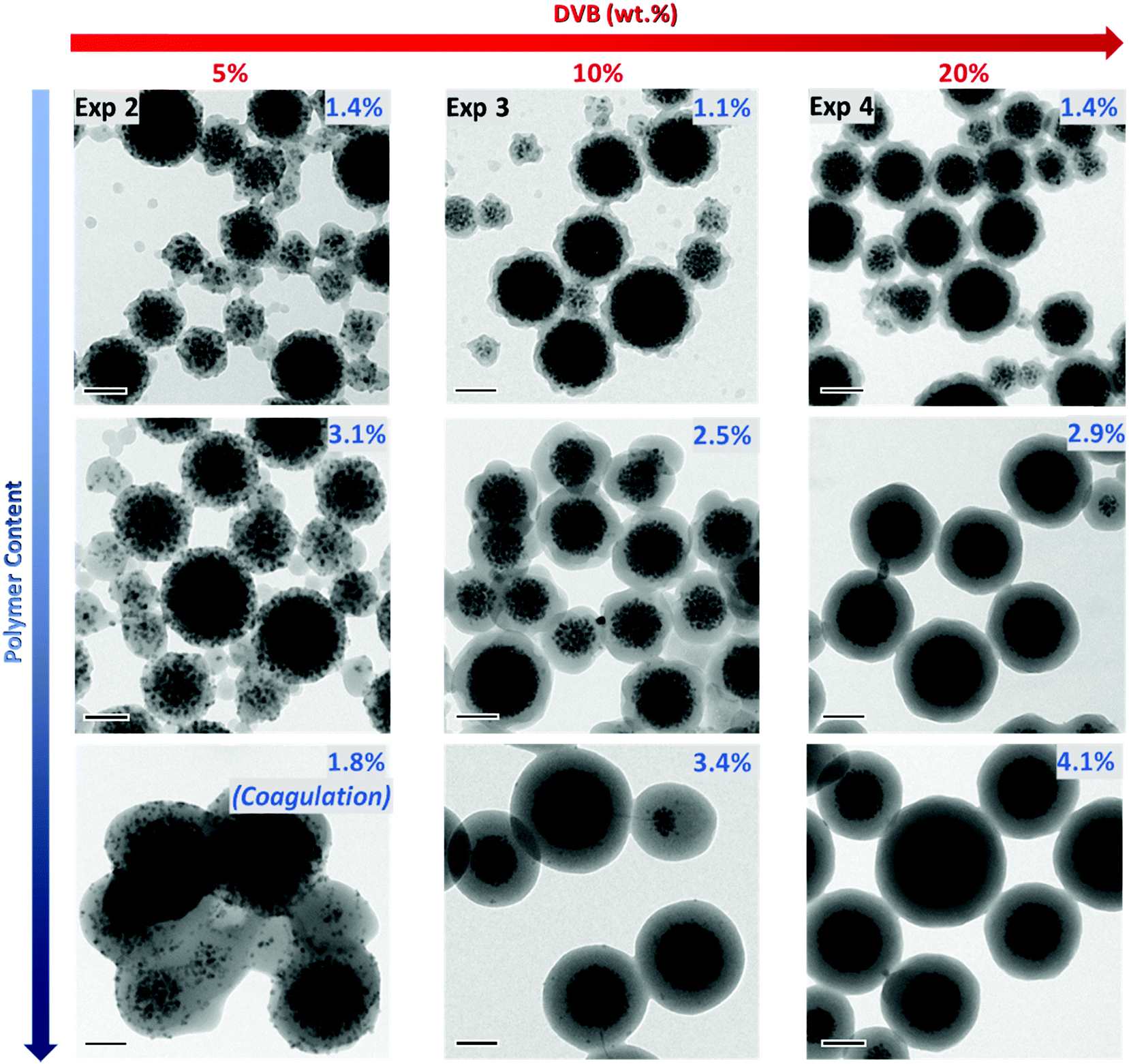
To gain further insight into the effect of DVB content on the particle size and morphology, the samples were observed using TEM (Fig. 3 and Fig. S8–S10, ESI†). In addition, samples were periodically collected during the emulsion polymerization reaction to see the influence of the polymer content on the morphology of the resulting PDMAEMA@MLPs. The first interesting observation is that the particles have diameters mostly below 250 nm (with the exception of Exp. 2, which was unstable and contained aggregated particles as seen in Fig. S8, ESI†). This result, associated with the visual inspection of the hybrid latexes, indicates that colloidally stable systems were obtained for DVB contents above 10 wt%, therefore confirming that the DLS data should be considered with care in the present system. As can be clearly seen in the TEM images, the DVB content had a strong influence on the particle morphology at the initial stages of the polymerization (i.e. for low PCs of 1.2–1.4%). The integrity of the iron oxide clusters tends to be better preserved when the DVB content increases in agreement with previous studies.61,62 As explained above, cross-linking increases the viscosity of the inner cluster core, which strongly restricts the movement of the IONPs within the monomer-swollen particles. In addition, the polymer shell formed around these clusters is different according to the initial DVB amount. A quite rough (raspberry-like) surface can be observed at low PC (1.2–1.4%) in the three experiments. Nonetheless, as the PC increases, a smoother and more homogeneous shell was obtained, in particular for the two stable systems: Exp. 3 and 4 (10 and 20 wt% DVB, respectively).
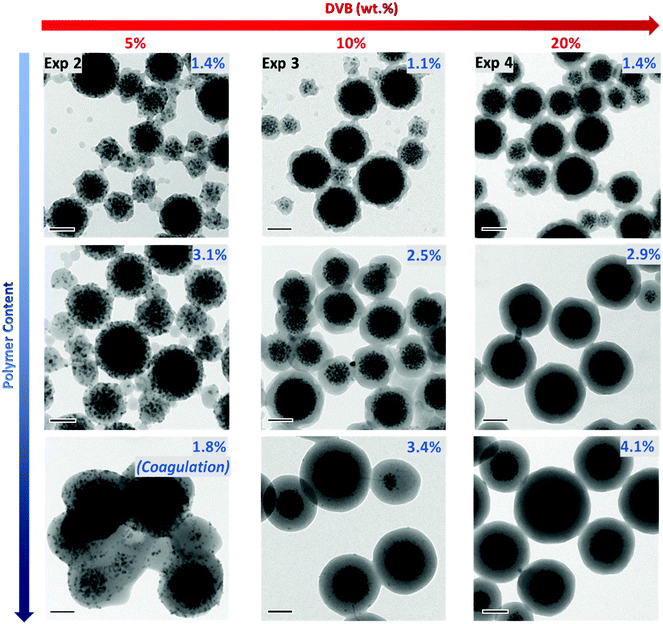 |
| | Fig. 3 TEM micrographs of PDMAEMA@MLPs (PCtheo = 10%) synthesized in the presence of increasing amounts of DVB (based on overall monomer mass): 5 wt%, 10 wt% and 20 wt% (Exp. 2, 3 and 4, respectively, Table 1). The images from top to bottom correspond to samples taken at different reaction times and therefore, to increasing polymer contents. Scale bar: 100 nm. | |
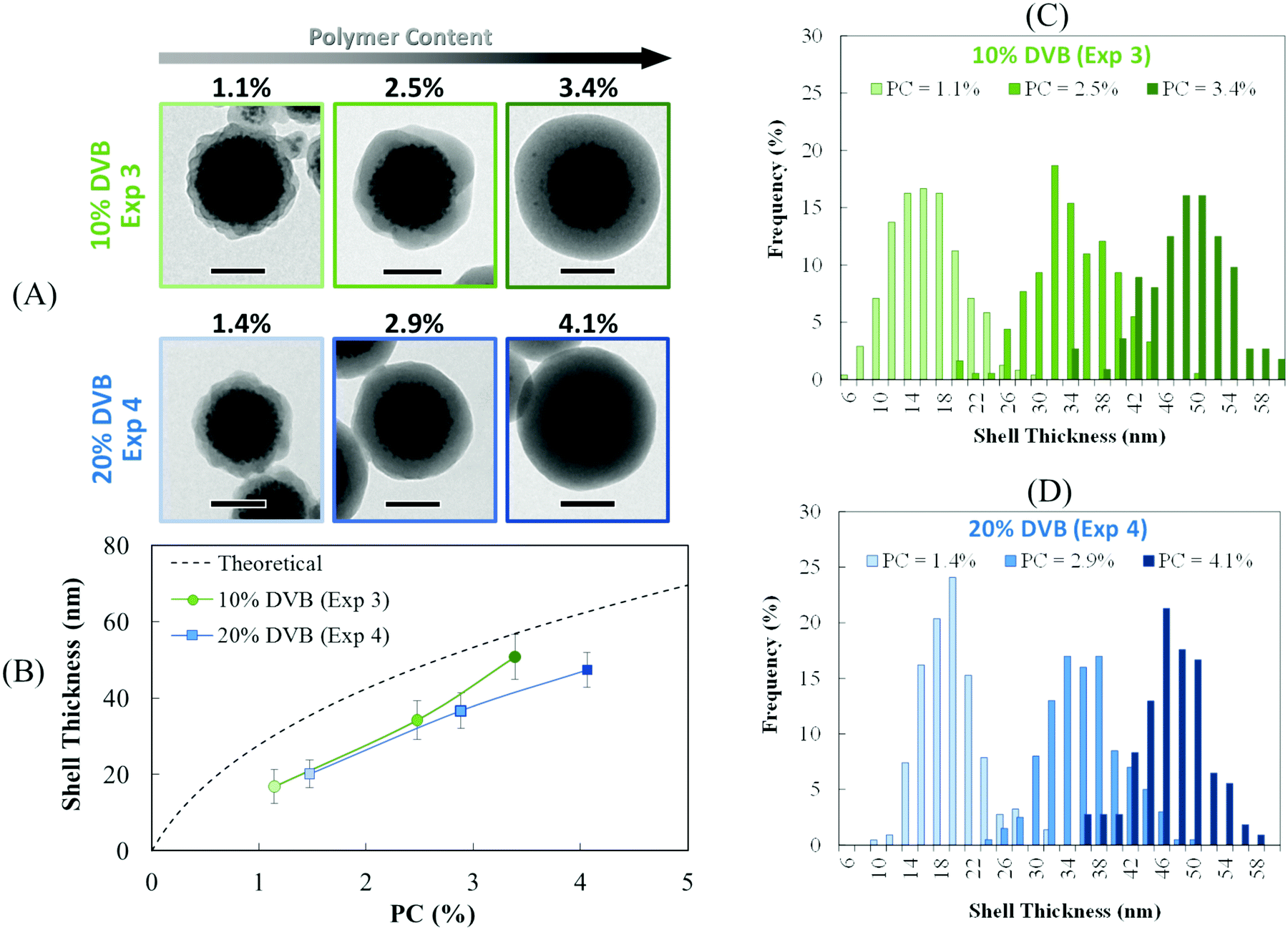
In order to determine the thickness of the polymer shell, a statistical analysis of the TEM images on ca. 300 particles was performed on these two samples. The histograms are presented in Fig. 4C and D. As expected, the shell thickness steadily increased with the increasing amount of polymer (Fig. 4A and B). However, the experimental values were lower than the theoretical ones (Fig. 4B), which may be due to three factors: (i) uncertainties in the estimation of the density of the cluster, which can affect the initial number of clusters; (ii) the assumption that all the monomer added is consumed to form the polymeric shell, whereas some of the polymer is obviously located in the inner core; and (iii) uncertainties in the polymer density used in the calculation (i.e. the PS value, 1.05 g cm−3), whereas this value might be slightly different due to the presence of DVB. Although the experimental data did not perfectly match the theoretical values, our results nonetheless show that the shell thickness can be easily modulated by quenching the polymerization at intermediate global conversions. Based on these observations, it becomes clear that a minimal amount of cross-linker (10 wt%) is necessary to obtain a well-defined core–shell morphology with an IO-rich core and a smooth polymeric shell, even for very low PC (ca. 1.1%).
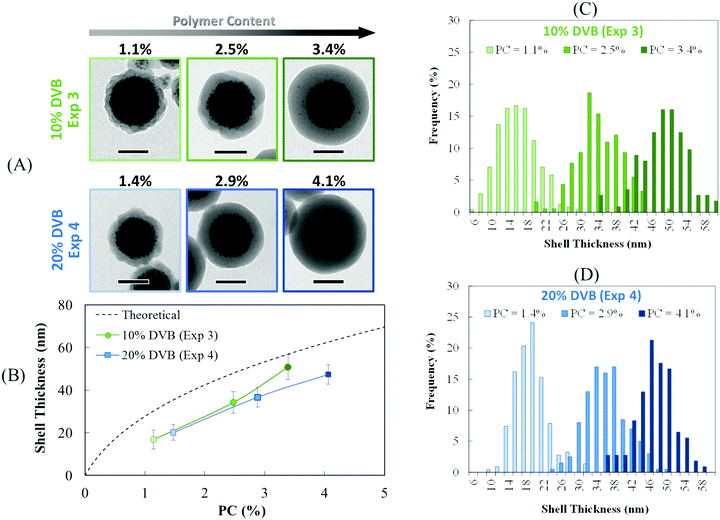 |
| | Fig. 4 Evolution of the shell thickness with the polymer content for PDMAEMA@MLPs prepared by seeded emulsion polymerization of styrene in the presence of 10 and 20 wt% of DVB (Exp. 3 and 4, respectively, Table 1). The theoretical shell thickness was calculated according to eqn (5) and (6). | |
As mentioned in the introduction, the iron oxide content is a key factor for the preparation of efficient magnetic carriers. Therefore, the protective shell should be the thinnest possible in order to maximize the IO content in the hybrid material. Such a thin shell was in fact obtained for a low PC of ∼1.1% and 10 wt% of DVB (Exp. 3 in Fig. 3). As a consequence, in the next series of experiments, the theoretical polymer content was decreased from 10 to 2.5 wt% with the aim of decreasing the shell thickness, while maintaining the same IO concentration in water (Exp. 5–8, Table 1). A first experiment was performed targeting the same global amount of DVB as that in Exp. 3 (ca. 10 wt%) but changing the feeding conditions (Exp. 5). Indeed, all the DVB (12 wt% based on overall monomer mass) was introduced in the initial monomer load followed by the feeding of pure styrene only (Table S3, ESI†). The amount of DVB initially introduced into the reactor was consequently increased by a factor 7 in comparison to that in Exp. 3. In addition to lowering the PC, our objective here was also to optimize the monomer conversion. Indeed, the formation of a highly cross-linked polymer network in the initial stages of the polymerization should guarantee a high internal viscosity of the monomer-swollen particles, rapidly entrapping the IO cluster, while the low cross-linking density of the polymeric shell should result in a higher conversion as the particles will have a better capacity to be swollen with the monomer. As shown in Table 1, the conversion increased from 26 to 66% indicating that this strategy was indeed effective. However, the particle morphology was also affected, and a raspberry-like morphology was observed instead of core–shell particles (Fig. 5). As a consequence, the resulting particles displayed a BET surface area of 27 m2 g−1, three times higher than that of the core–shell particles of Exp. 3 (SBET = 9 m2 g−1). Actually such an increase of specific surface area can be very advantageous for magnetic separation involving small biotargets (<3 nm), such as peptides and proteins, as more adsorption sites will be available on the magnetic carriers, which could increase the efficiency of the magnetic separation process.
 |
| | Fig. 5 TEM and SEM micrographs of PDMAEMA@MLPs synthesized by seeded semi-batch emulsion polymerization of styrene (PCtheo = 2.5%) in the presence of increasing amounts of DVB: 12 wt%, 23 wt% and 38 wt% (based on overall monomer mass) (Exp. 5, 6 and 7, respectively, Table 1), introduced either in the initial load or in both the initial load and the feed (see Table S3, ESI† for detailed experimental conditions). Scale bar: 100 nm. Additional micrographs are presented in the ESI.† | |
With the aim of further controlling the particle morphology, a second reaction was carried out by almost doubling the amount of DVB in the initial load (Exp. 6, DVB = 23 wt%) (see Table S3, ESI† for experimental details), but again a raspberry-like morphology was obtained, with however a smoother shell (Fig. 5). Finally, a higher total amount of DVB was used (38 wt%, Exp. 7) divided into 23 wt% introduced in the initial load and the rest added in the feed, leading to the formation of well-defined core–shell particles as evidenced using TEM and SEM (Fig. 5). At low DVB contents (Exp. 5 and 6), the growing polymer chains tend to form the raspberry-like morphology, which is most likely driven by the interfacial tension. Indeed, hydrophobic small polymer nodules are formed around the seed surface due to the inherent hydrophilicity of the MLP seed clusters. However, as the DVB content increases, the formation of cross-linked polymer chains restricts their motion (due to high internal viscosity) hindering the phase separation process, which results in a relatively smooth polymeric outer shell (Exp. 7, Fig. 5). The morphological transition can be thus rationalized by considering the viscosity of the monomer-swollen polymer domains formed in the early stages of the polymerization hindering the phase separation process. More images of the three samples (including cryo-TEM images) can be seen in Fig. S11–S13 (ESI†), and they all confirm that it is possible to modulate the morphology of the magnetic carrier by varying the amount of DVB introduced at the beginning of the reaction or in the feed leading to particles with variable surface areas, thereby opening the door to the magnetic separation of a wide range of biotargets, from small peptides to micron-size cells.
3.2 Synthesis of PAA-stabilized magnetic latex particles (PAA@MLPs)
To assess the versatility of our strategy for the synthesis of MLPs with tunable surface functionalities, the preparation of magnetic carriers decorated with PAA-segments was next investigated. For that purpose, a PAA-b-PS amphiphilic block copolymer was first synthesized, and used to form PAA@clusters (Table S2 and Fig. S6, ESI†) following the same method as described above for the PDMAEMA@clusters (see Scheme 1). Semi-batch emulsion copolymerization of styrene and DVB using the PAA@clusters as seeds was then carried out under the experimental conditions previously optimized with the PDMAEMA@clusters (PCtheo = 2.5%, IOCtheo = 55% and DVB = 36%, Exp. 8, Table 2 and Table S3, ESI†), leading however to an unstable latex. This is likely due to the nature of the initiator, VA-044, which is cationic at pH = 7.5, and thus of opposite charge of the PAA chains that are partially deprotonated at this pH (∼45 mol%).63 In the next experiments, VA-044 was thus substituted for anionic initiators (i.e., ACPA and APS, Table 2). The polymerization conducted at 70 °C with ACPA at pH 7.5 (Exp. 9) also resulted in an unstable latex (some TEM images of the coagulated particles are presented in Fig. S14, ESI†). The initial pH was therefore subsequently increased to 9.6 to ensure that ∼92% of the carboxylic acid groups of PAA were deprotonated,63 resulting in a stable latex with almost no coagulum, and an average particle diameter of 203 nm (Exp. 10, Table 2 and Fig. S15, ESI†). The monomer conversion was slightly lower (X = 41%, Table 2) compared to the previous experiments involving the PDMAEMA@clusters as seeds (Exp. 5–7, X = 55–66%, Table 1). In addition, there was a certain amount of free latex particles, which obviously contributed to a decrease of the polymer shell thickness, resulting in a poorly defined morphology (Fig. S15, ESI†). In order to increase the conversion, the reaction temperature was set at 80 °C (Exp. 11) leading to a slight improvement from 41 to 47% (Table 2). Again, the resulting PAA decorated magnetic particles were colloidally stable but displayed a more defined core–shell morphology with almost no free latex particles (Fig. 6 and Fig. S16, ESI†).
 |
| | Fig. 6 (A) TEM and (B) SEM micrographs of PAA@MLPs synthesized by seeded semi-batch emulsion polymerization of styrene with 23 wt% DVB (Exp. 11, Table 2). Scale bar = 100 nm. | |
APS was also tested as the initiator at 75 °C (Exp. 12). This experiment however led to an unstable latex most likely due to the decrease of pH induced by persulfate decomposition. NaHCO3 was thus used to keep the pH basic during the polymerization ensuring the colloidal stability (Exp. 13 in Table 2), for a conversion comparable to that previously attained using ACPA as the initiator. However, the encapsulation failed for this system (Fig. S17, ESI†). Therefore, the experimental conditions of Exp. 11 (ACPA/80 °C) were selected as the optimized conditions for the encapsulation of the PAA@clusters.
3.3 Magnetic properties of the PDMAEMA@MLPs and PAA@MLPs
As mentioned in the introduction, magnetic nanoparticles can be used as carriers in separation processes for biomedical diagnostic applications, in order to isolate and pre-concentrate a target analyte from a complex fluid. To ensure a fast and efficient separation, the magnetic response has to be maximized. The rate of magnetic separation [vx(x)] is given by the equation:64| | 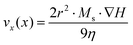 | (8) |
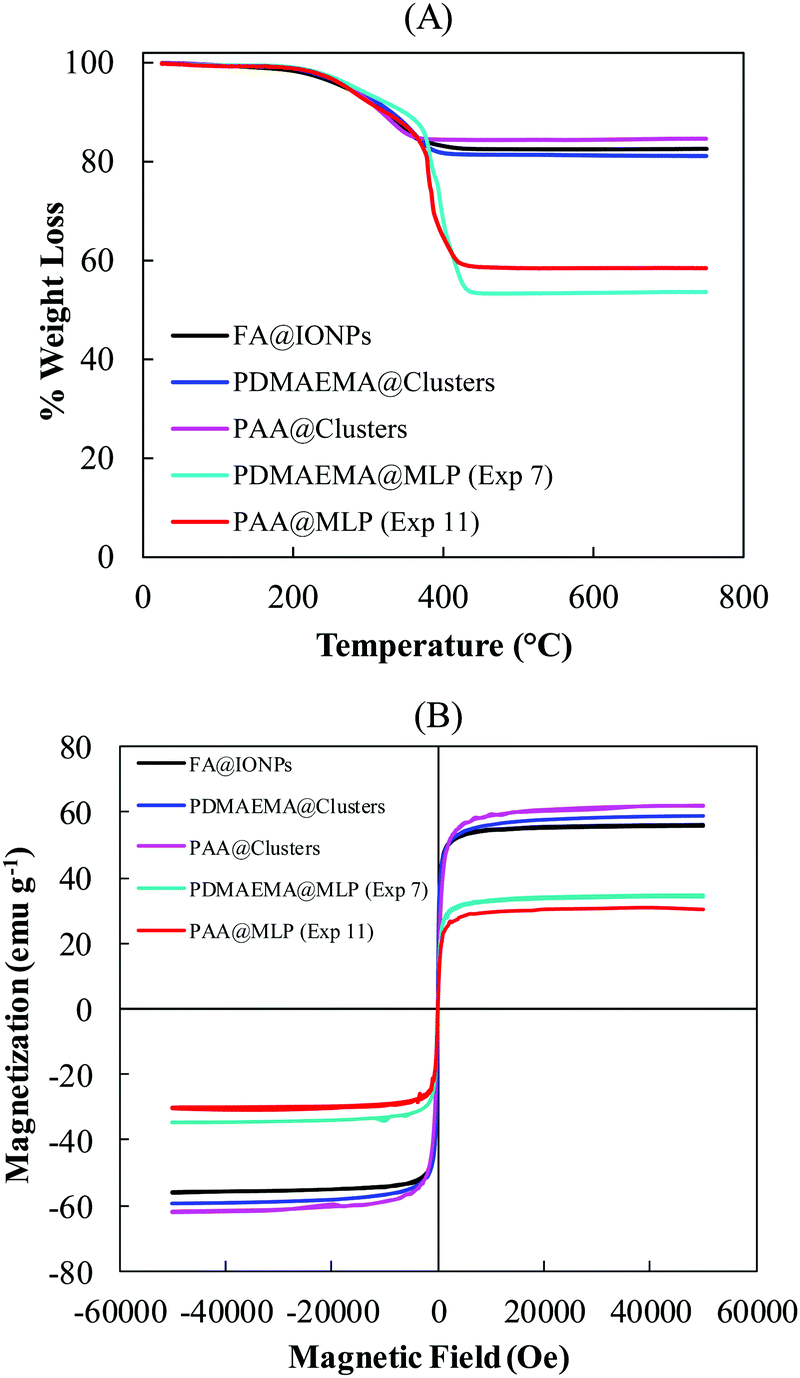
where r is the radius of the composite particles, Ms is the magnetization at saturation, ∇H is the magnetic gradient field, and η is the viscosity of the medium. To obtain magnetic particles with a fast response to an external magnetic field (∇H), it is necessary to prepare relatively large particles (diameter >100 nm) with high IO contents (and thus high Ms). The magnetic properties of the two optimized magnetic carriers with different surface functionalizations (PDMAEMA@MLPs and PAA@MLPs, Exp. 7 and 11, respectively), were investigated. As indicated in Tables 1 and 2, both samples contained a high fraction of magnetic particles (ωmag = 94 and 97 wt%, respectively), indicating that less than 6 wt% of the particles were generated by secondary nucleation, and/or contained a low amount of IO. In addition, a high yield of iron oxide incorporation in the magnetic fraction was obtained for both systems (YIO = 82 and 100% for Exp. 7 and 11, respectively). TGA analysis (Fig. 7A) revealed a very high IO content in the magnetic particles (IOCTGA = 54 and 59 wt% for Exp. 7 and 11, respectively, Tables 1 and 2), resulting in high magnetizations at saturation of, respectively, 35 and 31 emu g−1 (Fig. 7B).
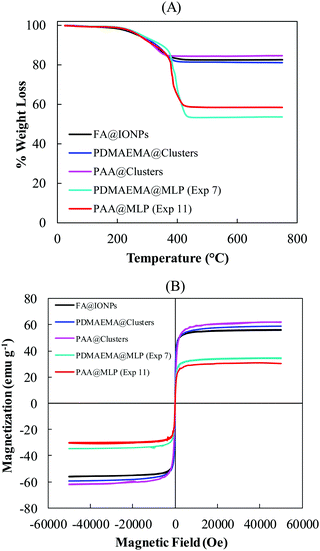 |
| | Fig. 7 (A) TGA and (B) SQUID analyses of the commercial fatty acid-modified IO nanoparticles (FA@IONPs), the PDMAEMA@clusters, the PAA@clusters, and the magnetic fraction (ωmag) of the PDMAEMA@MLP and PAA@MLP samples (Exp. 7, Table 1 and Exp. 11, Table 2). | |
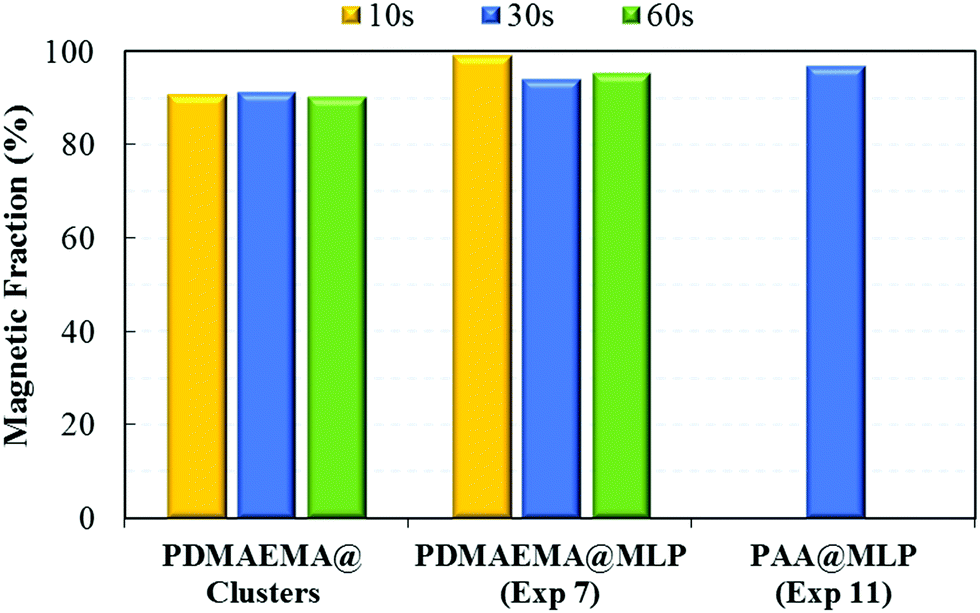
As expected, the high iron oxide content associated with relatively big particle sizes (TEM images of Fig. 5 and 6) resulted in an almost instantaneous magnetic separation with more than 90 wt% of the magnetic carriers being collected after 30 s exposure to the magnetic field (Fig. 8). Furthermore, the superparamagnetic properties of the small FA@IONPs contained in the IO clusters were maintained after the encapsulation process (Fig. 7B), making the resulting MLPs excellent candidates as magnetic carriers with tailored surface functionalization.
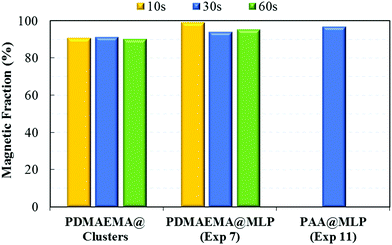 |
| | Fig. 8 Magnetic fraction (ωmag) obtained at different times of magnetic separation for the PDMAEMA@clusters, and the PDMAEMA@MLP (Exp. 7) and PAA@MLP (Exp. 11) samples obtained by seeded emulsion polymerization of styrene (Tables 1 and 2). | |
4. Conclusions
In conclusion, we have reported the successful preparation of magnetic carriers with encapsulated morphologies and tunable surface functionalizations. Our approach relies on the use of an amphiphilic PDMAEMA-b-PS macroRAFT block copolymer for the formation of stable IO clusters that were subsequently incorporated into polymer particles of various morphologies by seeded emulsion polymerization. The use of a cross-linker, DVB, together with the slow addition of styrene was key to preserving the cluster integrity during the polymerization. The amount of cross-linker and the feeding conditions had a direct impact on the hybrid particles leading to core–shell morphologies with either a smooth or a raspberry-like surface. The latter MLPs displayed a higher surface area than the former, making them excellent candidates to be utilized as magnetic carriers for the separation of small biotargets (<3 nm), such as peptides and proteins. The amphiphilic macroRAFT not only provides colloidal stability to the MLPs but also offers a versatile platform for the design of composite particles with tailored surface properties through the appropriate choice of the hydrophilic block. We demonstrated this versatility by the development of a second system based on the use of a PAA-b-PS macroRAFT. In both cases, efficient magnetic carriers were formed with relatively big particle sizes (ca. 140 nm), high iron oxide content and superparamagnetic properties (magnetizations at saturation of 35 and 31 emu g−1 for PDMAEMA@MLPs and PAA@MLPs, respectively). Therefore, fast magnetic responses to a magnetic field were obtained for both systems, with 90% of the carriers being collected after only 30 s exposure to an external magnetic field. These IO-encapsulated particles could thus find interesting applications as magnetic carriers in the biological field due their thermo- (for PDMAEMA) and pH- (for PDMAEMA and PAA) responsive properties. The magnetic separation of bacteria using these magnetic carriers is indeed currently under investigation and will be reported in future work.
Conflicts of interest
The authors declare no conflicts of interest.
Acknowledgements
The authors acknowledge the Brazilian funding agency National Council for Scientific and Technological Development (CNPq) (project no: 249808/2013-7). Pierre-Yves Dugas (C2P2) is thanked for the SEM observations, and Laurent Veyre (C2P2) for his help with the BET analyses. The SQUID measurements were performed with the kind support of Ruben Checa and Dominique Luneau from the Laboratoire des Multimatériaux et Interfaces, UMR CNRS 5615, Claude Bernard University (Villeurbanne, France).
References
- Q. Liu, J. Shi, M. Cheng, G. Li, D. Cao and G. Jiang, Chem. Commun., 2012, 48, 1874–1876 RSC.
- M. Shao, F. Ning, J. Zhao, M. Wei, D. G. Evans and X. Duan, J. Am. Chem. Soc., 2012, 134, 1071–1077 CrossRef CAS PubMed.
- W.-F. Ma, Y. Zhang, L.-L. Li, L.-J. You, P. Zhang, Y.-T. Zhang, J.-M. Li, M. Yu, J. Guo and H.-J. Lu, ACS Nano, 2012, 6, 3179–3188 CrossRef CAS PubMed.
- Z. Zou, M. Ibisate, Y. Zhou, R. Aebersold, Y. Xia and H. Zhang, Anal. Chem., 2008, 80, 1228–1234 CrossRef CAS PubMed.
- R. Veyret, A. Elaissari, P. Marianneau, A. A. Sall and T. Delair, Anal. Biochem., 2005, 346, 59–68 CrossRef CAS PubMed.
- A. Arkhis, A. Elaissari, T. Delair, B. Verrier and B. Mandrand, J. Biomed. Nanotechnol., 2010, 6, 28–36 CrossRef CAS PubMed.
- H. Dong, J. Huang, R. R. Koepsel, P. Ye, A. J. Russell and K. Matyjaszewski, Biomacromolecules, 2011, 12, 1305–1311 CrossRef CAS PubMed.
- K. Yang, D. M. Jenkins and W. W. Su, J. Microbiol. Methods, 2011, 86, 69–77 CrossRef CAS PubMed.
- K. El-Boubbou, C. Gruden and X. Huang, J. Am. Chem. Soc., 2007, 129, 13392–13393 CrossRef CAS PubMed.
- M. Kubota, M. Matsui, H. Chiku, N. Kasashima, M. Shimojoh and K. Sakaguchi, Appl. Environ. Microbiol., 2005, 71, 8895–8902 CrossRef CAS PubMed.
- J.-C. Liu, P.-J. Tsai, Y. C. Lee and Y.-C. Chen, Anal. Chem., 2008, 80, 5425–5432 CrossRef CAS PubMed.
- M. M. Rahman and A. Elaissari, Sep. Purif. Technol., 2011, 81, 286–294 CrossRef CAS.
- A. P. Majewski, U. Stahlschmidt, V. Jérôme, R. Freitag, A. H. Müller and H. Schmalz, Biomacromolecules, 2013, 14, 3081–3090 CrossRef CAS PubMed.
- N. C. Bigall, A. Curcio, M. P. Leal, A. Falqui, D. Palumberi, R. Di Corato, E. Albanesi, R. Cingolani and T. Pellegrino, Adv. Mater., 2011, 23, 5645–5650 CrossRef CAS PubMed.
- A. P. Philipse, M. P. B. van Bruggen and C. Pathmamanoharan, Langmuir, 1994, 10, 92–99 CrossRef CAS.
- A. M. Munshi, V. Agarwal, D. Ho, C. L. Raston, M. Saunders, N. M. Smith and K. S. Iyer, Cryst. Growth Des., 2016, 16, 4773–4776 CrossRef CAS.
- E. Bourgeat-Lami and M. Lansalot, Adv. Polym. Sci., 2010, 233, 53–123 CrossRef CAS.
- M. M. Rahman and A. Elaissari, Adv. Polym. Sci., 2010, 233, 237–281 CrossRef CAS.
- V. Agarwal, D. Ho, D. Ho, Y. Galabura, F. Yasin, P. Gong, W. Ye, R. Singh, A. Munshi and M. Saunders, ACS Appl. Mater. Interfaces, 2016, 8, 4934–4939 CrossRef CAS PubMed.
- H. Lee, T.-H. Shin, J. Cheon and R. Weissleder, Chem. Rev., 2015, 115, 10690–10724 CrossRef CAS PubMed.
- O. Olsvik, T. Popovic, E. Skjerve, K. S. Cudjoe, E. Hornes, J. Ugelstad and M. Uhlen, Clin. Microbiol. Rev., 1994, 7, 43–54 CrossRef CAS PubMed.
- K. B. Kockler, F. Fleischhaker and C. Barner-Kowollik, Macromolecules, 2016, 49, 8572–8580 CrossRef CAS.
- K. Chen, Y. Zhu, Y. Zhang, L. Li, Y. Lu and X. Guo, Macromolecules, 2011, 44, 632–639 CrossRef CAS.
- D. Hoffmann, K. Landfester and M. Antonietti, Magnetohydrodynamics, 2001, 37, 58 Search PubMed.
- K. Landfester and L. P. Ramirez, J. Phys. Condens. Matter, 2003, 15, S1345 CrossRef CAS.
- H. Xu, L. Cui, N. Tong and H. Gu, J. Am. Chem. Soc., 2006, 128, 15582–15583 CrossRef CAS PubMed.
- L. P. Ramirez and K. Landfester, Macromol. Chem. Phys., 2003, 204, 22–31 CrossRef CAS.
- C. Paquet, L. Page, A. Kell and B. Simard, Langmuir, 2010, 26, 5388–5396 CrossRef CAS PubMed.
- H. Deng, X. Li, Q. Peng, X. Wang, J. Chen and Y. Li, Angew. Chem., Int. Ed., 2005, 117, 2842–2845 CrossRef.
- B.-S. Kim, J.-M. Qiu, J.-P. Wang and T. A. Taton, Nano Lett., 2005, 5, 1987–1991 CrossRef CAS PubMed.
- C. Paquet, H. W. de Haan, D. M. Leek, H.-Y. Lin, B. Xiang, G. Tian, A. Kell and B. Simard, ACS Nano, 2011, 5, 3104–3112 CrossRef CAS PubMed.
- J.-F. Berret, N. Schonbeck, F. Gazeau, D. El Kharrat, O. Sandre, A. Vacher and M. Airiau, J. Am. Chem. Soc., 2006, 128, 1755–1761 CrossRef CAS PubMed.
- S. Xu, W. F. Ma, L. J. You, J. M. Li, J. Guo, J. J. Hu and C. C. Wang, Langmuir, 2012, 28, 3271–3278 CrossRef CAS PubMed.
- S. Braconnot, M. M. Eissa and A. Elaissari, Colloid Polym. Sci., 2012, 291, 193–203 CrossRef.
- H. Chen, M. Dong, Y. Li, J. Kong, Z. Chai and G. Fu, React. Funct. Polym., 2013, 73, 18–22 CrossRef CAS.
- E. Bourgeat-Lami, F. D’Agosto and M. Lansalot, Adv. Polym. Sci., 2016, 270, 123–161 CrossRef CAS.
- P. B. Zetterlund, S. C. Thickett, S. Perrier, E. Bourgeat-Lami and M. Lansalot, Chem. Rev., 2015, 115, 9745–9800 CrossRef CAS PubMed.
- J. Zhou, H. Yao and J. Ma, Polym. Chem., 2018, 9, 2532–2561 RSC.
- D. Nguyen, H. S. Zondanos, J. M. Farrugia, A. K. Serelis, C. H. Such and B. S. Hawkett, Langmuir, 2008, 24, 2140–2150 CrossRef CAS PubMed.
- S. I. Ali, J. P. Heuts, B. S. Hawkett and A. M. van Herk, Langmuir, 2009, 25, 10523–10533 CrossRef CAS PubMed.
- N. Zgheib, J.-L. Putaux, A. Thill, E. Bourgeat-Lami, F. D'Agosto and M. Lansalot, Polym. Chem., 2013, 4, 607–614 RSC.
- E. Bourgeat-Lami, A. J. P. G. França, T. C. Chaparro, R. D. Silva, P.-Y. Dugas, G. Alves and A. Martins dos Santos, Macromolecules, 2016, 49, 4431–4440 CrossRef CAS.
- A. Cenacchi, S. Pearson, D. Kostadinova, F. Leroux, F. D’Agosto, M. Lansalot, E. Bourgeat-Lami and V. Prevot, Polym. Chem., 2017, 8, 1233–1243 RSC.
- S. Pearson, M. Pavlovic, T. Augé, V. Torregrossa, I. Szilagyi, F. D’Agosto, M. Lansalot, E. Bourgeat-Lami and V. Prévot, Macromolecules, 2018, 51, 3953–3966 CrossRef CAS.
- F. Dalmas, S. Pearson, B. Gary, J.-M. Chenal, E. Bourgeat-Lami, V. Prévot and L. Chazeau, Polym. Chem., 2018, 9, 2590–2600 RSC.
- R. D. Silva, T. C. Chaparro, I. S. Monteiro, P.-Y. Dugas, F. D’Agosto, M. Lansalot, A. Martins dos Santos and E. Bourgeat-Lami, Macromolecules, 2019, 52, 4979–4988 CrossRef CAS.
- K. Li, P.-Y. Dugas, E. Bourgeat-Lami and M. Lansalot, Polymer, 2016, 106, 249–260 CrossRef CAS.
- D. Nguyen, B. T. Pham, V. Huynh, B. J. Kim, N. T. Pham, S. A. Bickley, S. K. Jones, A. Serelis, T. Davey and C. Such, Polymer, 2016, 106, 238–248 CrossRef CAS.
- T. R. Guimarães, M. Lansalot and E. Bourgeat-Lami, Polym. Chem., 2020, 11, 648–652 RSC.
- T. Boursier, I. Chaduc, J. Rieger, F. D'Agosto, M. Lansalot and B. Charleux, Polym. Chem., 2011, 2, 355–362 RSC.
- L. Couvreur, C. Lefay, J. Belleney, B. Charleux, O. Guerret and S. Magnet, Macromolecules, 2003, 36, 8260–8267 CrossRef CAS.
- E. Kühnel, D. D. Laffan, G. C. Lloyd-Jones, T. Martínez del Campo, I. R. Shepperson and J. L. Slaughter, Angew. Chem., Int. Ed., 2007, 46, 7075–7078 CrossRef PubMed.
- C. Zhang, Y. Zhou, Q. Liu, S. Li, S. Perrier and Y. Zhao, Macromolecules, 2011, 44, 2034–2049 CrossRef CAS.
-
J. Texter, Reactions and synthesis in surfactant systems, CRC Press, 2001 Search PubMed.
- M. Morton, S. Kaizerman and M. W. Altier, J. Colloid Sci., 1954, 9, 300–312 CrossRef CAS.
-
P. J. Flory, Principles of polymer chemistry, Cornell University Press, 1953 Search PubMed.
- L. Errede, J. Appl. Polym. Sci., 1986, 31, 1749–1761 CrossRef CAS.
- D. Y. Chung, M. Bartholin and A. Guyot, Angew. Makromol. Chem., 1982, 103, 109–123 CrossRef CAS.
- P. Kwant, J. Polym. Sci., Polym. Chem., 1979, 17, 1331–1338 CrossRef CAS.
- G. Schwachula, J. Polym. Sci., Polym. Symp., 1975, 53, 107–112 CrossRef CAS.
- S. Xu, W.-F. Ma, L.-J. You, J.-M. Li, J. Guo, J. J. Hu and C.-C. Wang, Langmuir, 2012, 28, 3271–3278 CrossRef CAS PubMed.
- J. Ge, Y. Hu, T. Zhang and Y. Yin, J. Am. Chem. Soc., 2007, 129, 8974–8975 CrossRef CAS PubMed.
- T. Swift, L. Swanson, M. Geoghegan and S. Rimmer, Soft Matter, 2016, 12, 2542–2549 RSC.
- J. Watson, IEEE Trans. Magn., 1975, 11, 1597–1599 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0tb00384k |
|
| This journal is © The Royal Society of Chemistry 2020 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  ,
Muriel
Lansalot
,
Muriel
Lansalot