
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Influence of molecular weight on transdermal delivery of model macromolecules using hydrogel-forming microneedles: potential to enhance the administration of novel low molecular weight biotherapeutics
Aaron R. J.
Hutton
,
Maelíosa T. C.
McCrudden
,
Eneko
Larrañeta
and
Ryan F.
Donnelly
 *
*
School of Pharmacy, Queen's University Belfast, 97 Lisburn Road, Belfast BT9 7BL, UK. E-mail: r.donnelly@qub.ac.uk
First published on 15th April 2020
Abstract
With a view to improve the current monoclonal antibody-based therapies dominating the pharmaceutical market, low molecular weight (MW) protein-based macromolecules, such as recombinant antibody fragments, typically within the range of 10–70 kDa, have been developed. Previously, our group successfully delivered Avastin®, a monoclonal antibody (mAb) across the skin using hydrogel-forming microneedles (MN). However, it is thought that this delivery system can be further enhanced using novel, lower MW biomolecules. To address this perception, in the current study, FITC–dextran of different MWs (10, 70 and 150 kDa) was used to model the transdermal delivery of low MW biotherapeutics and mAbs with MWs of approximately 150 kDa. Conversely, fluorescein sodium was the compound selected to model hydrophilic, low MW drugs. As expected, fluorescein sodium produced the greatest cumulative permeation (637.4 ± 42.69 μg). The amounts of FITC–dextran 10 kDa and 150 kDa which permeated across neonatal porcine skin in vitro were 462.17 ± 65.85 μg and 213.54 ± 15.19 μg after 24 h, respectively. The results collated here suggest that the delivery of emerging novel biotherapeutics, via ‘super swelling’ hydrogel-forming MNs, have the potential to result in greater permeation across human skin, compared to the delivery of mAbs delivered via the same route.
Introduction
During the last 30 years, significant research and development funds and resources have focused on antibody-based therapies.1 This field of drug treatment, driven by monoclonal antibodies (mAbs), has resulted in biotherapeutic compounds controlling the top 10 globally best-selling drugs. To use one biological drug class as an example, tumour necrosis factor-α (TNF-α) inhibitors have been proven to effectively induce remission in immunological disorders such as rheumatoid arthritis, Crohn's disease, ulcerative colitis and psoriasis.2–4 To this end, the most successful drug on the market is Humira® (Adalimumab), an anti TNF-α mAb, which provided over $19 billion in revenue in 2018.5 Evidently, this is a financially lucrative field of development for drug manufacturers and its success has led to the development of improved treatment strategies. One such strategy involves the development of lower molecular weight (MW) protein-based macromolecules.6–10 These alternatives, which include smaller recombinant antibody fragments such as Fab and scFv molecules, have been exploited in autoimmune disorders and certain cancers.11 By reducing the MW, these proteins are no longer restricted to systemically accessible targets, a considerable limitation with cancer-based mAb therapeutics. This gives these low MW biotherapeutics the ability to access recessed epitopes in cancer tumours.12 Furthermore, the reduced complexity of low MW proteins makes their pharmacokinetic properties easier to optimise, in addition to reducing manufacturing costs.11Currently, mAbs are limited to high dose administration using syringe and needles, due to their poor stability and high cost.1,13,14 With the potential for needle stick injuries, needle phobia and the necessity for dedicated sharps disposal, there is a need to develop new delivery systems to eliminate these costly obstacles.15 A possible alternative are hydrogel-forming microneedles (MN) which consist of drug-free, micron scale polymeric needles situated in perpendicular orientation on a base plate to which a separate drug containing reservoir is attached. Following pain-free application to the skin, these MNs imbibe interstitial skin fluid and swell, creating micro-conduits within the skin layers to permit drug delivery into the dermal micro-circulation.16 After application, needles become soft and are incapable of reinsertion. This is a significant advantage as it prevents needle stick injury and removes the need for dedicated sharps disposal.
Previously, Singh et al. (2010) modelled the delivery of small, hydrophilic molecules (<500 Da) using hydrogel-forming MNs. This study, which proved that hydrogel-forming MNs enhanced the transdermal delivery of small molecules, subsequently led to the successful delivery of a number of low MW therapeutics across the skin, including theophylline, metronidazole and ibuprofen-sodium.17–19 Surprisingly however, despite the emergence of biotherapeutics and the advantages offered by hydrogel-forming MNs, there has been very little research carried out to assess the capabilities of this delivery system to enhance the delivery of such molecules. Courtenay et al. (2018) were the first to report the transdermal delivery of a therapeutic antibody using hydrogel-forming MNs. Although Avastin® (150 kDa) was successfully stabilised in a lyophilised wafer and delivered across neonatal porcine skin in vitro, due to its high MW, a large volume of drug was trapped within the cross-linked network of the MN array.20 Furthermore, when applied in vivo, Avastin® was found to drain into the lymphatic system.20 The authors of this paper suggested that hydrogel-forming MNs could therefore be used to target cancer metastases within the lymphatic system. To develop this strategy further in a bid to enhance the systemic delivery of biomolecules using hydrogel-forming MNs, it seems logical to assess the capabilities of this delivery system using novel low MW biotherapeutics. By using FITC–dextran compounds of varying MWs as model compounds, the work presented here mimics the transdermal delivery of low and high MW biomolecules, via hydrogel-forming MNs. Specifically, FITC–dextran 10 kDa was chosen to model an unaltered form of a low MW biotherapeutic. FITC–dextran 70 kDa was chosen to model the half-life extended alternative of these compounds, with FITC–dextran 150 kDa modelling mAbs. As Singh et al. (2010) proved that low MW drugs can be successfully delivered across the skin using hydrogel-forming MNs, fluorescein sodium (376 Da) was utilised in this work as a model hydrophilic drug. Therefore, leading on from the original studies carried out in our research group and outlining the delivery capabilities of hydrogel-forming MNs, this work was designed with a view to informing upon the potential enhanced permeation of novel low MW biotherapeutics when compared to mAbs tested previously.
Materials and methods
Chemicals
Fluorescein isothiocyanate–dextran (FITC–dextran 10, 70 and 150, MW = 8000–12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 Da, MW = 63000–77000 Da and MW = 140000–160000 Da) were purchased from TdB Consultancy AB (Uppsala, Sweden). Fluorescein sodium (MW = 376 Da) and poly(ethylene glycol) (PEG) with MW = 10000 Da were purchased from Sigma-Aldrich, Dorset, UK. Gantrez® S-97, a co-polymer of methyl vinyl ether and maleic acid (PMVE/MA) was a gift from Ashland, Kidderminster, UK. Sodium carbonate (Na2CO3) was purchased from BDH Laboratory Supplies, London, UK. Cryogel SG3 was provided by PB Gelatins, Pontypridd, UK and Pearlitol® 50C-Mannitol was supplied by Roquette, Lestrem, France. All other chemicals were of analytical grade.
000 Da, MW = 63000–77000 Da and MW = 140000–160000 Da) were purchased from TdB Consultancy AB (Uppsala, Sweden). Fluorescein sodium (MW = 376 Da) and poly(ethylene glycol) (PEG) with MW = 10000 Da were purchased from Sigma-Aldrich, Dorset, UK. Gantrez® S-97, a co-polymer of methyl vinyl ether and maleic acid (PMVE/MA) was a gift from Ashland, Kidderminster, UK. Sodium carbonate (Na2CO3) was purchased from BDH Laboratory Supplies, London, UK. Cryogel SG3 was provided by PB Gelatins, Pontypridd, UK and Pearlitol® 50C-Mannitol was supplied by Roquette, Lestrem, France. All other chemicals were of analytical grade.
Manufacture of hydrogel-forming MNs
‘Super swelling’ hydrogel-forming MNs were prepared using a formulation published previously.17 Due to the low cross-linked density, this has been described as the optimum hydrogel formulation for rapid drug delivery.17,21 In short, approximately 500 mg of an aqueous blend of 20% w/w Gantrez® S-97, 7.5% w/w PEG 10000 and 3% w/w Na2CO3 was poured into laser engineered silicone moulds, composed of 121 (11 × 11) conical holes, 600 μm in depth, interspacing 300 μm and base widths of 300 μm (Fig. 1). Each mould was centrifuged at 3500 rpm for 15 min and allowed to dry under ambient conditions for 48 h. Following this, MNs were placed in an oven at 80 °C for 24 h to facilitate crosslinking. MNs were then demoulded and sidewalls removed using a heated scalpel.
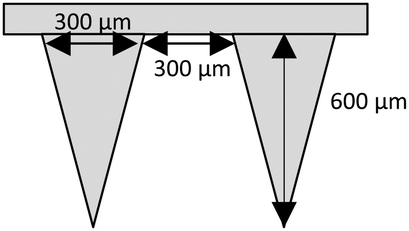 | ||
| Fig. 1 Schematic representation of a ‘super-swellable’ hydrogel-forming MN with conical shaped needles, 600 μm in height, interspacing 300 μm and base widths of 300 μm. | ||
Mechanical testing of hydrogel-forming MNs
Compression and insertion characteristics of ‘super swellable’ MNs were assessed using a TA.XT2 Texture Analyser. Before compression, MN heights were measured using a Leica EZ4W stereo microscope. Previous studies have suggested that 8 layers of Parafilm® M exhibit excellent similarity to porcine skin.21–24 Therefore, to artificially simulate MN insertion in a biological membrane, each MN array was placed above the eight layered polymeric film on the Texture Analyser platform. A previous study using human volunteers showed that when applying MNs, forces were exerted within the range of 0–50 N, with an average maximal force of 30 N.21 For this reason, 30 N was selected as the application force within this study. Taking this into account, a probe was lowered at a speed of 1.19 mm s−1 and a force of 30 N, held for 30 s was applied to each MN array. Following insertion, MNs were removed from the Parafilm® M and needle height remeasured to determine compression, expressed as percentage height reduction. MN morphology was also visually assessed before and after compression using scanning electron microscopy (SEM) (TM 3030 Tabletop Microscope, Hitachi, Japan). To assess insertion, each Parafilm® M layer was examined under the microscope and the number of holes counted. Insertion in each layer is regarded as successful if the number of holes created in each layer is greater than 20%.21 To confirm insertion into a biological membrane, ‘super-swellable’ hydrogel-forming MNs were placed above full thickness neonatal porcine skin and a 30 N force was applied for 30 s. Inserted MNs were immediately viewed in situ using an EX1301 optical coherence tomography (OCT) microscope (Michelson Diagnostics, Kent, UK).Fabrication of FITC–dextran and fluorescein sodium lyophilised wafers
FITC–dextran 10 kDa, 70 kDa, 150 kDa and fluorescein sodium (376 Da) lyophilised wafers were prepared using different combinations of gelatin, mannitol and deionised water (Table 1). To prepare each formulation, the fluorescent compound was first dissolved in deionised water and heated to 37 °C. Mannitol and gelatin were added, followed by hand mixing to facilitate dissolution. The formulations were then sonicated at 37 °C for 60 min before casting into open-ended cylindrical moulds with diameters of 11.8 mm and depths of 2 mm. Each formulation was frozen at −80 °C for 60 min and was then placed in a freeze dryer. Using an adapted version of a previously described method, the lyophilisation process involved; primary drying for 90 min at a shelf temperature of −40 °C, 90 min at a shelf temperature of −30 °C, 90 min at a shelf temperature of −20 °C, 530 min at a shelf temperature of −10 °C and 90 min at a shelf temperature of 0–10 °C.17 The secondary drying phase was performed over 660 min at a shelf temperature of 25 °C and a vacuum pressure of 50 mTorr.| ID | Gelatin (% w/w) | Mannitol (% w/w) | Analyte (% w/w) | Water (% w/w) |
|---|---|---|---|---|
| F101 | 3 | 3 | 0.5 | 93.5 |
| F102 | 5 | 3 | 0.5 | 91.5 |
| F103 | 7 | 3 | 0.5 | 89.5 |
| F104 | 10 | 5 | 0.5 | 84.5 |
| F705 | 10 | 5 | 0.5 | 84.5 |
| F1506 | 10 | 5 | 0.5 | 84.5 |
| FNa7 | 10 | 5 | 0.5 | 84.5 |
Dissolution of FITC–dextran and fluorescein sodium loaded lyophilised wafers
To determine the dissolution time of each lyophilised wafer, 20 mL of phosphate buffered saline (PBS) (pH 7.4) solution was added to a glass vial, heated to 37 °C and stirred at 200 rpm. Lyophilised wafers were then added to the PBS (pH 7.4) and the dissolution time was recorded, based on visual inspection.Determination of FITC–dextran and fluorescein sodium recovery from lyophilised wafers
FITC–dextran and fluorescein sodium recovery was assessed through a process of dissolution and quantification using fluorescence spectroscopy. Following complete dissolution, samples were removed, diluted appropriately in PBS (pH 7.4) and quantified using fluorescence spectroscopy.Saturation solubility of FITC–dextran and fluorescein sodium
To determine saturation solubility, the four compounds were added in excess into separate glass vials containing PBS (pH 7.4) and stirred at 200 rpm for 24 h at 37 °C. The saturated solution was then filtered using Minisart® 0.2 μm filters, diluted appropriately and analysed using fluorescence spectroscopy.In vitro permeation of FITC–dextran and fluorescein sodium using modified Franz cells
FITC–dextran and fluorescein sodium permeation through dermatomed (350 μm) neonatal porcine skin, which was acquired from stillborn piglets, was quantified using modified Franz diffusion cells. Skin integrity was confirmed using transepidermal water loss (TEWL). Each receiver compartment was filled with PBS (pH 7.4), stirred at 600 rpm using a metal stirring bar and left to equilibrate at 37 ± 1 °C for 12 h. Following the 12 h period, skin samples were shaved and fixed to the donor compartment using cyanoacrylate glue. MN arrays were then placed into the centre of each donor compartment and manual thumb pressure was applied for 30 s. An aliquot of water (20 μL) was then dispensed onto the MN baseplate, after which the lyophilised wafer containing the fluorescent compound was placed on top. A 5.0 g stainless steel weight was placed above the wafer to ensure MN insertion was maintained throughout the 24 h period. The donor compartment was then carefully placed on top of the receiver compartment and securely clamped into place. The sampling arm was occluded with Parafilm® M to prevent evaporation. Samples (200 μL) were removed at specified time points and diluted appropriately in PBS (pH 7.4). An equal volume of fresh pre-warmed PBS (pH 7.4) was added to the receiver vessel to replace this.Pharmaceutical analysis of FITC–dextran and fluorescein sodium
Fluorescence analysis was carried out using a BMG FLUOstar Omega Fluorescence Microplate Reader (BMG LabTech, Ortenberg, Germany) at an excitation wavelength 485 nm and emission wavelength 520 nm. Method validation was performed according to guidelines detailed by the ICH of Technical Requirements for Registration of Pharmaceuticals for Human use Validation of Analytical Procedures Q2 (R1) 2005. FITC–dextran stock solutions (3 mg mL−1) were prepared in phosphate buffered saline (PBS) (pH 7.4). Working standards of each stock solution were prepared by diluting with appropriate volumes of PBS (pH 7.4). Through a series of serial dilutions, standards were prepared in the range of 0.78 μg mL−1 to 100 μg mL−1. To produce a stock solution of fluorescein sodium (10 mg mL−1), fluorescein sodium was dissolved in PBS (pH 7.4). Through serial dilution, standards were prepared in the range of 0.08 μg mL−1 to 2.5 μg mL−1. Least squares linear regression analysis and correlation analysis over three consecutive days was used to calculate the coefficient of determination (R2) and line equation. Limit of detection (LoD) and limit of quantification (LoQ) were calculated using eqn (1) and (2), with S defined as the gradient of the calibration curve and σ the standard deviation of the response, determined using the standard error of the y-intercepts on the regression line.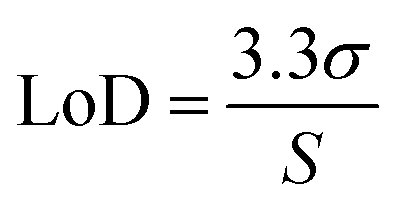 | (1) |
 | (2) |
Statistical analysis
Statistical analysis was performed using GraphPad Prism version 6.07 (GraphPad Software Inc., San Diego, California). This included calculation of means and standard deviations. Paired and unpaired t-tests, in addition to Mann Whitney U and Kruskal Wallis tests, were implemented where appropriate, with p < 0.05 denoting statistical significance.Results
Mechanical testing of ‘super swelling’ hydrogel-forming MNs
To investigate the effect of application force on MN compression, needle height was first measured using a Lecia EZ4W stereo microscope. A 30 N force was then applied to each ‘super swellable’ hydrogel-forming MN for 30 s using a TA.XT2 Texture analyser and MN height remeasured as shown in Fig. 2a. Scanning electron microscopy (SEM) showed that there was no visible difference in needle morphology after compression (Fig. 2b). | ||
| Fig. 2 (a) Compression analysis of 20% w/w Gantrez® S-97, 7.5% w/w PEG 10000 + 3% w/w Na2CO3 (‘super swelling’) MNs. Means ± S.D., n = 10. (b) SEM images of a ‘super-swelling’ MN before and after the application of a 30 N force for 30 s. | ||
Before compression, ‘super swellable’ MNs had mean needle heights of 0.523 ± 0.017 mm. Following compression, needle heights measured a mean of 0.511 ± 0.042 mm. There was no significant difference in needle height following the application of the documented compression force (p = 0.8457).
To determine insertion characteristics, ‘super swelling’ hydrogel-forming MNs were first examined under a Lecia EZ4W stereo microscope to confirm complete needle formation (Fig. 3a and b). Each MN was then placed on 8 layers of Parafilm® M. Following the application of a 30 N force, needle insertion was visually assessed using an OCT microscope (Fig. 3c). To enable a direct comparison between insertion into an artificial and a biological membrane, ‘super-swellable’ MNs were placed above full thickness porcine skin and the same force (30 N) was applied for 30 s. Using OCT, it was visually confirmed that this MN formulation has the strength to insert into both membrane types (Fig. 3c and d). To measure the depth of insertion, the number of holes in each Parafilm® M layer were counted using a Lecia EZ4W stereo microscope. As shown in Fig. 3e, more than 20% of the needles penetrated through the third layer of Parafilm® M and less than 20% through the fourth layer of Parafilm® M. For penetration to be regarded as successful, there must be greater than 20% needle penetration into a specific layer of the Parafilm® M.21 Therefore, as each layer is 126 μm thick, the insertion depth was in the range of 378–504 μm.
 | ||
| Fig. 3 (a) Light microscope image of a 20% w/w Gantrez® S-97, 10% w/w PEG 10000 + 3% w/w Na2CO3 (super swelling) hydrogel-forming MN. (b) Light microscope image confirming the heights (600 μm) of a ‘super swelling’ hydrogel-forming MN. (c) Optical coherence tomography (OCT) image showing the insertion of a ‘super swelling’ hydrogel-forming MN into 8 layers of Parafilm® M following the application of a 30 N force for 30 s. (d) OCT image showing the insertion of a ‘super swelling’ hydrogel-forming MN into full thickness porcine skin following the application of a 30 N force for 30 s. (e) Graphical representation of the number of Parafilm® M layers penetrated, and percentage holes created within each layer following a 30 N force applied for 30 s to ‘super swelling’ hydrogel-forming MNs. Means ± S.D., n = 3. | ||
Fabrication of lyophilised wafers containing FITC–dextran and fluorescein sodium
Using FITC–dextran 10 kDa, four different gelatin to mannitol ratios were used in the production of a range of lyophilised wafers, using an iterative approach (Table 1). Each formulation was poured into 250 mg open-ended cylindrical moulds, frozen at −80 °C for 1 h and then freeze dried over a 25 h cycle. As shown in Fig. 4, F101 and F102 were homogenous but possessed poor structural integrity, both crumbling during demoulding. F103 and F104 were homogenous and displayed good structural integrity. Consequently, the gelatin to mannitol ratio used to produce F104 was tested with the remaining three analytes. All three other formulations (F705, F1506 and FNa7) produced homogenous, intact wafers. | ||
| Fig. 4 Light microscope images of FITC–dextran 10 kDa, 70 kDa, 150 kDa and fluorescein sodium lyophilised wafers as detailed in Table 1. | ||
Pharmaceutical analysis of FITC–dextran and fluorescein sodium
Quantification of FITC–dextran and fluorescein sodium was determined using fluorescence spectroscopy (λex = 480 nm and λem = 520 nm). Calibration curves were run in triplicate over a three-day period to adhere to ICH standards. The limits of detection (LoD) and quantification (LoQ) are presented in Table 2.| Analyte | Concentration range (μg mL−1) | R 2 | Equation of regression line | LoD (μg mL−1) | LoQ (μg mL−1) |
|---|---|---|---|---|---|
| FITC–dextran 10 kDa | 0.78–100 | 0.997 | y = 1.4489x − 0.632 | 0.95 | 2.87 |
| FITC–dextran 70 kDa | 0.78–100 | 0.999 | y = 1.0224x − 0.6506 | 1.61 | 4.88 |
| FITC–dextran 150 kDa | 0.78–100 | 0.997 | y = 1.3352x − 1.5646 | 1.00 | 3.02 |
| Fluorescein sodium | 0.08–2.50 | 0.998 | y = 41.54x + 1.5603 | 0.09 | 0.28 |
FITC–dextran and fluorescein sodium dissolution and recovery
As F101 and F102 wafers displayed poor structural integrity, they were not considered for further studies. When placed into 20 mL of PBS (pH 7.4), F103 and FNa7 underwent complete dissolution after 8 min. F1506 had a dissolution time of 6 min 30 s, whereas F104 and F705 had dissolution times of 5 min. To determine the percentage analyte recovery, F103, F104, F705, F1506 and FNa7 solutions were diluted and quantified using fluorescence spectroscopy. The dissolution of F103, F104 F705, F1506 & FNa7 resulted in percentage recoveries of 80 ± 6.9%; 95.9 ± 4.0%; 93.0 ± 0.60%; 95.7 ± 5.4% and 96.0 ± 5.7% respectively. Assessing structural integrity, dissolution time and percentage recovery, F103 was excluded from in vitro studies due a slow dissolution time and low analyte recovery.Saturation solubility of FITC–dextran and fluorescein sodium
The saturation solubility of each fluorescent compound was calculated in PBS (pH 7.4) to ensure sink conditions were not exceeded (Table 3). Maintaining sink conditions in vitro is important in ensuring the rate of diffusion is not affected throughout the study. Fluorescein sodium, with a saturation solubility value of 466 mg mL−1, displayed the greatest solubility. Defining sink conditions as 10 times the volume needed to completely solubilise the analyte, it is apparent that the concentrations of all four analytes in vitro did not exceed sink conditions.25| Analyte | Mean saturation concentration ± S.D. (mg mL−1) |
|---|---|
| FITC–dextran 10 kDa | 91.93 ± 4.22 |
| FITC–dextran 70 kDa | 88.13 ± 3.26 |
| FITC–dextran 150 kDa | 61.76 ± 0.86 |
| Fluorescein sodium | 466 ± 51.61 |
FITC–dextran and fluorescein sodium in vitro permeation study
FITC–dextran permeation through porcine skin in vitro using ‘super-swelling’ hydrogel-forming MNs was quantified using Franz diffusion cells over 24 h (Fig. 5a and b).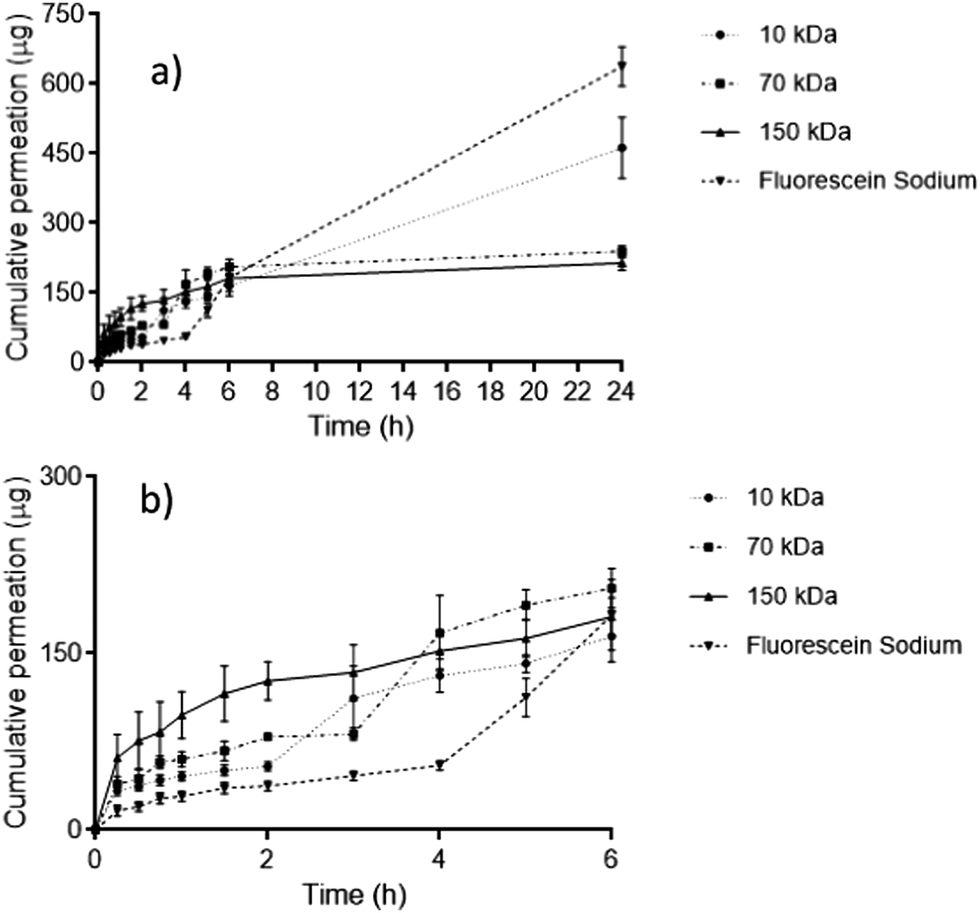 | ||
| Fig. 5 (a) In vitro permeation profiles of FITC–dextran 10 kDa, 70 kDa, 150 kDa and fluorescein sodium through dermatomed (350 μm) neonatal porcine skin over 24 h using ‘super swellable’ hydrogel-forming MNs. Means ± S.D., n = 5. (b) In vitro permeation profiles between 0–6 h within the same study. | ||
Between 0–6 h, there was no significant difference in cumulative permeation between all four fluorescent compounds (p = 0.1242). However, over a 24 h period, fluorescein sodium produced the greatest cumulative permeation, equivalent to 637.4 ± 42.69 μg (46.55 ± 4.67%). FITC–dextran 10 kDa produced a cumulative permeation of 462.17 ± 65.85 μg, equivalent to 35.28 ± 3.71% of the incorporated fluorescent compound over 24 h. Comparing both compounds, the cumulative permeation of fluorescein sodium and FITC–dextran 10 kDa was deemed to be statistically significant (p = 0.0011). With reference to FITC–dextran 10 kDa, FITC–dextran 150 kDa resulted in a significantly lower permeation over 24 h (213.54 ± 15.19 μg), equivalent to only 12.14 ± 2.73% release, (p = 0.0079). Interestingly, FITC–dextran 150 kDa displayed a similar permeation profile to FITC–dextran 70 kDa. In addition, there was no statistical difference in percentage permeation after 24 h between FITC–dextran 70 kDa and 150 kDa (p = 0.1349).
Discussion
High MW biomolecules, particularly mAbs (∼150 kDa), are now the most successful drug class on the market, with six of the top 10 best selling drugs derived from mAbs.5 Despite this success, mAbs are typically administered using a needle and syringe. Although needle and syringe administration is one of the most commonly used methods of drug delivery, fear of needles can result in therapy refusal. A meta-analysis report on needle phobia by McLenon et al. (2019), found that needle phobia in adolescents ranged from 20–50% and 20–30% in young adults.26 Another issue with this form of administration is that non-compliance is a key contributor in needle stick injuries. Alarmingly, between 2012–2017, the number of successful claims for needle stick injuries was 1213, costing the National Health Service (NHS) in excess of £4 million.27 Therefore, there is a need to develop alternative methods of administration to help alleviate these issues.Microneedles have the potential to offer a viable alternative. The field of microneedle (MN) drug delivery has expanded considerably in the last decade, with advances in technology enabling several types of MNs to be exploited. The first modalities developed were solid and hollow MNs, fabricated from silicon, metals and glass.28–32 In particular, hollow MNs have been tested previously in the delivery of biologics. Harvey et al. (2011) used a delivery device consisting of three 1 mm 34G steel needles incorporated into a fluid-disrupting hub to deliver etanercept (132 kDa) intradermally. This method was compared with intravenous (IV) and subcutaneous (SC) injections. As expected, the IV injection resulted in immediate delivery to the peripheral vasculature. The authors reported that intradermal (ID) administration resulted in rapid uptake into the lymphatic system, a feature not observed with SC delivery.33 It was concluded that MN facilitated drug delivery would be valuable in direct lymphatic targeting, with particular benefits in cancer therapy and diagnostic imaging.
Dissolving MNs have been tested with therapeutics possessing a wide range of solubilities and MWs.15,34–37 This delivery system has shown to be of considerable interest in bolus dosing due to the short dissolution time of the needle tips. In particular, corium have manufactured a drug-in tip dissolving MN containing teriparatide, a recombinant human parathyroid hormone indicated in osteoporosis which has successfully completed Phase 2a clinical trials.38 At present, this treatment regime consists of a low dose (20 micrograms) once daily SC injection. Therefore, despite the limited drug loading associated with dissolving MNs, this particular delivery system has shown considerable promise.
Unfortunately, therapeutic antibodies require sustained releases of higher doses to exert their therapeutic effect. Therefore, as drug loading is no longer restricted to what can be loaded into the MN, hydrogel-forming MNs offer a viable delivery option. Our group has tested this alternative method of delivery with Avastin®, a mAb, however, the high MW of this biotherapeutic caused entrapment within the cross-linked network of the MN resulting in sub-therapeutic dosing.20 Additionally, the large MW of this protein resulted in drainage into the lymphatic system following ID administration, leading to low concentrations in the systemic circulation.20,33 To this end, it is envisaged that low MW antibody-based alternatives could overcome these potential delivery issues.
In this study, ‘super-swellable’ hydrogel-forming MNs were used to facilitate delivery of FITC–dextran of three different MWs and fluorescein sodium in vitro. The experiments were designed to mimic the delivery of novel low MW biomolecules to allow for a direct comparison with mAbs tested previously. Currently, there are five different types of MNs, the most recent addition being hydrogel-forming MNs. These MNs have been shown to be safe to use. Following repeat MN application, results have shown that skin barrier function is not compromised, hereby minimising the possibility of inflammation or infection. Another advantage is that as drugs are not incorporated into or onto the MN, delivery is no longer limited to the loading capacity of the array, a considerable drawback associated with dissolving and coated MNs. Accordingly, hydrogel-forming MNs are of particular interest in the delivery of high dose macromolecules. Although they have been tested using a number of low MW compounds, each with different physical and chemical properties, to date there has been very little work carried out utilising higher MW compounds.
To begin, four FITC–dextran 10 kDa formulations, consisting of different gelatin and mannitol ratios were fabricated into lyophilised wafers. Following a 25 h freeze drying cycle, F104 produced a homogenous wafer with good structural integrity. This was considered a suitable formulation to develop the remaining lyophilised wafers containing FITC–dextran 70 kDa, 150 kDa and fluorescein sodium. Similar to FITC–dextran 10 kDa, the three other formulations (FITC–dextran 70 kDa, 150 kDa and fluorescein sodium) had short dissolution times and high percentage recoveries. Currently, there are no regulated dissolution tests with hydrogel-forming MNs, however, within this study, a suitable dissolution time for each lyophilised wafer was considered to be <10 min. Nevertheless, it is important that there is a trade-off between dissolution time and structural integrity. For this reason, the wafers chosen for further in vitro testing were homogenous, had good structural integrity and an appropriate dissolution time.
After the successful incorporation of all four compounds into separate lyophilised wafers, in vitro permeation through porcine skin using ‘super-swellable’ hydrogel-forming MNs was assessed. As the name suggests, a ‘super-swelling’ hydrogel-forming MN results in a significantly greater swelling capacity compared to hydrogel formulations reported previously by our group.17,19,39 This in turn causes the creation of larger micro-conduits within the skin.17 Additionally, the lower density and greater space within the polymer itself, permits the permeation of higher MW FITC–dextrans from their hygroscopic wafers, through the MN and into the receiver vessel of the Franz cell. It was interesting to note that there was no significant difference in permeation between all four compounds during 0–6 h period. During this period, the MN itself is imbibing the receiver fluid and swelling, before reaching its maximum swelling capacity at 6 h. After this period, it was observed that increasing the MW of the fluorescein-based compounds decreased the permeability across porcine skin between 6–24 h period in vitro. A plausible reason for this could be due to FITC–dextran 70 kDa and 150 kDa becoming trapped within the cross-linked network of the hydrogel, as observed with Avastin® which has been tested previously.20
An interesting study by Ambati et al. (2000) assessed the in vitro permeability of FITC–dextrans and FITC–IgG in the range of 4–150 kDa across rabbit sclera. The authors found that the sclera was more permeable to globular proteins than to linear dextrans of similar molecular weight.40 In this case, hydrodynamic radius was a better predictor of scleral permeability than MW. Considering this phenomenon, in relation to transdermal drug delivery using hydrogel-forming MNs, a comparison can be drawn between the FITC–dextran 150 kDa used in this report and Avastin® used previously.20 Although different compositions of mannitol and gelatin were used to yield homogeneous, intact structures, FITC–dextran and Avastin® wafers displayed similar percentage recoveries and dissolution times. Avastin®, a globular protein has a hydrodynamic radius of 46 Å.41 Although FITC–dextran 150 kDa has the same MW, it has a radius of 85 Å due to its linear structure.42 Comparing both compounds, the permeation of Avastin® across porcine skin in vitro, was approximately two-fold greater than FITC–dextran 150 kDa. This difference is consistent with that found by Ambati et al. (2000), therefore, it is possible that the use of FITC–dextran 10 kDa in this paper could lead to an underestimation in the permeability of low MW protein-based therapeutics across porcine skin in vitro. For this reason, hydrodynamic radius may be a better predictor of transdermal delivery of model compounds using MNs and will form the basis for future studies. Nevertheless, the results obtained within this study certainly suggest that the incorporation of low MW biotherapeutics will result in greater skin permeation when compared to the current mAb based therapeutics.43 As such, this study provides a basis and proof of concept for the incorporation of novel low MW protein-based therapeutics into lyophilised wafers for transdermal delivery using hydrogel-forming MNs.
Conclusions
Advances in antibody-based engineering have led to the production of low MW biotherapeutics. As these novel proteins are cheaper to manufacture and are not restricted to systemically accessible targets, they are now beginning to be considered as potential alternatives to mAbs in autoimmune and cancer-based therapies. At present, most protein-based therapeutics are administered using a syringe and needle, but it is thought that MNs can offer a viable alternative means of delivery. Previously, Avastin®, a mAb, was successfully delivered across porcine skin using hydrogel-forming MNs, however, lower MW biomolecules are thought to further improve transdermal delivery to anatomical sites. Consequently, this study aimed to model the skin permeation of low and high MW biotherapeutics using hydrogel-forming MNs as the delivery vehicle. To achieve this, FITC–dextran of different MWs and fluorescein sodium were delivered across neonatal porcine skin in vitro using hydrogel-forming MNs. The amount of FITC–dextran 10 kDa that permeated across the skin membrane was significantly greater than that achieved for FITC–dextran 150 kDa. Therefore, this model study has shown that low MW biomolecules will permeate through porcine skin in greater amounts than mAbs tested previously using MNs.Author contributions
A. R. J. H. and R. F. D. contributed to the conceptualization of this study. A. R. J. H. contributed to the methodology, validation, formal analysis, investigation, data curation and writing—original draft preparation. E. L., R. F. D. contributed to the resources. M. T. C. M., E. L., R. F. D. contributed to the writing—review and editing. M. T. C. M. and R. F. D. contributed to the supervision, project administration and funding acquisition.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
This work was supported in part by the Wellcome Trust (WT094085MA).Notes and references
- J. K. H. Liu, Ann. Med. Surg., 2014, 3, 113–116 CrossRef PubMed.
- NICE 2010, available at: https://www.nice.org.uk/guidance/ta195, accessed on 15 April 2019.
- NICE 2015, available at: https://www.nice.org.uk/guidance/ta329, accessed on 15 April 2019.
- NICE 2010, available at: https://www.nice.org.uk/guidance/ta199/evidence, accessed on 15 April 2019.
- A. Philippidis, Genet. Eng. Biotechnol. News, 2019, 39, 16–17 Search PubMed.
- D. L. Ludwig, et al. , J. Biol. Chem., 2005, 280, 19665–19672 CrossRef PubMed.
- B. Schlereth, et al. , Cancer Res., 2005, 65, 2882–2889 CrossRef CAS PubMed.
- X. Jimenez, et al. , Mol. Cancer Ther., 2005, 4, 427–434 CAS.
- J. S. Haas, Ann. Intern. Med., 2005, 142, 891 CrossRef PubMed.
- P. Å. Nygren, FEBS J., 2008, 275, 2668–2676 CrossRef CAS PubMed.
- P. Holliger and P. J. Hudson, Nat. Biotechnol., 2005, 23, 1126–1136 CrossRef CAS PubMed.
- A. Thakur, M. Huang and L. G. Lum, Blood Rev., 2018, 32, 339–347 CrossRef CAS PubMed.
- P. Chames, M. Van Regenmortel, E. Weiss and D. T. Baty, Br. J. Pharmacol., 2009, 157, 220–233 CrossRef CAS PubMed.
- R. Rouet, D. Lowe and D. Christ, FEBS Lett., 2014, 588, 269–277 CrossRef CAS PubMed.
- A. R. J. Hutton, et al. , Int. J. Pharm., 2018, 541, 56–63 CrossRef CAS PubMed.
- R. F. Donnelly, T. R. Raj Singh and D. A. M. Woolfson, Drug Delivery, 2010, 17, 187–207 CrossRef CAS PubMed.
- R. F. Donnelly, et al. , PLoS One, 2014, 9, 1–12 Search PubMed.
- R. F. Donnelly, et al. , Adv. Funct. Mater., 2012, 22, 4879–4890 CrossRef CAS PubMed.
- T. R. Raj Singh, D. A. Woolfson and R. F. Donnelly, J. Pharm. Pharmacol., 2010, 62, 829–837 CrossRef PubMed.
- A. J. Courtenay, M. T. C. McCrudden, K. J. McAvoy, H. O. McCarthy and R. Donnelly, Mol. Pharmaceutics, 2018, 15, 3545–3556 CrossRef CAS PubMed.
- E. Larrañeta, et al. , Int. J. Pharm., 2014, 472, 65–73 CrossRef PubMed.
- M. S. Lhernould, M. Deleers and A. Delchambre, Int. J. Pharm., 2015, 480, 8–15 CrossRef PubMed.
- S. N. Economidou, et al. , Mater. Sci. Eng., C, 2019, 102, 743–755 CrossRef CAS PubMed.
- E. Larrañeta, et al. , Int. J. Pharm., 2016, 497, 62–69 CrossRef PubMed.
- B. R. Rohrs, Dissolution Technol., 2001, 8, 6–12 CrossRef CAS.
- J. McLenon and M. A. M. Rogers, J. Adv. Nurs., 2019, 75, 30–42 CrossRef PubMed.
- NHS Resolution, 2017, available at: https://www.resolution.nhs.uk, accessed on 19 June 2019.
- P. Khanna, K. Luongo, J. A. Strom and S. Bhansali, J. Micromech. Microeng., 2010, 20, 1–8 CrossRef.
- P. Khanna, K. Luongo, J. A. Strom and S. Bhansali, Microsyst. Technol., 2010, 16, 973–978 CrossRef CAS.
- C. O’Mahony, Biomed. Microdevices, 2014, 16, 333–343 CrossRef PubMed.
- F. J. Verbaan, et al. , J. Controlled Release, 2007, 117, 238–245 CrossRef CAS PubMed.
- P. M. Wang, M. Cornwell, J. Hill and M. Prausnitz, J. Invest. Dermatol., 2006, 126, 1080–1087 CrossRef CAS PubMed.
- A. J. Harvey, et al. , Pharm. Res., 2011, 28, 107–116 CrossRef CAS PubMed.
- J.-Y. Kim, et al. , Eur. J. Pharm. Biopharm., 2016, 105, 148–155 CrossRef CAS PubMed.
- J. Mönkäre, et al. , J. Controlled Release, 2015, 218, 53–62 CrossRef PubMed.
- K. Lee, C. Y. Lee and H. Jung, Biomaterials, 2011, 32, 3134–3140 CrossRef CAS PubMed.
- H. L. Quinn, L. Bonham, C. M. Hughes and R. Donnelly, J. Pharm. Sci., 2015, 104, 3490–3500 CrossRef CAS PubMed.
- A. C. Anselmo, Y. Gokarn and S. Mitragotri, Nat. Rev. Drug Discovery, 2018, 18, 19–40 CrossRef PubMed.
- T. R. Raj Singh, P. A. McCarron, A. D. Woolfson and R. F. Donnelly, Eur. Polym. J., 2009, 45, 1239–1249 CrossRef CAS.
- J. Ambati, et al. , Invest. Ophthalmol. Visual Sci., 2000, 41, 1181–1185 CAS.
- L. M. Hirvonen, et al. , Pharm. Res., 2016, 33, 2025–2032 CrossRef CAS PubMed.
- A. Ekani-Nkodo and D. K. Fygenson, Phys. Rev., 2003, 67, 1–7 Search PubMed.
- K. T. Xenaki, S. Oliveira and P. M. van Bergen en Henegouwen, Front. Immunol., 2017, 8, 1–6 Search PubMed.
| This journal is © The Royal Society of Chemistry 2020 |