
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Auto-catalytic redox polymerisation using nanoceria and glucose oxidase for double network hydrogels
Ali A.
Mohammed
 ,
Alessandra
Pinna
,
Siwei
Li
,
Tian
Sang
and
Julian R.
Jones
*
,
Alessandra
Pinna
,
Siwei
Li
,
Tian
Sang
and
Julian R.
Jones
*
Department of Materials, Imperial College London, SW7 2AZ, London, UK. E-mail: julian.r.jones@imperial.ac.uk
First published on 6th March 2020
Abstract
A novel auto-catalytic reaction, a combination of naturally occurring enzyme glucose oxidase (GOx) and amine-functionalised cerium oxide nanoparticles (nanoceria), was employed for open vessel free radical polymerisation of double network hydrogels (DNHGs). The nanoceria also incorporated into the gels to enhance mechanical strength. GOx reduces atmospheric O2 to H2O2, causing a cyclic change of cerium ion states, resulting in propagating free radicals in the carbon group in the amino functionalised nanoceria surface. We synthesised novel nanocomposite DNHGs by grafting polymers onto amine-functionalised nanoceria (ANC), with poly(2-acrylamido-2-methylpropanesulfonic acid), PAMPS, and polyacrylamide (PAAm) in the first and second networks respectively. The graft polymerisation was initiated using the alternating cerium states on the ANC. GOx held two major roles within the reaction: to provide an oxygen free system, without any other form of degassing, and to provide cyclical cerium ion states between Ce4+ and Ce3+, creating new free radicals for polymerisation. Polymer conversion using ANC as the sole initiator in the presence of GOx resulted in 83% conversion for PAMPS and 64% PAAm. Polymers degassed only with argon resulted in less than 55% conversion for both PAAm and PAMPS, proving that the addition of GOx enhanced the reaction. The new gels (1.76 MPa) showed an order of magnitude improvement in mechanical properties compared to DNHG made without ANC/GOx (0.10 MPa).
Introduction
Hydrogels have many applications in the field of tissue engineering. They are 3D polymer networks that hold 90–99 wt% water, mimicking the extracellular matrix (ECM) of tissues such as articular cartilage.1 Articular cartilage is an important load-bearing tissue that exhibits cyclical loading on daily basis. It has limited self-regenerative properties due to its avascular and non-lymphatic nature.2 One of the main drawbacks of many traditional (single network) hydrogels is poor mechanical properties and their inability to sustain cyclic loads, for example, articular cartilage has a compressive fracture stress of 36 MPa, posing a challenge for cartilage repair in tissue engineering.3,4Double network hydrogels (DNHG), developed by Gong et al.,5–10 have interpenetrating networks of contrasting polymers that allow for improved control over their mechanical properties. DNHGs are often synthesised using free radical polymerisation (FRP) with chemical covalent cross linkers and radical initiators.11 Gong and Katsuyama12 produced a hydrogel of poly(2-acrylamido-2-methylpropanesulfonic acid)/polyacrylamide (PAMPS/PAAm) with unusually high mechanical properties. PAMPS, a polyelectrolyte, was used as the first network where it was cross linked using N,N′-methylene bis acrylamide (MBAA). It was then soaked in an aqueous solution of PAAm, a neutral polymer, also cross linked with MBAA to form the second network. The DNHG had a compressive fracture stress of up to 20 MPa at 92% strain whilst holding 90 wt% water, whereas the single network gel components had sub-MPa fracture stresses at 80% strain for PAAm and 40% for PAMPS closely matching that of native articular cartilage.12 A study comparing DNHGs to tradition single-network hydrogels, in a rabbit model, provided evidence that a DNHG provoked hyaline-like cartilage production, whereas a mixture of fibrocartilage and hyaline cartilage was produced when a single network gel was used.13 Successful cartilage matrix production was also shown by encapsulation of human chondrocytes in DNHGs consisting of gelatin and hyaluronic acid.14
The limited mechanical stiffness of hydrogels create drawbacks for the progression of cartilage repair.15 Therefore nanocomposite hydrogels, particularly nanocomposite DNHGs, may help solve these problems.16 Recent studies have used nanostructures such as silica nanoparticles (SNPs) and LAPONITE® as cross linkers to further improve the mechanical properties.5,11,17–21 Fu et al. developed a “super-tough” DNHG with covalently bonded vinyl-functionalised SNPs (VSNPs).22 PAMPS/PAAm were synthesised in the usual two-step polymerisation reaction, initially forming the PAMPS network using MBAA with the addition of the vinyl SNPs forming a PAMPS/SNP composite gel. The VSNPs acted as macro cross linkers in the DNHG providing a denser network. VSNP grafted gels reached a strain of 0.98 without fracture, and up to 73.5 MPa fracture stress, a 16.5 MPa compressive strength and a 0.33 MPa elastic modulus for 300 nm VSNP (1 wt%) grafted gels.22 A thermoresponsive nanocomposite DNHG was synthesised using poly(N-isopropylacrylamide) (PNIPAAm) and 50 nm and 200 nm polysiloxane nanoparticles, resulting in a compressive stress of 175 kPa and 181 kPa respectively.
The addition of SNPs to the first network of DNHG can be used to achieve tight cross linking as a way to introduce tailorability into the materials properties, such as increased mechanical strength and toughness, better resistance to strain deformation and controllable swelling rates.22,23 This approach can be altered to enhance the covalent bonds between the polymer and nanostructures by using in situ surface graft polymerisation, which allows the polymers to covalently link to the surface of the nanoparticles.
Previous studies have successfully achieved polymer grafts on the surface of SNPs, for example: polystyrene with controlled/living radical polymerisation24 and with atom transfer radical polymerisation (ATRP);25 thermoresponsive poly-N-isopropylacrylamide (PNIPAM) brushes by ATRP;26 polymethyl methacrylate (PMMA) by graft photopolymerisation;27 polyacrylamido-2-methyl-1-propane sulfonic acid (PAMPS) and polystyrene sulfonic acid sodium salt (PSSA) by surface initiated redox polymerisation using cerium ammonium nitrate (CAN) as an initiator.28 PAMPS and PSSA were grafted on the surface of amine-functionalised SNP (ASNPs). The ASNPs were dispersed in acidic solution of AMPS monomers before CAN and stabiliser sodium dodecyl sulfate (SDS) were added. The system was purged with nitrogen gas and heated to 40 °C to commence the graft polymerisation of monomers to the ASNPs. The grafting was initiated by a redox pair of Ce(IV) (oxidant) and the alkyl amine (reductant), causing an intermediate free radical at the α-carbon atom of the alkyl amine group. The grafting percentage and polymer conversion were low at 46% and 4.6% for PAMPS, and 22% and 2.2% for PSSA, respectively. However, conversion was enough for successful grafting, resulting in a polymer nanocomposite that has the potential to be used as a first network in DNHGs.
Cerium oxide is known for its redox activity as a catalyst in many applications, such as chemical–mechanical polishing, UV filtering, fuel cells and polymer or ceramic nanocomposites.29,30 More recently, cerium oxide nanoparticles (nanoceria) have found promise in the biomedical field as therapeutic antioxidants against diseases associated with oxidative stress such as cancer, neurodegenerative and ocular diseases.31 Nanoceria has catalytic and antioxidant properties due to the coexistence of two oxidation states and to the presence of oxygen vacancies in the crystal lattice. The two valence states; Ce(III) and Ce(IV), provide efficient redox behaviour, allowing cerium atoms to cyclically change their oxidation states between reduced Ce3+ and oxidised Ce4+ states. In the form of nanoparticles at sizes less than 10 nm,32 nanoceria has an abundance of reduced Ce3+ atoms on the surface with a core consisting of mainly oxidised Ce4+ atoms.
Nanoceria has the ability to mimic different enzyme activity, such as superoxide dismutase (SOD) and catalase (CAT), in which H2O2 is involved. SOD catalyses the dismutation of superoxide anion (*O2) to hydrogen peroxide (H2O2) and molecular oxygen (O2) while CAT catalyses the degradation or reduction of H2O2 to water and O2.33 The latter property makes nanoceria a promising material in bio-catalysis. Several studies have shown that nanoceria is a highly biocompatible material with no systemic toxicity or genotoxicity, and reduces local inflammatory reactions when integrated into implants such as biomaterials.34–37 This makes nanoceria a viable candidate for this work, similar to other redox initiators such as the commonly used ammonium persulfate (APS).38,39
Here, we aim to optimise the use of redox initiator CAN to enhance polymer conversion, and to convert cerium into a multipurpose component within the reaction. Nanoceria's intrinsic oxygen vacancy system enables it to have an auto-catalytic mechanism.40 Our hypothesis was that this mechanism could be exploited for the formation of polymeric networks on the surface of the nanoceria. This has yet to be used in the formation of polymeric materials, or DNHGs. Our second hypothesis was that nanoceria's auto-catalytic properties and two valence states can provide efficient redox behaviour to increase reaction kinetics and conversion. Our strategy was to form nanoceria and add a naturally occurring enzyme (GOx) that acts as a reaction catalyst that also regenerates the redox valence states of the cerium ions.
The proposed mechanism is as follows. When nanoceria is active in aqueous solution, its surface ions reduce from Ce4+ to Ce3+ by interacting with reactive oxygen species (eqn (1)), releasing O2 as a by-product.41 However, in the presence of H2O2, Ce3+ can be re-oxidised to Ce4+ (eqn (2)).41
| O2˙− + Ce4+ → O2 + Ce3+ | (1) |
| O2˙− + Ce3+ + 2H+ → H2O2 + Ce4+ | (2) |
GOx was previously used as a degassing agent for O2 removal in an open vessel system for a reversible addition–fragmentation chain-transfer (RAFT) polymerisation of hydroxyethyl acrylate and dimethylacrylamide with non-cytotoxic by-products.43 A recent study showed that GOx concentrations as low as 200 nM were able to achieve 100% conversion for PAAm and PAMPS in open vessel synthesis, using 0.1 wt% photoinitiator.23 This study also demonstrated that there were no cytotoxic by-products from the polymers or GOx at these concentrations. The kinetics for both polymers improved significantly, resulting in faster reaction times (factor of 2) under the same conditions.23 The improved kinetics were a result of eliminating the interactions between the propagating free radicals and O2. By evading O2 inhibition, the reaction reaches full conversion with the use of lower concentrations of initiator.23
The aim here was to combine GOx and nanoceria, which has been proven to work effectively in previous studies aimed at glucose sensing.33,44 When GOx reacts with glucose (G) in the presence of O2 it will produce H2O2 as a by-product (eqn (3)). Therefore, during graft polymerisation, GOx can play the role of degassing agent and as a facilitator for H2O2 production, which revitalises the cerium based initiators at the nanoceria surface (eqn (4)–(6)):45
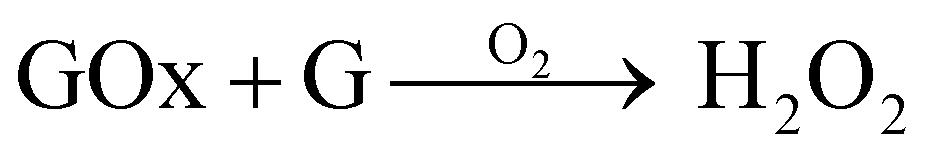 | (3) |
| Ce4 + H2O2 → Ce3+ + H+ + HO2 | (4) |
| Ce4+ + HO2 → Ce3+ + H+ + O2 | (5) |
| Ce3+ + H+ + HO2 → Ce4+ + H2O2 | (6) |
Here, we carry out the first graft polymerisation on the surface of nanoceria, investigating the impact of GOx compared to argon degassing. Novel DNHGs were then produced formed via graft polymerisation on nanoceria in the presence of GOx in both networks. ANC gels are the first of their kind, to have both networks formed through an autocatalytic redox surface graft polymerisation, with GOx as a degassing agent and reaction mediator.
Materials and methods
Acrylamide (AAm; ≥99%) and 2-acrylamido-2-methyl-1-propanesulfonic acid (AMPS; 99%); N,N′-methylenebisacrylamide (BIS; 99%); photoinitiator 2-hydroxy-4′-(2-hydroxyethoxy)-2-methylpropiophenone (Irgacure 2959; 98%), 1,3,5-trioxane (≥99%); (3-aminopropyl)triethoxysilane (APTES; ≥98%), deuterium oxide (D2O; 99.9 atom% D), cerium(IV) ammonium nitrate (CAN; ≥99.99% trace metals basis), cerium(III) nitrate hexahydrate (Ce(NO3)3·6H2O ≥ 99.99% trace metals basis), sodium dodecyl sulfate (SDS; ≥98.5%), ammonium hydroxide (NH4OH; 28–30% NH3 basis) 2-propanol (≥99.5%), hydrochloric acid (HCl; 1 M) and urea were purchased from Sigma-Aldrich (UK). No additional processing and/or purifications were performed. D-Glucose (G) and glucose oxidase (GOx; from Aspergillus niger as a lyophilised powder) were purchased from Sigma-Aldrich (UK) and stored in phosphate buffer saline (PBS) aliquots at −20 °C when received.Amine nanoceria synthesis
2 g of urea and 7 g of Ce(NO3)3·6H2O were dissolved separately in beakers with 20 ml of 2-propanol. 0.5 ml HCl was added to each solution they were fully dissolved. Urea was then added dropwise into the Ce(NO3)3 solution whilst stirring. 14 ml of aqueous NH4OH was then added and a precipitate was formed and stirred until it was homogenously mixed. The solution was exposed to 600 W microwaves, 4 times for 10 seconds each, or until a crackling sound was heard. Once the solution was removed from the microwave, 14 ml of deionised water was added to stop the reaction. The solution was then washed and centrifuged using the Eppendorf 5430 with water 3 times at 7830 rpm, and kept as a stock suspension in water with a concentration of 1.7 g CeO2/50 ml deionised water.46Nanoceria was functionalised by sonicating 340 mg of CeO2 in 150 ml EtOH until fully dispersed. 9 ml of APTES was then added while under stirring.47 The solution was left to stir at room temperature for 24 h, and then washed/centrifuged with EtOH 3 times at 7830 rpm. The final product was dried at 60 °C to obtain a powder of amine functionalised CeO2 (ANC). This process is represented in a schematic drawing in Fig. 1.
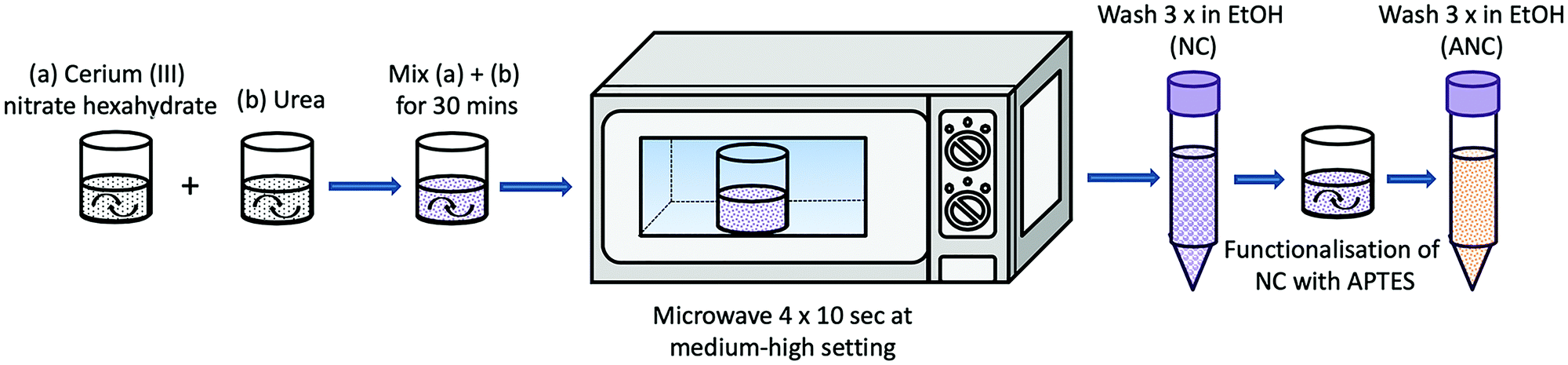 | ||
| Fig. 1 Schematic representation showing the synthesis and functionalisation of nanoceria with APTES. | ||
Nanoceria characterisation
Bare and amine nanoceria were diluted in ethanol, sonicated and then carefully dropped onto 300 mesh copper TEM grids coated with holey carbon film. A JEOL FX2000 was used for imaging with tungsten filament as the electron source at an operating voltage of 200 kV and a 10 μm objective aperture. Mean diameter was calculated by measuring 100 separate nanoceria particles.Bare NC and ANC were dried at 60 °C and ground to a fine powder for FTIR analysis. A Nicolet iS10 Thermo Scientific FTIR was used. Samples were scanned 32 times per run with a resolution of 6 in a wavenumber region of 4000–400 cm−1.
For UV-VIS analysis, samples were diluted to 3 mg in 1 ml deionised water solutions and sonicated until fully dispersed. Samples were then loaded into the PerkinElmer Lambda 25 UV-VIS spectrometer and run against a blank sample of deionised water.
Surface graft polymerisation on nanoceria
150 mg of ANC was fully dispersed and sonicated in 5 ml of deionised water. 5 mg of trioxane and 4.5 g of AMPS were dissolved in 35 ml deionised in a round bottom flask, with a final concentration of 0.24 M AMPS. The dissolved ANC was added to the AMPS monomer solution and mixed for 5 minutes. A 20 μl sample was taken and topped up with 500 ml of D2O, for 1H NMR analysis of the 0 h reference. The solution was degassed for 20 minutes in a hot bath set at 40 °C and left to stir for 24 hours. A 24 h sample was taken for 1H NMR analysis of the final polymer conversion. The final solution was wash/centrifuged, using the Eppendorf 5430, 3 times with water at 7830 rpm and dried at 60 °C. The process was repeated a second time with CAN 0.7 g being added to the monomer solution, and a third time with CAN 0.7 g and 1.5 g SDS being added to the monomer solution. The same process was repeated for AAm.Surface graft polymerisation on nanoceria in the presence of GOx
5 mg of trioxane and 4.5 g AMPS were dissolved in deionised water and titrated to pH 5.4 using NaOH (0.25 M). The monomer solution was topped up to 35 ml with deionised water with a final concentration of 0.24 M AMPS. 150 mg ANC was dispersed and sonicated in 5 ml H2O for 1 h then added to the monomer solution. The pH increased to 8–9 once the ANC was added; therefore HCl (1 M) was used to bring down the pH to 5.4. GOx was added at a final concentration of 200 nM to the monomer/ANC solution. The solution was transferred to a round bottom flask and the process continued as described above. Repeats were also done to include CAN and CAN/SDS. This was then repeated with 2×, 3× and 4× the concentration of ANC. The initial ANC concentration is referred to as ANC ×1.The entire process was repeated, without titration, for AAm with a final concentration of 1.41 M. The pH was checked once the ANC was added and if needed, brought down to 5.4 pH using HCl before adding GOx (Fig. 2). This was to ensure GOx was added at optimum pH conditions.
 | ||
| Fig. 2 Schematic showing the redox graft polymerisation of PAMPS and PAAm polymers on the surface of amine functionalised nanoceria (ANC), in the presence of GOx. | ||
Surface graft polymer characterisation
In addition to FTIR and UV-VIS, 1H NMR samples were prepared by diluting 1 drop of polymer solution and 5 mg of trioxane into 500 ml deuterated water (D2O) and analysed using a Bruker AV-400 spectrometer operating at 400 MHz. The resulting spectra were analysed using MestReNova software with a ‘Whittaker smoother’ baseline correction. All samples contained trioxane to aid in assessing monomer to polymer conversion. Trioxane peaks were integrated to represent a value of 1. Monomer peaks were integrated relative to trioxane.Thermogravimetric analysis (TGA) was conducted using a NETZSCH STA-449C Jupiter. Samples were prepared by being heat and vacuum drying, then grounded to a powder. Sample analysis was conducted from room temperature to 800 °C. A heating rate of 10 °C min−1 was applied to all samples.
Hydrogel synthesis
The first network was formed by fully dispersing and sonicating 300 mg of ANC in 5 ml H2O. 4.5 g AMPS was titrated to 5.4 pH using NaOH (0.25 M), to a final concentration of 0.24 M AMPS. ANC was then added to the AMPS monomer solution and the pH corrected to 5.4, if required. GOx and G was added to the monomer/ANC solution and mixed for 5 minutes. The solution was then put into aliquots of 2 ml in polystyrene moulds and placed in a sonication bath set to 40 °C to ensure homogenous ANC distribution. Once the solutions in the moulds had gelled, the moulds were placed in an oven at 40 °C for 24 h, and then for 6 h at 60 °C to denature GOx and dry the first network.The hydrogels were removed from the moulds and soaked in a monomer solution of 2.54 M AAm containing 150 mg ANC, 0.1 wt% BIS, GOx and G. Once the hydrogels were swollen, they were placed in moulds and put into a 40 °C oven to form the second network. The final hydrogels were then dried at 60 °C and, at their dry state, placed in water to swell for a week. This process is highlighted in Fig. 3.
 | ||
| Fig. 3 Schematic showing the synthesis route of nanoceria based double network hydrogels (ANC gels) in the presence of GOx. | ||
Hydrogel characterisation
Swelling studies were conducted on ANC gels that were dried for three days at 40 °C, and swollen in water until a water uptake plateau was reached. Swollen samples were used for mechanical analysis in compression, using a Zwick Roell z2.5 machine fitted with a load cell of 10 kN, and a strain rate of 1.5 mm min−1. Samples for SEM were frozen overnight in −80 °C and then freeze dried for 24 h. Samples were placed on SEM sample holders and held by carbon tape. Samples were then sputter coated for 2 minutes at 20 mA to form a 10 nm layer of chromium. The LEO Gemini 1525 FEG-SEM was used to image the surface morphology of freeze dried hydrogels using 3–5 kV and a working distance of 5–10 mm.Results and discussion
Amine functionalised nanoceria
Nanoceria was successfully synthesised and functionalised by APTES. Amine functionalisation was confirmed through FTIR and UV-VIS (Fig. 4a and b). FTIR Bands associated with Si–O–Si at 1160 cm−1, NH bending at 1156 cm−1, NH stretching 3324 cm−1, CH2 stretching 1450 cm−1 all indicate to successful surface modification of the nanoceria by APTES.48 Bands at 550 cm−1 represent Ce–O stretching from CeO2.49 UV-VIS shows the changes in cerium ion states within the bare NC and ANC. Ce4+ is found at wavelength 200 nm, whereas Ce3+ is found at 300 nm. Bare NC have a higher concentration of Ce4+ on the surface, and a lower concentration of Ce3+. Once functionalised with amine groups, the distribution of oxidation states reverses, resulting in an increased concentration of Ce3+ on the surface. Analysis through TEM imaging (Fig. 4c and d) shows ANC have an average diameter of 14 ± 3 nm. This value was calculated by taking the average diameter of 100 single ANC particles. | ||
| Fig. 4 (a) FTIR spectra and (b) UV-VIS spectra of bare nanoceria (NC) and amine functionalised nanoceria (ANC). (c and d) TEM images of ANC. | ||
Polymer grafts on ANC
The surface of ANC was used as a building block for grafting PAMPS and PAAm. The initial aim was to examine the possibility of synthesising polymeric networks from the surface of these nanostructures. Monomers were polymerised in situ with ANC, with the addition of CAN alone, as well as a combination of CAN and SDS, under inert argon atmospheres.UV-VIS analysis was conducted post synthesis on all samples to examine the success of polymer grafts on the surface of ANC. Surface grafts of AAm on ANC were successful using all chemical combinations, (Fig. 5). A disappearance in Ce4+ and a reduction in Ce3+ for all reactions with AAm provide evidence that grafts were present on the surface. SDS was not completely removed from the sample despite several washes, with a subtle peak present between 220–260 nm. SDS is suggested to have toxic effects, particularly in unreacted powder form and residues would have to be removed from the final material.50 AAm/ANC and AAm/ANC/CAN exhibited the least amount of Ce3+ remaining on the surface therefore suggesting high polymer graft coverage.
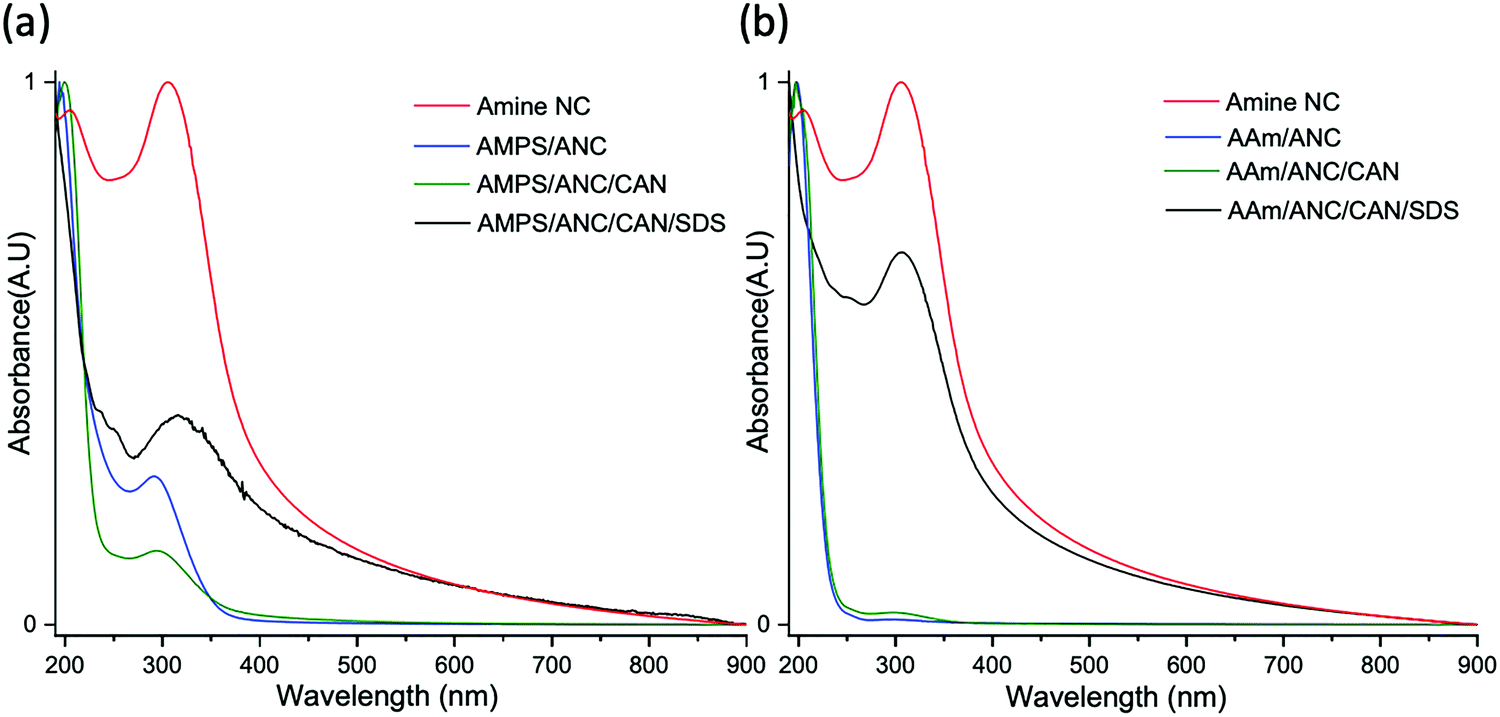 | ||
| Fig. 5 UV-VIS of amine nanoceria (ANC) and reactions with (a) AMPS and (b) AAm, with or without cerium ammonium nitrate (CAN) and stabiliser sodium dodecyl sulfate (SDS). | ||
UV-VIS for AMPS revealed successful graft polymerisations occurred on the surface of ANC, as seen in Fig. 5. Similar to AAm, a reduction in Ce4+ and Ce3+ revealed grafts were present on the surface of ANC. AMPS/ANC and AMPS/ANC/CAN had the clearest polymer peaks at 200 nm with the largest reductions in Ce3+ indicating they had the highest graft polymerisation. SDS was still present in the sample with small peaks at 220–260 nm.
Fig. 6 shows FTIR spectra for AAm and AMPS in combination with ANC/CAN/SDS, compared to their respective homopolymers and ANC. Table 1 provides a summary of the bands present in AMPS grafted polymers on the surface of ANC. Bands were present for all samples with AMPS/ANC, for sulfonic acid groups at 1040 cm−1, reflecting the PAMPS spectra. This suggests a large amount of polymer was grafted on the surface of ANC. Therefore, it was proven that ANC has the ability to initiate and propagate polymer grafts without the need of a chemical initiator such as CAN, or a stabiliser such as SDS. Similar bands with reduced intensity were seen for AMPS/ANC/CAN, suggesting lower graft amounts. Residual SDS was detected in AMPS/ANC/CAN/SDS samples, between 2950–2956 cm−1 (asymmetric and symmetric CH2 stretching), along with reduced polymer bands.51
 | ||
| Fig. 6 FTIR spectra of reactions of amine nanoceria (ANC) with (a) AMPS and (b) AAm, with or without cerium ammonium nitrate (CAN) and stabiliser sodium dodecyl sulfate (SDS). Band indexing shown in Tables 1 and 2. | ||
| Bond | Type | Wavenumber (cm−1) | |
|---|---|---|---|
| A | C–H | Stretching | 2956 |
| 2918 | |||
| 2850 | |||
| B | CH2 | Stretching | 2966 |
| CH3 | 2932 | ||
| C | C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O O |
Stretching, coupled with N–H bending | 1652–1659 |
| D | N–H | Primary amine | 1620 |
| E | CO2 | 1319 | |
| F | N–H | Bending | 1238 |
| G | Ce–O–Si | Stretching | 952 |
| H | R–S(O)–R′ |
1040 |
Table 2 gives a summary of the bands found in AAm grafted polymers on the surface of ANC. All samples showed polymer growth, suggesting ANC on its own was able to result in polymer growth. AAm/ANC gelled during experimentation and therefore the FTIR spectra do not reflect the band intensities relative to other samples. Gelling of the sample is a strong indicator that cross linking within the polymer grafted nanoceria had occurred. The disappearance of bands at 952 cm−1 in all samples, representing Ce–O–Si bonds from APTES on the surface of ANC, indicates thick polymer layers covering the surface formed. All spectra that represent grafted polymers exhibit bands at 1652 cm−1 and 1620 cm−1, indicating the presence of amide bonds from PAAm. Residual SDS was found in AAm/ANC/CAN/SDS in bands between 2850–2956 cm−1, despite several wash/centrifuge cycles.51 From the FTIR spectra provided, it can be assumed, based on band intensity and similarity to PAAm, that AAm/ANC/CAN was the most successful at grafting polymers on the surface of ANC.
| Bond | Type | Wavenumber (cm−1) | |
|---|---|---|---|
| A | C–H | Stretching | 2956 |
| 2918 | |||
| 2850 | |||
| B | CH2 | Stretching | 2966 |
| CH3 | 2932 | ||
| C | CO |
Stretching, coupled with N–H bending | 1652–1659 |
| D | N–H | Primary amine | 1620 |
| E | C–N | Stretching primary amide | 1420 |
| F | CO2 | 1319 | |
| F | N–H | Bending | 1238 |
| G | Ce–O–Si | Stretching | 952 |
| H | C–H | Out-of-plane rocking | 942 |
| I | Ce–O | Stretching | 550 |
Polymer conversion was measured using 1H NMR and calculated relative to trioxane peaks in 0 h and 24 h samples. ANC was proven to initiate the self-catalysis redox polymerisation for both PAMPS and PAAm, noted by polymer conversion in the AAm/ANC and AMPS/ANC systems (Fig. 7). Fig. 7 shows reactions degassed with argon resulted in lower conversions, when compared to the same reactions in the presence of GOx. This suggests that GOx is more efficient at degassing the monomer solution by reacting with oxygen species than argon purging. Under argon gas, AMPS samples resulted in conversion of approximately 50% for all cases (Fig. 7a). This suggests that either all combinations were equally effective, or that degassing with argon was insufficient and resulted in a rate limiting step. However, the addition of GOx causes a big increase in conversion.
 | ||
| Fig. 7 Conversion profiles for (a) AMPS and (b) AAm, using different combinations of reactions and degassed using either argon or GOx. | ||
Results for AAm (Fig. 7b) show the combination of AAm/ANC/CAN and AAm/ANC/CAN/SDS resulted in the highest conversion of 100%, when GOx was present. The addition of GOx also meant that the stabiliser SDS was no longer required for the reaction, which provides fewer purification steps to remove potentially toxic chemicals from the end product. Both AAm and AMPS exhibited an increase in conversion of approximately 30% when GOx was added to samples only containing ANC. This was likely due to a combination of two factors. First, the efficiency at which GOx degasses oxygen, creating a better kinetic profile for both polymers by eliminating the effect of O2 on free radicals. Secondly, the ability of GOx to deplete oxygen further benefited the redox reaction, through the activity of nanoceria, by impacting the Ce3+/Ce4+ valence states, through the release of GOx by-product H2O2. This re-oxidises Ce3+ to Ce4+ allowing the reaction to become faster and more efficient (eqn (7)). Hence, resulting in increased conversion in the presence of GOx.44 The difference in final conversion rates between both polymers under argon and GOx will also be due to their inherent specific kinetic profiles, which have been studied in previously.23
| 2Ce3+ + H2O2 → 2Ce4+ + 2OH− | (7) |
Further tests were conducted to find the highest polymer conversion based on varying ANC concentration. ANC concentration was adjusted to 2×, 3× and 4× the initial concentration. The results are displayed in Fig. 8. The highest conversion achieved for both AMPS and AAm was found to be at two times that of the original concentration of ANC (ANC ×2). At ANC ×3, the conversion for both polymers began to decrease, and at x 4 the conversion dropped significantly. At ANC ×2, more monomers come into contact with ANC, than the original concentration, therefore the polymerisation became more efficient with GOx. Oversaturation of the system began at ANC ×3, where the conversion began to decrease, making it more difficult for monomers to come in contact with active surfaces of ANC. The solution became notably denser as ANC concentration increased, likely making diffusion more difficult. It was also suspected that GOx became denatured at high ANC concentrations present in the solution.
 | ||
| Fig. 8 Conversion profiles for AAm and AMPS and with varying amine nanoceria (ANC) concentrations, in the presence of GOx (no CAN or SDS). | ||
A concentration of ANC × 2 provided AMPS/ANC with a conversion of 91% and AAm/ANC with a conversion of 84%. This means CAN and SDS are not be required for hydrogel synthesis, removing the subsequent purification steps required to remove those chemicals. More importantly, it showed the success of the combination of GOx working in unison with nanoceria to allow a self-catalysed system to form a polymeric nanocomposite network.
TGA analysis was conducted on samples degassed by GOx with varying ANC concentrations to examine the resulting graft mass on ANC. Fig. 9 shows AAm with ANC × 4 had the lowest mass loss, indicating the lowest graft mass on ANC, whereas AAm with ANC × 2 had the highest mass loss, confirming the conversion results from Fig. 8. This indicates that ANC × 2 is the best concentration for PAAm to achieve a combination of grafting mass on the surface of ANC whilst maintaining high conversion.
 | ||
| Fig. 9 TGA and DTG profiles for (a) AMPS and (b) AAm in the presence of GOx at varying amine nanoceria (ANC) concentrations. | ||
Similar results were for found for AMPS (Fig. 9).
Double network hydrogels
DNHGs were successfully synthesised with PAMPS/ANC (PAMPS 0.24 M) in the first network and PAAm/ANC (PAAm 2.54 M) in the second network. Both polymer–ANC networks were synthesised in the presence of GOx. The final materials are referred to as ANC gels. Fig. 10 shows PAMPS/ANC after synthesis and the ANC gel after sequential addition of PAAm, in its hydrated state. Residual amounts of ANC and monomer were left over after swelling the first network in a solution of AAm/ANC, due to the saturation of the first network. Residues on the surface were washed off post synthesis and during swelling. | ||
| Fig. 10 Images of (a) PAMPS/ANC first network hydrogel after synthesis, and (b) double network hydrogel after sequential synthesis containing PAMPS and PAAm. | ||
ANC gels were swollen from a dry state until a plateau in water uptake was reached. Although the samples were heavily cross linked, they were still able to achieve 87.38 ± 2.35% water uptake. A plateau was reached at 72 h with small incremental changes at 168 h. The majority of the water uptake occurred within the first 8 h. ANC gels exhibited swelling of 811 ± 151%. Swelling was considered relatively low in comparison with water uptake, confirming the tightly cross linked hydrophilic nature of the material.
ANC gels were freeze dried for 48 h and imaged using SEM (Fig. 11). SEM imaging revealed thick struts within the material that surrounded pore-like structures. Further examination of the struts and inner walls of the material showed that ANC particles were present within the structure. ANC was predominantly found within exposed structures rather on the surface of the material. This suggests that ANC particles were covered with polymers due to the graft polymerisation during synthesis.
 | ||
| Fig. 11 SEM images of amine nanoceria (ANC) hydrogels with PAMPS and PAAm polymer grafts at varying magnifications. ANC are highlighted with white arrows. | ||
Gels with perfectly flat surfaces were difficult to synthesise due to the swelling nature of the material, instead, concave surfaces were formed (Fig. 10). Mechanical testing was conducted on five ANC gels (Fig. 12) and the shape of the compression curves was typical for hydrogels. A fracture compressive stress of 1.76 ± 0.10 MPa was recorded with strain of 60.9 ± 1.6% (Fig. 12). These values are comparable to DNHGs with 50 nm and 150 nm diameter silica nanoparticles in their first network synthesised in the presence of GOx, with a range of compressive strength of 2.6–3.6 MPa.23 The ANC gels showed significant improved properties compared to the control DNHG (PAMPS/PAAm without ANC/GOx), which had a fracture compressive stress of 0.10 ± 0.06 MPa and strain of 45.9 ± 2.1%. This increase in mechanical integrity can be attributed to the formation of a more integrated nanoparticle–polymer matrix, within both networks, that is lacking in the control. The complex structure allows the material to withstand increased stress without breaking up to 60% strain. It is also more likely that the polymers are better integrated into the ANC due to surface graft polymerisation, as opposed to the previous studies with of polymer chains cross linked by chemical cross linkers.5–7,12,17,55,56 This provides stronger reinforcement in the material against compressive stresses. The ability of the material to withstand up to 60% strain is likely due to the dense polymeric networks sharing the load due to their covalent links with ANC, compared to control gels without ANC. This is likely due to the nanocomposite structure of the material where both networks are composed of polymer networks/ANC, as opposed to only the first network containing nanoparticles in previous studies, using silica nanoparticles, and compared to controls that are synthesised without nanoparticles.11 A higher density of ANC will also be in contact with the polymers due to the small diameter of ANC, resulting in increased specific surface area for polymer grafting. Physical entanglements or bridge cross linking within PAMPS/ANC and PAAm/ANC is also possible. These combinations would all have benefited the ANC gels resulting in the improved mechanical strength, and provide insight into the potential tailorability of this material.
 | ||
| Fig. 12 Compression curves for the double network amine nanoceria hydrogel (ANC gel) with PAAm (2.45 M) and PAMPS (0.24 M) polymer grafts. 5 repeats are shown (ANC gels 1–5), compared to a control DNHG without ANC/GOx at the same polymer concentrations of both networks. Insert shows ANC gel at 0% and 60% compression moments before fracture. | ||
Conclusion
A first of its kind nanocomposite double network hydrogel consisting of two contrasting polymers grafted on amine functionalised nanoceria was successfully synthesised. This was achieved through a novel autocatalytic redox polymerisation involving glucose oxidase and nanoceria. GOx was used as a degassing agent and nanoceria as a polymerisation mediator and enhancer. PAMPS and PAAm were both successfully grafted on the surface of ANC with significantly improved conversion profiles with the addition of GOx, reaching 90% conversion. Ultimately, CAN and SDS were made redundant in the synthesis route due to the combined efficacy of ANC and GOx at promoting graft polymerisation. A strong double network hydrogel with compressive fracture stress of 1.8 MPa at 60% strain, with 87% water content, was synthesised based on the optimal concentration of ANC for both polymers, in combination with GOx. The compressive strength was an order of magnitude higher than that of the equivalent gel made without ANC and GOx. The ANC gels fall into the range of compressive strengths of articular cartilage. As these gels are intended for cartilage repair applications, future work will explore compression under various cyclical strains using dynamic mechanical analysis for comparison with the cyclic loading exhibited by natural cartilage. This novel material and new synthesis method can pave the way to tailored biomaterials suitable for varying properties, by controlling ANC and monomer concentrations.Author contributions
The manuscript was written with contributions from all authors. Dr Alessandra Pinna assisted in nanoceria synthesis, Dr Siwei Li assisted in glucose oxidase aliquots, and Dr Tian Sang assisted in mechanical testing and TGA. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is supported by Qatar Foundation, and EPSRC grant (EP/I020861/1). Raw data can be obtained on request from http://rdm-enquiries@imperial.ac.uk.References
- M. L. Oyen, Int. Mater. Rev., 2014, 59, 44–59 CrossRef CAS.
- D. J. Huey, J. C. Hu and K. A. Athanasiou, Science, 2012, 338, 917–921 CrossRef CAS PubMed.
- N. K. Simha, C. S. Carlson and J. L. Lewis, J. Mater. Sci.: Mater. Med., 2004, 15, 631–639 CrossRef CAS PubMed.
- C. Madeira, A. Santhagunam, J. B. Salgueiro and J. M. Cabral, Trends Biotechnol., 2015, 33, 35–42 CrossRef CAS PubMed.
- M. A. Haque, T. Kurokawa and J. P. Gong, Polymer, 2012, 53, 1805–1822 CrossRef CAS.
- S. Liang, Q. M. Yu, H. Yin, Z. L. Wu, T. Kurokawa and J. P. Gong, Chem. Commun., 2009, 7518–7520, 10.1039/B916581A.
- W. Yang, H. Furukawa and J. P. Gong, Adv. Mater., 2008, 20, 4499–4503 CrossRef CAS.
- J. Hu, K. Hiwatashi, T. Kurokawa, S. M. Liang, Z. L. Wu and J. P. Gong, Macromolecules, 2011, 44, 7775–7781 CrossRef CAS.
- Y.-H. Na, T. Kurokawa, Y. Katsuyama, H. Tsukeshiba, J. P. Gong, Y. Osada, S. Okabe, T. Karino and M. Shibayama, Macromolecules, 2004, 37, 5370–5374 CrossRef CAS.
- R. Takahashi, K. Shimano, H. Okazaki, T. Kurokawa, T. Nakajima, T. Nonoyama, D. R. King and J. P. Gong, Adv. Mater. Interfaces, 2018, 5, 1801018 CrossRef.
- J. P. Gong, Soft Matter, 2010, 6, 2583–2590 RSC.
- J. P. Gong, Y. Katsuyama, T. Kurokawa and Y. Osada, Adv. Mater., 2003, 15, 1155–1158 CrossRef CAS.
- M. Ogawa, N. Kitamura, T. Kurokawa, K. Arakaki, Y. Tanaka, J. P. Gong and K. Yasuda, J. Biomed. Mater. Res., Part A, 2012, 100, 2244–2251 Search PubMed.
- P. A. Levett, D. W. Hutmacher, J. Malda and T. J. Klein, PLoS One, 2014, 9, e113216 CrossRef PubMed.
- N. Asadi, E. Alizadeh, R. Salehi, B. Khalandi, S. Davaran and A. Akbarzadeh, Artif. Cells, Nanomed., Biotechnol., 2018, 46, 465–471 CrossRef CAS PubMed.
- M. Biondi, A. Borzacchiello, L. Mayol and L. Ambrosio, Gels, 2015, 1, 162–178 CrossRef.
- L. Weng, A. Gouldstone, Y. Wu and W. Chen, Biomaterials, 2008, 29, 2153–2163 CrossRef CAS PubMed.
- G. Gao, G. Du, Y. Cheng and J. Fu, J. Mater. Chem. B, 2014, 2, 1539–1548 RSC.
- K. Haraguchi and H.-J. Li, Macromolecules, 2006, 39, 1898–1905 CrossRef CAS.
- C.-J. Wu, A. K. Gaharwar, P. J. Schexnailder and G. Schmidt, Materials, 2010, 3, 2986–3005 CrossRef CAS.
- A. K. Gaharwar, C. Rivera, C.-J. Wu, B. K. Chan and G. Schmidt, Mater. Sci. Eng., C, 2013, 33, 1800–1807 CrossRef CAS PubMed.
- Q. Wang, R. Hou, Y. Cheng and J. Fu, Soft Matter, 2012, 8, 6048–6056 RSC.
- A. A. Mohammed, J. A. Milan, S. Li, J. J. Chung, M. M. Stevens, T. K. Georgiou and J. R. Jones, J. Mater. Chem. B, 2019, 7, 4030–4039, 10.1039/C9TB00658C.
- T. von Werne and T. E. Patten, J. Am. Chem. Soc., 1999, 121, 7409–7410 CrossRef CAS.
- A. El Harrak, G. Carrot, J. Oberdisse, J. Jestin and F. Boué, Polymer, 2005, 46, 1095–1104 CrossRef CAS.
- T. Wu, Y. Zhang, X. Wang and S. Liu, Chem. Mater., 2008, 20, 101–109 CrossRef CAS.
- S. Kim, E. Kim, S. Kim and W. Kim, J. Colloid Interface Sci., 2005, 292, 93–98 CrossRef CAS PubMed.
- P. Salarizadeh, M. Abdollahi and M. Javanbakht, Iran. Polym. J., 2012, 21, 661–668 CrossRef CAS.
- E. Grulke, K. Reed, M. Beck, X. Huang, A. Cormack and S. Seal, Environ. Sci.: Nano, 2014, 1, 429–444 RSC.
- K. Reed, A. Cormack, A. Kulkarni, M. Mayton, D. Sayle, F. Klaessig and B. Stadler, Environ. Sci.: Nano, 2014, 1, 390–405 RSC.
- A. S. Karakoti, N. A. Monteiro-Riviere, R. Aggarwal, J. P. Davis, R. J. Narayan, W. T. Self, J. McGinnis and S. Seal, JOM, 1989, 2008(60), 33–37 Search PubMed.
- C. Loschen, A. Migani, S. T. Bromley, F. Illas and K. M. Neyman, Phys. Chem. Chem. Phys., 2008, 10, 5730–5738 RSC.
- R. Guo, Y. Wang, S. Yu, W. Zhu, F. Zheng, W. Liu, D. Zhang and J. Wang, RSC Adv., 2016, 6, 59939–59945 RSC.
- V. Kalyanaraman, S. V. Naveen, N. Mohana, R. M. Balaje, K. R. Navaneethakrishnan, B. Brabu, S. S. Murugan and T. S. Kumaravel, Toxicol. Res. Appl., 2019, 8, 25–37 CrossRef CAS PubMed.
- L. Falchi, L. Bogliolo, G. Galleri, F. Ariu, M. T. Zedda, A. Pinna, L. Malfatti, P. Innocenzi and S. Ledda, Theriogenology, 2016, 85, 1274–1281.e1273 CrossRef CAS PubMed.
- F. Ariu, L. Bogliolo, A. Pinna, L. Malfatti, P. Innocenzi, L. Falchi, D. Bebbere and S. Ledda, Reprod., Fertil. Dev., 2017, 29, 1046–1056 CrossRef CAS PubMed.
- L. Falchi, G. Galleri, G. M. Dore, M. T. Zedda, S. Pau, L. Bogliolo, F. Ariu, A. Pinna, S. Nieddu, P. Innocenzi and S. Ledda, Reprod. Biol. Endocrinol., 2018, 16, 19 CrossRef PubMed.
- J. S. Temenoff, H. Shin, D. E. Conway, P. S. Engel and A. G. Mikos, Biomacromolecules, 2003, 4, 1605–1613 CrossRef CAS PubMed.
- T. S. Wilems, X. Lu, Y. E. Kurosu, Z. Khan, H. J. Lim and L. A. Smith Callahan, J. Biomed. Mater. Res., Part A, 2017, 105, 3059–3068 CrossRef CAS PubMed.
- U. Taylor, D. Tiedemann, C. Rehbock, W. A. Kues, S. Barcikowski and D. Rath, Beilstein J. Nanotechnol., 2015, 6, 651–664 CrossRef CAS PubMed.
- I. Celardo, J. Z. Pedersen, E. Traversa and L. Ghibelli, Nanoscale, 2011, 3, 1411–1420 RSC.
- E. G. Heckert, A. S. Karakoti, S. Seal and W. T. Self, Biomaterials, 2008, 29, 2705–2709 CrossRef CAS PubMed.
- R. Chapman, A. J. Gormley, K.-L. Herpoldt and M. M. Stevens, Macromolecules, 2014, 47, 8541–8547 CrossRef CAS.
- C. Lv, W. Di, Z. Liu, K. Zheng and W. Qin, Analyst, 2014, 139, 4547–4555 RSC.
- T. J. Sworski, H. A. Mahlman and R. W. Matthews, J. Phys. Chem., 1971, 75, 250–255 CrossRef CAS.
- A. Pinna, C. Figus, B. Lasio, M. Piccinini, L. Malfatti and P. Innocenzi, ACS Appl. Mater. Interfaces, 2012, 4, 3916–3922 CrossRef CAS PubMed.
- A. Pinna, L. Malfatti, G. Galleri, R. Manetti, S. Cossu, G. Rocchitta, R. Migheli, P. A. Serra and P. Innocenzi, RSC Adv., 2015, 5, 20432–20439 RSC.
- F. Caputo, M. Mameli, A. Sienkiewicz, S. Licoccia, F. Stellacci, L. Ghibelli and E. Traversa, Sci. Rep., 2017, 7, 4636 CrossRef PubMed.
- A. A. Ansari, P. R. Solanki and B. D. Malhotra, J. Biotechnol., 2009, 142, 179–184 CrossRef CAS PubMed.
- C. S. Newby, R. M. Barr, M. W. Greaves and A. I. Mallet, J. Invest. Dermatol., 2000, 115, 292–298 CrossRef CAS PubMed.
- J. Zhang, X. Xie, C. Li, H. Wang and L. Wang, RSC Adv., 2015, 5, 29757–29765 RSC.
- N. Kitamura, K. Yasuda, M. Ogawa, K. Arakaki, S. Kai, S. Onodera, T. Kurokawa and J. P. Gong, Am. J. Sports Med., 2011, 39, 1160–1169 CrossRef PubMed.
- O. Stoilova, N. Koseva, N. Manolova and I. Rashkov, Polym. Bull., 1999, 43, 67–73 CrossRef CAS.
- S. R. S. Ting, J. M. Whitelock, R. Tomic, C. Gunawan, W. Y. Teoh, R. Amal and M. S. Lord, Biomaterials, 2013, 34, 4377–4386 CrossRef CAS PubMed.
- K. Yasuda, J. Ping Gong, Y. Katsuyama, A. Nakayama, Y. Tanabe, E. Kondo, M. Ueno and Y. Osada, Biomaterials, 2005, 26, 4468–4475 CrossRef CAS PubMed.
- A. Nakayama, A. Kakugo, J. P. Gong, Y. Osada, M. Takai, T. Erata and S. Kawano, Adv. Funct. Mater., 2004, 14, 1124–1128 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2020 |