
Stretchable and tough conductive hydrogels for flexible pressure and strain sensors
 ab
Jun
Fu
*a
ab
Jun
Fu
*a
Abstract
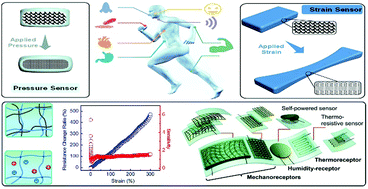
Flexible pressure and strain sensors have great potential for applications in wearable and implantable devices, soft robotics and artificial skin. Compared to flexible sensors based on filler/elastomer composites, conductive hydrogels are advantageous due to their biomimetic structures and properties, as well as biocompatibility. Numerous chemical and structural designs provide unlimited opportunities to tune the properties and performance of conductive hydrogels to match various demands for practical applications. Many electronically and ionically conductive hydrogels have been developed to fabricate pressure and strain sensors with different configurations, including resistance type and capacitance type. The sensitivity, reliability and stability of hydrogel sensors are dependent on their network structures and mechanical properties. This review focuses on tough conductive hydrogels for flexible sensors. Representative strategies to prepare stretchable, strong, tough and self-healing hydrogels are briefly reviewed since these strategies are illuminating for the development of tough conductive hydrogels. Then, a general account on various conductive hydrogels is presented and discussed. Recent advances in tough conductive hydrogels with well designed network structures and their sensory performance are discussed in detail. A series of conductive hydrogel sensors and their application in wearable devices are reviewed. Some perspectives on flexible conductive hydrogel sensors and their applications are presented at the end.
- This article is part of the themed collections: 2020 Journal of Materials Chemistry B most popular articles, Journal of Materials Chemistry B Lunar New Year collection 2021 and Biosensors
 Please wait while we load your content...
Please wait while we load your content...

