
Modulating the properties of flow-assembled chitosan membranes in microfluidics with glutaraldehyde crosslinking
Piao
Hu
a,
Christopher B.
Raub
 b,
John S.
Choy
c and
Xiaolong
Luo
*a
b,
John S.
Choy
c and
Xiaolong
Luo
*a
aDepartment of Mechanical Engineering, The Catholic University of America, Washington, D.C. 20064, USA. E-mail: luox@cua.edu; Tel: +1 202 319 6952
bDepartment of Biomedical Engineering, The Catholic University of America, Washington, D.C. 20064, USA
cDepartment of Biology, The Catholic University of America, Washington, D.C. 20064, USA
First published on 14th February 2020
Abstract
Flow-assembled chitosan membranes are robust and semipermeable hydrogel structures formed in microfluidic devices that have been used for important applications such as gradient generation and studying cell–cell signaling. One challenge, however, remains unresolved. When a polydimethylsiloxane (PDMS) microchannel with a flow-assembled, deprotonated chitosan membrane (DCM) is treated with anti-adhesion agents such as Pluronic F-127 to prevent biomolecular and cellular adsorption on PDMS, the interaction between DCM and PDMS is compromised and the DCM easily delaminates. To address this challenge, DCMs in microfluidics are crosslinked with glutaraldehyde to modulate their properties, and the altered properties of the glutaraldehyde treated chitosan membrane (GTCM) are investigated. First, the GTCM's acidic resistance was confirmed, its mechanical robustness against hydrostatic pressure was significantly improved, and it remained intact on PDMS after Pluronic treatment. Second, crystallization in DCM and GTCM was investigated with quantitative polarized light microscopy (qPLM), which revealed that GTCM's optical retardance and anisotropy were lower, implying less molecular alignment than in DCM. Finally, membrane permeability was tested with FITC-labeled dextran transport experiments, which showed that the transport across GTCM was slightly higher than that across DCM. Overall, glutaraldehyde-crosslinked chitosan membrane has better acidic resistance, higher strength under Pluronic treatment, and less molecular microalignment, while its semi-permeability is retained. This study demonstrates how glutaraldehyde crosslinking can be used to modify and improve biopolymer membrane properties for broader applications, such as in an acidic environment or when Pluronic passivation is needed.
Introduction
Chitosan is a linear cationic polysaccharide that can be industrially obtained by deacetylation of chitin. Chitin, a derivative of glucose and a primary component of exoskeletons of arthropods like crabs and shrimps, is the second most plentiful natural biopolymer after cellulose. The presence of active amine groups in chitosan on its deacetylated glucosamine units endues its solubility and reactivity greater than chitin and cellulose, and promotes its applications ranging from agricultural seed treatment1 to a biomedical drug carrier2 to a biological semi-permeable membrane.3 Chitosan as a good biomaterial has been widely used in the form of a membrane structure in medical and tissue engineering as a scaffold,4 wound dressing3 and bone regeneration guide5 among many others.6–8 The pKa value of chitosan is around 6.3, which means it is soluble in aqueous solution with pH lower than 6.3, while the amine groups will be deprotonated and it becomes insoluble at higher pH, as shown in Fig. 1(a-i). By taking this characteristic of chitosan, we have conveniently biofabricated deprotonated chitosan membrane (DCM), as depicted in Fig. 1(a-ii), with locally generated pH gradients between converging flows in polydimethylsiloxane (PDMS) microfluidic devices.9 In an improved membrane biofabrication strategy, a polyelectrolyte complex membrane (PECM) was spontaneously formed when the negatively charged carboxyl groups on alginate and positively charged amine groups on chitosan were brought into contact in a microchannel network.10 A chitosan membrane was then grown to the desired thickness with a local pH gradient between parallel flows along the PECM, as recapitulated in Fig. 2(b). The biofabrication process has been adopted by other researchers with extra care in pumping the solutions into microchannels.11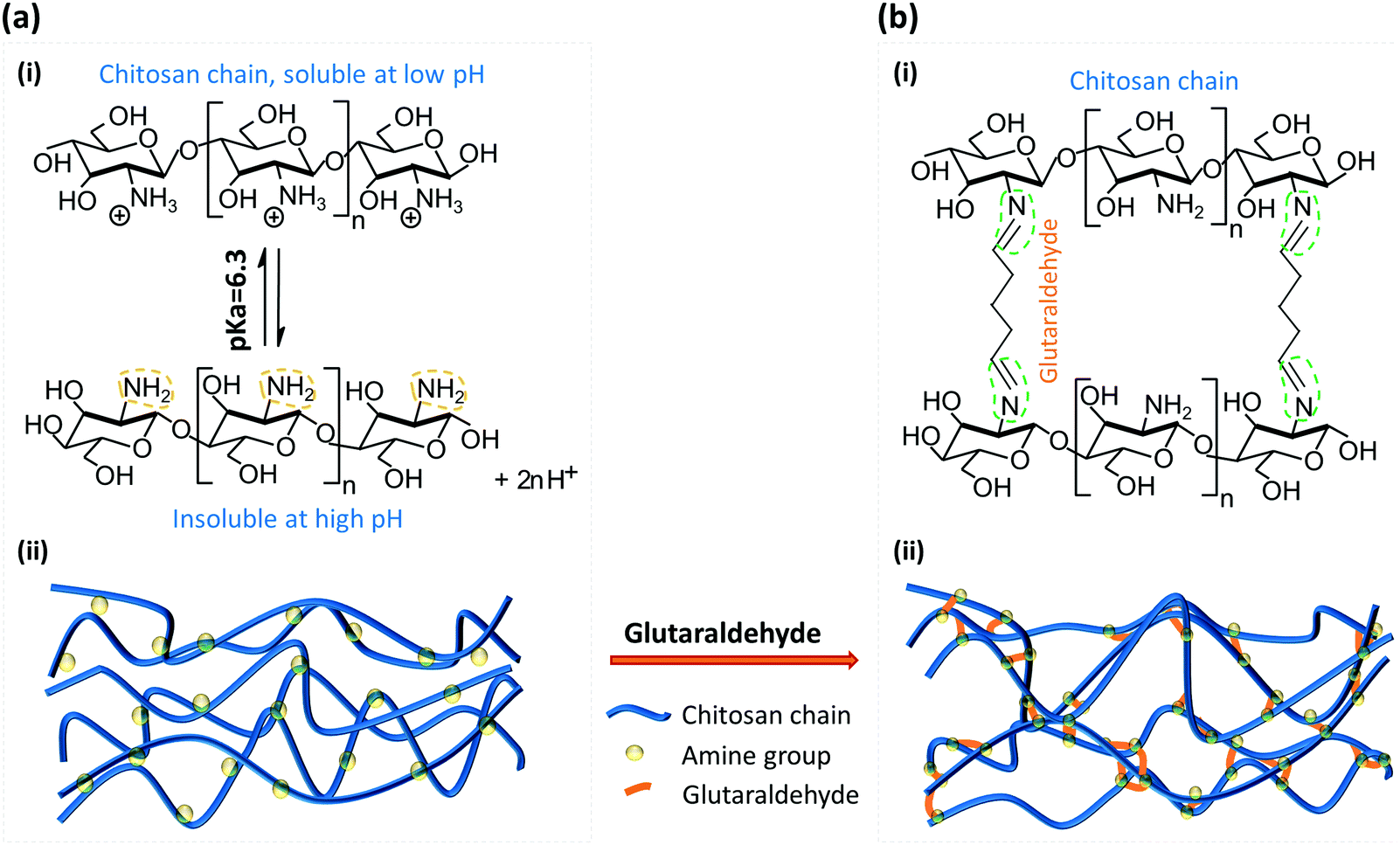 | ||
| Fig. 1 Molecular schematic of the deprotonated chitosan membrane (DCM) cross-linked with glutaraldehyde. (a)-(i) Transition between protonated (soluble) and deprotonated (insoluble) chitosan around its pKa value 6.3; (ii) molecular structure of DCM with deprotonated amine groups. (b)-(i) The most commonly recognized molecular reaction between amine groups on DCM and glutaraldehyde (GA) molecules; and (ii) molecular structure of glutaraldehyde treated chitosan membrane (GTCM) with GA linking between amine groups on nearby chitosan chains or within one chitosan chain. | ||
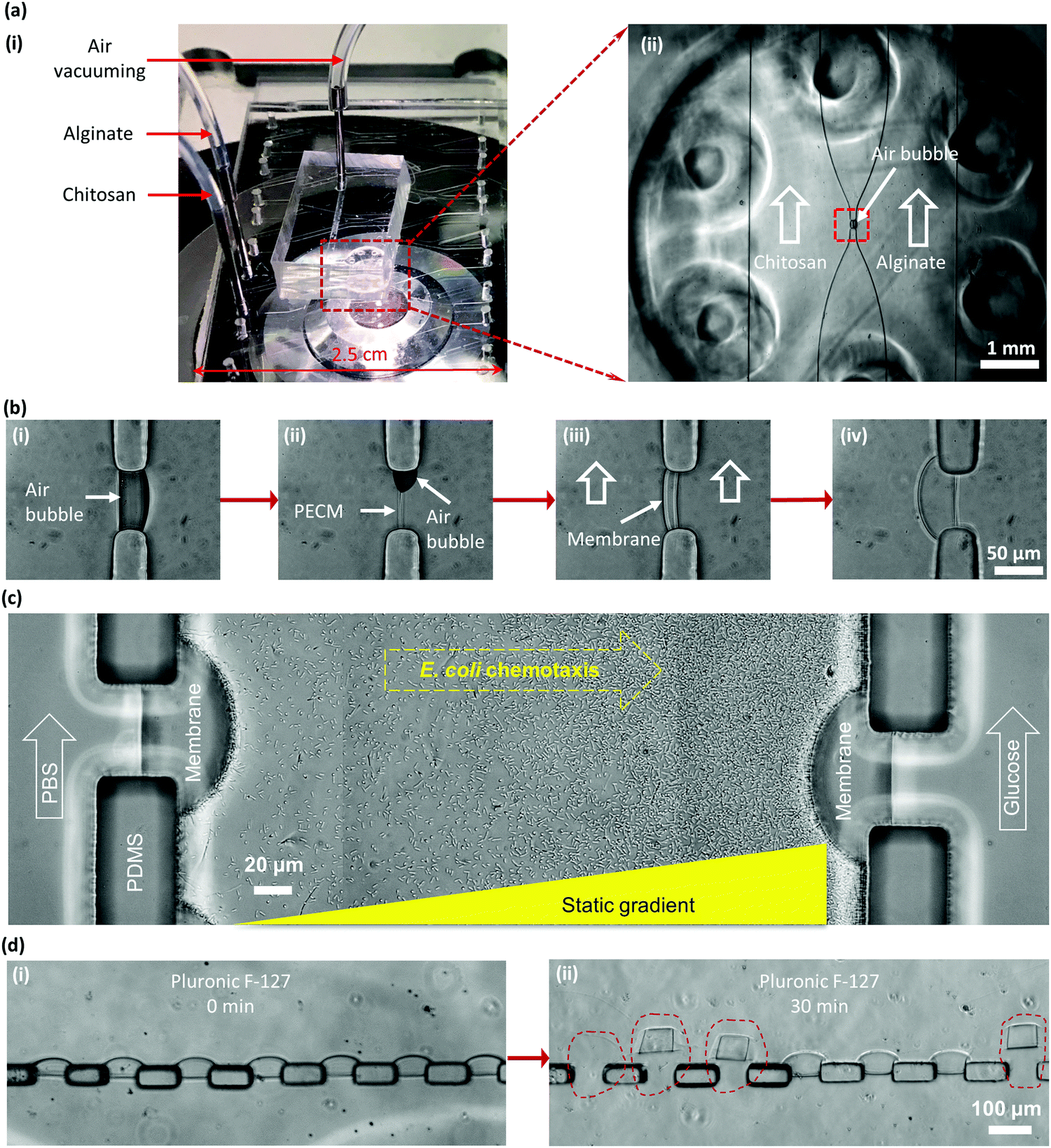 | ||
| Fig. 2 Biofabrication of the DCM in a PDMS microfluidic device and the complication of DCMs in PDMS microchannels with Pluronic treatment. (a)-(i) A PDMS microfluidic device with an add-on air vacuum chamber on an inverted microscope stage; and (ii) zoom-in view of the device with air bubbles naturally trapped in an aperture between chitosan and alginate solutions in two connected microchannels. (b)-(i) Trapped air bubbles; (ii) spontaneous formation of the polyelectrolyte complex membrane (PECM) between positively charged chitosan and negatively charged alginate; (iii) growth of the DCM on the chitosan side of the PECM with the continuous pumping of both solutions; and (iv) DCM grown to a desired thickness. (c) A representative application of DCMs to generate static gradients of glucose in the middle channel with continuous flows of sink and source solutions (1 mM) on the left and right microchannels, correspondingly, to study E. coli chemotaxis. With appropriate treatment such as with Pluronic F-127 of the PDMS microchannel, bacteria did not adhere to the channel surfaces and swam toward a glucose source on the right. (d)-(i) Soaking a device with an array of 7 DCMs in 0.1% (w/v) Pluronic F-127 in PBS for 30 minutes to minimize protein and cell adhesion; and (ii) DCMs easily detached from PDMS pillars after Pluronic treatment. | ||
The biofabricated chitosan membranes in microfluidics are freestanding, robust and semipermeable with both sides being accessible to flows.10 The average pore size of the biofabricated chitosan membrane was found to be around 7–10 nanometers that allows small molecules to diffuse freely through while stopping big macromolecules such as antibodies. These unique features have presented the biofabricated chitosan membrane as an important platform for applications including static gradient generation,9 yeast chemotropism,12 and cell–cell signalling in constructed synthetic ecosystems.13 Bacterial chemotaxis shown in Fig. 2(c) is another representative application of the semi-permeable DCMs. Here, a static gradient of glucose was generated in the middle microchannel separated by DCMs from the left and right microchannels with continuous flows of sink and source solutions, respectively. In a recent report, chitosan membranes connecting individually addressable side microchannels to a common space facilitated the construction of synthetic biofilms to study multi-kingdom interactions.14
One challenge, however, remains unresolved. When the PDMS microchannels were treated with anti-adhesion agents such as Pluronic F-127 for passivation to prevent biomolecular and cellular adsorption, for example in chemotaxis study, as shown in Fig. 2(c), the originally strong interaction between the chitosan membrane and the PDMS surface was greatly compromised and the DCM easily delaminated from PDMS, as shown in Fig. 2(d). Apparently, the amphiphilic tri-block copolymer Pluronic F-127 disrupted the adhesion of the deprotonated chitosan on the PDMS surface. This research aims to address this challenge by improving DCM's properties with glutaraldehyde (GA) crosslinking to broaden the use of the freestanding, semipermeable biopolymer membranes.
Glutaraldehyde was first used to immobilize proteins such as enzymes, where the crosslinking was achieved between the aldehyde groups on GA and amine groups on proteins.15,16 As a common crosslinking agent, GA has been used to cross-link chitosan for various applications,17,18 and the mechanism and properties of GA cross-linked chitosan have been a subject of great interest. Fig. 1(b) depicts one of the most recognized molecular reaction between the amine groups and aldehyde ends. Most studies focused on the cross-linking of chitosan with hydrogels or beads and the corresponding properties.17–19 The cross-linked chitosan was homogeneous with different porosity. Few researchers, however, have studied the mechanism of GA reacting with solidified chitosan chains.20 The flow-assembled DCM was molecularly well-aligned in the direction of flow, as demonstrated in our previous report with a high birefringence signal using quantitative polarized light microscopy (qPLM).21 The crosslinking of well-aligned chitosan membranes assembled with flow is yet to be investigated.
This paper examines the change in properties of the flow-assembled DCM when it is cross-linked with GA in order to broaden its applications in microfluidics. The membrane characterization was performed from the aspects of acid resistance, mechanical robustness, molecular microalignment and semi-permeability. The study sheds light on the GA-chitosan reaction mechanism and provides insight to modulate the biopolymer membrane properties for broader applications.
Results and discussion
Acidic resistance
After soaking DCM in 10% GA in PBS solution for one hour (Fig. 3(a)) to convert DCM into GTCM, the first obvious property change is its improved acidic resistance. Apparently, both DCM formation and maintenance are sensitive to the solution pH since chitosan is soluble in solutions with pH lower than its pKa around 6.3, as shown in Fig. 1(a). The fact that DCM cannot be sustained in an acidic environment is shown in Fig. 3(b)-(i) and (ii). When a HCl solution of pH 2 was introduced into the microchannel with DCM at a flow rate of 10 μL min−1, the DCM quickly dissolved and lost more than half of its original thickness within 20 seconds. On the other hand, there was no thickness reduction or observable swelling in GTCM after one hour under the same conditions, as shown in Fig. 3(b)-(iii) and (iv), and this remained the same for hours (results not shown). These results suggest that GA crosslinked the amine groups on the deprotonated chitosan chains and made the GTCM resistant to acidic erosion, which can broaden the applications of chitosan membrane to tolerate acidic environments.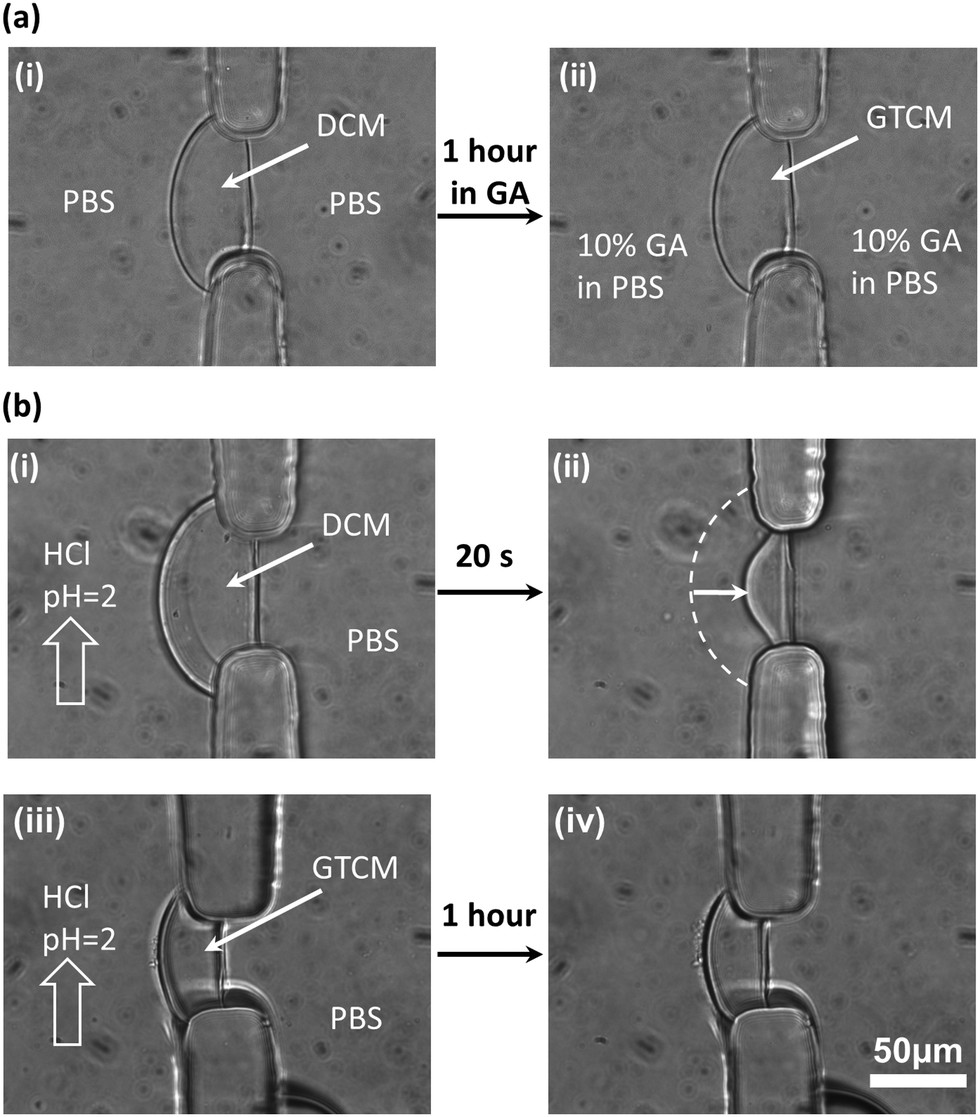 | ||
| Fig. 3 GA crosslinking and acidic resistance of DCM and GTCM. (a) No obvious morphological changes were observed after soaking DCM in 10% GA in PBS for one hour to convert it into GTCM. (b)-(i) & (ii) DCM was dissolved by HCl (pH = 2) solution in seconds, while (iii) & (iv) no change in the GTCM was observed under the same condition for one hour. | ||
Most previous studies17–19 have focused on the mechanism, properties and applications of crosslinking between GA and chitosan under a homogeneous condition, by dropping GA into a chitosan solution to form chitosan beads. However, no research has explored how GA alters the properties of a well-aligned chitosan membrane prefabricated in a microchannel. The equilibrium structures of GA in aqueous solution are complicated and strongly depend on manufacturing and environmental conditions such as temperature and pH,19 because of which the GA structure is still under debate. It is likely that there are several crosslinking mechanisms between GA and chitosan membranes. One of the most acknowledged crosslinking mechanisms22 between GA and chitosan is shown in Fig. 1(b)-(i), and it is the conjugation between two reactants: aldehyde from GA and amine from chitosan. The observation that there is no obvious swelling or thickness reduction of GTCM under the acidic condition confirms that the aldehyde-linked amine groups [–NH2] are no longer free to convert back to the protonated form [–NH3+] in the presence of [H+]. Although a Schiff base is unstable under acidic conditions, the linkage between aldehyde and amine has been reported with exceptional stability at both extreme pH and temperature, which means that the Schiff base can resist hydrolysis as long as both ends of GA have reacted with amines.23 Therefore, the GTCM can last for a long time under the acidic condition because of the chemically stable bond, and it can be used in an acidic environment such as that when creating an acidic chemorepellent gradient for chemotaxis study.
Mechanical robustness
The mechanical robustness of GTCMs was quantified with a simple pressure measurement strategy based on ideal gas law, as shown in Fig. 4(a). The critical pressure (CP) of a membrane refers to the maximum gage pressure that the membrane withstands before bursting. Fig. 4(b) shows a membrane before, during and immediately after the burst moment in a robustness test. The CP values measured in atmospheric pressure (atm) of individual DCM and GTCM with different thicknesses before and after Pluronic treatment are shown in the scatter plot in Fig. 4(c). We observed that membranes within each of the four categories have similar pressures with slight or no dependence on membrane thickness: (1) the CP of DCMs without Pluronic treatment (open circles) was 1.02 ± 0.07 atm; (2) the CP of GTCMs without Pluronic treatment (open triangles) was 1.07 ± 0.12 atm; (3) the CP of DCMs after Pluronic treatment (solid circles) was 0.16 ± 0.03 atm; and (4) the CP of GTCMs after Pluronic treatment (solid triangles) was 0.58 ± 0.08 atm. Pluronic treatment had obvious negative impacts on both DCMs and GTCMs. The CP values of DCMs decreased from 1.02 to 0.16 atm, while those of GTCMs decreased from 1.07 to 0.58 atm. Therefore, the GTCMs were much better at withstanding Pluronic treatment since their CP values decreased by 46% compared to a decrease of 84% for DCMs. Practically, a reduction of roughly half of the originally strong mechanical robustness of the GTCM after the Pluronic treatment does not compromise its use in PDMS microchannels, while a major reduction as for DCMs after Pluronic treatment almost destroys its use in microfluidic experiments.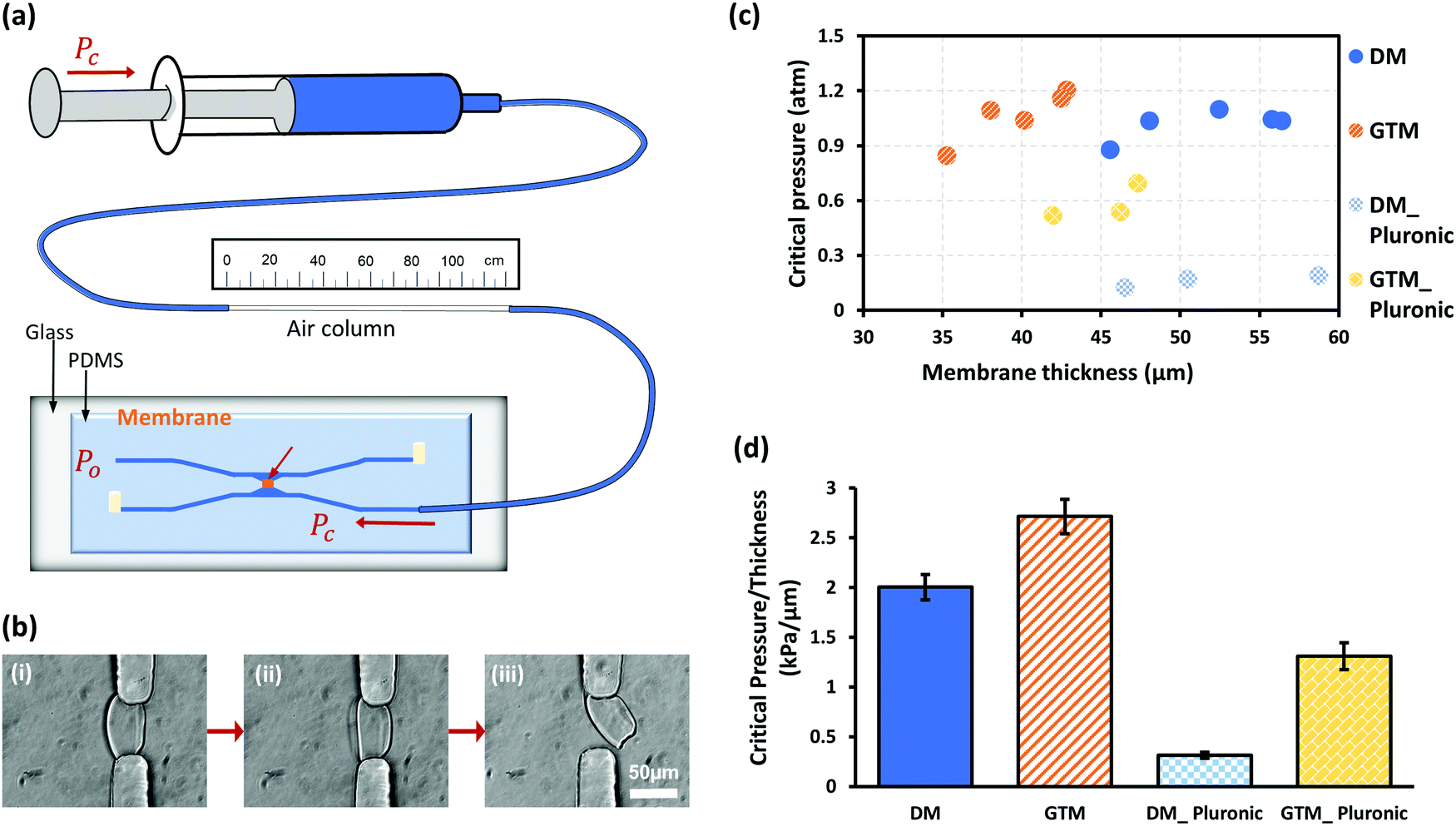 | ||
| Fig. 4 Mechanical robustness of DCM and GTCM. (a) Schematic of measurement setup based on Ideal Gas Law to measure the critical pressure to detach membranes from PDMS pillars. (b) Membrane (i) before, (ii) during and (iii) right at the burst moment during a robustness test. (c) Critical pressure of membranes when burst and those after Pluronic treatment. (d) Normalized critical pressure per μm thickness for different membranes. | ||
The slight or no dependence of CP on membrane thickness close to or higher than 40 μm can probably be explained by the anchoring area of membranes on PDMS. The width, length and height of the aperture where the DCM was biofabricated has the same dimension of about 50 μm. The anchoring area of membranes within the aperture presumably contributes most to the mechanical robustness. Considering the aperture dimension, membranes thicker than 40 μm probably have a similar anchoring area on PDMS as thicker membranes do not have a considerably bigger anchoring area on PDMS within the aperture.
The CP values in Fig. 4(c) were further normalized to their corresponding thickness and plotted in Fig. 4(d). The average strength of DCM was 2.00 kPa μm−1 while it was 2.71 kPa μm−1 for GTCM. After Pluronic F-127 passivation, the CP value of DCMs dropped to 0.31 kPa μm−1, a decrease of 85% compared to the untreated DCMs. The CP values of GTCMs decreased to 1.31 kPa μm−1 after Pluronic F-127 passivation, a decrease of 52% compared to the untreated GTCMs. Significantly, the normalized CP value of the Pluronic-treated GTCM is more than four times that of the Pluronic-treated DCM. Furthermore, the normalized CP value of the Pluronic treated GTCM is only 35% less than that of the untreated DCM. These results suggest that GA strengthens the robustness of DCM and offsets the weakening effect of Pluronic F-127 treatment.
Distribution of Pluronic F-127 in membranes
To better understand the large difference in the mechanical robustness between DCM and GTCM, the distribution of Pluronic F-127 in the chitosan membrane was investigated. Previous studies have reported that Pluronic F-127 coats on the surface of PDMS microfluidic channels,24 but the distribution of Pluronic F-127 in chitosan membranes is still unknown. Therefore, 5-DTAF labeled Pluronic F-127 was used as a probe to determine Pluronic F-127 distribution in DCMs and GTCMs. In Fig. 5(c), the bright field images (i) & (iii) on the left show the shape of DCM and GTCM, while the fluorescent images (ii) & (iv) on the right show the fluorescence distribution in DCM and GTCM after being soaked with 0.1% (w/v) fluorescent Pluronic F-127 for 1 hour and then rinsing with PBS. The results show a strong and relatively uniform fluorescence distribution in the DCM, while little fluorescence signal is observed in the GTCM. The fluorescence intensity profiles through the DCM and GTCM were analyzed and are plotted in Fig. 5(d). This result confirmed that fluorescence intensity through the DCM was much higher than the background (PDMS channels) and relatively uniform, which suggests that Pluronic F-127 molecules abundantly filled the DCM. Because the DCM was still partially positively charged with free amine groups on chitosan chains, negatively charged Pluronic F-127 molecules are likely attracted into the DCM. However, after GA treatment, most amine groups in the DCM are predicted to be crosslinked and neutralized by GA molecules, thus few Pluronic F-127 molecules are retained inside GTCM. Therefore, GTCM, as shown in Fig. 4(c)-(iv), is dim overall except for one bright spot presumably due to a fabrication defect.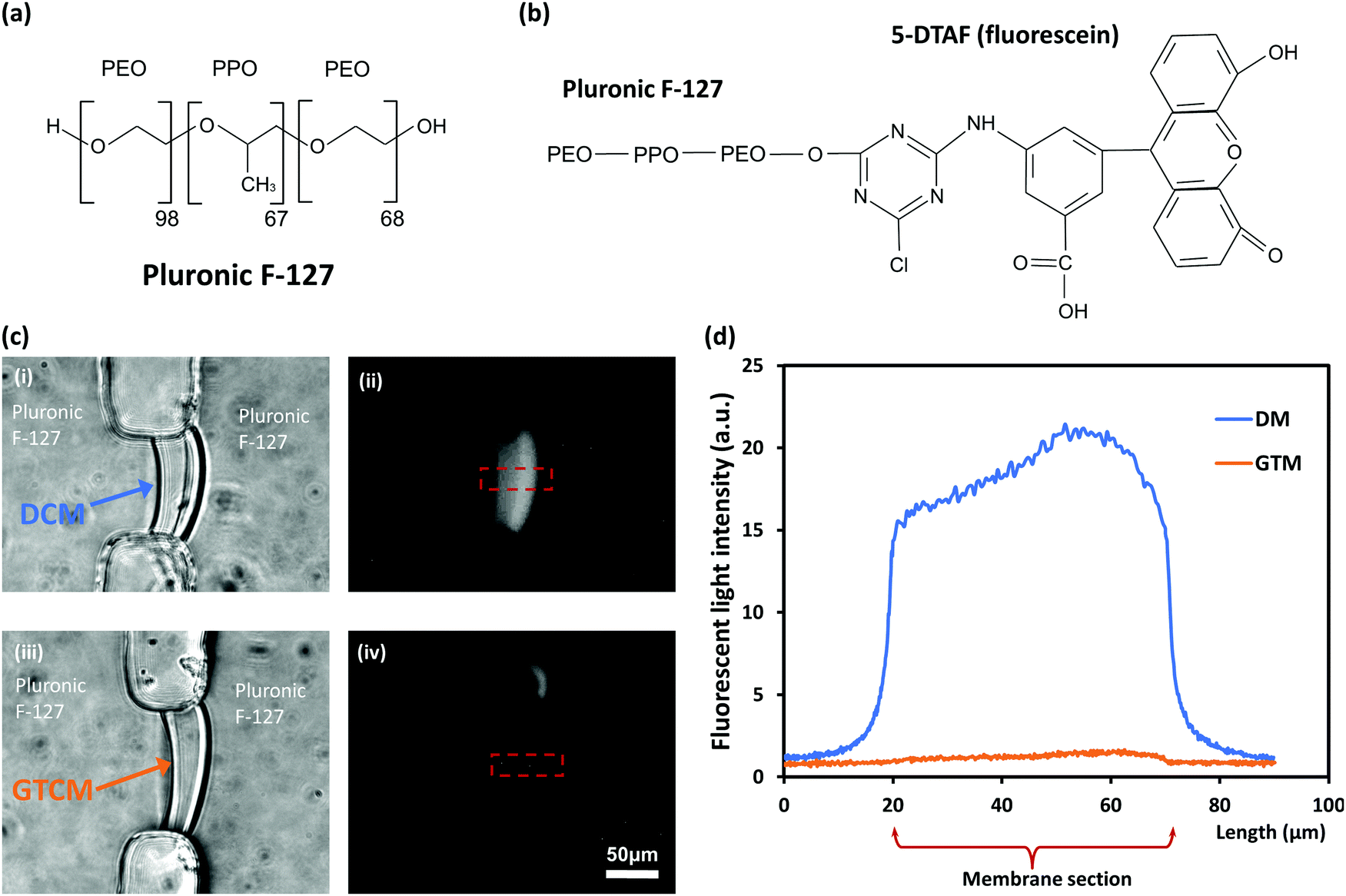 | ||
| Fig. 5 Interactions of Pluronic F-127 molecules with DCM and GTCM. (a) Chemical structures of tri-block copolymer Pluronic F-127. (b) Chemical structure of 5-DTAF (fluorescein) labeled Pluronic F-127. (c) After treating DCM (i) and GTCM (iii) with fluorescein-tagged Pluronic F-127 for one hour and thorough rinsing in PBS, a strong fluorescence signal was observed in DCM (ii) while minimum fluorescence signal was observed in GTCM (iv). (d) Profile plots of fluorescence intensity in DCM vs. GTCM of the plotted areas in (c)-(ii) & (iv). | ||
The PDMS surface is hydrophobic, while chitosan membranes in PBS are highly deprotonated, although a small percent of the amine groups are still charged, rendering the DCM slightly hydrophilic. The adhesion of DCM on PDMS is likely due to the high level of molecular micro-alignment within the DCM biofabricated using the flow assembly method. However, when abundant hydrophilic Pluronic F-127 populates the DCM, the interaction between PDMS and DCM is severely compromised. This may be a result of the amphiphilic tri-block copolymer Pluronic F-127 molecules disrupting the adhesion of the deprotonated chitosan on PDMS surfaces. GA crosslinking converts DCM into GTCM, which significantly alleviates the presence of Pluronic F-127 inside the membrane and thus the negative impact on the membrane's mechanical robustness. The crosslinked DCM retains the mechanical robustness of the membrane for practical applications, which are under investigation and will be reported in the near future.
Crystallization
The impact of GA crosslinking on the flow assembled chitosan membranes was further investigated regarding their molecular microstructural change. Birefringence and parallelism index (PI) measurements of DCM have previously been reported.21 The optical retardance (Γ) was determined from the birefringence signal for each pixel of the images by fitting the pixel signal versus analyzer angle to a second-order polynomial, determining the minimum and then generating an optical retardance map of the membrane. A higher optical retardance signal is related to a higher crystalline order in the membrane, with contributions from intrinsic and form birefringence. The optical retardance maps of a DCM and its corresponding GTCM after GA crosslinking are shown in Fig. 6(a)-(i) & (ii), while profiles of optical retardance across the membranes are plotted in Fig. 6(b). Net optical retardance was calculated with 5 trials of both DCM and GTCM, as shown in Fig. 6(d). These results show that the average net optical retardance for the DCM was 40 ± 7.8 nm, but it dropped to 22 ± 5.4 nm, which was a 45% drop in retardance after GA treatment. The paired sample t-test was used to find the statistically significant difference between DCM and GTCM in optical retardance and PI values. The p-value 6.7 × 10−5 in Fig. 6(d) referred to a significant difference between DCM and GTCM. The significant reduction of optical retardance confirmed that GA crosslinking altered the microstructure of the highly micro-aligned flow-assembled chitosan membrane, with less crystalline order than before crosslinking.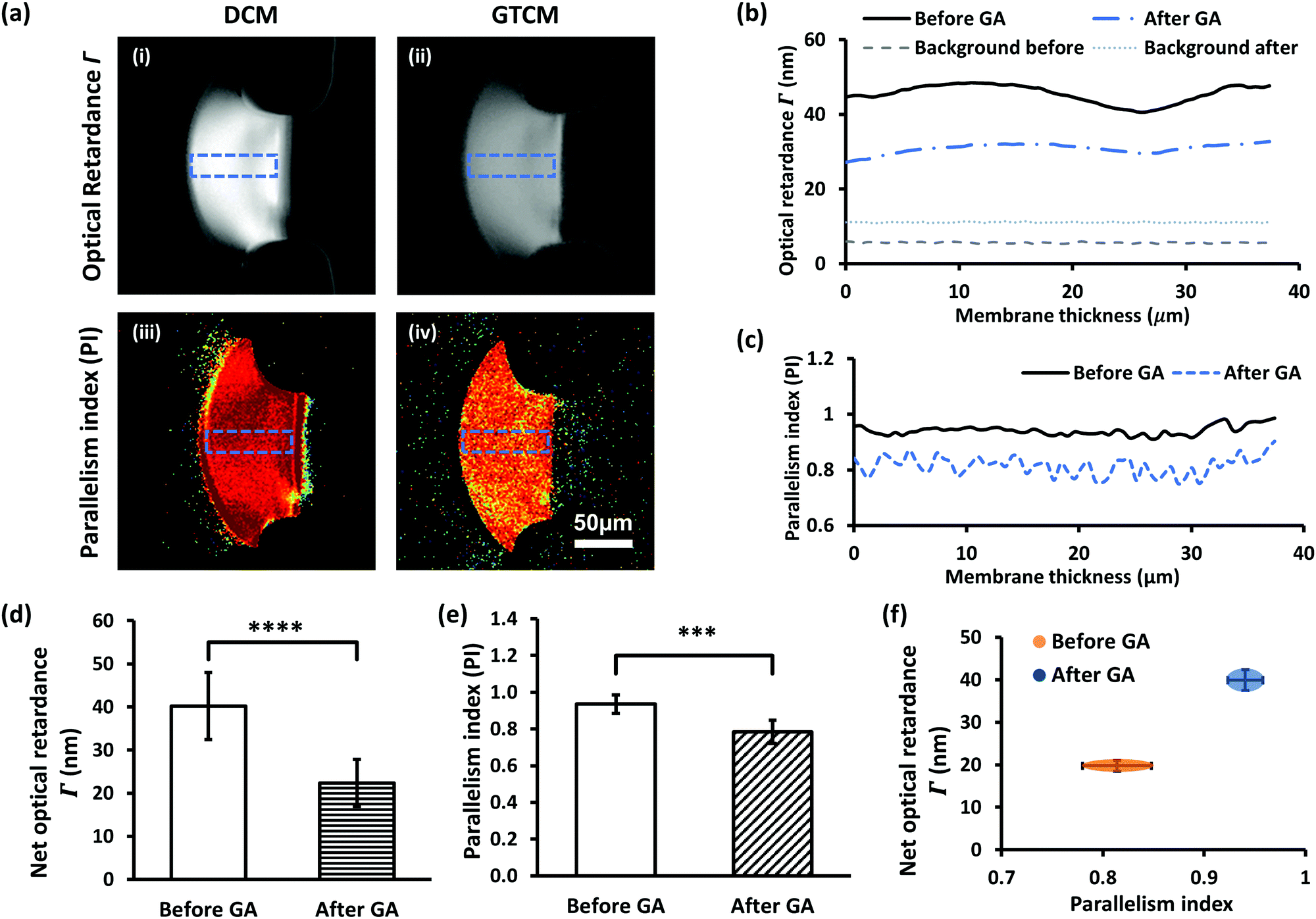 | ||
| Fig. 6 Birefringence of DCM and GTCM indicating different molecular micro-alignment levels inside the membranes. (a) Optical retardance Γ map of DCM (i) and GTCM (ii), and the parallelism index (PI) map of DCM (iii) and GTCM (iv) obtained via quantitative Polarized Light Microscopy (qPLM). (b) Optical retardance of DCM and GTCM through the indicated sections in (a). (c) Parallelism index of DCM and GTCM as in (a). (d) Optical retardance through DCM and GTCM. (e) Average PI through DCM and GTCM. (f) Separation of DCM and GTCM of birefringence on the Γ-PI map. *** indicates p < 0.05, and **** indicates p < 0.01. | ||
Furthermore, both the PI maps of DCM and GTCM, as shown in Fig. 6(a)-(iii) & (iv), and the profile of PI across the membranes, as shown in Fig. 6(c), showed obvious lower values in the membrane after GA treatment, a trend similar to retardance. The mean PI from 5 trials dropped from 0.94 ± 0.05 for DCMs to 0.78 ± 0.06 for the corresponding GTCMs after GA crosslinking, which is a significant change (p = 6.3 × 10−3) before and after GA treatment. The separation of DCM and GTCM in the two parameters Γ and PI, as shown in Fig. 6(f), clearly shows the effect of GA crosslinking on the membrane's molecular microstructure. Optical retardance and the parallelism index depend on the intrinsic and form birefringence of single chitosan chains and inter-chain networks, respectively.25 The lack of volumetric change in the GTCM plus the likely mode of interaction of GA as a bifunctional crosslinker supports a local, intrachain conformational change among deacetylated glucosamine units, rather than a change in overall micro-alignment between macromolecular chains initially co-aligned by the flow assembly process.
Semi-permeability
Many applications of microfluidic platforms with membranes are based on membrane permeability, which can generate gradients and selective barriers for certain molecules or ions. Because the molecular structure of the chitosan membrane has been altered by GA crosslinking, there is a need to examine whether the membrane's permeability is changed. Membrane permeability was investigated with transport experiments using FITC-labelled dextran macromolecules of various sizes. Fluorescence intensity across membranes was recorded every 5 minutes for 30 minutes with continuous flow at 0.5 μL min−1 in the left channel to ensure a consistent fluorescence background, while the right channel across the membrane was filled with PBS in stop flow, as indicated in Fig. 7(a).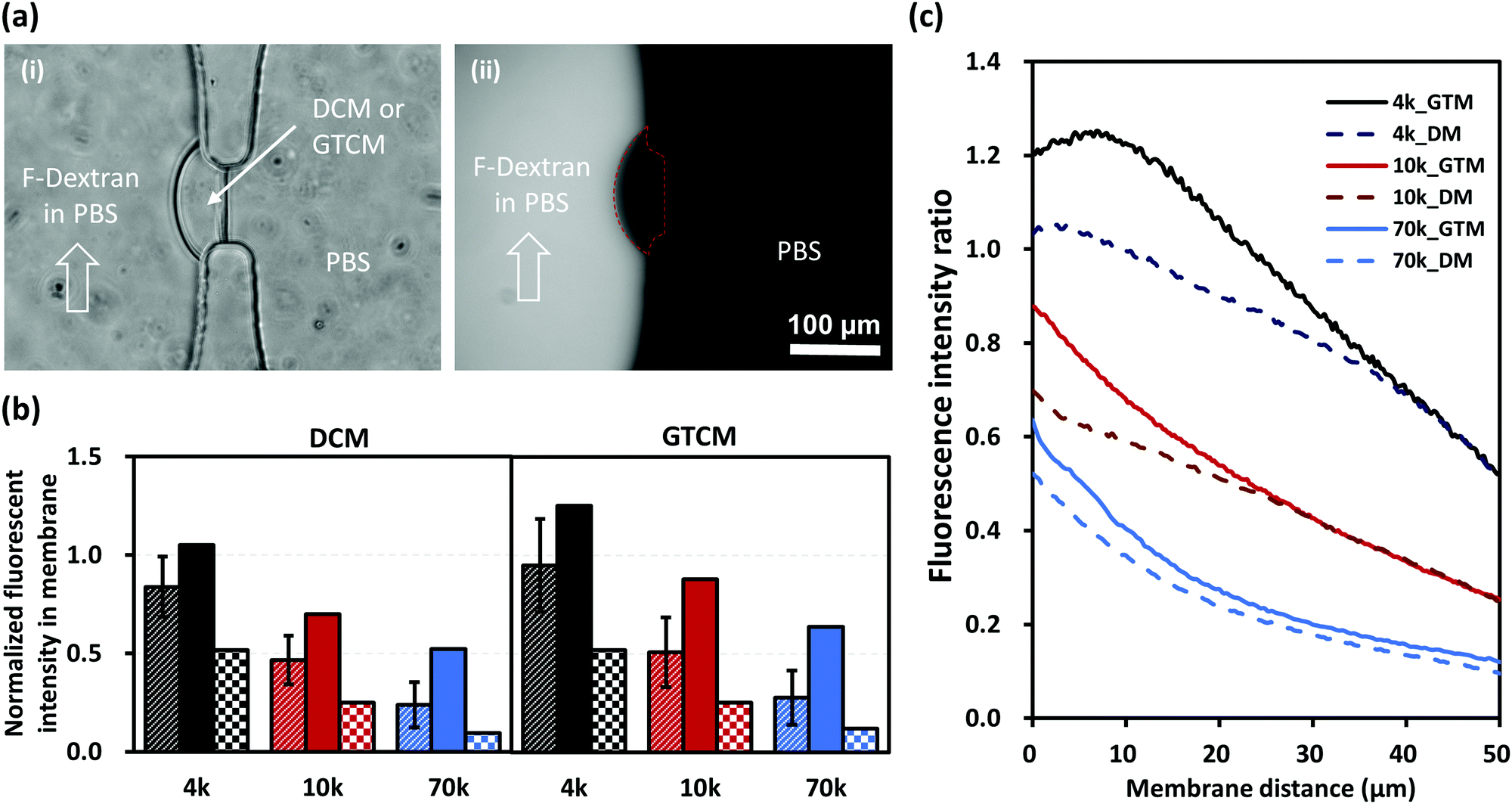 | ||
| Fig. 7 Semi-permeability of DCM and GTCM as confirmed by transport experiments with FITC-labelled dextran of different molecular weights. (a) Bright field (i) and fluorescence (ii) images of membrane in a PDMS microfluidic device during a transport test for one hour with fluorescein-5-isothiocyanate (FITC)-labelled dextran of 70 kDa. (b) Normalized fluorescence intensity (stripes – average, solid – maximum, checkers – minimum) inside the DCM and GTCM after transport test for one hour with FITC-labelled dextran of 4, 10 and 70 kDa. (c) Normalized fluorescence intensity profile across membranes, which shows that GTCM has slightly better semi-permeability and thus a slightly bigger pore size than DCM. | ||
Fig. 7(b) shows the average (stripes), maximum (solid) and minimum (checkers) fluorescence intensities in membranes that were normalized to the corresponding fluorescence intensities in the left channel under continuous flow. Fig. 7(c) shows the normalized fluorescence signals with respect to the membrane thickness from the left to the right of the membranes. These results show that the fluorescence intensity within and along the membranes decreased with the increasing size of dextrans as expected. Furthermore, the increase in fluorescence intensity from DCM to GTCM was subtle in all three cases with 4, 10 and 70 kDa dextran molecules labeled with FITC, although the change was slightly higher for the 4 and 10 kDa cases. The transport of the three tested dextran macromolecules with hydrodynamic radii of 2–7 nm26 across the membranes further confirms that the average pore sizes of DCMs and GTCMs are similar. Overall, the semi-permeability remained similar between DCM and GTCM. We conclude that GA crosslinking would not affect applications where the membrane permeability is a key factor, while its mechanical robustness in a microfluidic network is dramatically improved.
Conclusions
We have studied glutaraldehyde crosslinking on freestanding and semipermeable chitosan membranes (DCM) assembled with flow in single-layer microfluidic networks. We intend to modulate DCM's properties for broader applications, and examine the changes in terms of acidic resistance, mechanical robustness, molecular structure and permeability. After GA crosslinking, the chitosan membrane resisted acidic erosion, which enables the membrane microfluidic platform to be used in acidic environments. The mechanical robustness of the DCM in PDMS microchannels is strong but easily compromised by the Pluronic F-127 coating that is a passivation step necessary for many cell and protein studies in microfluidics. After GA crosslinking, the retention of Pluronic F-127 in the membrane is minimized, and the mechanical robustness of the GTCM in PDMS microchannels is significantly strengthened for practical applications of the membranes in microfluidics. To characterize the GA crosslinking at the molecular level, the crystallization levels of DCM and GTCM were quantified in terms of birefringence and parallelism index through a membrane with a quantitative Polarized Light Microscope. We observed that both optical retardance and PI drop after GA treatment, but the decrease in PI is much smaller than that in optical retardance, which indicates that the microstructural change of well micro-aligned chitosan chains was mostly local among the deacetylated glucosamine units as expected. From the transport experiment through the membrane with FITC-labeled dextran of various molecular weights, it was found that there was little change in the membrane's semi-permeability. Overall, this report reveals new insights into the effects of glutaraldehyde crosslinking of a biofabricated chitosan membrane and demonstrates how GA can be used to modulate the biopolymer membrane properties for broader applications.Experimental
Materials
Glutaraldehyde solution (25% in H2O, molecular weight 100.12 g mol−1), chitosan flakes (85% deacetylated, medium molecular weight), sodium alginate powder (extracted from brown algae with medium viscosity), phosphate buffered saline (PBS) tablets, Pluronic F-127 powder, and fluorescein isothiocyanate (FITC)-dextran with molecular weights of 4, 10 and 70 kilo Dalton (FITC![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :glucose = 1:250), sodium bicarbonate power and dimethyl sulfoxide (DMSO) were bought from Sigma-Aldrich. Sylgard 184 and its curing agent for PDMS device fabrication were purchased from Ellsworth Adhesives. Disposable syringes of 1 mL volume were purchased from Becton, Dickinson and Company. NE-1000 syringe pumps were purchased from New Era Pump Systems, Inc. The microbore PTFE thin wall plastic tubing, 0.022′′ID × 0.042′′OD, 100 ft per roll, was bought from Cole-Parmer. 5-(4,6-Dichlorotriazinyl)aminofluorescein (5-DTAF) was bought from Thermo Fisher Scientific. Bio-Spin® Columns with Bio-Gel® P-6 were bought from Bio-Rad Laboratories, Inc.
:glucose = 1:250), sodium bicarbonate power and dimethyl sulfoxide (DMSO) were bought from Sigma-Aldrich. Sylgard 184 and its curing agent for PDMS device fabrication were purchased from Ellsworth Adhesives. Disposable syringes of 1 mL volume were purchased from Becton, Dickinson and Company. NE-1000 syringe pumps were purchased from New Era Pump Systems, Inc. The microbore PTFE thin wall plastic tubing, 0.022′′ID × 0.042′′OD, 100 ft per roll, was bought from Cole-Parmer. 5-(4,6-Dichlorotriazinyl)aminofluorescein (5-DTAF) was bought from Thermo Fisher Scientific. Bio-Spin® Columns with Bio-Gel® P-6 were bought from Bio-Rad Laboratories, Inc.
GA solution of 10% w/w was prepared by mixing 25% GA with deionized (DI) water. A PBS solution of pH 7.4 was made with PBS tablets in DI water. An alginate solution of 0.5% w/w, pH 11.5 was prepared by dissolving sodium alginate powder in DI water, followed by stirring on a stirring plate overnight, and then dropwise addition of 1 M NaOH to adjust its pH to 11.5. The chitosan solution of 0.5% w/w, pH 5.5 was prepared by dissolving the chitosan flakes in DI water, with 1 M HCl added dropwise to obtain pH 2, and then leaving stirring on a stirring plate overnight, followed by the dropwise addition of 1 M NaOH to adjust its pH to 5.5. The solution was filtered, and DI water was added to bring the final concentration to 0.5% w/w. Both the prepared alginate and chitosan solutions can be stored at 4 °C for future use for up to a year.
Microfluidic device fabrication
The microchannels and an add-on air vacuum chamber,27 as shown in Fig. 2(a), were designed in AutoCAD, transferred to silicon wafers with negative photoresist SU-8 using conventional photolithography, and fabricated using the soft lithography technique. A mixture of Sylgard 184 and its curing agent in a 10:1 weight ratio was poured over the patterned silicon wafer, cured at 65 °C on a hotplate for 4 hours, peeled off from the mold, and punched with microfluidic inlets and outlets. Oxygen plasma (200 mTorr, 10 psi, medium RF level) and Plasma Cleaner PDG-32G (Harrick plasma) were used to bond PDMS microchannels to glass slides. The bonded PDMS devices were put in an oven set at 150 °C for at least 24 hours to restore their hydrophobicity before they were ready for membrane biofabrication.
Deprotonated chitosan membrane biofabrication
Chitosan membranes were biofabricated following a recently reported procedure by steering air bubbles in PDMS microchannels with an add-on vacuuming layer.27,28 Briefly, the positively charged chitosan solution (pH 5.8) and the negatively charged alginate solutions (pH 11.5) were separately pumped into two microchannels, while an air bubble was spontaneously trapped in the aperture due to the natural hydrophobicity of PDMS, as shown in Fig. 2(a)-(ii) and (b)-(i). The flows were stopped, and the add-on PDMS vacuum layer was placed on top of the device to vacuum the air bubble out simply by withdrawing a connected syringe on a syringe pump. Upon the dissipation of air bubbles within 5 to 8 minutes, the negatively charged alginate and the positively charged chitosan solutions came into contact and instantaneously formed a thin polyelectrolyte complex membrane (PECM) layer due to electrostatic interactions (Fig. 2(b)-ii). Both flows were restarted at 1 μL min−1, and the chitosan membrane was grown on the PECM with hydroxyl ions continuously diffused from the alginate channel. Once a desired thickness of DCM around 20–40 μm was achieved within minutes, flows were stopped, all channels were manually rinsed with PBS, and the device was stored at 4 °C in a Petri dish with DI water for future use.Pluronic F-127 treatment
Pluronic F-127, as shown in Fig. 4(a), is a hydrophilic non-ionic surfactant commonly used to coat PDMS microfluidic devices to minimize protein and cell adhesion on the PDMS surface.29 For this purpose, Pluronic F-127 at 0.1% (w/v) in PBS was introduced into the PDMS microchannels with biofabricated DCMs, the flow was stopped, and the membranes were soaked in Pluronic solution for 30 minutes. It was found that after coating PDMS channels with Pluronic F-127 in PBS, the interaction between the biofabricated DCMs and PDMS was severely compromised and the DCMs easily detached from PDMS apertures, as shown in Fig. 2(d). This limits the applications of microfluidic devices with biofabricated membranes for protein and cell studies where the passivation coating such as Pluronic treatment in PDMS microchannels might be needed. For the E. coli chemotaxis study, as shown in Fig. 2(c), Pluronic F-127 treatment of microchannels was necessary to ensure the free swimming of bacteria. Because Pluronic F-127 compromised the adhesion of the chitosan membrane to PDMS microchannels, the static gradient of the chemoattractant11 or chemorepellent could not be established to induce bacterial chemotaxis. To address this challenge, GA as a cross-linker was used to enhance membrane robustness.Glutaraldehyde treatment on DCM
Glutaraldehyde (GA) of 10% w/w in water has been reported as the effective concentration to crosslink chitosan solution into a hydrogel,19,28 and it was used here to modulate the properties of the biofabricated DCM (Fig. 3(a)). GA solution was introduced into both microchannels to soak the DCM for one hour to allow for full reaction before being rinsed with PBS. The treatment converted a DCM into a GTCM, which was further analyzed for its property changes.Mechanical robustness test
A simple strategy based on the ideal gas law was previously reported to measure pressure inside microchannels10 and fluid viscosity through connecting tubing.30 Inspired by this method, a modified measurement setup to quantify the robustness of membranes in microfluidics, with no hydrostatic pressure and more accurate change of air volume, was used here, as shown in a non-proportional schematic of Fig. 4(a). An air column in tubing was trapped between PBS solution segments and marked with the original length Lo. Two of the four outlets/inlets of the PDMS microfluidic device were sealed with metal plugs, and one input was connected to the syringe and tubing for pressure input Pc, while the rest outlet was left open to atmospheric pressure Po. Because PBS as a liquid is incompressible and air is compressible, Pc built up in the air column compressed by the syringe piston. The membrane deformed under pressure till it burst, as shown in Fig. 4(b). The final air column length Lf was recorded at the time when the membrane burst to determine the bursting pressure Pc as the membrane robustness. Assuming a constant temperature during the two minutes for measuring, the Ideal Gas Law PV = nRT = constant is valid. Then, the pressure change is in an inverse ratio to air volume change, which refers to the ratio Pc: Po= Lo: Lf. Thus, membrane robustness is represented as the critical pressure (Pc − 1) that is the gage pressure to the burst membrane.Fluorescein labeling of Pluronic F-127
To better understand the effects of Pluronic F-127 molecules (Fig. 5(a)) on the interaction between DCM and PDMS, the distribution of Pluronic molecules in the DCM and GTCM was investigated. For this purpose, Pluronic F-127 was conjugated with 5-(4,6-dichlorotriazinyl)aminofluorescein (5-DTAF) at room temperature under aqueous conditions,31 and the resulting 5-DTAF labeled Pluronic F-127 is shown in Fig. 5(b). Pluronic F-127 stock solution at 6% (w/v) was prepared by dissolving Pluronic F-127 powder in 0.1 M sodium bicarbonate, and 5-DTAF at 2% (w/v) was dissolved in dimethyl sulfoxide (DMSO) and diluted into 0.1 M sodium bicarbonate at 0.5% (w/v) as the stock solution. Pluronic F-127 and 5-DTAF solutions were mixed in a molar ration of 0.5:1 and the concentration of Pluronic F-127 was 0.1% (w/v) in 0.1 M sodium bicarbonate. The reacting solution was retained in the dark at room temperature for 5 h before purifications. The Bio-Spin® column with Bio-Gel® P-6 was used for size exclusion chromatography to separate 5-DTAF labeled Pluronic F-127 from free 5-DTAF. The reacted solution was filled into the column, and by gravity, large molecules like free Pluronic F-127 and 5-DTAF labeled Pluronic F-127 made their way through the column to the collector, while small molecules like free 5-DTAF were trapped in beads in the columns. This purification process was operated three times to get rid of free 5-DTAF in the final collected solution. The final working solution of Pluronic F-127 was fluorescently labeled with 5-DTAF at 0.1% (w/v) in 0.1 M sodium bicarbonate with minimum free 5-DTAF, which was introduced into microchannels to investigate the penetration of Pluronic F-127 into the DCM and GTCM.
Optical retardance and parallelism index
By using quantitative polarized light microscopy (qPLM) with a de Sénarmont compensator as previously reported,21,32,33 optical retardance, Γ, and parallelism index, PI, of the chitosan membranes were investigated before and after GA treatment. Briefly, the extinction axes of the polarizer and the analyzer of the qPLM instrument were first rotated to 0° and 90°, respectively. Next, a de Sénarmont (546 nm) compensator was placed beneath the analyser with fast and slow axes aligned with the polarizer and analyser extinction axes. Then, the brightest signal from the central region of the membrane was found by rotating the sample stage, which was subsequently locked. Last, a series of images of the membrane was taken at every 1° of analyzer rotation until every pixel within the central region of the membrane passed the minimum brightness (the extinction angle, α). The optical retardance was determined from the extinction angle for each pixel, determined as the minimum of a least squares, second-order polynomial fit to intensity versus analyser angle, according to Γ = α(λ/π), where λ = 546 nm is the wavelength of light used for imaging. An optical retardance map valid for the central region of the membrane was then generated from pixel calculations, as shown in Fig. 6(a). The parallelism index was calculated as PI = (IMAX − IMIN)/(IMAX + IMIN), where IMAX and IMIN are determined for every pixel by removing the compensator from the light path, rotating the polarizer and analyser with 15° increments over 90°, and fitting the signal, I, versus rotation angle, θ, to the function I = A + B![[thin space (1/6-em)]](https://www.rsc.org/images/entities/i_char_2009.gif) sin2(2θ + ϕ), where A, B, and ϕ are parameters determined from a nonlinear least squares fit.
sin2(2θ + ϕ), where A, B, and ϕ are parameters determined from a nonlinear least squares fit.
Transport experiments
Fluorescein isothiocyanate (FITC)-labelled dextran molecules with different molecular weights from 4k to 10k to 70k Da were used as probes of varied sizes to investigate if there is any difference in the semi-permeability of DCM and GTCM. FITC-dextran of 0.5 mg mL−1 in PBS was run through the left side channel at 1 μL min−1 to maintain the fluorescence concentration, while the right channel was filled with PBS in stop flow. The shelter of fluorescent light was opened every 5 minutes to take images in order to avoid photo bleach. The light intensity of the fluorescent light source was consistent due to the continuous feeding of fluorescein. The semi-permeability of the membrane was quantified by the amount of FITC-dextran traveled after 30 min, which was determined by fluorescence light intensity through the membranes.Microscopy and analysis
Bright field and fluorescent images were taken with either a Nikon TS100 or a Ludesco EXI-310 inverted microscope. Fig. 6(a) shows the images taken with quantitative polarized light microscopy (qPLM) (Meiji Techno America, MT9930). ImageJ (NIH) was used to measure membrane thickness in the microscopy images, and to quantify the light intensities of optical retardance maps and fluorescence by pixels. One-factor analysis of variance (ANOVA) was performed on the net optical retardance and the parallelism index of DCMs and GTCMs.Conflicts of interest
The authors declare that there is no conflict of interest.Acknowledgements
This work was funded by the National Science Foundation (CAREER #1553330) and the School of Engineering at the Catholic University of America.Notes and references
- D. Zeng, X. Luo and R. Tu, Int. J. Carbohydr. Chem., 2012, 2012, 104565 CrossRef.
- S. A. Agnihotri, N. N. Mallikarjuna and T. M. Aminabhavi, J. Controlled Release, 2004, 100, 5–28 CrossRef CAS PubMed.
- A. K. Azad, N. Sermsintham, S. Chandrkrachang and W. F. Stevens, J. Biomed. Mater. Res., Part B, 2004, 69, 216–222 CrossRef PubMed.
- L. Ma, C. Gao, Z. Mao, J. Zhou, J. Shen, X. Hu and C. Han, Biomaterials, 2003, 24, 4833–4841 CrossRef CAS PubMed.
- Z. Li, H. R. Ramay, K. D. Hauch, D. Xiao and M. Zhang, Biomaterials, 2005, 26, 3919–3928 CrossRef CAS PubMed.
- V. K. Thakur and S. I. Voicu, Carbohydr. Polym., 2016, 146, 148–165 CrossRef CAS PubMed.
- A. Rafique, K. M. Zia, M. Zuber, S. Tabasum and S. Rehman, Int. J. Biol. Macromol., 2016, 87, 141–154 CrossRef CAS PubMed.
- C. Jia, F. Jiang, P. Hu, Y. Kuang, S. He, T. Li, C. Chen, A. Murphy, C. Yang and Y. Yao, ACS Appl. Mater. Interfaces, 2018, 10, 7362–7370 CrossRef CAS PubMed.
- X. Luo, D. L. Berlin, J. Betz, G. F. Payne, W. E. Bentley and G. W. Rubloff, Lab Chip, 2010, 10, 59–65 RSC.
- X. Luo, H.-C. Wu, J. Betz, G. W. Rubloff and W. E. Bentley, Biochem. Eng. J., 2014, 89, 2–9 CrossRef CAS.
- X. Luo, T. Vo, F. Jambi, P. Pham and J. S. Choy, Lab Chip, 2016, 16, 3815–3823 RSC.
- T. Vo, S. B. Shah, J. S. Choy and X. Luo, Biomicrofluidics, 2020, 14, 014108 CrossRef PubMed.
- X. Luo, C.-Y. Tsao, H.-C. Wu, D. N. Quan, G. F. Payne, G. W. Rubloff and W. E. Bentley, Lab Chip, 2015, 15, 1842–1851 RSC.
- P. L. Pham, S. A. Rooholghodos, J. S. Choy and X. Luo, Adv. Biosyst., 2018, 2, 1700180 CrossRef.
- P. Monsan, G. Puzo and H. Mazarguil, Biochimie, 1975, 57, 1281–1292 CrossRef CAS.
- D. R. Walt and V. I. Agayn, TrAC, Trends Anal. Chem., 1994, 13(10), 425–430 CrossRef CAS.
- S. He, W. Zhang, D. Li, P. Li, Y. Zhu, M. Ao, J. Li and Y. Cao, J. Mater. Chem. B, 2013, 1, 1270–1278 RSC.
- W. W. Ngah, C. Endud and R. Mayanar, React. Funct. Polym., 2002, 50, 181–190 CrossRef CAS.
- O. A. Monteiro Jr and C. Airoldi, Int. J. Biol. Macromol., 1999, 26, 119–128 CrossRef CAS.
- M. Beppu, R. Vieira, C. Aimoli and C. Santana, J. Membr. Sci., 2007, 301, 126–130 CrossRef CAS.
- K. Li, S. Correa, P. Pham, C. Raub and X. Luo, Biofabrication, 2017, 9, 034101 CrossRef CAS PubMed.
- N. Kildeeva, P. Perminov, L. Vladimirov, V. Novikov and S. Mikhailov, Russ. J. Bioorg. Chem., 2009, 35, 360–369 CrossRef CAS.
- I. Migneault, C. Dartiguenave, M. J. Bertrand and K. C. Waldron, Biotechniques, 2004, 37, 790–802 CrossRef CAS PubMed.
- J. Zhou, D. A. Khodakov, A. V. Ellis and N. H. Voelcker, Electrophoresis, 2012, 33, 89–104 CrossRef CAS PubMed.
- R. Oldenbourg and T. Ruiz, Biophys. J., 1989, 56, 195–205 CrossRef CAS PubMed.
- J. K. Armstrong, R. B. Wenby, H. J. Meiselman and T. C. Fisher, Biophys. J., 2004, 87, 4259–4270 CrossRef CAS PubMed.
- P. Pham, T. Vo and X. Luo, Lab Chip, 2017, 17, 248–255 RSC.
- L. Ma, C. Gao, Z. Mao, J. Shen, X. Hu and C. Han, J. Biomater. Sci., Polym. Ed., 2003, 14, 861–874 CrossRef CAS PubMed.
- J. L. Tan, J. Tien, D. M. Pirone, D. S. Gray, K. Bhadriraju and C. S. Chen, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 1484–1489 CrossRef CAS PubMed.
- L. H. Phu Pham, L. Bautista, D. C. Vargas and X. Luo, RSC Adv., 2018, 8(53), 30441–30447 RSC.
- F. Ahmed, P. Alexandridis and S. Neelamegham, Langmuir, 2001, 17, 537–546 CrossRef CAS.
- K. Kocsis, M. Hyttinen, H. J. Helminen, M. B. Aydelotte and L. Módis, Microsc. Res. Tech., 1998, 43, 511–517 CrossRef CAS PubMed.
- J. Rieppo, J. Hallikainen, J. S. Jurvelin, I. Kiviranta, H. J. Helminen and M. M. Hyttinen, Microsc. Res. Tech., 2008, 71, 279–287 CrossRef PubMed.
| This journal is © The Royal Society of Chemistry 2020 |