
A perylenetetracarboxylic dianhydride and aniline-assembled supramolecular nanomaterial with multi-color electrochemiluminescence for a highly sensitive label-free immunoassay†

Abstract
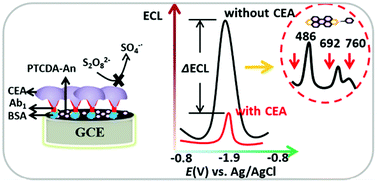
Most electrochemiluminescence (ECL) studies focus on the single emission of luminophores, which severely limits the development of the multi-color ECL fundamental theory and applications. Herein, we prepared a multi-color ECL supramolecular nanomaterial self-assembled by 3,4,9,10-perylenetetracarboxylic dianhydride (PTCDA) and aniline (An) through hydrogen bonding. This PTCDA–An supramolecular nanomaterial simultaneously produced multi-color emissions peaking at 486, 692 and 760 nm with K2S2O8 as a coreactant. These multi-color emissions were assigned to the excited PTCDA monomer (486 nm), H-dimer (692 nm) and J-dimer (760 nm). The simultaneously increased dual-color ECL intensity significantly enhanced the total ECL intensity of PTCDA–An. Furthermore, this highly efficient ECL nanomaterial was used as an ECL platform to construct a label-free immunosensor for tumor marker carcinoembryonic antigen (CEA) detection. The total ECL intensity of the immunosensor sensitively decreased due to the simultaneously decreased ECL of multiple emissions. Also, this immunosensor exhibited a wide linear range from 1 pg mL−1 to 10 μg mL−1 with a low detection limit of 0.23 pg mL−1. The multi-color ECL from the same luminophore PTCDA in this work also provides a new perspective for multi-color ECL biomaterial design.
- This article is part of the themed collection: Biosensors
 Please wait while we load your content...
Please wait while we load your content...

