
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEffect of boron incorporation on the bioactivity, structure, and mechanical properties of ordered mesoporous bioactive glasses†
Leonie
Deilmann
a,
Oliver
Winter
a,
Bianca
Cerrutti
b,
Henrik
Bradtmüller
b,
Christopher
Herzig
c,
Andreas
Limbeck
 c,
Olaf
Lahayne
d,
Christian
Hellmich
d,
Hellmut
Eckert
*be and
Dominik
Eder
*a
c,
Olaf
Lahayne
d,
Christian
Hellmich
d,
Hellmut
Eckert
*be and
Dominik
Eder
*a
aInstitut für Materialchemie, Technische Universität Wien, Getreidemarkt 9/BC/02, 1060 Wien, Austria. E-mail: dominik.eder@tuwien.ac.at
bInstituto de Física de Sao Carlos, Universidade de Sao Paulo, CEP 369, Sao Carlos SP 13566-590, Brazil
cInstitut für Chemische Technologien und Analytik, Technische Universität Wien, Getreidemarkt 9/164, 1060 Wien, Austria
dInstitut für Mechanik der Materialien und Strukturen (IMWS), Technische Universität Wien, Karlsplatz 13/202, 1040 Wien, Austria
eInstitut für Physikalische Chemie, WWU Münster, Corrensstr. 28/30, 48149 Münster, Germany. E-mail: eckerth@uni-muenster.de
First published on 9th January 2020
Abstract
B2O3 doped (0.5–15 mol%) ordered mesoporous bioactive glasses (MBG) with the composition 80% SiO2–15% CaO–5% P2O5 were synthesized via a sol–gel based evaporation-induced self-assembly process using the block-copolymer P123 as a structure directing agent and characterized by biokinetic, mechanical and structural investigations. Nitrogen physisorption isotherms and electron microscopy indicate no detrimental effect of B2O3 on the ordered hexagonal pore structure. Boron incorporation increases both the bulk modulus and hardness of the glasses. In vitro bioactivity tests reveal a rapid initial release of Ca2+ and PO43− ions, followed by formation of hydroxyapatite carbonate within a few hours. Contrary to the tight incorporation of Al in Al-doped MBGs, the rapid release of borate species into simulated-body-fluid suggests loosely bound species localized at the internal surfaces of the mesopores. 29Si, 11B, 31P, and 1H solid state NMR spectroscopy reveal that the majority of the borate is present as anionic BO4/2− species. The need for charge compensation leads to an increase in the average degree of polymerization of the phosphate species for high boron contents. 11B{31P} rotational echo double resonance NMR results reveal the absence of B–O–P linkages. This structural model explains the rapid release of borate and the enhanced dissolution kinetics of the Ca2+ and phosphate species.
Introduction
In the late 1960s, Hench et al. introduced Bioglass® as a new class of biocompatible materials.1 Contact with human blood plasma or simulated body fluid (SBF) readily creates a connective layer between the inorganic surface and the soft and/or hard tissue. This layer is typically made of crystalline hydroxyapatite, which is the main component of the inorganic part of bone.2 Bioactive glasses are a later addition to this materials class and considered next-generation. According to Cao and Hench,3 they are Class A biomaterials that show both osteoconductive and osteoproductive behavior as well as an enhanced bone proliferation. The ability to directly bond to bone without the danger of an inflammatory response renders these materials promising candidates for bone tissue engineering.The most common Bioglass®, 45S5, has a composition of 45% SiO2, 24.5% Na2O, 24.5% CaO and 6% P2O5 (wt%) and was first produced in 1971 by Hench et al. via the melt-quenching method.2 The sol–gel derived bioactive glass was introduced 20 years later by Li et al.4 Sol–gel glasses offer many advantages, including a larger surface area due to inherent porosity, which can significantly increase the number of binding sites and hence the bioactivity. In addition, sol–gel glasses offer lower synthesis and processing temperatures and often exhibit higher purity and homogeneity. Furthermore, Na2O can be eliminated from the glass system, and the compositional range can be expanded up to silica-rich compositions.
In 2004 Yan et al.5 introduced hexagonally ordered mesopores into bioactive sol–gel glasses by using block-copolymers as structure directing agents in combination with the evaporation-induced self-assembly (EISA) process.6 In short, the inorganic precursors for the oxide components in the glass system (e.g. tetraethyl orthosilicate, TEOS, for SiO2) and a block-copolymer (e.g. the commercially available Pluronics® P123) are mixed into a solvent at c < cmc (critical micelle concentration). Upon evaporation of the solvent the surfactant concentration increases beyond cmc, which initiates spontaneous organization through non-covalent interactions and micelle formation at the mesoscale. Concurrently with the self-assembly process, hydrolysis and condensation reactions of the precursors lead to the formation of amorphous pore walls that separate the mesopores. The arrangement of the pores depends on various synthesis conditions, such as choice of polymer, temperature, pH, and reactant ratios.7,8 So far, mesoporous glasses with two-dimensional (2D) hexagonal or three-dimensional (3D) cubic pore structures have been obtained.5,9
Due to their tunable porosity and large surface area, mesoporous bioactive glasses (MBGs) are ideal for various applications in biomaterials science. They can be filled either with pharmaceuticals for drug delivery or in vivo with osteogenetic cells/proteins for bone tissue engineering.10 Above all, MBG are ideal candidates for bone tissue regeneration, because they show a much faster and more intense hydroxycarbonate apatite (HCA) formation compared to the traditional sol–gel or melt-quenched glasses.5 However, the low mechanical strength of MBG still poses a major challenge in applications.11,12 To alter the structural and textural properties, additional ions or oxides can be incorporated into the basic SiO2–CaO–P2O5-system of the bioactive glass.
One potential candidate to strengthen the mechanical properties of MBG is B2O3 due to the abrasion resistance and hardness that it imparts upon silicate and phosphate glasses; hence it has been introduced as an additional component into melt-derived glasses.13 Boron doped bioactive glasses are currently attracting interest also owing to their improved biocompatibility and antibacterial function.14,15 Boron can influence vital processes, including embryogenesis, bone growth and psychomotor skills.16,17 Additionally, it has been recognized to preserve bone health, and a diet containing adequate amounts of boric acid enhances bone strength under compression.16,18 It is also an essential mineral in the conversion of vitamin D to its active form. Moreover, boron-containing bioglass has demonstrated increased proliferation of osteoblasts in cell cultures,16,17,19 and the controlled release of boron from bioglass can favor the regeneration of bone tissues. So far, however, there has been no comprehensive study that correlated the structural information of boron incorporation in MBGs with their mechanical properties and bioactive response.
In this work we doped MBG of the system SiO2–CaO–P2O5, (80–x![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :15:5 mol%) with different amounts of boron trioxide ranging from x = 0.5 to 15 mol%. We thoroughly analyzed the textural properties of MBG, such as pore geometry, porosity and surface area. Moreover, we performed in vitro bioactivity tests and nanoindentation studies and correlated the changes in bioactivity and mechanical properties with the changes in glass structure revealed by in-depth solid-state nuclear magnetic resonance (NMR) spectroscopic studies. While solid state NMR has been widely used for the structural characterization of bioglasses,20 to date the literature on boron-containing systems is rather limited. Work by the Eden group stands out, in particular reporting detailed compositional dependences and advanced connectivity information on the basis of two-dimensional dipolar NMR spectroscopy in traditional melt-quenched bioglasses.21,22 To date, however, no experimental data on boron-containing MBGs are available. In the present contribution we develop a quantitative structural description of these materials based on a comprehensive solid-state NMR investigation, providing a suitable structural rationale for the composition–property relationship.
:15:5 mol%) with different amounts of boron trioxide ranging from x = 0.5 to 15 mol%. We thoroughly analyzed the textural properties of MBG, such as pore geometry, porosity and surface area. Moreover, we performed in vitro bioactivity tests and nanoindentation studies and correlated the changes in bioactivity and mechanical properties with the changes in glass structure revealed by in-depth solid-state nuclear magnetic resonance (NMR) spectroscopic studies. While solid state NMR has been widely used for the structural characterization of bioglasses,20 to date the literature on boron-containing systems is rather limited. Work by the Eden group stands out, in particular reporting detailed compositional dependences and advanced connectivity information on the basis of two-dimensional dipolar NMR spectroscopy in traditional melt-quenched bioglasses.21,22 To date, however, no experimental data on boron-containing MBGs are available. In the present contribution we develop a quantitative structural description of these materials based on a comprehensive solid-state NMR investigation, providing a suitable structural rationale for the composition–property relationship.
Materials and methods
Sample preparation
MBGs with glass compositions of 80–x mol% SiO2, 15 mol% CaO, 5 mol% P2O5, and x = 0–15 mol% B2O3 were prepared according to the synthesis reported by Yan et al.,5 using commercial Pluronic® block-copolymers as structure-directing agents. The corresponding glass compositions are listed in Table 1. The overall experimental procedure is analogous to that previously used by us to obtain Al2O3-doped MBGs23 and the details are summarized in ESI.†| Sample | Composition in mol% | |||
|---|---|---|---|---|
| SiO2 | CaO | P2O5 | B2O3 | |
| MBG | 80 | 15 | 5 | 0 |
| 0.5 B-MBG | 79.5 | 15 | 5 | 0.5 |
| 1.0 B-MBG | 79 | 15 | 5 | 1 |
| 3.0 B-MBG | 77 | 15 | 5 | 3 |
| 5.0 B-MBG | 75 | 15 | 5 | 5 |
| 10.0 B-MBG | 70 | 15 | 5 | 10 |
| 15.0 B-MBG | 65 | 15 | 5 | 15 |
In short, Pluronic® P123 (2 g, Mw = 5800, BASF) was dissolved in ethanol (38 mL) under magnetic stirring followed by the addition of the respective amounts of the precursors. The precursors were added in the following sequence and each precursor was completely dissolved before the addition of the next one. Tetraethyl orthosilicate (TEOS, 99% ABCR), calcium nitrate tetrahydrate (Ca(NO3)2 × 4H2O, 99% ACS), triethyl phosphate (TEP, 99% Merck), and boric acid (H3BO3, 99%, ABCR) were used as precursors for SiO2, CaO, P2O5, and B2O3, respectively. Hydrochloric acid (0.5 mL, 0.5 M) was added to catalyze the hydrolysis reaction. The final solution was stirred at room temperature (RT) for 24 h, then transferred into a Petri dish and stored at RT for 6 days to initiate the EISA process. The resulting gels were dried and calcined in a muffle oven at 600 °C for 2 h (heating/cooling rate 1 °C min−1) to remove the polymer.
Materials characterization
The structure of the bioactive glasses was characterized by high resolution transmission electron microscopy (HRTEM), using a Libra 200 microscope (Carl Zeiss Microscopy GmbH), as well as Tecnai G20 and F20 (FEI TecnaiTM), all operated with an acceleration voltage of 200 keV. The powders were dispersed in ethanol by ultrasonication and drop-casted on a copper 200 mesh Lacey carbon coated grid (Plano).Surface area and pore size distribution were analyzed by N2 physisorption (ASAP 2010 Micromeritics GmbH), using the Brunauer–Emmett–Teller (BET) and Barret–Joyner–Halenda (BJH) method,24 respectively. To remove moisture and adsorbed gases the samples were heated at temperatures up to 150 °C under vacuum for several hours.
Mechanical properties
Elasticity modulus and hardness were measured by the nanoindentation method, following the procedure of Oliver and Pharr.25 A TI-900 TriboIndenter® (Hysitron Incorporated, Minneapolis, USA) was used. The diamond tip had the Berkovich geometry with half angle of 65.27°, included angle of 142.30°, Young's modulus of 1140 GPa and Poisson's ratio of 0.07. The tip diameter was 50 nm. Each sample was measured in the quasi-static mode which is a standard measuring option for all indentation tests. Small pieces of calcined glass were glued on a silicon wafer on a standard sample holder that is usually used for SEM. This sample holder was placed onto the magnetic nanoindenter sample stage and samples were investigated with a pre-defined measuring program. 225 unique single indents on a 15 × 15 grid and with 20 μm distance between the single indents were taken for every sample, resulting in an area of 300 × 300 μm in size. The load function or maximum force was increased from 1000 to 12000 μN in steps of 53.125 μN to obtain load vs. displacement curves.
In vitro bioactivity tests
A simulated body fluid (SBF) solution was prepared according to Kokubo et al.26 The bioactive glass samples (40 mg) were soaked in SBF (40 mL) at 36.5 °C for time periods between 10 min to 7 days and subsequently filtrated and dried in order to terminate the formation of hydroxycarbonate apatite (HCA). The ratio of bioactive glass to SBF was 1 mg mL−1.The concentrations of the solute species (calcium, phosphorus, silicon, and boron) were measured in regular time intervals by inductively coupled plasma emission spectroscopy (ICP-OES). Prior to analysis samples were diluted (1:10) with 1% (v/v) nitric acid and indium was added as internal standard with a final concentration of 1 μg mL−1. Matrix matched standard solutions required for external calibration were prepared just before use by appropriate combination of single element standards (Carl ROTH® Single-Element ICP-Standard-Solution Roti® Star) with 1:10 diluted SBF, followed by the addition of the internal standard. Samples and standards were analyzed with an iCAP 6500 ICP-OES spectrometer (Thermo Scientific, USA) equipped with Mira Mist nebulizer (Burgener Research Inc., Ontario, Canada) and a Teflon cyclonic spray chamber to keep Si background as low as possible. Sample-uptake was achieved with the peristaltic pump of the instrument. Background-corrected emission signals were recorded in the radial viewing mode and processed using Qtegra software (Thermo Scientific, USA). Four replicates with an integration time of 5 s each were measured for samples as well as standard solutions. The optimized ICP parameters are summarized in Table S1 (ESI†). Observed signal intensities were normalized using the signal response for the internal standard (In), and finally converted into concentration units by means of external aqueous calibration. Derived In signals were constant over each measurement session (less than 5% relative standard deviation for the whole measurement period, indicating the absence of temporal trends), and no significant difference in In-response between samples and calibration standards was observed.
Mineralization of HCA of the identical samples was monitored by X-ray powder diffraction (XRD), using a Bruker ASX D8 diffractometer, equipped with a Cu-Kα source. Detected scattering angles ranged from 20° to 80°.
Solid state NMR studies
Solid state NMR experiments were performed on 5.7 T Varian/Agilent 240-MR DD2 and 14.1 T Bruker Advance Neo spectrometer systems. The spectrometers were equipped with a 4 mm double resonance probe (Agilent) and a 2.5 mm triple resonance probe (Bruker). 29Si MAS-NMR spectra were obtained on the 5.7 T magnet using the following conditions: 90° pulses near 5.0 μs at spinning speeds near 5.0 kHz, and a relaxation delay of 180 s. Spectra were deconvoluted into various Gauss–Lorentz components using the DMFIT software27 and referenced to tetramethylsilane using solid tetrakis (trimethylsilyl) silane as a secondary reference. 31P MAS-NMR spectra were recorded at 5.7 T, using 90° pulses near 5.0 μs, at spinning speeds in the range of 12.0 kHz, and relaxation delays up to 1200 s. Chemical shifts are reported relative to an 85% H3PO4 solution. 1H MAS-NMR spectra were measured at 600 MHz, with a sample spinning speed of 25.0 kHz. 11B MAS-NMR spectra were recorded at both 5.7 and 14.1 T using short pulses (<30°) of 0.5 μs length and a relaxation delay of 5 s. Chemical shifts are reported relative to a BF3·OEt2 solution, using solid BPO4 (−3.5 ppm) as a secondary standard. Spectra were simulated using the DMFIT program, with two Gaussian components for the B(4) and a second-order quadrupolar line shape for the B(3) species. 11B{31P} rotational echo double resonance (REDOR) measurements28 were conducted at 14.1 T with a rotor spinning frequency of 15.0 kHz. Solid 11B 180° pulse lengths were typically 2.8 μs for measurements observing three-coordinate boron and 5.3 μs for measurements observing four-coordinate boron. 31P recoupling π pulses were consistently employed with a length of 3 μs. Recycle delays were between 1 s and 2 s, followed by a pre-saturation train of 20π/2 pulses. The normalized signal attenuation data obtained with the initial decay regime (ΔS/S0 ≤ 0.2) as a function of dipolar evolution time (NTr = number of rotor cycles times rotor period) were fitted to a parabola according to the expression29| ΔS/S0 = (4/3π2)(NTr)2M2(B–P) | (1) |
Results
Textural characterization
XRD confirms the amorphous nature of all glasses and the absence of any crystalline impurities, although the presence of nanosized species below the detection limit cannot be excluded (ESI,† Fig. S1).Typical TEM images of the samples are shown in Fig. 1 and reveal that all the samples consist of well-ordered mesopores with 2D hexagonal pore arrangement with average pore diameters of around 5 nm. There was no noteworthy effect of the incorporation of boron on the pore architecture or ordering (Fig. S2 and S3, ESI†).
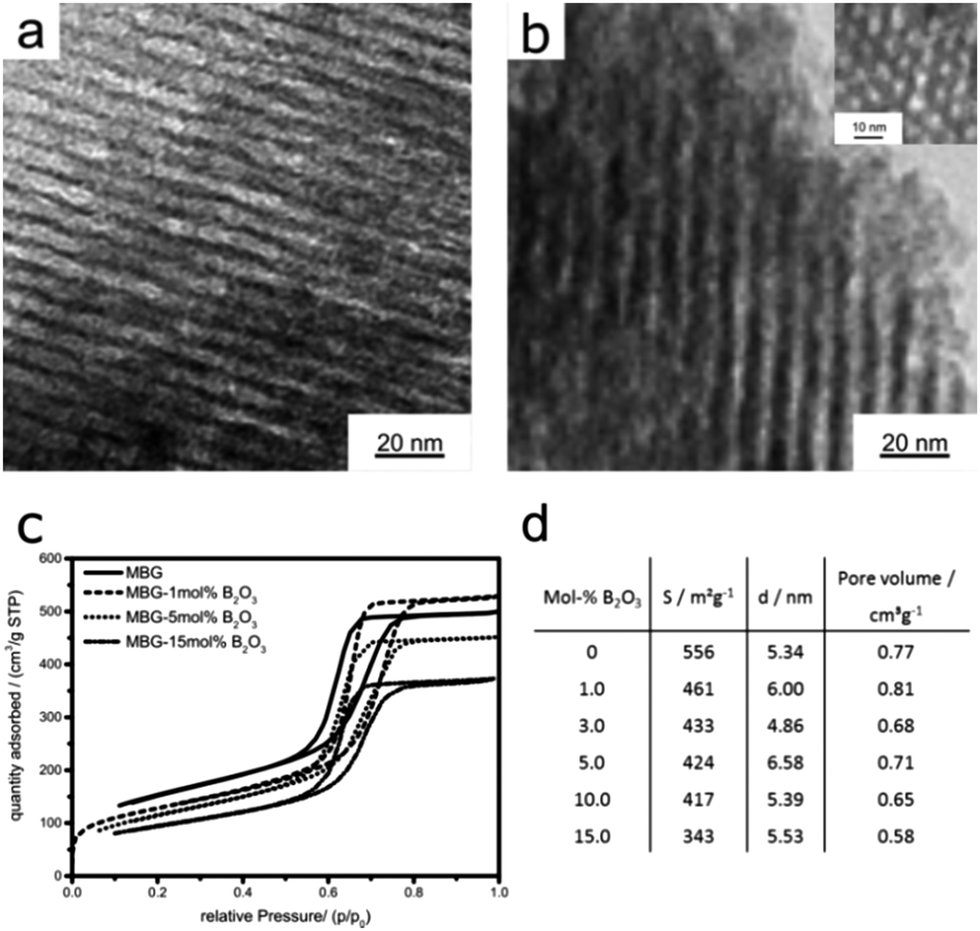 | ||
| Fig. 1 TEM images of boron-doped MBGs with concentrations of (a) 0.5 and (b) 15 mol% B2O3, showing 2D hexagonally-ordered mesopores, (c) nitrogen adsorption–desorption isotherms from 0, 1, 5 and 15 mol% B2O3-doped MBGs and (d) surface areas, average pore diameters and pore volumes extracted from N2 sorption data of B2O3-doped MBGs. | ||
The nitrogen adsorption–desorption isotherms in Fig. 1c show for all samples type IVa isotherms with H1 hysteresis, which is typical for mesoporous materials with uniform pores and narrow pore size distributions.24 The BJH analysis further confirms the narrow pore size distributions with maxima between 4–6 nm, in good agreement with the TEM results. In contrast to the pore characteristics, the boron incorporation has a considerable impact on the BET specific surface areas (see Fig. 1d). Remarkably, already the addition of 1 mol% B2O3 results in a strong decrease of the surface area by about 20% (461 m2 g−1) compared to the pure MBG (556 m2 g−1). The surface area continues to decrease with increasing B2O3 content, albeit to a lower extent. The addition of 10 and 15 mol% B2O3 further leads to a significant loss in surface area from 417 to 343 m2 g−1. Considering that neither the size nor the arrangement of the mesopores is affected by the incorporation of B2O3; the decrease in surface area is likely associated with a partial collapse of micropores within the glass's walls or a pore blocking enacted by the presence of B2O3.
Mechanical properties
Fig. 2 summarizes the bulk modulus and hardness data of the MBG samples obtained by nanoindentation. The maximum indentation forces implied indentation depths ranging from 35 nm to 4600 nm. The indentation depths are separated by a factor of six from the size of the material phase inside the investigated material volume (representative volume element – RVE)31 and this phase size typically measures one third to one half of the size of the RVE.32 Hence, our indentations probe material volumes measuring one third to one half of the indentation depth, i.e. RVEs of 100 nm to 2300 nm size. Such RVEs obviously refer to the porous material encompassing several porous cells as seen in Fig. 1. Given a surface roughness of 10–30 nm, which is less than one fifth of the herein recorded indentation depths,33 the nanoindentation-probed surfaces can be regarded as sufficiently flat so as to allow for the employment of the classical Oliver–Pharr solution for the determination of hardness and elastic modulus. Corresponding sample-specific statistical distributions can be well fitted by means of one single Gaussian probability distribution function each; with expected values and standard deviations found in Fig. 2b. Thereby, deviations from the expected values may stem from deviations of patches of material with the textures seen in Fig. 1, from the loading direction of the indenter. Both, modulus and hardness increased considerably with the incorporation of increasing amounts of boron into MBG. The addition of 0.5 mol% led to an increase of nearly 50% compared with the pure MBG, while an additional 50% increase required the addition of 10% B2O3. Interestingly, the samples with B2O3 contents of 3% and larger exhibit modulus values comparable to that of cortical bone (16.6 to 21.2 GPa),34 which make them suitable candidates for load-bearing applications.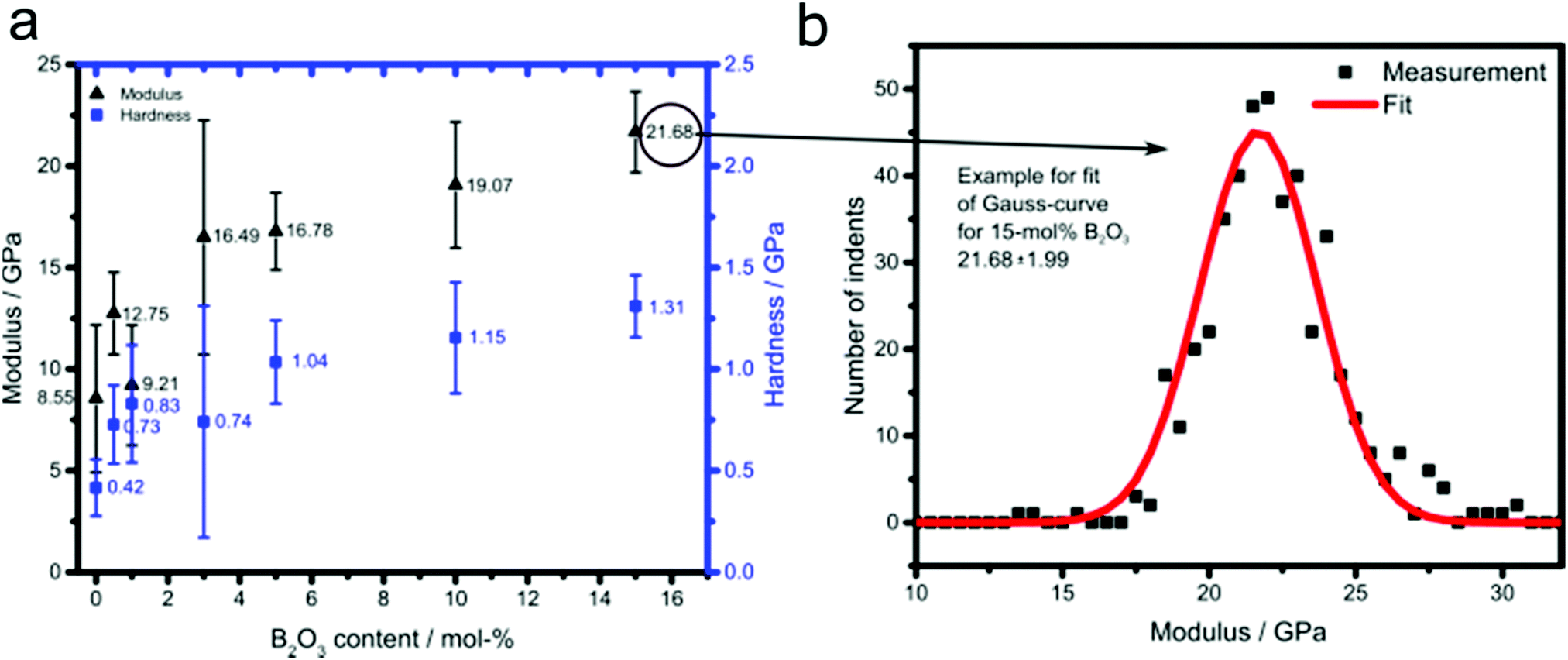 | ||
| Fig. 2 (a) Dependence of modulus and hardness for B2O3-doped MBG on boron oxide content and (b) example for fit of Gauss-curve for 15 mol% B2O3. | ||
In vitro bioactivity studies
All the samples were soaked in SBF for different times up to 7 days, filtered and the solute analyzed by ICP-OES. The data are summarized in Fig. 3 and Fig. S4 (ESI†).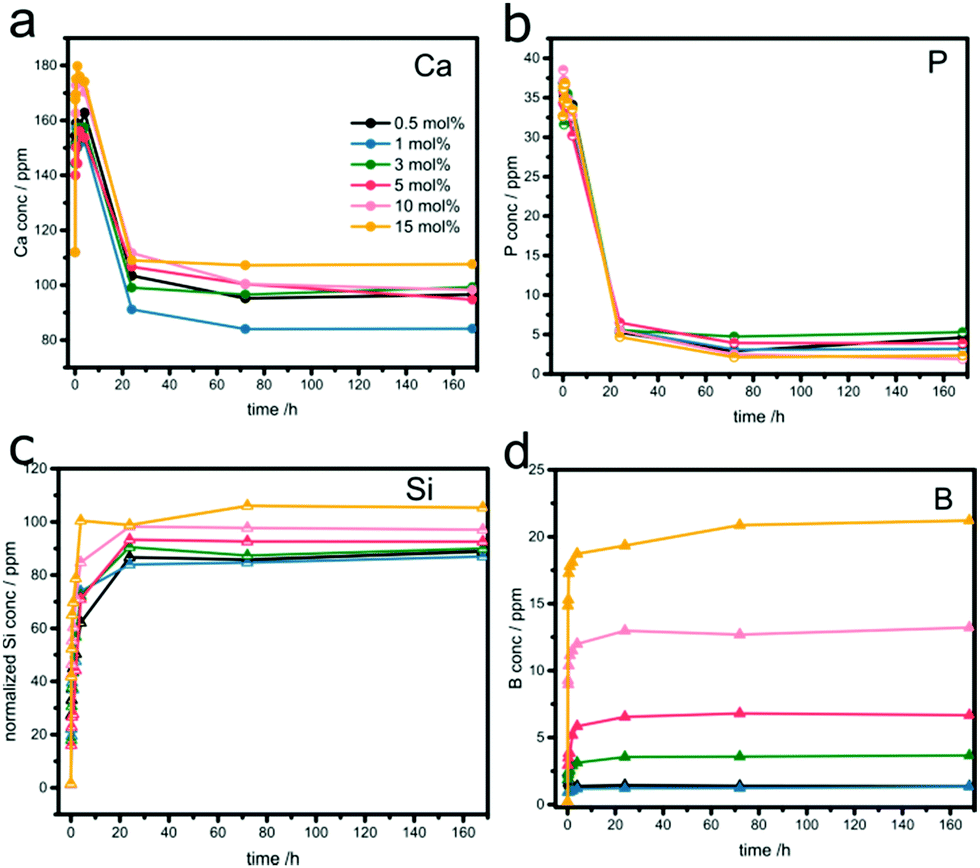 | ||
| Fig. 3 Temporal evolutions of (a) Ca, (b) P, (c) normalized Si, and (d) B concentrations in SBF after soaking boron-doped MBGs. | ||
The temporal evolutions of the calcium (Fig. 3a) and phosphate ion concentrations (Fig. 3b) show the typical profile for bioactive glasses: an increase in concentration within the first 4 hours, followed by a considerable decrease upon re-uptake, due to the formation of Ca-phosphates (e.g. hydroxycarbonate apatite, HCA) on the samples’ surfaces. The concentrations reached saturation levels after approximately one day. The phosphate release and uptake kinetics seem to be independent of the sample composition (Fig. 3b). In contrast, both release and re-uptake of Ca2+ ions varied with B2O3 concentration. Generally, it appears that high concentrations of boron facilitate the release of Ca2+ but suppress its re-uptake and the subsequent formation of HCA, e.g. residual concentration of 110 ppm (μg mL−1) for 15% B. The best performance was found with 1% B, which showed a moderate ion release, but the strongest uptake profile (residual concentration of 85 ppm).
Fig. 3c summarizes the Si release profiles, normalized to the initial Si content in the samples. In all samples, the majority of Si was released within the first few hours, reaching a saturation level of around 70 ppm after one day. It is important to note than this final level is significantly lower than the maximum amount of Si present in the glasses, indicating that part of it remained in the sample, presumably blocked from dissolution by the HA layer formed on the surface. The relative amounts of released Si increase slightly with increasing boron concentration. For example, the percentage of released Si in the sample with 15% B is about 24% higher than in the sample with 0.5% B. Furthermore, there is a significant effect of boron on the release kinetics of Si. The initial rates, taken from the first 4 hours, increased from ∼5.2 ppm h−1 to ∼9 ppm h−1. Both results suggest that boron facilitates the release of Si from the glass network.
The boron release profiles, summarized in Fig. 3d, are similar for all samples. The majority of boron gets released within the first few hours before reaching saturation after one day. The concentrations in the solution correspond well with the respective glass composition, as seen in the linear increase in Fig. S4 (ESI†). This suggests a complete boron release and indicates a “loose” incorporation, likely in the form of species attached on the interior surface.
The formation of crystalline HCA was confirmed by XRD (see Fig. S1, ESI†). The characteristic (002), (211) and (222) reflections for HCA become visible at 2θ values of 25.8°, 31.8° and 46.7°, respectively, for all samples after 4 hours. Importantly, the 1% B sample shows the most prominent peaks, which is in line with the ICP studies.
Structural characterization by solid-state NMR
Fig. 4 summarizes the 29Si MAS-NMR spectra and Table 2 specifies the line shape parameters of their deconvolution components. The spectrum of the B-free sample comprises three principal contributions: the dominant one near −110 ppm can be assigned to Si(4) units, the shoulder near −100 ppm to some residual Si(3)OH groups and the weak feature near −90 ppm to a few Si(2)(OH)2 units. While the intensities of these resonances are subject to some variations, they do not show a systematic dependence on B2O3 content. Note, however, that the Si(4) component can be fitted to two distinct contributions near −108 and −112 ppm. As the peak ratio for these two components is independent of borate content, the two components cannot be attributed to Si(4)0B and Si(4)1B species in a straightforward manner. Rather, the fitting of this Si(4) spectral feature to two Gaussians should be considered an artificial approach to mimic the more realistic scenario that a continuous distribution of isotropic chemical shifts is present. The 29Si spectra also give no indication of any low-frequency component, suggesting the absence of Si–O–P linkages. Overall, inspection of Table 2 reveals that the silicon speciation is more or less independent of the boron content. The data are most consistently interpreted in terms of a fully polymerized Si(4) network with silanol defects. There is no evidence that the introduction of boron into these glasses (and the associated demand for cations to charge compensate anionic borate species) affects the connectivity of the silicate network. There is also no strong evidence for the incorporation of B(4) units into the silicate framework from these data, even though some subtle changes in the overall appearance of the line shapes in the glasses with higher boron contents may point into this direction.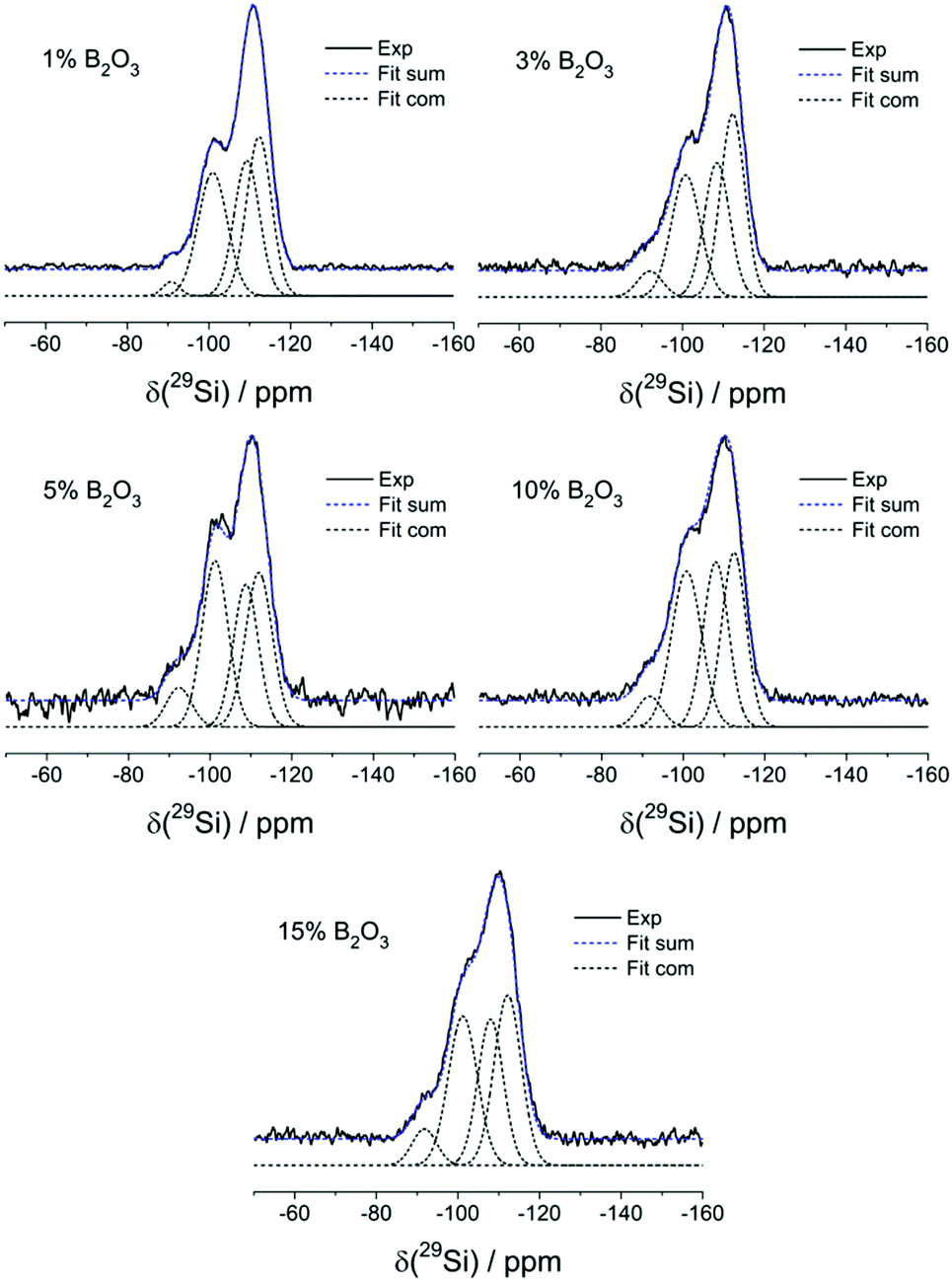 | ||
| Fig. 4 Experimental and simulated 29Si MAS-NMR spectra of B2O3 doped MBGs. | ||
| mol% B2O3 | Component | δ csiso/ppm ± 0.5 | FWHM/ppm ± 0.5 | Area/% ± 2% |
|---|---|---|---|---|
| 15 | 1 | −112.2 | 7.8 | 34 |
| 2 | −108.0 | 7.3 | 27 | |
| 3 | −101.2 | 8.4 | 32 | |
| 4 | −91.8 | 7.1 | 7 | |
| 10 | 1 | −112.4 | 7.0 | 31 |
| 2 | −108.0 | 7.3 | 30 | |
| 3 | −100.8 | 8.6 | 34 | |
| 4 | −91.8 | 7.1 | 5 | |
| 5.0 | 1 | −112.0 | 7.8 | 32 |
| 2 | −109.0 | 7.2 | 27 | |
| 3 | −101.2 | 7.5 | 33 | |
| 4 | −92.4 | 7.3 | 8 | |
| 3.0 | 1 | −112.3 | 7.0 | 37 |
| 2 | −108.5 | 7.4 | 28 | |
| 3 | −100.8 | 8.4 | 29 | |
| 4 | −92.0 | 7.2 | 6 | |
| 1.0 | 1 | −112.3 | 7.2 | 35 |
| 2 | −109.3 | 7.3 | 31 | |
| 3 | −101.0 | 8.4 | 32 | |
| 4 | −91.0 | 4.8 | 2 | |
Fig. 5 shows the 11B MAS-NMR spectra of the present samples. All the glasses containing 5% B2O3 or less show slightly asymmetric Gaussian line shapes, as characterized by an isotropic chemical shift near −1.1 ± 0.5 ppm. Based on its chemical shift and the absence of a nuclear electric quadrupolar effect upon the line shape, we can assign this signal to four-coordinated boron (B(4)) species. To account for the asymmetry, two closely spaced Gaussian components are used in the fitting of the B(4) spectral line shape for all the samples. In the two samples with the highest B2O3 contents (10 and 15 mol%), respectively, the B(4) peak is substantially broader and its isotropic chemical shift moves towards −0.5 ppm. In addition, there is clear evidence of another spectral component, whose line shape can be fitted to a spectrum influenced by quadrupolar interaction within the regime of second-order perturbation theory. The parameters, isotropic chemical shift, δcsiso = 19.0 ± 0.5 ppm, nuclear electric quadrupolar coupling constant, CQ = 2.60 ± 0.05 MHz, and electric field gradient asymmetry parameter, ηQ = 0.35 ± 0.05 do not allow one to decide unambiguously whether this species is a symmetric B(3) unit or an anionic three-coordinate borate species bound to a non-bridging oxygen, i.e. a B(2) unit. The chemical shift suggests that the B(3) units are predominantly linked to other B(3) or B(4) units via B–O–B linkages. We find no evidence for B(3)–O–Si linkages which would give rise to line shape components with lower chemical shifts, near 10–12 ppm.35 While the value of the asymmetry parameter appears rather large for a B(3) species, a line shape of this kind will also arise if there is a distribution of quadrupolar coupling constants and/or chemical shifts. Due to the underdetermined character of this fitting problem it is generally not possible to extract unique distribution functions of these parameters from such spectra (Table 3).
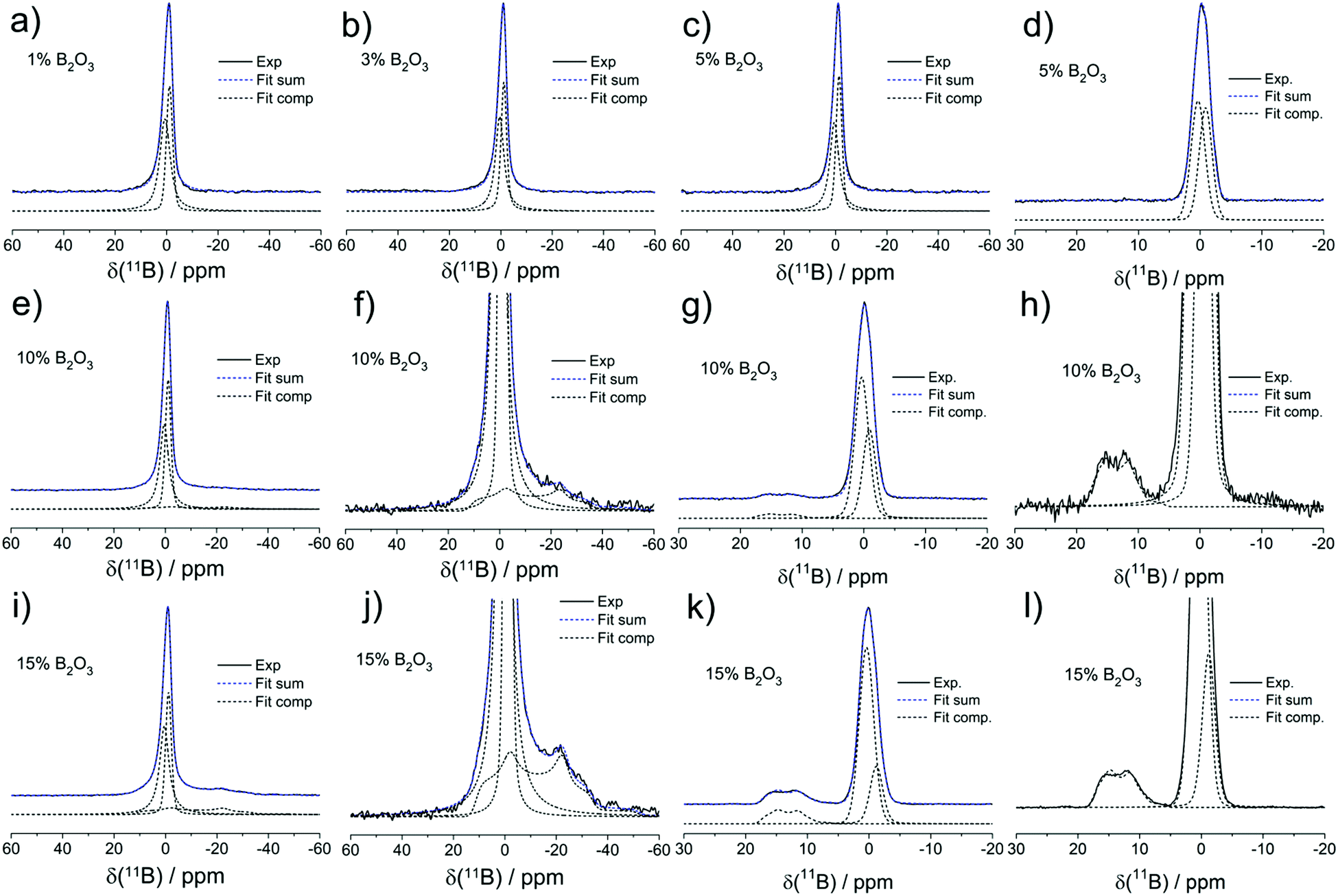 | ||
| Fig. 5 Field-dependent experimental and simulated 11B MAS-NMR spectra of boron oxide modified MBGs with different B2O3 contents: (a) 1 mol%, 5.64 T; (b) 3 mol%, 5.64 T; (c) 5 mol%, 5.64 T; (d) 5 mol%, 14.1 T; (e) 10 mol%, 5.64 T, (f) 10 mol%, 5.64 T, vertically expanded; (g) 10 mol% 14.1 T; (h) 10 mol% 14.1 T, vertically expanded, (i) 15 mol%, 5.64 T, (j) 15 mol%, 5.64 T, vertically expanded, (k) 15 mol%, 14.1 T, (l) 15 mol%, 14.1 T, vertically expanded. | ||
| mol% B2O3 | Component | δ csiso/ppm ± 0.5 ppm | C Q/MHz ± 0.1 MHz | η Q | FWHM/ppm ± 0.5 ppm | Area fractions/% ± 1% |
|---|---|---|---|---|---|---|
| (A) | ||||||
| 15 | 1 | 15.2 | 2.5 | 0.35 | 18 | |
| 2 | 0.5 | 4.4 | 49 | |||
| 3 | −1.2 | 2.7 | 33 | |||
| 10 | 1 | 15.4 | 2.6 | 0.35 | 8 | |
| 2 | 0.4 | 4.0 | 52 | |||
| 3 | −1.1 | 2.6 | 40 | |||
| 5.0 | 1 | 0.4 | 4.2 | 56 | ||
| 2 | −1.4 | 2.7 | 44 | |||
| 3.0 | 1 | 0.4 | 4.0 | 56 | ||
| 2 | −1.2 | 2.6 | 44 | |||
| 1.0 | 1 | 0.7 | 4.9 | 60 | ||
| 2 | −1.1 | 2.9 | 40 | |||
| (B) | ||||||
| 15 | 1 | 18.1 | 2.7 | 0.39 | 12 | |
| 2 | 0.4 | 4.7 | 67 | |||
| 3 | −1.2 | 2.4 | 21 | |||
| 10 | 1 | 18.5 | 2.7 | 0.37 | 4 | |
| 2 | 0.4 | 2.6 | 60 | |||
| 3 | −0.9 | 2.5 | 35 | |||
| 5.0 | 1 | 0.4 | 2.6 | 53 | ||
| 2 | −0.9 | 2.5 | 47 | |||
Thus, the high value of ηQ might be an artifact created by our fitting approach of using a singular value of CQ when there is really a distribution present. Based on the spectra observed, as well as on charge balance considerations discussed further below, we attribute the trigonal borate species to uncharged B(3) units (BO3/2 groups) linked predominantly to other borate species within cyclic or oligomeric borate clusters. The formation of such species, which would tend to clog the micropores may explain the sudden drop in surface area in going from 10 to 15 mol% B2O3.
To probe connectivity and spatial proximity of these boron species with phosphorus, 11B{31P} REDOR experiments were conducted on two representative glasses. The results are shown in Fig. 6 along with data for the model compound BPO4.
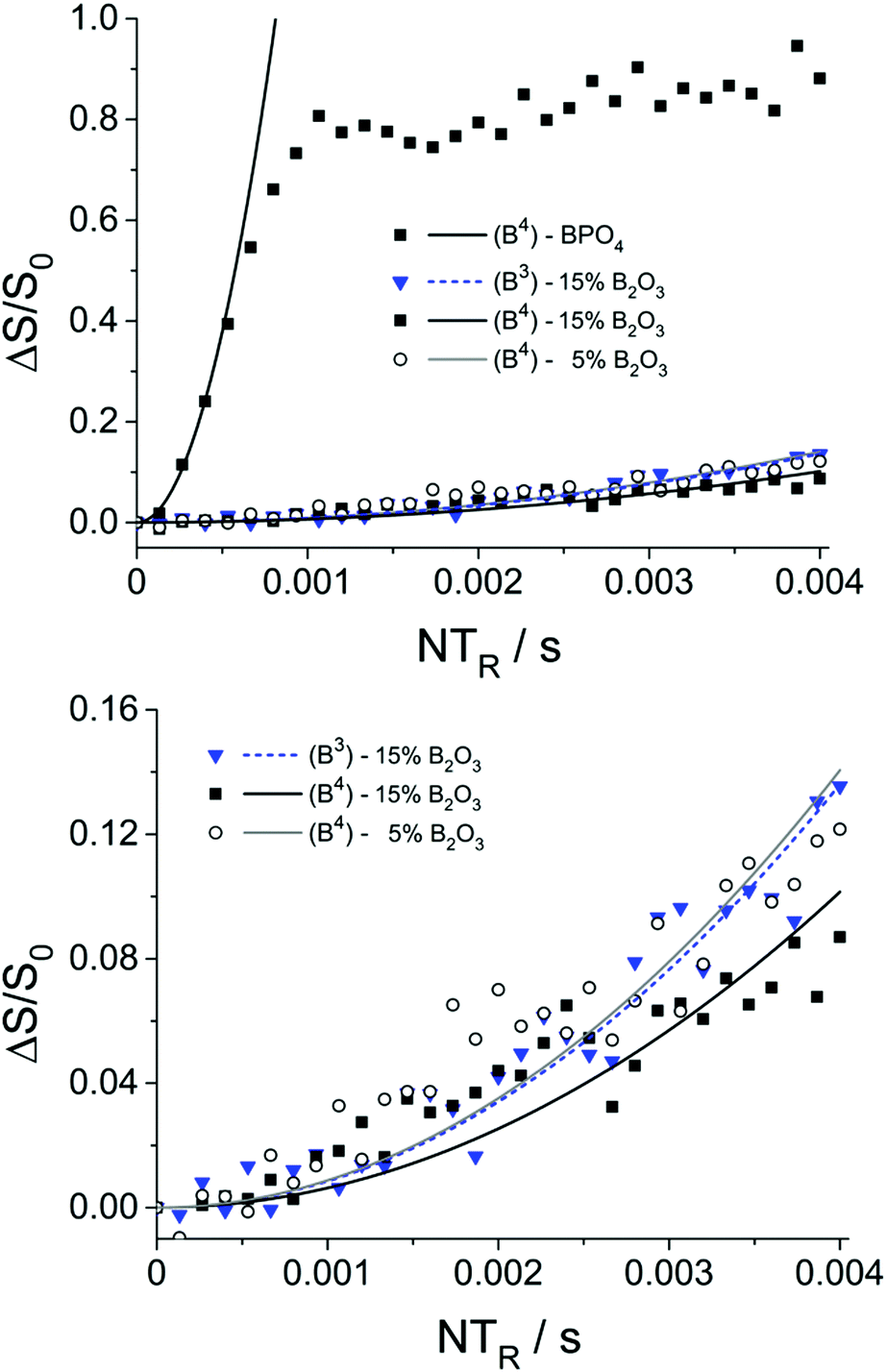 | ||
| Fig. 6 11B{31P}REDOR results and parabolic analyses (solid curves) of boron-doped MBGs containing 5 and 15 mol% B2O3. Data for the model compound BPO4 are shown for comparison. Due to the limited signal-to-noise ratio of the data at longer evolution times, the expected oscillatory behavior of the REDOR curve of BPO436 is not clearly evident in this data. | ||
The M2(B–P) values extracted from these plots, using eqn (1), are very small for both tetrahedral boron (0.05 × 106 and 0.07 × 106 rad2 s−2 for glasses with 15 and 5 mol% B2O3, respectively) and trigonal boron (0.06 × 106 rad2 s−2 for glass with 15 mol% B2O3), compared to BPO4 (11.2 × 106 rad2 s−2). Considering that the B⋯P distances across a B–O–P linkage in the glasses are of similar magnitude as those in the crystalline model compound BPO4 (4B⋯P distances at 277 pm) the result shows that the number of B–O–P linkages in the present glasses is negligible. Nevertheless, based on the observation of a non-zero REDOR effect (Fig. 6, bottom) we can conclude a spatial proximity between the boron and the phosphorus species, indicating that both components coexist within the same phase and are not separated in different phases.
Fig. 7 summarizes the results from 31P MAS-NMR. Peak deconvolutions and assignments are summarized in Table 4. In the absence of boron, one dominant peak at 3.4 ± 0.2 ppm is observed, which can be assigned to the expected orthophosphate (P(0)) species. In glasses having B2O3 contents >5 mol% a minor component near −2.9 ppm is observed, suggesting the formation of a P(1) – type species. Here n, the superscript of the P(n) notation, is the number of bridging oxygen atoms (rather than the number of P–O–P linkages that would be specified in the frequently used Q(n) nomenclature). As the B content is increased, the feature near −2.9 ppm gradually becomes stronger, suggesting a systematic increase of the P(1) species concentration with increasing B2O3 content. Both signals show minimal effects in 31P{11B} REAPDOR experiments (data not shown), again confirming the absence of significant P–O–B connectivity.
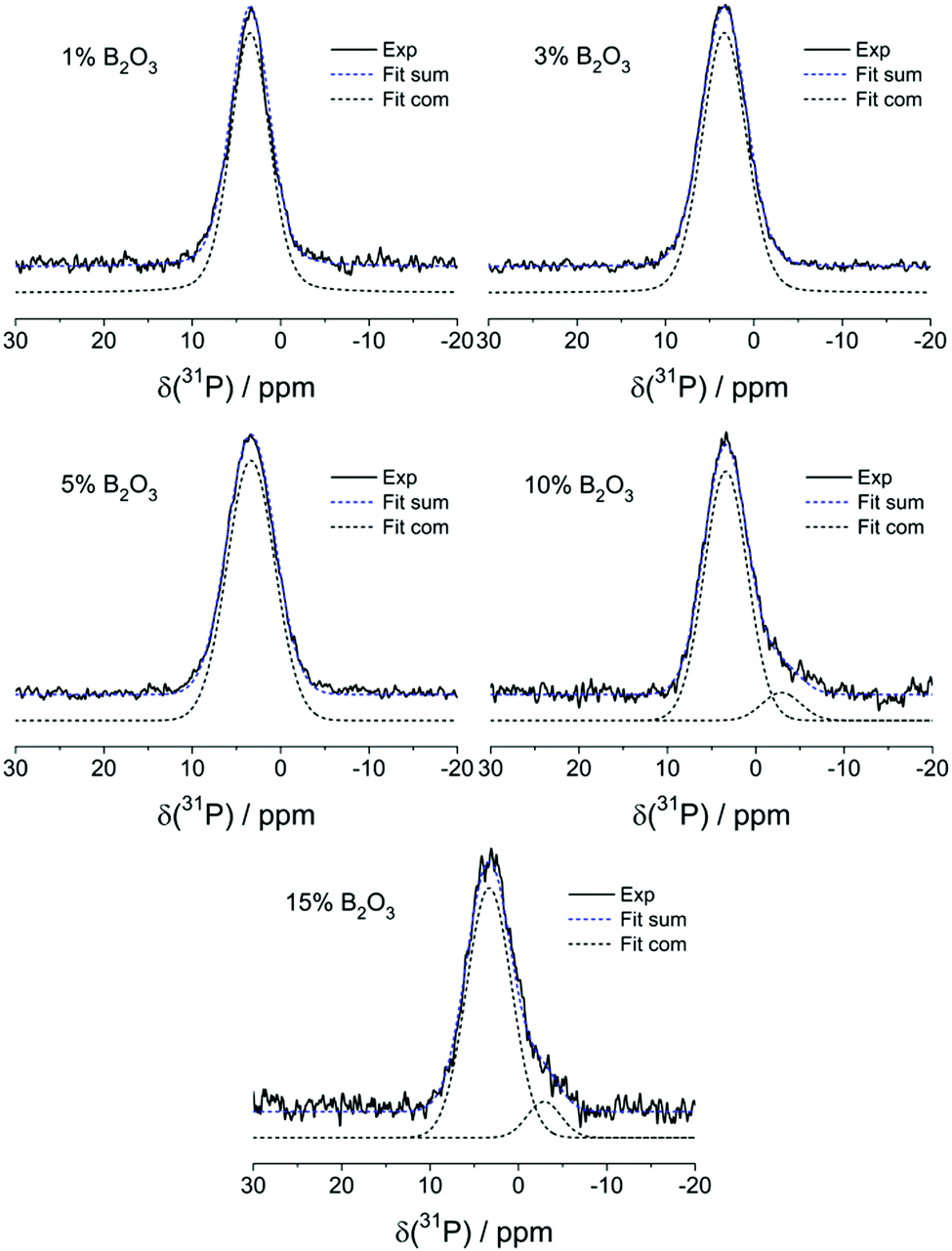 | ||
| Fig. 7 Experimental and simulated 31P MAS-NMR spectra of B2O3 doped MBGs. | ||
| mol% B2O3 | Component | δ csiso/ppm ± 0.5 ppm | FWHM/ppm ± 0.5 ppm | Area fractions/% ± 2% |
|---|---|---|---|---|
| 15 | 1 | 3.3 | 6.0 | 90 |
| 2 | −2.9 | 4.5 | 10 | |
| 10 | 1 | 3.4 | 5.8 | 91 |
| 2 | −2.9 | 5.1 | 9 | |
| 5.0 | 1 | 3.4 | 6.3 | 100 |
| 3.0 | 1 | 3.4 | 5.9 | 100 |
| 1.0 | 1 | 3.5 | 5.2 | 100 |
Fig. S5 (ESI†) shows 1H MAS-NMR spectra, revealing a dominant broad line near 5.0 ppm and two distinct sharp components near 1.0 and 3.4 ppm. While the signal near 5.0 ppm shows the typical signature of water adsorbed in the internal pores, the 1.0 ppm peak can be attributed to Si–OH groups. The peak near 3.4 ppm represents a distinct and well-defined hydrogen species, and could possibly be assigned to a Broensted site, a HPO42− species or an OH group linked to a B(4) unit. The latter two possibilities could be ruled out by 1H{31P} REDOR and 1H{11B} REAPDOR experiments, which showed no dipolar recoupling effects. While a definitive assignment of this signal is not possible at the moment, we note their undiminished presence in boron-free Al-containing MBG samples.
Discussion and conclusions
The results of the present study indicate that borate modification of mesoporous bioactive glasses can enhance their mechanical properties as well as their bioactivity in SBF solutions.TEM and BJH analysis confirm that, even at high concentrations, the incorporation of borates has no significant effect on the pore ordering or on the average pore diameter. The observed gradual decrease in specific surface area with increasing boron concentration is thus attributable to a diminished contribution by micropores within the glass network, such as through pore blocking.
ICP results reveal a quick and quantitative boron release into the SFB solution in direct correlation between the boron release and the initial boron concentration in the glasses and confirm that the boron species are quickly and quantitatively released into the SBF solution. This suggests that the boron species are not incorporated within the glass network, but rather weakly bound on the interior surface of the mesopores, resulting in the aforementioned blocking of micropores. The presence of boron accelerates the release of Si from the silicate network. The increasing amounts of Si being released with higher boron concentrations may facilitate a collapse of the pore network and loss of internal surface area. This loss in porosity may restrict the re-uptake of Ca in the form of HCA. Consequently, there is a compromise between Si release/pore collapse and Ca release/uptake, which appears to be optimal at boron concentrations between 1% and 3%.
The mechanical properties improve considerably with boron incorporation. Again, it appears that the samples with 1–3% B offer the best results, i.e. a modulus (13–16 GPa) that is just slightly lower than that of cortical bone, and hardness (about 1 GPa), which is not too strong for bone transplant applications.34
The NMR results indicate that – contrary to the situation in 45S5 bioglasses – the phosphorus speciation in the present samples is significantly influenced by the B2O3 component. While at low boron content only P(0) species can be detected, there is clear evidence for P(1) species at higher borate contents. This can be explained by the fact that the anionic B(4) units formed are competing with the phosphate anions for charge compensation by Ca2+ and possibly protonic species. Whether these P(1) units are pyrophosphate groups or end groups linking to silica with P–O–Si linkages cannot be determined at the present stage. There is no clear evidence that the B(4) units are integrated into the silica framework. Both, changes in 11B chemical shift and the appearance of B(3) species at B2O3 contents ≥10 mol%, suggest the appearance of B(4)–O–B(3) linkages, which is consistent with previous results in the literature obtained on bioglasses.21,22 The phosphate species in bioactive glasses is predominantly of the orthophosphate (P(0)) type, if the inventory of modifier cations is sufficient to balance the total charge of all of the anionic species present.14 Regarding this model (and not considering the presence of protonic species), the B-free glass of the present study was designed to be perfectly charge-balanced as the amount of Ca2+ ions (15 mol%) present is sufficient to compensate the charge of the 5 mol% of orthophosphate (PO43−) ions. For borate containing glasses, the total concentration A of accumulated negative (anionic) charges is given by:
| A = 10 × [3 × f(P(0)) + 2 × f(P(1))] + 2x × f(B(4)) mol% | (2) |
| C = 2 × 15 mol% + z | (3) |
Finally, the structural effects of boron-doping of MBGs are distinctly different from those observed for aluminum doping.23 In the latter materials, there is definitive evidence for a significant amount of Al–O–P linkages associated with six-coordinated Al species; furthermore, the anionic Al(4) species are integrated in the silicate network, resulting in strong Al–O–Si linkages charge compensated by Ca2+ ions. While both of these structural features have the desired effect to increase the mechanical strength of these materials, they also impede the disintegration of the silicate network, as well as the release of phosphate and calcium, leading to significantly decreased bioactivities. In contrast, borate incorporation results in the desired combination of increased mechanical stability and bioactivity. This result is attributed to (1) a borate distribution dominantly at the pore surface, thus maintaining the highly ordered mesoporous structure of the material; (2) the absence of B–O–P connectivity as indicated by the REDOR results; (3) a weaker Coulombic interaction between the tetrahedral BO4 units and the charge-compensating Ca2+ ions; (4) at high borate contents the formation of hydrolytically unstable B–O–B linkages involving B(3) units. The concomitant reduction in surface area at these high borate concentrations can be understood in terms of the formation of polymeric or cyclic borate ring structures containing B(3) and B(4) units, which clog the mesopores and/or their openings. Overall, the results of the present study suggest that borate-modified MBGs at the level of 1–3 mol% B2O3 are a promising class of bioactive materials with improved mechanical properties, which warrant further study. In view of the remarkably different structural modification mechanisms observed in the B2O3 and Al2O3 containing systems it will be of interest to study the competition of both processes in co-doped systems. These studies are currently being pursued in our laboratories.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
TEM analysis was carried out using the facilities at the University Service Centre for Transmission Electron Microscopy, TU Wien, Austria. H. E. would like to acknowledge the Brazilian funding agencies FAPESP (CEPID Project 2013/07793-6) and CNPq. H. B. also thanks for support from the Deutsche Forschungsgemeinschaft.References
- L. L. Hench, J. Mater. Sci.: Mater. Med., 2006, 17, 967–978 CrossRef CAS.
- L. L. Hench, R. J. Splinter, W. C. Allen and T. K. Greenlee, J. Biomed. Mater. Res., 1971, 5, 117–141 CrossRef.
- W. P. Cao and L. L. Hench, Ceram. Int., 1996, 22, 493–507 CrossRef CAS.
- R. Li, A. E. Clark and L. L. Hench, J. Appl. Biomater., 1991, 2, 231–239 CrossRef CAS PubMed.
- X. Yan, C. Yu, X. Zhou, J. Tang and D. Zhao, Angew. Chem., Int. Ed., 2004, 43, 5980–5984 CrossRef CAS PubMed.
- C. J. Brinker, Y. Lu, A. Sellinger and H. Fan, Adv. Mater., 1999, 11, 579–585 CrossRef CAS.
- I. Izquierdo-Barba and M. Vallet-Regi, Solid State Sci., 2011, 13, 773–783 CrossRef CAS.
- M. Cicuéndez, M. T. Portolés, I. Izquierdo-Barba and M. Vallet-Regí, Chem. Mater., 2012, 24, 1100–1106 CrossRef.
- H. S. Yun, S. E. Kim and Y. T. Hyeon, Mater. Lett., 2007, 61, 4569–4572 CrossRef CAS.
- A. López-Noriega, D. Arcos and M. Vallet-Regí, Chem. – Eur. J., 2010, 16, 10879–10886 CrossRef PubMed.
- C. T. Wu, Y. F. Zhang, Y. F. Zhu, T. Friis and Y. Xiao, Biomaterials, 2010, 31, 3429–3438 CrossRef CAS.
- C. Kohlhauser, C. Hellmich, C. Vitale-Brovarone, A. R. Boccaccini, A. Rota and J. Eberhardsteiner, Strain, 2009, 45, 34–44 CrossRef.
- N. Sharmin, A. J. Parsons, C. D. Rudd and I. Ahmed, J. Biomater. Appl., 2014, 29, 639–653 CrossRef CAS PubMed.
- P. Balasubramanian, T. Buttner, V. M. Pacheco and A. R. Boccaccini, J. Eur. Ceram. Soc., 2018, 38, 855–869 CrossRef CAS.
- C. T. Wu and J. Chang, J. Controlled Release, 2014, 193, 282–295 CrossRef CAS PubMed.
- R. Patricia, R.-N. Aleixandre and S.-S. Manuel, Tissue Eng., Part A, 2015, 21, 2662–2672 CrossRef.
- O. Sych, O. Gunduz, N. Pinchuk, G. E. Stan and F. N. Oktar, J. Aust. Ceram. Soc., 2016, 52, 103–110 CAS.
- F. Baino, M. Marshall, N. Kirk and C. Vitale-Brovarone, Ceram. Int., 2016, 42, 1482–1491 CrossRef CAS.
- H. B. Pan, X. L. Zhao, X. Zhang, K. B. Zhang, L. C. Li, Z. Y. Li, W. M. Lam, W. W. Lu, D. P. Wang, W. H. Huang, K. L. Lin and J. Chang, J. R. Soc., Interface, 2010, 7, 1025–1031 CrossRef CAS PubMed.
- H. Eckert, J. Sol-Gel Sci. Technol., 2018, 88, 263–295 CrossRef CAS.
- Y. Yu and M. Edén, RSC Adv., 2016, 6, 101288–101303 RSC.
- Y. Yu, B. Stevensson and M. Edén, J. Phys. Chem. B, 2017, 121, 9737–9752 CrossRef CAS PubMed.
- S. Melchers, T. Uesbeck, O. Winter, H. Eckert and D. Eder, Chem. Mater., 2016, 28, 3254–3264 CrossRef CAS.
- M. Thommes, K. Kaneko, A. V. Neimark, J. P. Olivier, F. Rodriguez-Reinoso, J. Rouquerol and K. S. W. Sing, Pure Appl. Chem., 2015, 87, 1051–1069 CAS.
- G. M. Pharr and W. C. Oliver, MRS Bull., 1992, 17, 28–33 CrossRef.
- T. Kokubo and H. Takadama, Biomaterials, 2006, 27, 2907–2915 CrossRef CAS PubMed.
- D. Massiot, F. Fayon, M. Capron, I. King, S. Le Calvé, B. Alonso, J.-O. Durand, B. Bujoli, Z. Gan and G. Hoatson, Magn. Reson. Chem., 2002, 40, 70–76 CrossRef CAS.
- T. Gullion and J. Schaefer, J. Magn. Reson., 1989, 81, 196–200 CAS.
- M. Bertmer and H. Eckert, Solid State Nucl. Magn. Reson., 1999, 15, 139–152 CrossRef CAS.
- J. H. Van Vleck, Phys. Rev., 1948, 74, 1168–1183 CrossRef.
- G. Constantinides and F. J. Ulm, J. Mech. Phys. Solids, 2007, 55, 64–90 CrossRef CAS.
- W. J. Drugan and J. R. Willis, J. Mech. Phys. Solids, 1996, 44, 497–524 CrossRef CAS.
- M. Miller, C. Bobko, M. Vandamme and F.-J. Ulm, Cem. Concr. Res., 2008, 38, 467–476 CrossRef CAS.
- V. A. Koleganova, S. M. Bernier, S. J. Dixon and A. S. Rizkalla, J. Biomed. Mater. Res., Part A, 2006, 77A, 572–579 CrossRef CAS PubMed.
- D. Möncke, D. Ehrt, H. Eckert and V. Mertens, Phys. Chem. Glasses, 2003, 44, 113–116 Search PubMed.
- H. Eckert, Bunsenmagazin, 2008, 5, 159–179 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9tb01805k |
| This journal is © The Royal Society of Chemistry 2020 |