
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSurface modification effects on nanocellulose – molecular dynamics simulations using umbrella sampling and computational alchemy
Pan
Chen
 a,
Giada
Lo Re
bd,
Lars A.
Berglund
cd and
Jakob
Wohlert
*cd
a,
Giada
Lo Re
bd,
Lars A.
Berglund
cd and
Jakob
Wohlert
*cd
aBeijing Engineering Research Center of Cellulose and its Derivatives, School of Materials Science and Engineering, Beijing Institute of Technology, Beijing 100081, China
bDepartment of Industrial and Materials Science, Chalmers University of Technology, SE-41296 Gothenburg, Sweden
cDepartment of Fiber and Polymer Technology, KTH Royal Institute of Technology, SE-10044 Stockholm, Sweden. E-mail: jacke@kth.se
dWallenberg Wood Science Center, KTH Royal Institute of Technology, SE-10044 Stockholm, Sweden
First published on 2nd November 2020
Abstract
Topochemical modification of nanocellulose particles, in particular acetylation, is commonly used to reduce hygroscopicity and improve their dispersibility in non-polar polymers. Despite enormous experimental efforts on cellulose surface modification, there is currently no comprehensive model which considers both (a) the specific interactions between nanocellulose particles and the surrounding liquid or polymer matrix, and (b) the interactions between the particles themselves. The second mechanism is therefore frequently ignored. The present approach is based on atomistic molecular dynamics (MD) simulations, where computational alchemy is used to calculate the changes in interactions between nanocellulose and the surrounding medium (liquid or polymer) upon modification. This is combined with another method, based on potential of mean force, to calculate interactions between particles. Results show that both contributions are of equal importance for nanoparticle surface acetylation effects. The proposed method is not restricted to either cellulose or acetylation, and has the prospect to find application in a broad context of nanomaterials design.
Introduction
Topochemical surface modification is a common strategy to fine-tune properties of solid particles. As opposed to liquid and gas phases, for which a uniform probability for the course of a reaction can be assumed, solid-phase reactivity depends strongly on topographical features,1 which restrict molecular mobility. In the case of nanoparticles, the high specific surface area means that topographical features dominate interfacial interactions and surface group reactivity. Therefore, the chemistry of the building blocks needs to be controlled down to the nano-scale.2 Surface modification of nanocellulose (Fig. 1) is widely used for cellulosic plant fibers,3 cellulose nanofibrils (CNF),4 and cellulose nanocrystals (CNC).5 In this case, topochemical modification begins and further progresses in connection with distinct sites in the crystal accessible surfaces.6 These surfaces have a large abundance of highly reactive hydroxyl groups, which constitutes an opportunity to introduce specific chemical functionalities for the use of nanocellulose in novel applications.7–9 The possibility to tune characteristics such as adhesion properties10 and self-assembly11 makes topochemical surface modification one of the most exploited routes towards nanostructural control.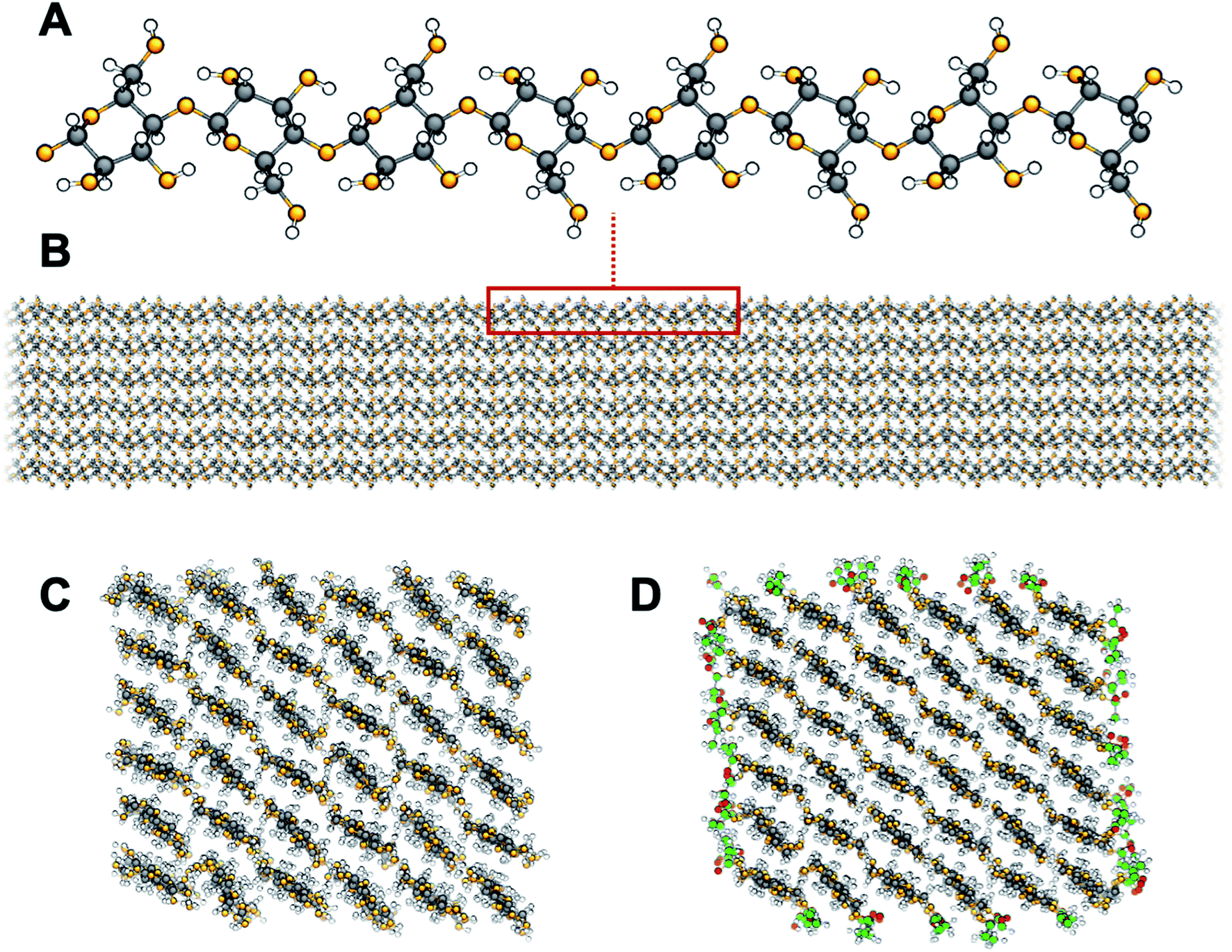 | ||
| Fig. 1 Cellulose structure: the glucan chain (A) and a model cellulose nanofibril (B). Cross sections showing the crystalline Iβ structure of unmodified (C) and surface acetylated (D) cellulose. Carbon atoms are shown in black, oxygen atoms in orange, and hydrogen atoms in white. Carbon and oxygen atoms of surface acetyl groups are shown in green and red, respectively. | ||
Chemically, cellulose is the homopolymer of 1,4 linked β-D-glucopyranose units. Axially oriented, extended glucan chains pack in a mixture of two native crystal allomorphs, of which cellulose Iβ is the most abundant in cellulose from higher plants, to form fibrils of nanoscale lateral dimensions (Fig. 1A and B). The nanofibrillar arrangement leads to a large accessible surface area with a high density of polar hydroxyl groups (∼5 OH per nm2, Fig. 1C). This becomes a problem in engineering applications of nanocellulose-based materials as it leads to high moisture sorption with poor dimensional stability and reduced mechanical properties.12–15 It is also a problem during materials processing, since nanocellulose is difficult to disperse in non-polar media, such as most matrix polymers, leading to agglomeration and ultimately poor performance.16 One phenomenon of large practical importance is hornification, which takes place among cellulose fibrils when dried from wet environment.17,18 As a consequence, the fibrils become more or less permanently aggregated to each other and are difficult to re-disperse in aqueous media. This means that nanocellulose needs to be kept in its dispersed form in water before use. From an industrial viewpoint this is problematic since it leads to large storage and transportation costs.
Topochemical modification of surface hydroxyls to more hydrophobic moieties is a common strategy to overcome the drawback of hydrophilic nanocellulose. Among the large variety of possible surface modifications, acetylation (Fig. 1D) increases the hydrophobic character of nanocellulose and decreases the self-agglomeration of cellulose fibrils, while preserving fibrillar morphology. Acetylation has been an industrially relevant chemical modification method since early 1950s for the production of modified wood and cellulosics with improved dimensional stability, reduced water sensitivity, and resistance to microbial decay.19 Interestingly, acetylation is also a strategy commonly adopted by nature to fine-tune molecular structure and function. Plants exploit acetylation of polysaccharides as a mean to control both structure and function of the cell wall. The degree of acetylation of xylan, an abundant hemicellulose found primarily in the secondary cell wall of plants, has been correlated to the resistance to cellulolytic enzyme degradation.20 Possibly the strength and elasticity of the cell walls are modulated as acetylation is directly affecting the bonding between xylan chains and cellulose nanofibrils.21 Since industrial processing of cellulosics commonly results in substantial removal of the hemicelluloses, some their induced functions may be regained by reintroducing the acetyl functionality through synthetic means.
Yano et al. reported reduced hygroscopicity and thermal expansion coefficient, and increased optical transmittance for bacterial cellulose nanofiber composites after surface acetylation.22,23 In particular, these studies indicate reduced agglomeration of the nanofibrils in the composite, which shows that surface acetylation affects the interactions between individual fibrils. Significantly reduced moisture sorption was also demonstrated for acetylated nanopapers based on both wood and bacterial cellulose, also for their nanocomposites after impregnation by acrylic or epoxy resins.24 Furthermore, surface acetylation of nanocellulose increases its dispersibility in apolar solvents, and allows the stabilization of “water in oil” Pickering emulsions, by tailoring the wettability at the oil/water interfaces.25 Also, the adsorption of pollutants can be tuned by controlled surface acetylation in nanocellulose membranes and filters for water purification.26 The enhanced hydrophobicity has also been exploited as a compatibilization strategy for nanocellulose mixing with more hydrophobic thermoplastic polymers. In this context, controlled acetylation resulted in increased cellulose thermostability and melt-processed biocomposites with improved cellulose dispersion and thermomechanical performance.27–29
The purpose of the present study is to investigate some of the mechanisms for acetylation effects in the context of cellulose nanocomposites and nanomaterials. The possibilities to predict the outcome of topochemical modification are limited due to the inherent nanoscale of the problem. This leads to the breakdown of common models for the physical behavior of these material systems, such as colloidal properties.30 A common approach to predict the compatibility of nanoparticles is to use Hansen solubility parameters,31 which are based on the similarity of the cohesive energy components (dispersive, polar, and hydrogen bonding) between the different phases. Solubility parameters has been determined experimentally for, e.g., bare32 and surface coated33 carbon nanotubes, as well as hydroxyapatite and titanium dioxide nanoparticles.34 Another method for obtaining solubility parameters is to use one of several group contribution methods (GCM).35,36 These methods treat the solubility parameter as an additive quantity, which can be determined by summing up contributions from all constituent chemical motifs in a complex molecule. For instance, Gårdebjer et al.37 used Hansen solubility parameters based on the group contribution approach for cellobiose that was chemically modified in the C6 position to predict the dispersion of surface grafted CNC, and could observe a correlation between solubility parameter and dispersibility in a range of solvents, and, interestingly, also properties of their nanocomposites with a polyethylene matrix. The compatibility of nanocellulose with polyurethane polymer matrices has also been discussed on the basis of the solubility parameters of the individual surface polymers.38
Since group contributions are developed for small solutes or low molecular weight polymers, it is, however, questionable whether they are applicable to the case of nanoparticles. Moreover, to explain increased compatibility between an acetylated surface and a non-polar medium in terms of hydrophobicity alone, based on “like dissolves like”, is at best only part of the story. When surfaces of nanocellulose particles are modified, it also changes inter-particle interactions, and particle–particle interphases on the molecular level, which is anticipated to affect aggregation. Such effects are not captured by GCM.
For the reasons discussed, molecular dynamics (MD) computer simulations on the atomistic scale are used in the present study. Effect of surface acetylation on the surface free energies of cellulose in the presence of water are quantified, for which experimental data are hard to access. The aim is twofold. Firstly, it is to improve the current understanding of the physics of acetylated cellulose fibrils by investigating molecular scale interactions. As part of this aim, changes in surface free energies of model fibrils are also compared to the solubility of small model solutes. Those are relevant for solubility parameters derived using GCM. Secondly, the aim is to introduce and validate a computational framework designed to study effects of topochemical modification of nanoparticles in general, which is based on a combination of potential of mean force calculations and computational alchemy. The method probes the change in work of adhesion from surface modification, and it permits decomposition into separate contributions from particle–particle and particle–liquid interactions. It is not limited to acetylation of cellulose in aqueous media.
Theory
Chemical surface modification of nanoparticles is used to tailor interparticle interactions, or between nanoparticles and surrounding media, to reduce aggregation and improve the dispersibility. A question often disregarded is the reason for improved dispersion. It could be from enhanced specific interactions between nanoparticles and surrounding liquid, from decreased interparticle affinity, or a combination of both. The atomistic potential of mean force (PMF) between nanoparticles in the presence of liquid is the total free energy as a function of separation distance, and thus it contains both contributions. Although it can be calculated from MD, it cannot be decomposed in a trivial way. Here, we propose a strategy for such a decomposition. The method relies on a few theoretical assumptions, discussed below.When the particle surface area is large compared to the size of the liquid molecules, both particle–particle and particle–liquid interactions essentially scale with the particle surface area. For water, this assumption is valid down to as small particle diameters as 1 nm.39 In this case the interactions can be expressed on the basis of the work of adhesion, W, which is the free energy needed to separate two phases (in the presence of a third phase), per unit contact surface area. Generally, W, can be written as a combination of surface (free) energies. For particle–particle interactions in a liquid, the work of adhesion can be expressed as the free energy change of removing one solid–solid interface, and replacing it with two new solid–liquid interfaces:
| W11(2) = 2γ12 − γ11 | (1) |
| W(0)11 = 2γ1 − γ11 | (2) |
| W(0)12 = γ1 + γ2 − γ12 | (3) |
| W11(2) = −2W(0)12 + W(0)11 + 2γ2. | (4) |
| ΔW11(2) = −2ΔW(0)12 + ΔW(0)11 | (5) |
Assuming that surface modification leaves the entropy of mixing of nanoparticles unchanged, ΔW11(2) is directly related to dispersibility: if ΔW11(2) < 0 the dispersibility is improved, if ΔW11(2) > 0 dispersibility is decreased. This shows that the effect of chemical modification on dispersibility cannot be assessed by only considering the particle–liquid interface, since this is only one contribution. It is equally important to investigate modification effects on the interactions between the nanoparticles themselves. In the present study, both these contributions are assessed using MD simulations. Since there is no single MD-based method where both terms can be determined simultaneously, we calculate them by using a combination of two independent methods. Here, ΔW11(2) can be identified as the total change in the particle–particle PMF between contact and large separations. It is thus not influenced by the functional form of the distance dependence. This term is calculated using umbrella sampling with the inter-fibril separation as reaction coordinate. The term that contains the particle–liquid adhesion, ΔW(0)12, can be calculated separately using several different methods, for instance the phantom wall method of Leroy and Müller-Plathe,40 or by simulation of nanodroplets on solid substrates.41 Here it is calculated using the method of computational alchemy, as described below, which directly probes the change in free energy from a chemical modification. Finally, the first two terms combined gives ΔW(0)11 from eqn (5).
Methods
Molecular dynamics simulations
MD simulations were performed using GROMACS 2016 (ref. 42) with a basic time step of 1 fs. The non-bonded interactions used a straight cutoff of 1.2 nm, and the long-range electrostatics was included using PME.43,44 Bonds were constrained to their equilibrium values using P-LINCS.45 Pressure was maintained at 1 atm using a Parrinello–Rahman barostat46 with semi-isotropic coupling for simulations of surfaces and fibrils, and isotropic coupling for simulations of oligomers (see below), while a constant temperature of 298 K was maintained using a Nosé–Hoover thermostat.47,48 The simulation employed the GLYCAM06 parameter set49 with the TIP3P water model.50The aim of the present study was to calculate both the surface free energy of cellulose exposed to liquid, and the particle–particle energy between fibrils. Therefore, two model systems were used, where the cellulose was either non-acetylated or acetylated. Here, cellulose was partially acetylated in the C6 position (Fig. 2A). In reality, acetylation of cellulose is heterogeneous, and other hydroxyls may be acetylated. However, since the C6 hydroxyl is the most reactive one51 this hydroxyl group only was selected. The first model was in the form of a crystalline cellulose surface in contact with water or vacuum (Fig. 2B), to specifically target the cellulose surface energy. The second model, which was used to quantify particle–particle interaction, consisted of a pair of interacting cellulose fibrils in water, that were either non-acetylated or fully C6-acetylated (Fig. 2C).
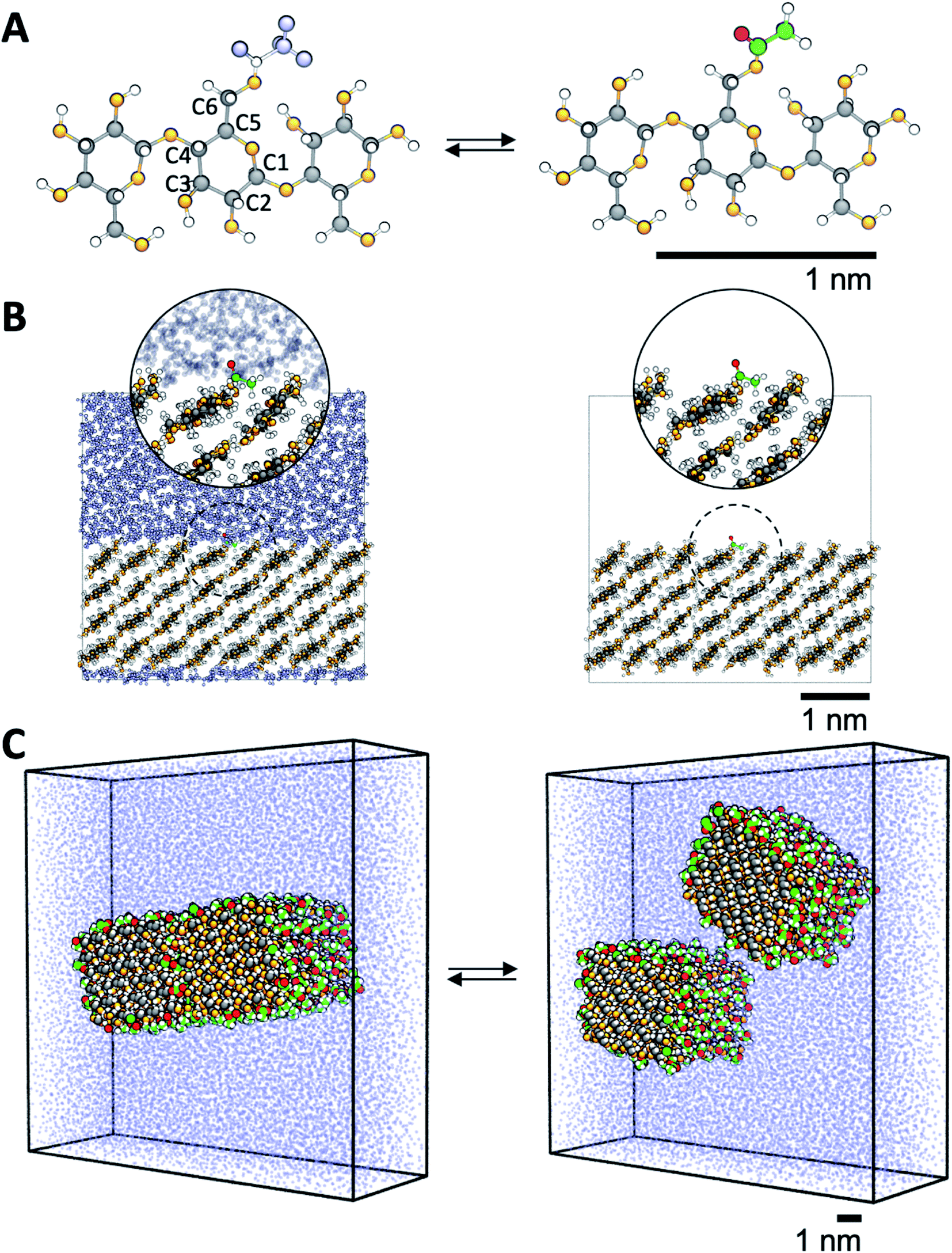 | ||
| Fig. 2 Description of the model systems. (A) Details of cellulose structure; cellotriose with the carbon numbering used. C6 hydroxyls are transformed into acetyl groups by computational alchemy, which uses dummy atoms in the unmodified state. (B) Surface acetylation is modeled using infinite slabs of cellulose in contact with both water and vacuum. (C) The cellulose–cellulose work of adhesion is calculated from atomistic PMFs in water where model cellulose nanofibrils are pulled apart using umbrella sampling simulations. Colors are the same as in Fig. 1. | ||
Both surfaces and fibril models were built from the native cellulose Iβ structure.52 The surfaces were built as one slab, four layers thick, where each chain was 8 anhydroglucose units long, exposing the ![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) 10 crystallographic plane. The model fibrils consisted of 36 chains in a 6 by 6 arrangement exposing both the 10 and 110 crystallographic planes, each chain 10 anhydroglucose units long. The fibrils were aligned parallel to each other. In both surfaces and fibrils, the cellulose chains were covalently bonded to its periodic image, thereby mimicking chains of infinite length. In addition, simulations were also performed for a single cellotriose molecule in water and vacuum, where the central residue was acetylated in the C6 position (Fig. 2A).
10 crystallographic plane. The model fibrils consisted of 36 chains in a 6 by 6 arrangement exposing both the 10 and 110 crystallographic planes, each chain 10 anhydroglucose units long. The fibrils were aligned parallel to each other. In both surfaces and fibrils, the cellulose chains were covalently bonded to its periodic image, thereby mimicking chains of infinite length. In addition, simulations were also performed for a single cellotriose molecule in water and vacuum, where the central residue was acetylated in the C6 position (Fig. 2A).
Cellulose–cellulose work of adhesion from potential of mean force calculations
The PMF between two model cellulose fibrils (acetylated and non-acetylated) in water was calculated using umbrella sampling. This gives the reversible work (free energy) for separating the two model fibrils, and can be converted into work of adhesion by dividing the result by the initial contact area. The simulations used the center-of-mass distance perpendicular to the axial direction between the fibrils as reaction coordinate. In total, 38 intermediate states were used, from fully aggregated (3.6 nm for the native CNF, 3.8 nm for the acetylated case) up to 5.6 nm (fully separated). Each state was simulated for 30 ns and used a constraining potential with a force constant of 3000 kJ mol−1 nm−2. The Full PMF was constructed using the weighted histogram analysis method (WHAM),53 and the error was estimated using bootstrapping.54Cellulose–water work of adhesion from computational alchemy
For cellulose–water interactions, a different approach is needed. To quantify the effect of acetylation, the difference in surface energy is calculated and two states are compared: (A) without acetylation and (B) with acetylation. The background is the following. The free energy difference between two states (A and B) can be calculated using thermodynamic integration, where the two states are interpolated using a single coupling parameter λ such that λ = 0 corresponds to state A, and λ = 1 to state B. By performing simulations at both the end points as well as at several intermediate values of λ (in which the derivative of the total Hamiltonian with respect to the coupling parameter is sampled), the free energy difference between A and B can be calculated using Bennet's acceptance ratio.55 Since the free energy is a state function, the actual path between the states is not important as long as the relevant thermodynamic variables can be accurately sampled. Thus, going between two chemically different states using interpolation of the interaction potentials will give the correct free energy difference, despite the actual path being non-physical. Hence, this method is termed computational alchemy.56In this work the introduction of chemical modification was inspired by studies of mutations in proteins.57 The system was constructed such that state A corresponded to the case where a specific cellulose surface C6 group was non-acetylated (i.e. chemically attached to a hydroxyl group), and state B to the case when the same group was acetylated (Fig. 2A). The acetyl group has more atoms than the hydroxyl group, and these extra atoms have to be represented also in state A. Therefore, they were included as dummy atoms, which have the same mass and bonded interaction potentials as in state B, but with the Lennard-Jones (LJ) interactions coupled to λ such that they were zero in state A, but fully turned on in state B. The hydroxyl proton was similarly turned into a carbonyl carbon, which had consequences on both bonded and non-bonded LJ parameters. Partial charges were also redistributed, but as a separate process after the change in LJ potentials. The systems were simulated at the two end points, as well as at 30 intermediate states, 20 of which were dedicated to the decoupling of LJ and bonded potentials, and 10 to the redistribution of charge. Each state was subjected to 5 ns of MD. Using this method, the absolute free energy difference, ΔG, between the acetylated and non-acetylated states was calculated. This was done in the presence of both solvent (water) and in vacuum (Fig. 2B). Utilizing that any closed thermodynamic cycle must add up to zero (Fig. 3), the change in work of adhesion from a single mutation is calculated from the double difference:
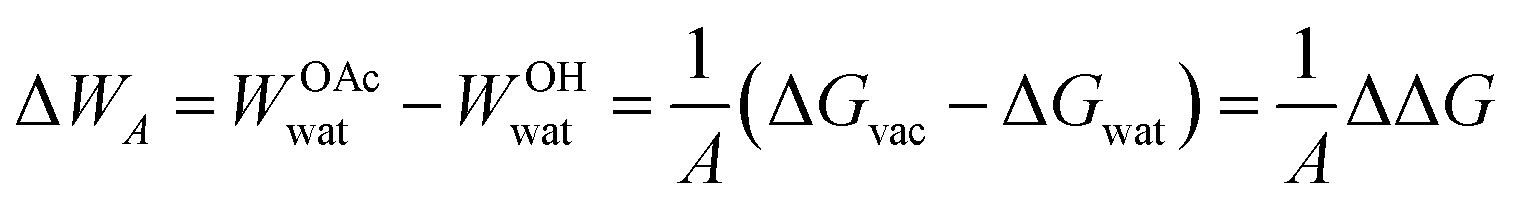 | (6) |
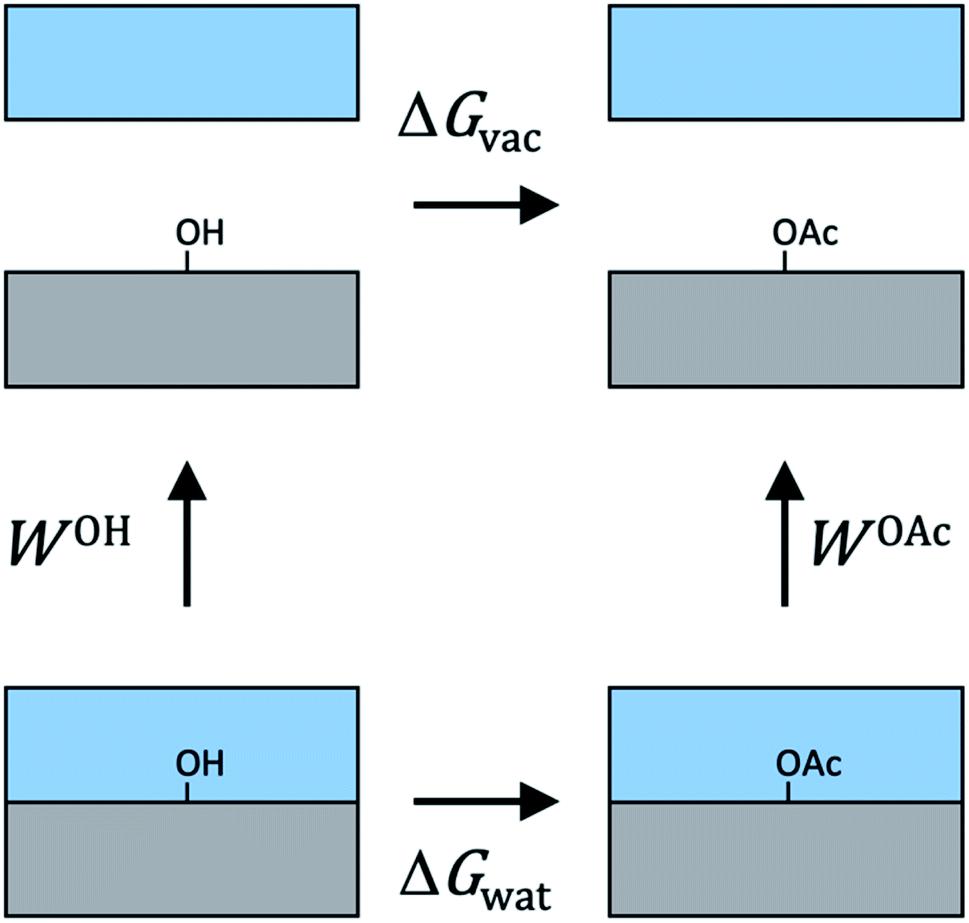 | ||
| Fig. 3 Illustration of the thermodynamic cycle used to relate works of adhesion to alchemical free energies. Gray color depicts the cellulose phase, and blue the water phase. Since free energy is a function of state, the whole cycle must add up to zero. | ||
Results
Work of adhesion between fibrils from the potential of mean force
Two cellulose model fibrils adhering to each other in water are pulled apart in order to calculate the thermodynamic work of adhesion. As starting point, the surfaces are brought in contact. This is the most favorable cellulose–cellulose interaction between two fibrils, and this configuration is stable in water on simulation time scales. The PMF as a function of separation distance was then calculated for the native as well as for the fully acetylated fibrils (Fig. 4) and normalized by initial contact area to predict work of adhesion. The PMF includes both direct fibril interaction, as well as contributions from the liquid water. The value where the PMFs level off is the total work of adhesion for complete separation. Fig. 4 shows that the adhesion energy is much higher for the native fibrils than for the acetylated ones. The difference between the two curves in Fig. 4 represents ΔW11(2), i.e. the left-hand side in eqn (5), where the superscript (2) represents liquid water. This value is −14 mJ m−2, which means that acetylation greatly reduces the affinity between fibrils in water. | ||
| Fig. 4 The potential of mean force as a function of separation distance scaled by initial contact area between non-acetylated (black curve) and acetylated (blue curve) cellulose fibrils in water. The gray areas represent standard errors calculated using bootstrapping. The plateau values represent the total work of adhesion. Images to the right show the separation of the non-acetylated fibrils at selected points along the reaction coordinate. | ||
Similar PMFs for separating two cellulose fibrils in water has been published before. Oehme et al. used the CHARMM force field to calculate the desorption energies for different cellulose surfaces,58 and Garg et al. have recently published similar simulations with the OPLS-AA force field.59 Note that in neither case they normalize the surface energies by area – the values given here are based on our estimates of the initial fibril–fibril contact. Their PMFs for the hydrophilic (10 and 110) surfaces are highly similar to the present one, although the values are slightly smaller with CHARMM parameters (approx. 12 to 19 mJ m−2 depending on surface), and higher with OPLS-AA (around 40 mJ m−2). Reasons for the discrepancy may be different interaction potentials and different choices of reaction coordinates, but also very likely different fibrillar geometries. These results, however, are in sharp contrast to the work by Paajanen et al.,60 who reports overall repulsion, also between unmodified fibrils. This is difficult to rationalize since native CNF is known to form stable aggregates. It is possible that the conflicting results in Paajanen et al. stem from the use of a finite fibril model, whereas in the present work periodic (hence infinite) fibrils are used. Without constraints from periodic boundary conditions, finite models develop substantial twisting around the chain axis, which consequently leads to reduced fit between parallel fibrils. Although twisting of CNF is possible,61 the importance of the phenomenon is unclear. If twisting takes place, the present results may be viewed as an upper limit of forces involved in aggregation.
An interesting question is by which mechanism acetylation decreases the fibril–fibril interactions. Using the formalism of eqn (5), is it due to ΔW(0)11, i.e. the specific fibril–fibril interactions, or ΔW(0)12, i.e. an effect from a change in the affinity to the liquid? The importance of the cellulose–cellulose interface was discussed in the introduction. Fig. 5 shows that the interface between acetylated fibrils has a more irregular and disordered structure at the molecular scale, compared with the seamless transition between the two native cellulose fibrils. This indicates that acetyl groups on the surface sterically hinders aggregation, which is also apparent from the smaller equilibrium separation for the non-acetylated fibrils (Fig. 4). The increased disorder of the interface also leads to visible interpenetration of water molecules. This reduced contact between the acetylated cellulose fibrils translates into a quantitative effect on ΔW(0)11.
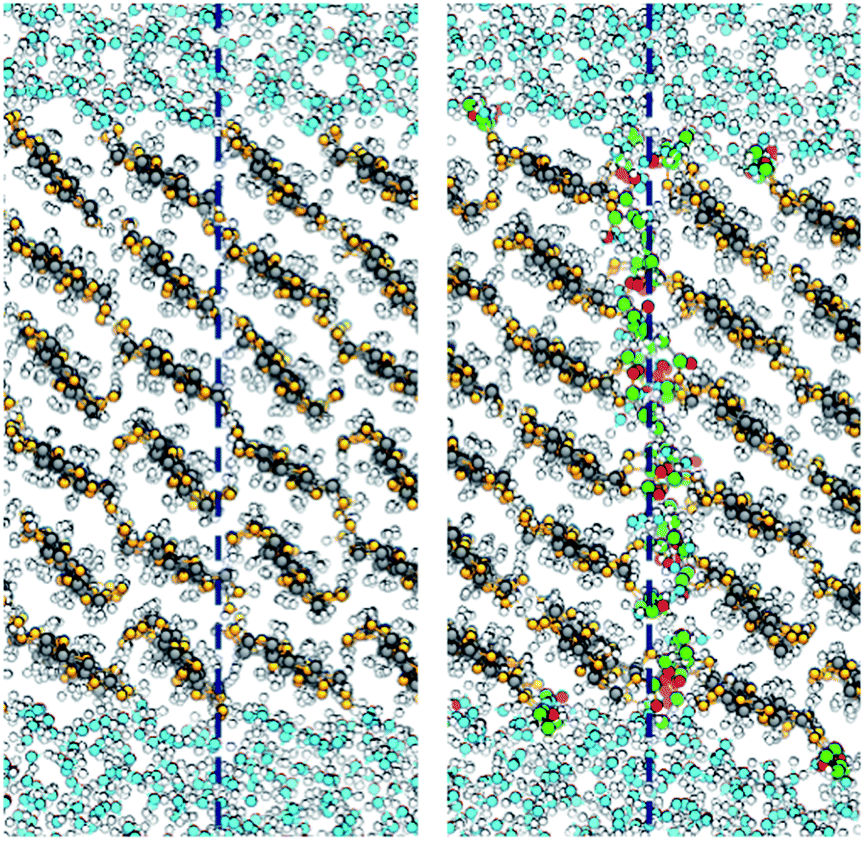 | ||
| Fig. 5 Simulation snapshots showing the interface between the aggregated fibrils: native (left) and acetylated (right). The dashed lines indicate the position of the fibril–fibril interface. Colors are the same as in Fig. 1, using green and red for surface acetyl groups. | ||
Work of adhesion between fibrils and water from computational alchemy
Computational alchemy gives the change in total free energy from a chemical modification. Here, ΔG from acetylation was calculated in simulations where surface hydroxyl groups in the C6 position were “mutated” into OAc-groups (Fig. 2A). This was done in the presence of both the liquid medium (water), and in vacuum (Fig. 2B), which permitted the change in work of adhesion to be calculated from the double difference ΔΔG (eqn (6), see Methods section). Furthermore, ΔG was evaluated for the two environments in which the “mutation” was introduced: on an otherwise (i) totally unmodified surface (DS 0) and (ii) fully (except one single site) acetylated surface (DS 1). The calculated absolute free energy changes pertaining to one mutation are shown in Table 1. Since these values are calculated from the substitution of one single surface group, the change in work of adhesion for an arbitrary degree of substitution can be calculated from| ΔW(0)12 = DS × n(ΔGvac − ΔGwat) = DS × nΔΔG, | (7) |
| Cellulose surface (DS 0) | Cellulose surface (DS 1) | Cellotriose | |
|---|---|---|---|
| ΔGwat | −84.5 (0.2) | −83.9 (0.2) | −76.7 (0.3) |
| ΔGvac | −82.2 (0.3) | −84.3 (0.2) | −92.7 (0.4) |
| ΔΔG | −1.3 (0.3) | −4.8 (0.4) | −16.0 (0.5) |
In eqn (7), ΔΔG is a measure of the change in work of adhesion from the transformation of one single hydroxyl group into an acetyl group. However, from Table 1 it is observed that ΔΔG depends on the overall DS, i.e. the free energy cost of introducing one acetyl group depends on how many groups that were already modified. Effects of non-additivity have been seen before in MD simulations of chemically modified model surfaces.62 Specifically, it has been noted that a hydrophilic substitution on a predominantly hydrophobic surface perturbs the proximal water structure to a larger extent than the opposite case when a hydrophobic substitution is performed on a hydrophilic surface.63 This is consistent with a larger ΔΔG at DS 1 than at DS 0. A varying ΔΔG is a complication for the present case. If one imagines that the surface acetyl groups are placed one by one, the ΔΔG for each of them depends on the placement of all the previous ones. We address this by introducing an average ΔΔG per surface acetyl group, <ΔΔG>, as the mean of the two extreme cases (DS 0 and DS 1) given in Table 1. The change in work of adhesion is computed from
| ΔW(0)12 = DS × n<ΔΔG>, |
10) plane. With DS = 1 (full acetylation), this results in (after unit conversion) ΔW(0)12 = −9.3 mJ m−2, which is a measure of the acetylation effect on fibril–water interaction. The value is negative; so when surface hydroxyl groups are transformed into acetyl groups, the work of adhesion between the modified cellulose surface and water is decreased. The cellulose surface thus becomes more hydrophobic by acetylation.
To further investigate acetylation effects at the cellulose–water interface, mass distribution profiles of water are displayed in Fig. 6, as a function of distance from the cellulose surface. Data were calculated from equilibrium simulations at both DS 0 (native cellulose) and DS 1 (fully acetylated). The difference between the two cases is striking. Water accumulates at the native surface, which gives rise to a strong peak (solid blue line) where the water density is larger than the average density of bulk water (a value greater than one in Fig. 6). Such structures are typical for solid–water interfaces where average interactions are attractive, i.e. hydrophilic, and this behavior is a very important characteristic of native cellulose fibrils.
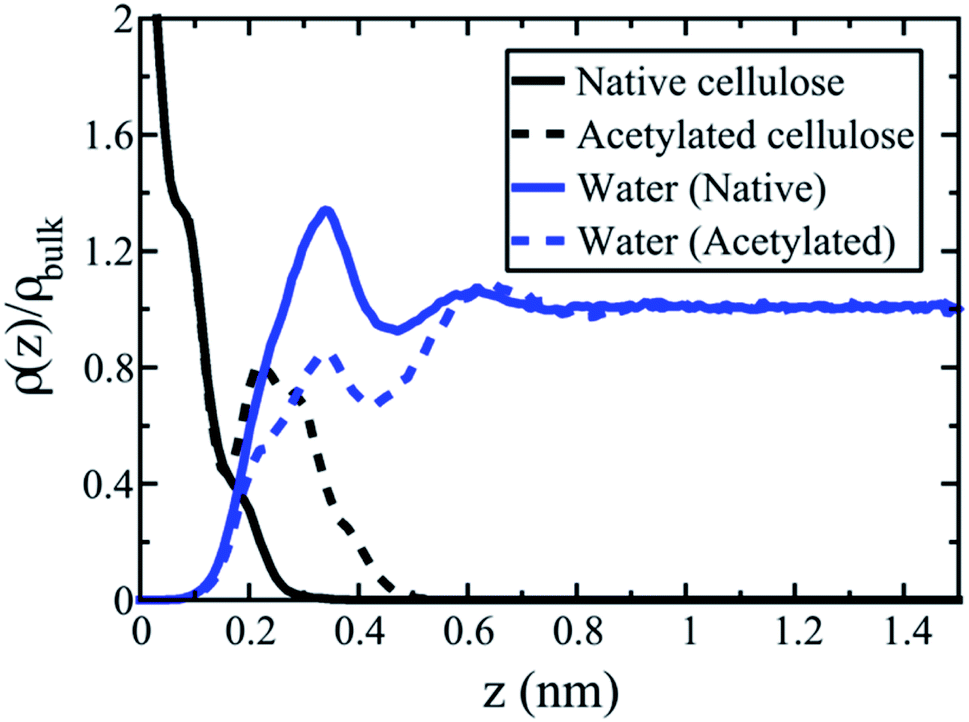 | ||
| Fig. 6 The interphase region in cellulose–water. Mass distribution profiles of water are presented perpendicular to the cellulose surface at DS 0 (native cellulose, solid lines) and DS 1 (acetylated cellulose, dashed lines). The y-axis shows the density of cellulose (black) and water (blue) relative to that of bulk water density (horizontal line at larger distances z). The x-axis is the distance z from the cellulose surface, measured (quite arbitrarily) from the first cellulose peak. | ||
Acetylation of the cellulose surface (DS 1) changes the water density profile significantly. The first peak remains but is substantially reduced, and the average water density is no longer larger than in bulk state. The relatively bulky acetyl groups induce a depletion of water molecules close to the interface, which makes the density profile approach the state of a hydrophobic surface. In this sense, the surface is dewetted.39 The acetylated surface is indeed more hydrophobic than native cellulose, although there are still a significant number of remaining surface hydroxyls. At molecular scale, the surface is therefore better described as patchy, with alternating hydrophobic and hydrophilic regions. Hydrophobicity is not a question of either or, it is rather a continuous scale. So the statement “acetylation makes cellulose hydrophobic”, is moderated; acetylation makes the average behavior of cellulose more hydrophobic. The simulations support this statement through the data for ΔW(0)12. Thus, the method can quantify to what extent acetylation leads to hydrophobization of the cellulose surface. It can obviously also be used to quantify effects from other types of topochemical cellulose modification.
Solubility parameters
As already mentioned, one commonly used approach to predict dispersibility of nanoparticles is to use solubility parameters31 based on group contribution methods GCM.35,36 Therefore, it is of interest to investigate the effects of changing functional groups in small solutes (the basis for group contribution parameters) and compare to effects from surface modification. To this end, the free energy of transforming the C6 hydroxyl in the central residue in cellotriose into an O-acetyl group was calculated in water and in vacuum. The corresponding ΔΔG is a measure of the “hydrophobization effect” and can be interpreted as the (negative) change in hydration free energy of the whole molecule. This can be directly compared to changes in free energy in Table 1 for surface modifications of fibrils. In Table 1, the calculated ΔΔG for the cellotriose is −16.0 kJ mol−1, which is significantly larger in magnitude than for the fibril surfaces, both DS 0 (−1.3 kJ mol−1) and DS 1 (−4.8 kJ mol−1). The free cellotriose is apparently hydrophobized to a much larger extent than a cellulose nanoparticle. The reason is that a free cellotriose molecule is more solvated than a glucan chain at the surface of a fibril. As a consequence, acetylation in C6 perturbs the local water structure much more for cellotriose.This highlights a problem with the use of group contribution methods for solubility parameters in the context of nanoparticle aggregation. Although the approach is sensitive to chemical structure, it does not explicitly take the surface topography of the cellulose nanoparticles into account. It is concluded that the use of solubility parameters from small cellulose fragments is likely to greatly overestimate the degree of hydrophobization of nanocellulose fibrils. Here molecular simulation is advantageous for quantitative assessment of surface energies on the nanoparticle scale.
The net effect of cellulose acetylation
The starting point of this investigation was that topochemical surface acetylation of nanocellulose affects both the interactions with the liquid medium, and the interactions between the nanofibrils themselves. The objective was to investigate both effects separately. The results presented clarify to what extent the acetylation of a cellulose fibril surface decreases its affinity for water, and makes it more hydrophobic. Indeed, the simulations show that the cellulose–water work of adhesion decreases, ΔW(0)12 < 0, as expected. If this was the only effect from acetylation, then neat cellulose fibrils surrounded by water would tend to aggregate more when acetylated.Complimentary simulations, however, contradict increased aggregation tendency. The adhesion calculated from the PMF between acetylated fibrils (Fig. 3) is much lower than that between native cellulose fibrils, which can be expressed as ΔW11(2) < 0. As a consequence, acetylation will lower the tendency for aggregation. For eqn (5) to hold, this means that the fibril–fibril adhesion in the absence of water decreases by more than twice the amount of the fibril–water adhesion, |ΔW(0)11| > 2|ΔW(0)12|. With numbers from the simulations (ΔW11(2) = −14 mJ m−2 from the PMFs, and ΔW(0)12 = −9.3 mJ m−2 from computational alchemy), we obtain ΔW(0)11 = −32.6 mJ m−2. Thus, it is concluded that the hydrophobization effect from acetylation is cancelled by the fact that the direct cellulose–cellulose work of adhesion is decreased even more. The net result is, against intuition, that cellulose “compatibility” with water is improved in the acetylated cellulose–water system, although cellulose–cellulose aggregation is hindered.
The model, however, is idealized compared with complex experimental nanocellulose material systems. The presence of hemicelluloses in wood-based cellulose systems will for instance make aggregation weaker.64 Further, in the model used here cellulose nanofibrils were perfectly aligned. This gives the possibility for the fibrils to fuse upon contact and form one continuous crystalline phase. In reality, the cellulose–cellulose contact points may be less perfect due to other configurations (anti-parallel or at an angle), nanofibril twisting,61,65 or contacts between different crystallographic planes.58 Such imperfections reduce the possibility to fuse completely, and make individual fibrils maintain their integrity as separate entities while still having direct fibril–fibril contact.18,66,67 Nevertheless, the present model is relevant for any material system where cellulose nanoparticles tend to aggregate, such as in suspensions of bacterial cellulose, tunicate whiskers, CNF or CNC. It is also relevant to material applications such as melt processing of cellulose nanocomposites, or those based on delignified wood templates, and cellulose microfibrils in the cell wall of chemical wood pulp fibers, where the hemicellulose content is low.
Conclusion
In this study a methodology to quantitatively investigate the effects of topochemical surface modification of nanoparticles was introduced. In particular, both the effects on specific particle–particle interactions and the effects on the interactions with the liquid medium was considered. As a model system, surface acetylation of nanocellulose in aqueous environment was used.It was found that acetylation in the C6 position leads to hydrophobization of cellulose fibrils, in the sense that the work of adhesion between an acetylated model cellulose surface and water is decreased compared to the unmodified one. In addition, the simulations showed that acetylation in the C6 position of cellotriose in water lead to a free energy change that greatly exceeded that of surface acetylation. This has implications for the commonly used approach to use solubility parameters of the individual surface polymers to assess dispersibility of cellulose nanoparticles.
At the same time, introduction of surface acetyl groups disrupts the near-crystalline order at the interface between two aggregated cellulose nanoparticles, negatively affecting direct short-range inter-fibril interactions such as hydrogen bonding and van der Waal's interaction. Energetically, this effect is larger than the hydrophobization, so that the overall work of adhesion between cellulose surfaces in aqueous environment decreases significantly. This means that the net effect from acetylation is that it reduces the tendency for cellulose aggregation in water. These findings show that when compatibility of nanoparticles in liquid environment is assessed, consideration of solute–solvent interactions only may lead to erroneous conclusions.
The present methodology is neither limited to acetylation of cellulose, nor to aqueous environments, but can be expanded to include any type of chemical modification, solid substrate, and liquid environment (e.g. solvent or polymer melt), in a straightforward manner. This opens for the use of MD simulations as a screening tool for tailoring interfacial interactions and fine-tuning compatibility in the materials development process; at the same time providing insights of molecular-level phenomena involved.
Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
PC thanks Beijing Municipal Natural Science Foundation (2204096) and Beijing Institute of Technology Research Fund Program for young scholars. GLR acknowledges the support of Genie and Areas of Advance at Chalmers. This work was supported by the European Research Council (ERC) under the European Union's Horizon 2020 research and innovation program (grant agreement no. 742733). Computational resources were provided by the Swedish National Infrastructure for Computing (SNIC) at the PDC Center for High Performance Computing, KTH, partially funded by the Swedish Research Council through grant agreement no. 2016-07213.References
- V. V. Boldyrev, React. Solid., 1990, 8, 231–246 CrossRef CAS.
- L. Piot, D. Bonifazi and P. Samori, Adv. Funct. Mater., 2007, 17, 3689–3693 CrossRef CAS.
- M. N. Belgacem and A. Gandini, Compos. Interfaces, 2005, 12, 41–75 CrossRef CAS.
- F. Rol, M. N. Belgacem, A. Gandini and J. Bras, Prog. Polym. Sci., 2019, 88, 241–264 CrossRef CAS.
- S. Eyley and W. Thielemans, Nanoscale, 2014, 6, 7764–7779 RSC.
- J.-F. Sassi and H. Chanzy, Cellulose, 1995, 2, 111–127 CrossRef CAS.
- C. Moreau, A. Villares, I. Capron and B. Cathala, Ind. Crops Prod., 2016, 93, 96–107 CrossRef CAS.
- J. A. Sirviö, M. Visanko, J. P. Heiskanen and H. Liimatainen, J. Mater. Chem. A, 2016, 4, 6368–6375 RSC.
- E. Lizundia, T.-D. Nguyen, J. L. Vilas, W. Y. Hamad and M. J. MacLachlan, J. Mater. Chem. A, 2017, 5, 19184–19194 RSC.
- D. J. Gardner, G. S. Oporto, R. Mills and M. A. S. A. Samir, J. Adhes. Sci. Technol., 2008, 22, 545–567 CrossRef CAS.
- I. Capron, O. J. Rojas and R. Bordes, Curr. Opin. Colloid Interface Sci., 2017, 29, 83–95 CrossRef CAS.
- L. A. Berglund and T. Peijs, MRS Bull., 2010, 35, 201–207 CrossRef CAS.
- R. J. Moon, A. Martini, J. Nairn, J. Simonsen and J. Youngblood, Chem. Soc. Rev., 2011, 40, 3941–3994 RSC.
- A. Walther, F. Lossada, T. Benselfelt, K. Kreichbaum, L. Berglund, O. Ikkala, T. Saito, L. Wågberg and L. Bergström, Biomacromolecules, 2020, 21, 2536–2540 CrossRef CAS.
- A. J. Benítez and A. Walther, J. Mater. Chem. A, 2017, 5, 16003–16024 RSC.
- K. Oksman, Y. Aitomäki, A. P. Mathew, G. Siqueira, Q. Zhou, S. Butylina, S. Tanpichai, X. Zhou and S. Hooshmand, Compos. Part A Appl. Sci. Manuf., 2016, 83, 2–18 CrossRef CAS.
- C. Moser, G. Henriksson and M. Lindström, Nord. Pulp Paper Res. J., 2018, 33, 647–650 CAS.
- H. Nilsson, S. Galland, P. T. Larsson, K. Gamstedt, T. Nishino, L. A. Berglund and T. Iversen, Compos. Sci. Technol., 2010, 70, 1704–1712 CrossRef CAS.
- R. M. Rowell, Chemical modification of wood, in Handbook of wood chemistry and wood composites, CRC Press, 2005 Search PubMed.
- D. J. Mitchell, K. Grohmann, M. E. Himmel, B. E. Dale and H. A. Schroeder, J. Wood Chem. Technol., 1990, 10, 111–121 CrossRef CAS.
- M. Busse-Wicher, T. C. F. Gomes, T. Tryfona, N. Nikolovski, K. Stott, N. J. Grantham, D. N. Bolam, M. S. Skaf and P. Dupree, Plant J., 2014, 79, 492–506 CrossRef CAS.
- S. Ifuku, M. Nogi, K. Abe, K. Handa, F. Nakatsubo and H. Yano, Biomacromolecules, 2007, 8, 1973–1978 CrossRef CAS.
- M. Nogi, K. Abe, K. Handa, F. Nakatsubo, S. Ifuku and H. Yano, Appl. Phys. Lett., 2006, 89, 233123 CrossRef.
- A. G. Cunha, Q. Zhou, P. T. Larsson and L. A. Berglund, Cellulose, 2014, 21, 2773–2787 CrossRef CAS.
- K. Y. Lee, J. J. Blaker, R. Murakami, J. Y. Heng and A. Bismarck, Langmuir, 2014, 30, 452–460 CrossRef CAS.
- H. Voisin, L. Bergström, P. Liu and A. Matthew, Nanomaterials, 2017, 7, 57 CrossRef.
- S. Spinella, G. L. Re, B. Liu, J. Dorgan, Y. Habibi, P. Leclere, J. M. Raguez, P. Dubois and R. A. Gross, Polymer, 2015, 65, 9–17 CrossRef CAS.
- G. Lo Re, S. Spinella, A. Boujemaoui, F. Vilaseca, P. T. Larsson, F. Adås and L. A. Berglund, ACS Sust. Chem. Eng., 2018, 6, 6753–6760 CrossRef CAS.
- N. Lin, J. Huang, P. R. Chang and J. Yu, Carbohydr. Polym., 2011, 83, 1834–1842 CrossRef CAS.
- C. A. Silvera Batista, R. G. Larson and N. A. Kotov, Science, 2015, 350, 1242477 CrossRef.
- C. M. Hansen, Hansen Solubility Parameters, CRC Press, Boca Raton, FL, 2007 Search PubMed.
- S. D. Bergin, Z. Sun, D. Rickard, P. V. Streich, J. P. Hamilton and J. N. Coleman, ACS Nano, 2009, 3, 2340–2350 CrossRef CAS.
- S. Mathioudaki, B. Barthélémy, S. Detriche, C. Vandenabeele, J. Delhalle, Z. Mekhalif and S. Lucas, ACS Appl. Nano Mater., 2018, 1, 3464–3473 CrossRef CAS.
- J. U. Wieneke, B. Kommoß, O. Gaer, I. Prykhodko and M. Ulbricht, Ind. Eng. Chem. Res., 2012, 51, 327–334 CrossRef CAS.
- D. W. van Krevelen and K. te Nijenhuis, Properties of Polymers: Their Correlation with Chemical Structure; their Numerical Estimation and Prediction from Additive Group Contributions, Elsevier, 4th edn, 2009 Search PubMed.
- K. L. Hoy, J. Paint Technol., 1970, 42, 76–80 CAS.
- S. Gårdebjer, M. Andersson, J. Engström, P. Restorp, M. Persson and A. Larsson, Polym. Chem., 2016, 7, 1756–1764 RSC.
- K. Benhamou, H. Kaddami, A. Magnin, A. Dufresne and A. Ahmad, Carbohydr. Polym., 2015, 122, 202–211 CrossRef CAS.
- D. Chandler, Nature, 2005, 437, 640–647 CrossRef CAS.
- F. Leroy and F. Müller-Plathe, J. Chem. Phys., 2010, 133, 044110 CrossRef.
- P. Johansson and B. Hess, Phys. Rev. Fluids, 2018, 3, 074201 CrossRef.
- M. J. Abraham, T. Murtola, R. Schultz, S. Páll, J. C. Smith, B. Hess and E. Lindahl, SoftwareX, 2015, 1–2, 19–25 CrossRef.
- T. Darden, D. York and L. Pedersen, J. Chem. Phys., 1993, 98, 10089–10092 CrossRef CAS.
- U. Essmann, L. Perera, M. L. Berkowits, T. Darden, H. Lee and L. G. Pedersen, J. Chem. Phys., 1995, 103, 8577–8592 CrossRef CAS.
- B. Hess, J. Chem. Theory Comput., 2008, 4, 116–122 CrossRef CAS.
- M. Parrinello and A. Rahman, J. Appl. Phys., 1981, 52, 7182–7190 CrossRef CAS.
- S. Nosé, Mol. Phys., 1984, 52, 255–268 CrossRef.
- W. G. Hoover, Phys. Rev. A, 1985, 31, 1695–1697 CrossRef.
- K. N. Kirschner, A. B. Yongye, S. M. Tschampel, J. González-Outeiriño, C. R. Daniels, L. Foley and R. J. Woods, J. Comput. Chem., 2008, 29, 622–655 CrossRef CAS.
- W. L. Jorgensen, J. Chandrasekhar, J. D. Madura, R. W. Impey and M. L. Klein, J. Chem. Phys., 1983, 79, 926–935 CrossRef CAS.
- C. J. Malm, L. J. Tanghe, B. C. Laird and G. D. Smith, J. Am. Chem. Soc., 1953, 75, 80–84 CrossRef CAS.
- Y. Nishiyama, P. Langan and H. Chanzy, J. Am. Chem. Soc., 2002, 124, 9074–9082 CrossRef CAS.
- S. Kumar, J. M. Rosenberg, D. Bouzida, R. H. Swendsen and P. A. Kollman, J. Comput. Chem., 1992, 3, 1011–1021 CrossRef.
- J. Hub, B. L. de Groot and D. van der Spoel, J. Chem. Theory Comput., 2010, 6, 3713–3720 CrossRef CAS.
- C. H. Bennet, J. Comput. Phys., 1976, 22, 245–268 CrossRef.
- T. P. Straatsma and J. A. McCammon, Annu. Rev. Phys. Chem., 1992, 43, 407–435 CrossRef CAS.
- V. Gapsys, S. Michielssens, D. Seeliger and B. L. de Groot, Angew. Chem., Int. Ed., 2016, 55, 7364–7368 CrossRef CAS.
- D. P. Oehme, M. S. Doblin, J. Wagner, A. Bacic, M. T. Downton and M. J. Gidley, Cellulose, 2015, 22, 3501–3520 CrossRef CAS.
- M. Garg, M. Linares and I. Zozoulenko, Biomacromolecules, 2020, 21, 3069–3080 CrossRef CAS.
- A. Paajanen, Y. Sonavane, D. Ignasiak, J. A. Ketoja, T. Maloney and S. Paavilainen, Cellulose, 2016, 23, 3449–3462 CrossRef CAS.
- Y. Ogawa, Nanoscale, 2019, 11, 21767–21774 RSC.
- J. Wang, D. Bratko and A. Luzar, Proc. Natl. Acad. Sci. U.S.A, 2011, 108, 6374–6379 CrossRef CAS.
- N. Giovambattista, P. J. Rossky and P. G. Debenedetti, Annu. Rev. Phys. Chem., 2012, 63, 179–200 CrossRef CAS.
- X. Yang, M. S. Reid, P. Olsén and L. A. Berglund, ACS Nano, 2020, 14, 724–735 CrossRef CAS.
- A. Paajanen, S. Ceccherini, T. Maloney and J. A. Ketoja, Cellulose, 2019, 26, 5877–5892 CrossRef CAS.
- L. H. Thomas, A. Martel, I. Grillo and M. C. Jarvis, Cellulose, 2020, 27, 4249–4254 CrossRef CAS.
- E.-L. Hult, P. T. Larsson and T. Iversen, Polymer, 2001, 42, 3309–3314 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2020 |